

Abstract

Photodecarboxylation of biomass-derived fatty acids to alkanes offers significant potential to obtain hydrocarbons and economic benefits due to the mild conditions and high activity. Herein, the photodecarboxylation of hexanoic acid into alkanes using TiO2-supported monometallic Au or Pd and bimetallic Au–Pd catalysts is reported. It was found that bimetallic Au–Pd catalysts, featuring a core–shell structure evidenced by EDX-mapping and element line profile, show better photocatalytic performance, achieving 94.7% conversion of hexanoic acid and nearly 100% selectivity to pentane under UV–vis irradiation in the absence of H2 than the monometallic Au analogue. This remarkable enhancement in activity compared to its TiO2 supported monometallic Au or Pd analogues can be attributed to the synergistic effect between Au and Pd within the nanostructured Au(core)-Pd(shell) alloy for achieving more efficient charge-separation efficiency upon visible light excitation. This photocatalyst exhibits a wide scope converting multiple fatty acids into hydrocarbons. Moreover, it can even photocatalyze the conversion of raw bio-oils into alkanes directly. No obvious activity loss was observed during the reusability tests, demonstrating the good stability of the present catalyst. Density functional theory (DFT) calculations indicate that oxidation of carboxylates on TiO2 leads to alkyl radicals that become bound to metal nanoparticles. The superior catalytic performance of Au(core)-Pd(shell)/TiO2 is derived from the weaker adsorption for H on the alloy and the lower hydrogen evolution reaction overpotential. Our research can result in an efficient bio-oil upgrading, resulting in the synthesis of biofuels from biomass under mild conditions.
Keywords: photodecarboxylation, fatty acid into biofuels, Au−Pd, core−shell, TiO2
1. Introduction
Biomass-derived oils, such as inedible vegetable oils (e.g., jatropha oil) or wasted cooking oils, are suitable feedstocks for the production of biofuels. These oils contain large amounts of carboxylic acids and esters.1−3 Decarboxylation of fatty acids derived from biomass results in hydrocarbons that can be used as synthetic fuels with zero CO2 footprint.4,5 The process has attracted much current interest to provide synthetic gasoline, jet-fuel and gas oil.6−9 Among the various decarboxylation processes, photocatalytic decarboxylation is very convenient and appealing due to the mild conditions, high selectivity and the possibility to use natural solar light as the ultimate energy source.7,10,11 Catalytic thermal fatty acid hydrogenation usually requires harsh reaction conditions to occur (T ≥ 250 °C, P ≥ 2 MPa H2).12−14
Carboxylic acid photodecarboxylation requires the development of efficient photocatalysts, and considerable research has been conducted in this area trying to find more suitable materials.7,8,15 In a semiconducting photocatalyst, photon absorption promotes electrons from the valence band to the conduction band, generating an electron hole in the valence band. This elementary process generates a transient charge separated state with negative electrons (e–) and positive holes (h+) pairs.16 The photon energy has to be equal to or higher than the bandgap of the semiconductor so that light can be effectively absorbed by the photocatalyst. TiO2, having a bandgap of about 3.4 eV, absorbs photons of wavelength shorter than 380 nm, but other semiconductors with narrower bandgap can be excited by visible light.17,18 TiO2 possesses several advantages, including high activity under UV irradiation, high chemical and photochemical stability against photocorrosion, high relative abundance, low toxicity, and large scale affordability. These advantages have motivated the interest for TiO2 in photocatalytic processes such as solar fuels production,19,20 environmental remediation,21,22 and photodecarboxylation.7,15
The photocatalytic activity of TiO2 is known to increase with deposition on its surface of metal and metal oxide nanoparticles, acting as cocatalysts.23,24 Although there is ample evidence that these metal nanoparticles can increase the photocatalytic activity of TiO2, the current data is mostly limited to monometallic nanoparticles, bimetallic nanoparticles having been considerably much less studied.25,26 Bimetallic nanoparticles, particularly those exhibiting unique core–shell structures, have aroused enormous interest because of their fascinating optical, electronic, sensing, and improved catalytic properties. These distinctive physicochemical and catalytic properties evolved in bimetallic nanoparticles derive from the new electronic states of the alloy which are different from individual metals.27−30 Furthermore, besides adaptation of the d band energy, the lattice strain created between the core and shell region as well as the tuning of the electron density at the shell also contribute to the enhanced catalytic properties of core–shell nanoparticles.31 Considering the numerous advantages of TiO2 as decarboxylation photocatalyst and the fact that the catalytic capability of core–shell metal nanoparticles remain mostly unexplored, it appears of interest to study the activity of a core–shell bimetallic catalyst deposited on TiO2 for the photodecarboxylation of fatty acids to obtain hydrocarbons.
In a recent publication, we have reported the photocatalytic decarboxylation activity of a series of nickel supported TiO2 photocatalysts for the conversion of octanoic acid into heptane and tetradecane.15 An octanoic acid conversion of 25% with a selectivity to heptane of about 70% was achieved at 3 h irradiation with a 300 W Xe lamp under 0.2 bar of hydrogen pressure, there being room for further improvement. Continuing with this line of research, it would be important to develop other TiO2 photocatalysts, showing an enhanced activity and selectivity. Herein, we report the photocatalytic activity of core–shell Au–Pd alloy nanoparticles supported on P25 TiO2. The results show that performance of TiO2 having core (Au)-shell (Pd) alloy nanoparticles exhibit a synergy in comparison with analogous TiO2 materials of the individual metals, giving an almost complete decarboxylation selectivity at total carboxylic acid conversion in the absence of hydrogen gas in the system. The present catalyst is not only effective in converting model carboxylic acids but also shows suitability for the conversion of crude bio-oils. This catalytic activity has been explained by DFT calculations to understand the role of the metals in the system.
2. Experimental Section
2.1. Chemicals
All chemicals were used directly as provided commercially without additional purification. Milli Q water was obtained using an IQ 7000 purifying system. P25 TiO2, HAuCl4·3H2O, PdCl2, acetone (99.9%, CH3COCH3), sodium hydroxide (reagent grade, 97%, powder, NaOH), 1-phenylethanol (97%), hexanoic acid (99%), pentane (99%), lauric acid (97%), palmitic acid (99%), stearic acid (98%), oleic acid (97%), undecane (99%), pentadecane (99%), heptadecane (99%), p-xylene (99.5%, as internal GC standard), different solvents including dodecane (99%), PhMe (99%), THF (99%), DMF (99%) and dichloroethane (99%) were purchased from Sigma-Aldrich. Jatropha oil was purchased from the Shenyu company in Yunnan Province, China. Wasted cooking oil and wasted hot-pot oil were collected from Sichuan Province, China.
2.2. Preparation of Catalysts
A series of Au, Pd and Au–Pd bimetallic nanoparticles supported on P25 TiO2 were prepared by impregnation P25 TiO2 with HAuCl4.3H2O and/or PdCl2 under basic condition for Au/TiO2 or in water and acetone mixture for Pd/TiO2. After impregnation, the solid was dried in an oven at 100 °C and finally submitted to calcination at 400 °C (Au/TiO2) or reduced by treatment with 1-phenylethanol at reflux temperature (160 °C) for those samples containing Pd.32 The preparation procedure is illustrated in Scheme 1. Au–Pd bimetallic catalysts with different Au/Pd atomic ratios were prepared by adding in all cases the same amount of HAuCl4·3H2O, but a different amount of PdCl2.
Scheme 1. Illustration of the Preparation Procedure of TiO2-Based Photocatalysts. (a) Au/TiO2, (b) Pd/TiO2 and (c) Au–Pd/TiO2.

For comparison, a AuPd/TiO2 alloy catalyst in which Au and Pd are randomly distributed in the nanoparticle was also prepared by the coimpregnation method, at an Au and Pd loading of 1.5 and 0.8 wt %, respectively. After impregnation and calcination, the sample was reduced at 400 °C for 2 h under a H2 flow (30 mL/min). The obtained catalyst was denoted as a 1.5Au0.8Pd alloy/TiO2.
2.3. Catalyst Characterization
Transmission electronic microscopy (TEM), EDX-Mapping and EDX element line profile were conducted at Philips CM300 FEG electron microscope with an accelerating voltage of 200 kV. The samples were first dispersed in ethanol and then treated with ultrasound (440 W) for 15 min, to obtain a homogeneous suspension. A drop of this suspension was deposited on a copper grid coated with carbon films. XRD was recorded by using a Shimadzu XRD-7000 diffractometer with CuKα radiation. The XRD patterns were recorded from 10 to 80° at a scanning speed of 10°·min–1. The actual loadings of Au and Pd were determined by ICP-AES using a Varian 715-ES instrument. Diffuse reflectance UV/vis spectra were recorded on a Varian Cary 5000 spectrophotometer in the wavelength range 200–800 nm. The samples were also analyzed by XPS (SPECS spectrometer with monochromated Al radiation). XPS data were calibrated taking the C 1s at 284.5 eV using Casa XPS software. The experimental high-resolution XPS peaks were fitted by using XPSPEAK1 software. The in situ IR spectra reported here were taken by BRUKER VERTEX 70 FT-IR spectrometer equipped with a high sensitivity MCT detector at a resolution of 4 cm–1 in the range of 1000–4000 cm–1 with a cell holder having KBr windows. Propionic acid was chosen as the probe molecule for IR spectroscopy measurements due to its higher vapor pressure. The detailed experimental procedure is as follows: the samples were first treated with an initial pretreatment by heating them to 150 °C under vacuum to remove adsorbed water or gases. Subsequently, the sample was cooled to room temperature. Then, propionic acid was adsorbed on the catalyst surface from the vapor phase. Following this step, the catalyst was immediately subjected to irradiation under UV–visible light.
2.4. Theoretical Basis
Periodic DFT calculations were conducted using the Cambridge Serial Total Energy Package (CASTEP) module33 with the exchange-correlation functional described by Perdew–Burke–Ernzerhof within the generalized gradient approximation (GGA-PBE).34 Tkatchenko and Scheffler (TS) dispersion corrections scheme35 was incorporated along with the exchange and correlation functional to improve the structural and vibrational properties. The anatase TiO2 (101) surface was presented by a three-layer slab. A 3 × 2 supercell along the [010] and [101] directions with a vacuum layer of 20 Å perpendicular to the surface was used. The system contains 130 atoms, and only the Γ point was included for sampling of the Brillouin zone. A self-consistent field method (tolerance 5.0 × 10–7 eV/atom) was employed in conjunction with plane-wave basis sets with a cutoff energy of 460 eV in reciprocal space. All structures are geometry-optimized until energy is converged to 5.0 × 10–6 eV/atom, maximum force to 0.01 eV/Å and maximum displacement to 5.0 × 10–4 Å. In order to avoid metal layers becoming corrugated due to geometry optimization leading toward stable nanoparticle shapes indicated above, we have constrained all the metal layers and focused on the geometry optimization into different adsorbates considered. The adsorption energy of species on the catalyst surface was calculated as Eads = Etotal – EA – Esur, where Etotal represents the total energy of the catalytic surface with an adsorbed molecule, and EA and Esur are the energies of isolated adsorbate molecule and the clean surface, respectively. The energy of an isolated molecule (EA) was computed by placing it in the same lattice box (about 10.2 × 11.3 × 29.3 Å3). The energy of the free radical is computed by a spin-polarized calculation with one unpaired electron (spin: 1).
2.5. Analysis of Products
The products were detected by GC with an HP-5 column. The detection program was set from 50 to 280 °C with a heating rate of 10 °C· min–1. The conversion, products yield and selectivity of each component were calculated by the following eqs 1–3:
| 1 |
where C0 and C represent the contents of substrate in the reactant and product, respectively.
| 2 |
where np and nm represent the moles of product and added substrate, respectively.
| 3 |
where np and nc represent the mole of product and converted substrate, respectively.
The initial reaction rates based on the metal amount were calculated by using eq 4:
 |
4 |
3. Results and Discussion
3.1. Characterizations of the Catalyst
The metal loadings of the samples prepared in the present work are provided in Table S1 of the Supporting Information. In general, the Au loading was fairly constant about 1.5 wt %, while Pd percentage increased from 0 to about 5 wt %.
The presence of the metal nanoparticles on TiO2 was determined by TEM. High-resolution images show that P25 is predominately constituted by anatase phase with less abundant rutile domains (seen in Figure S1),36 which is also confirmed by the XRD patterns commented later. The average particle size of TiO2 was in all cases about 20 nm (Figure 1). The presence of Au and Pd is observed in these images by the presence of darker, higher contrast, and smaller particles with average particle sizes about 5 nm. Figure 1d,e present selected high-resolution TEM images of some of the samples prepared in the present study. In the case of Au/TiO2 or Pd/TiO2, the interplanar distances (2.41 and 2.14 Å) of the metal nanoparticles measured by high-resolution TEM indicate that they correspond in both cases to the (111) facet of metallic Au or Pd.
Figure 1.

TEM images with particle size distributions (brown columns represent TiO2, and blue columns represent metal particles) of (a) Au/TiO2, (b) Pd/TiO2 and (c) 1.5Au-0.8Pd/TiO2. High resolution TEM images of (d) Au/TiO2 and (e) Pd/TiO2. The insets indicate the lattice fringe distance. EDX-mappings of Au and Pd elements of (f) 1.5Au-0.8Pd/TiO2 catalyst. (g1-g3) STEM images, STEM-EDX elemental profile and EDX elemental line scanning profile of 1.5Au-0.8Pd/TiO2 catalyst. Note that Au is located mostly in the central part of the nanoparticle.
Interestingly, for the 1.5Au-0.8Pd/TiO2 sample, elemental mapping of Au and Pd at nanometric resolution indicates that Pd is preferentially located at the external part of the Au–Pd nanoparticle, therefore having a structure of a core (Au)/shell (Pd) nanoparticle. The EDX elemental line profile (Figure 1g1-g3) across the nanoparticles further confirm the core–shell structure features of 1.5Au-0.8Pd/TiO2 catalyst, which would determine the electron density of the external Pd atoms and may create lattice strain. Both factors can result in a synergistic effect between these two metals. The elemental EDX-mapping of other metal/TiO2 photocatalysts are also provided in Figure S2 of Supporting Information. As shown in Figure S3, peaks located at 2 theta = 25.3, 36.9, 37.8, 38.6, 48.0, 53.9, 55.1, and 62.7° are ascribed to anatase TiO2 (PDF#21-1272), while those at 27.4, 36.1, 41.2, and 56.6° are ascribed to rutile TiO2 (PDF#21-1276). The anatase/rutile composition of TiO2 is in accordance with the images taken by high resolution TEM. No obvious peaks corresponding to metallic Au or Pd are found on these samples, a fact that is probably due to the low metal content and the small sizes of the Au or Pd nanoparticles.
The surface chemical states of each element on the different catalysts were evaluated by XPS. Figure 2 presents the experimental XPS peaks for each element as well as their best deconvolutions to individual components. The Au 4f peaks were deconvoluted into two peaks including Au 4f 5/2 with a binding energy around 84.0 eV and Au 4f 3/2 near 87.6 eV, both corresponding to the metallic Au state.37−40 For the XPS Pd 3d spectra, the bands were also deconvoluted into two peaks corresponding to the spin–orbit splitting of the Pd (3d 3/2) and Pd (3d 5/2) lines (Δ = 5.26 eV), appearing at a binding energy around 335.0 and 340.3 eV, respectively.40,41 Compared with the monometallic catalysts, the binding energy of Au 4f 5/2 shifts to lower energy (from 84.0 to 83.6 eV), while Pd 3d 5/2 shifts to higher energy (from 335.0 to 335.5 eV) for the bimetallic 1.5Au-0.8Pd/TiO2 catalyst, indicating electron density transfer from Pd to Au.22,42 The presence of a strong electronic effect between Au and Pd metals in a bimetallic 1.5Au-0.8Pd/TiO2 catalyst can significantly improve the capability to capture electrons. The XPS Ti 2p peaks were also deconvoluted, and the peaks with binding energy values at 458.8 eV are ascribed to Ti4+.43−45 No component corresponding to Ti3+ was found, indicating that Ti4+ is the only state of Ti on the surface of these catalysts. For the O 1s spectra, they were deconvoluted into two peaks centered at 529.4 and 521.4 eV, which are ascribed to the lattice oxygen for Ti–O and chemisorbed oxygen such as −OH, respectively.46−48 No significant differences in binding energy are observed for both Ti 2p and O 1s between monometallic and bimetallic catalysts.
Figure 2.

Experimental high resolution XP spectra and the corresponding deconvolution to individual components for: (a) Au 4f, (b) Pd 3d (c) Ti 2p and (d) O 1s XPS spectra of Au/TiO2, Pd/TiO2 and 1.5Au-0.8Pd/TiO2.
Diffuse reflectance UV–vis spectroscopy is a useful tool for evaluating light absorption and electronic characteristics of photocatalytic materials. Compared with the pure P25 TiO2, the maxima observed in the visible range at 500–600 nm can be ascribed to the localized surface plasmon resonance (LSPR) arising from the supported Au and Pd metals (Figure 3a).49−51 The bandgaps were calculated using eq (5): αhv1/n = A(hv – Eg) (5), where α is the absorption coefficient, A is a constant, h is Planck constant, Eg is the band energy, v is the incident light frequency, and n is associated with the type of semiconductor.52 TiO2 is regarded as an indirect semiconductor, wherein n = 2. The Tauc plots used to estimate the optical bandgaps are presented in Figure 3 b,c.
Figure 3.

(a) UV–vis spectra of the as-prepared samples as indicated by the color codes, (b) Tauc plots of (αhv)2 versus photon energy and (c1-c4) bandgap of the samples obtained by magnification of plot (b).
The bandgap energies (Eg) of these samples (Figure 3c1-c4) were obtained from the extrapolated intercept on the abscissa (Figure 3b). The estimated bandgap energies of pure P25 TiO2, Au/TiO2, Pd/TiO2 and 1.5Au-0.8Pd/TiO2 are 3.54, 3.50, 3.49, and 3.45 eV, respectively. Obviously, the bandgap of the photocatalysts after metal doping is shorter than the pure P25 TiO2, wherein the bimetallic Au–Pd/TiO2 has the shortest bandgap. This red shift could be attributed to the metal doping and LSPR effect. Although this effect is small, shorter bandgap is beneficial for improving the efficiency of the photocatalyst by harvesting more photons.
In situ IR spectra were collected to detect the adsorption of carboxylic acids on the photocatalyst surface and the formation of intermediates upon irradiation. The spectra recorded are shown in Figure 4 and Figures S4–S5. The peak at 1130 cm–1 is attributed to P25 TiO2, while the peak at around 1620 cm–1 is attributed to the adsorbed water on the surface, and the one located at 3670 cm–1 is ascribed to the adsorbed acid over the TiO2 (seen in Figure 4A).53−55 The intensity of peaks attributed to propionic acid increases with the prolonged adsorption time, indicating that more propionic acid becomes adsorbed on the surface (Figure 4A (a,b). No differences were found among the IR spectra before and after irradiation for the TiO2 sample. As for the 1.5Au-0.8Pd/TiO2 catalyst (Figure 4B), the peaks at 1440 and 1540 cm–1 are attributed to the symmetric stretching vibration (vs) and asymmetric stretching vibration (vas) of COO- bands of propionic acid, respectively.53,56 This indicates the formation of a chemical bond between the carboxylic group and the Brønsted acid site (Ti–OH) of the TiO2 surface.57 Noteworthy, new peaks located at 1300 cm–1, 1470 cm–1, 2940 and 2980 cm–1 are observed, while the peak at 3670 cm–1 disappeared upon irradiation. Disappearance of the 3670 cm–1 peak agrees with the occurrence of photodecarboxylation. Peaks at 1300, 1470, 2940 and 2980 cm–1 are attributed to the bending vibration of −CH2–, vs and vas of alkyl radicals, respectively.58 This demonstrates that alkyl radicals were formed on the surface of 1.5Au-0.8Pd/TiO2 after UV–vis light irradiation. Meanwhile, it was found that the intensity of these peaks increased along with the irradiation time (Figure 4B (c,d)). Similar results were also obtained over Au/TiO2 and Pd/TiO2 (Figures S4–S5).
Figure 4.

IR spectra of (A) TiO2 and (B) 1.5Au-0.8Pd/TiO2 with propionic acid adsorbed for (a) shorter and (b) longer time and then irradiated with UV–vis light for (c) 30 and (d) 60 min.
3.2. Photocatalytic Activity
Photocatalytic experiments were performed initially in dodecane (10 mL) dissolving hexanoic acid (30 mg) and using 20 mg of photocatalyst, irradiating with the full output of a 300 W Xe lamp through quartz. Prior to the irradiation, the suspension was deaerated with Ar, and the reactor was sealed with an overpressure of 0.5 bar Ar. In normal cases, the formation of pentane was observed. The results after 4 and 11 h irradiation are presented in Table S2 and Table 1. As can be seen in Table 1, those bimetallic samples with Pd loading equal to or higher than 0.8 wt % perform better than the photocatalysts without metal nanoparticles or containing individual Au or Pd nanoparticles. The 1.5Au-0.2Pd/TiO2 catalyst with lesser Pd loading performs even worse than Pd/TiO2 with individual Pd loading. This can be caused by the much lower loading of Pd (0.2 wt %) in 1.5Au-0.2Pd/TiO2 compared to the Pd/TiO2 (0.8 wt %). The other bimetallic catalysts with different Au–Pd ratios show similar catalytic performance, wherein the 1.5Au-0.8Pd/TiO2 catalyst combines a low Pd content and high photodecarboxylation activity. In fact, 1.5Au-5Pd/TiO2 with much higher Pd content and also a core (Au)@shell (Pd) Au–Pd structure exhibits similar photocatalytic performance but at much higher Pd cost. From this initial screening, the samples Au/TiO2, Pd/TiO2 and 1.5Au-0.8Pd/TiO2 were selected for further study because of their performance, while 1.5Au-0.8Pd/TiO2 contains the lowest Pd content. For comparison, the performance of 1.5Au0.8Pd alloy/TiO2, a 1:1 mechanical mixture of 1.5Au/TiO2 and 0.8Pd/TiO2 was also examined. The results are listed in Table S2. In all cases, the catalytic performance was much worse than that of 1.5Au(core)-0.8Pd(shell)/TiO2, which demonstrates that the synergistic effect endowed by the Au(core)-Pd(shell) structure is essential for the efficient photodecarboxylation of hexanoic acid.
Table 1. Photocatalytic Data at 11 h Irradiation of Hexanoic Acid Photodecarboxylation in the Presence of Different Photocatalysts.
| Catalysts | Conversion (%)a | Pentane Yield (%) | Pentane Selectivity (%) | Initial Rate (mol·gcat–1·h–1)b |
|---|---|---|---|---|
| P25 TiO2 | 16.9 | 15.9 | 94.1 | – |
| Au/TiO2 | 70.6 ± 2.9 | 67.9 | 96.2 | 0.63 |
| Pd/TiO2 | 90.0 ± 2.0 | 86.8 | 96.4 | 0.77 |
| 1.5Au-0.2Pd/TiO2 | 72.7 ± 3.1 | 69.9 | 96.3 | – |
| 1.5Au-0.8Pd/TiO2 | 96.3 ± 1.6 | 95.6 | 99.3 | 0.86 |
| 1.5Au-1.8Pd/TiO2 | 97.5 ± 0.9 | 96.6 | 99.1 | – |
| 1.5Au-5Pd/TiO2 | 97.5 ± 1.7 | 96.9 | 99.3 | – |
Data correspond to the average of three independent experiments.
The initial reaction rates of catalysts were calculated based on the metal amount and activity data at 0.5 h as indicated in eq 4 in the experimental section.
The temporal evolution of hexanoic acid decarboxylation for Au/TiO2, Pd/TiO2 and 1.5Au-0.8Pd/TiO2 is presented in Figure 5 that further illustrates the benefit of the Au(core)-Pd(shell)/TiO2 bimetallic catalyst in comparison to the individual metals. The superior activity of 1.5Au-0.8Pd/TiO2 can be explained by a combination of its narrower bandgap energy and the more efficient catalytic activity of the Au(core)-Pd(shell) nanoparticle.
Figure 5.

Temporal evolution of hexanoic acid decarboxylation for Au/TiO2, Pd/TiO2 and 1.5Au-0.8Pd/TiO2. (Inset figure is the effect of 1.5Au-0.8Pd/TiO2 catalyst amounts on the catalytic results). Reaction conditions: 30 mg of hexanoic acids, 10 mL of dodecane, room temperature, 300 W Xe lamp, 2 h, 20 mg of catalysts, 0.5 bar H2.
From this temporal profile, irradiation time for 4 h was selected, since using the presented conditions, high conversion values were achieved at this time. The initial reaction rates of different catalysts were calculated based on the metal amounts, and the corresponding values are given in Table 1. It is found that the bimetallic Au(core)-Pd(shell) catalyst shows higher initial reaction rate, indicating its higher intrinsic reactivity toward the photodecarboxylation of hexanoic acid than other catalysts with only mono metal loaded. The influence of photocatalyst weight on hexanoic acid conversion and pentane selectivity was also studied under the same conditions using 5 to 80 mg of 1.5Au-0.8Pd/TiO2 in 10 mL dodecane (inset of Figure 5). Photocatalytic results show that conversion increases notably from 5 to 10 mg, becoming then stable even up to a considerable increase in mass excess of 80 mg. From this experiment, an optimal 20 mg photocatalyst mass in 10 mL of dodecane was used for all the experiments. Noteworthy is the fact that decane was not detectable even though the reaction was carried out in the absence of H2 as the H atom donor (see Figure S6). Also, an overpressure of Ar was not necessary, and identical conversion and pentane selectivity were observed for irradiations under atmospheric pressure.
The influence of the solvent on hexanoic acid photodecarboxylation was then studied, and the corresponding results are presented in Figure S7. The best result was obtained using dodecane as the solvent, while toluene as the solvent gave the worst result, probably due to an internal filter effect of this aromatic compound in the UV region. Photodecarboxylation seems to be quenched considerably using THF, DMF or dichloroethane (DCE) as solvents, reaching hexanoic acid conversions below 20%, which is probably due to the competition of this solvent with the substrate for trapping of holes (THF) or photogenerated electrons (DMF and DCE).
To gain some information about the reaction mechanism of the photodecarboxylation, additional experiments were carried out. Thus, no hexanoic acid conversion was observed upon irradiation of hexanoic acid in the absence of photocatalysts or in the presence of 1.5Au-0.8Pd/TiO2 but in the dark at 100 °C, meaning that both light and photocatalyst are necessary in the photodecarboxylation process (Table S3). The influence of the wavelength range on the photocatalytic activity was also conducted, as shown also in Table S3. Using cutoff filters for radiations of wavelengths shorter than 360 or 455 nm, both conversions and alkane yield decreased. Moreover, an experiment using TEMPO as a carbon radical trapping agent reveals the formation of pentyl radical as intermediate. This result (Figure 6) is in accordance with the in situ IR measurements that show the presence of alkyl groups during the photodecarboxylation process. These pentyl radicals would arise from the decarboxylation of hexanoate promoted by photogenerated holes.
Figure 6.

Radical trapping experiment. Products were observed in the photocatalytic decarboxylation of hexanoic acid in the presence of 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) in a H2 atmosphere. Reaction conditions: 30 mg hexanoic acids, 10 mL dodecane, room temperature, 300 W Xe lamp, 2 h, 20 mg of catalysts, 0.5 bar H2.
The stability of the 1.5Au-0.8Pd/TiO2 catalyst was also assessed, and the results are shown in Figure 7. In the recycling experiments, the solid catalyst was retrieved after each run from the reaction mixture through filtration. It was subsequently washed with dodecane and employed in the next run. The result reveals a slight, gradual decrease in the conversion of hexanoic acid, while the selectivity of the product remains almost constant for each run, at approximately 97.5%. These reusability data indicate the stability of the bimetallic 1.5Au-0.8Pd/TiO2 photocatalyst. Moreover, the initial reaction rates measured at 0.5 h across successive runs show nearly constant values, with only a slight decrease (from 0.86 to 0.72 mol. g cat–1. h–1), further exemplifying the stable nature of this Au (core)-Pd(shell)/TiO2 bimetallic catalyst. XRD and XPS results of the five times used 1.5Au-0.8Pd/TiO2 indicate almost no changes compared with the fresh one (Figures S8–S9). Particularly, no agglomeration or growth of the metal nanoparticles was found by TEM analysis of the used 1.5Au-0.8Pd/TiO2 photocatalyst. Moreover, the core–shell structure of the Au–Pd alloy nanoparticles remains (Figure S10). The stability of the photocatalyst nanostructure is closely related to the stable activity results.
Figure 7.
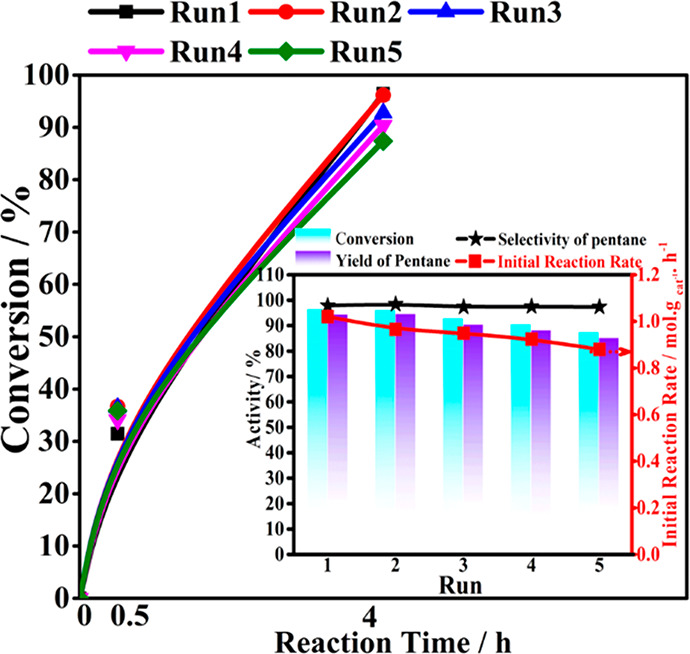
Stability tests for hexanoic acid conversion over 1.5Au-0.8Pd/TiO2 photocatalyst. Reaction conditions: 30 mg of hexanoic acids, 10 mL of dodecane, room temperature, 4 h; catalyst was recovered after the last run, 0.5 bar H2. The inset plot shows activity, CO2 conversion, pentane yield and initial reaction rate.
The main components in crude bio-oils include saturated fatty acids such as lauric acid, palmitic acid, stearic acid, unsaturated oleic acid, linoleic acid, and corresponding triglycerides. Therefore, we were interested in determining the scope of this core (Au)-Pd (shell)/TiO2 bimetallic photocatalyst to efficiently convert long-chain fatty acids that have to be the feedstocks in realizing the photocatalytic conversion of bio-oils into alkanes. Table 2 demonstrates that 1.5Au-0.8Pd/TiO2 exhibits a broad substrate scope, allowing for high conversions and yields of Cn–1 alkanes in the photodecarboxylation of various saturated fatty acids. As for the oleic acid, the somewhat lower Cn–1 alkane yield and selectivity are due to the presence of C=C double bonds in the molecule. In this case, the hole-induced alkyl radicals from photodecarboxylation should tend to attack the unsaturated C=C bonds of the substrate and induce polymerization. Complex raw bio-oils constituted by a mixture of fatty acids also undergo photodecarboxylation. Thus, crude bio-oils such as jatropha oil, wasted cooking oil and wasted hot-pot oil become also converted. Due to the complex composition of crude raw bio-oils, it is difficult to calculate the conversion, yield as well as alkanes selectivity. However, according to the GC-MS spectra of corresponding products in these three cases (Figure S11), various alkanes not present in the feedstock were detected after photocatalytic irradiation, implying the capability of the synthesized core (Au)-shell (Pd)/TiO2 bimetallic catalyst to photodecarboxylate fatty acids present in crude bio-oils, converting them into alkanes.
Table 2. Photodecarboxylation of Fatty Acids and Bio-oils over 1.5Au-0.8Pd/TiO2a.
| Feedstock | Products | Conversion (%) | Yield (%) | Selectivity (%) |
|---|---|---|---|---|
| Lauric acid | Undecane | 100 | 94.7 | 94.7 |
| Palmitic acid | Pentadecane | 100 | 97.7 | 97.7 |
| Stearic acid | Heptadecane | 100 | 94.3 | 94.3 |
| Oleic acid | Heptadecane | 95.5 | 77.5 | 81.2 |
These reactions are conducted in a H2 atmosphere (0.5 bar) under UV–visible light irradiation for 4 h.
3.3. Computational Results
To gain some insight into the reaction sites and mechanism of this photodecarboxylation reaction on 1.5Au-0.8Pd/TiO2 (denoted for the calculations as Au@Pd), periodic DFT calculations were performed. Here, we focus on the thermodynamic process of the surface reactions, including the dissociation of carboxylic acid and the formation of alkane. The photon absorption event or the kinetic process of the subsequent charge migration is not considered in the calculations, since the carrier dynamics of metals supported on titania and the Schottky barrier between metal titania have been extensively studied elsewhere.59−62 Herein, photoexcitation and charge migration are considered to generate electrochemical potentials for catalyzing carboxylic acid decomposition and transformation to alkane. Butyric acid is regarded as a model carboxylic acid for the DFT calculation. Based on the XRD pattern that shows that the main exposed lattice plane in P25 is anatase TiO2(101), calculations were made for a model of this 101 anatase plane.
Based on the calculated results, the TiO2 (101) facet and metal surfaces (Au@Pd, Au and Pd) show a strong adsorption energy of carboxylic acids (Figure S12). TiO2 (101) surface is considered as the oxidation reaction site due to its larger reaction surface area and higher hole concentration. The probable oxidation half-reaction mechanism steps are as follows:
| 6 |
| 7 |
where RCOOH is the carboxylic acid, the asterisk (*) sign denotes the anatase TiO2 (101) surface, RCOO* and COO* denote adsorbed intermediate species, and R• denotes the free alkyl radical. We obtain the energy of H+ + e– implicitly by referencing it to the energy of H2 using the computational values for the hydrogen electrode.63 In this oxidation reaction, the first step is the adsorption of carboxylic acid. The adsorption energy is calculated to be −0.71 eV (Figure S13), which indicates that the carboxylic acid molecule strongly interacts with the TiO2 surface. This interaction was also demonstrated by calculating Hirshfeld charge distribution, and it was found that the carboxylic acid molecule presents a more positive charge (0.26 |e|) when it adsorbs on the TiO2 surface. Projected density of states (PDOS) shows the Ti 3d and O 2p orbitals dominate the electrons density under the Fermi level (Figure S13 a), and the O 2p orbital of *RCOOH intermediate shows a high degree of overlap with TiO2 p and d-orbitals between 0 and −4 eV under the Fermi level (Figure S13 b). This overlap suggests a significant interaction between the O 2p orbital in RCOOH and Ti 3d and the O 2p orbitals in TiO2. All the above results reveal the strong interaction between carboxylic acid and the TiO2 surface, which benefits the subsequent reaction.
The next step is dissociative dehydrogenation of the carboxylic acid. Carboxylic acid is thought to be deprotonated by hole oxidation.64 In this work, we have considered overpotential as the important activity descriptor since it is of considerable interest from an experimental perspective. The overpotential obtained from the energy difference has been used as activity descriptor, because it follows the same trend to kinetic barriers owing to the Brønsted–Evans–Polanyi (BEP) relation between free energy and activation barriers.65,66 Here, we use reaction enthalpies instead of free energies to compute the reaction mechanism since frequency calculations for this model are computationally very expensive. In addition, for long-chain carboxylic acids and alkanes, the vibrational frequency is relatively less affected by the adsorption and reaction. Hence, we believe that our energy-profile-based analysis is reasonable to explain the activity of this system. As shown in Figure 8a, the calculated overpotential for RCOOH deprotonation is 2.26 eV (eq 6), subsequent C–C bond breaking to generate free radical R• is exergonic (eq 7), and the TiO2 is recovered by desorption of CO2 with moderate energy (0.39 eV). Therefore, photogenerated holes in the valence band (3.08 eV)67 provide enough driving force to compensate the RCOOH deprotonation overpotential.
Figure 8.

Potential energy diagram for the (a) oxidation half-reaction on the TiO2 (101) surface and (b) reduction half-reaction on Au@Pd, Au and Pd.
On the other hand, the probable reduction half-reaction mechanism steps have also been considered:
| 8 |
| 9 |
As shown in Figure S14, the adsorption and reduction of H+ on TiO2 (101) surface is endergonic (Eads = 0.61 eV), while this reduction process of the proton on Au@Pd (Eads= −0.95 eV), Au (Eads= −0.65 eV) or Pd (Eads= −1.58 eV) is thermodynamically favorable. Furthermore, it is commonly known that the metal has a higher work function; thus, the photogenerated electrons formed on the conduction band of TiO2 tend to migrate to the metal nanoparticle. In addition, the adsorption of •R on Au@Pd (Eads= −2.08 eV), Au (Eads= −2.38 eV) or Pd (Eads= −3.26 eV) is also more thermodynamically favorable than that on the TiO2 (101) surface (Eads= −0.68 eV) (Figure S15). Therefore, the metal nanoparticle is considered as the reduction reaction site. The reduction reactions to form HR on Au@Pd, Pd and Au are all considered. As shown in Figure 8b, Pd and Au exhibit much stronger adsorption of H• and R•, and the subsequent overpotential for H–R bond formation indicates that Au@Pd (0.25 eV) and Pd (0.16 eV) have a lower overpotential compared to only Au (1.27 eV). However, it is worth noting that Pd exhibits a higher H-R desorption energy (1.71 eV), which results in a higher initial reaction rate and conversion than that of Au, but still lower than that of Au@Pd. These findings confirm that Au@Pd should possess superior activity for alkane formation compared to Pd and Au.
Combining the experimental and DFT calculation results, we propose a plausible mechanism for the photodecarboxylation of carboxylic acid over 1.5Au-0.8Pd/TiO2 in Scheme 2.
Scheme 2. Diagram of the Proposed Reaction Mechanism for Photodecarboxylation of Carboxylic Acid over 1.5Au-0.8Pd/TiO2 Catalyst.
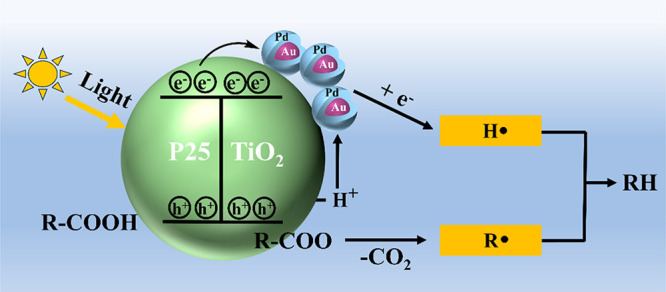
First, light irradiation over P25 TiO2 leads to charge separation with the generation of electrons and holes. Carboxylic acid molecules become dissociated, and the resulting carboxylate strongly adsorbed on the TiO2 surface. The photogenerated holes on the TiO2 surface will be quenched by the carboxylate groups in a kind of ligand to metal electron transfer to form RCOO• intermediates. RCOO• intermediates adsorbed strongly on the TiO2 surface undergo decarboxylation to form CO2 and alkyl radicals. H+ generated in the carboxylic acid dissociation would migrate to Au–Pd nanoparticles, combining with photoinduced electrons to form H•. Finally, the alkyl radical bonds with H• to form Cn–1 alkanes. Au–Pd nanoparticles would play several roles including: (i) the increase in the charge separation efficiency by accepting electrons from TiO2, (ii) the generation of highly mobile H• atoms through the photocatalyst surface by reducing H+ protons, and (iii) promote the evolution of pentane by coupling penntyl radical and H• atoms. The fact that the n-pentane selectivity does not decrease in the photocatalytic reactions in the absence of H2 as an additional source of hydrogen indicates that the protons derived from hexanoic acid dissociation can be efficiently reduced to H• atoms.
4. Conclusions
In summary, the synthesized Au (core)-Pd (shell)/TiO2 catalyst was proven to be a novel UV–vis light responsive photocatalyst, demonstrating excellent activity and stability for the photodecarboxylation of hexanoic acid (conversion for 94.7% and near 100% pentane selectivity) as well as other fatty acids and even raw crude bio-oils. The unique structure characteristics endow the Au–Pd bimetallic TiO2 photocatalyst with distinctive electronic effects and high charge-separation efficiency, thus exhibiting superior photocatalytic activity compared with pure TiO2, or monometallic Au and Pd catalysts. DFT calculations revealed that oxidative decarboxylation reactions of carboxylic acids and hydrogen bonding of alkyl radicals are likely to occur on TiO2 and metal nanoparticles, respectively. Meanwhile, it revealed that the adsorption for H is weaker and the overpotential of HR formation is lower on Au(core)-Pd(shell)/TiO2 catalysts, which is favorable for forming Cn–1 alkanes. The present study shows a promising way to upgrade the low-value biomass-derived carboxylic acids to high-value alkanes.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 21536007) and the 111 Project (B17030). Financial support by the Spanish Ministry of Science and Innovation (CEX-2021-001230-S and PDI2021-0126071-OB-CO21 funded by MCIN/AEI/10.13039/501100011033) and Generalitat Valenciana (Prometeo 2021/038 and Advanced Materials programme Graphica MFA/2022/023 with funding from European Union NextGenerationEU PRTR-C17.I1) is gratefully acknowledged. Huiru Yang also acknowledges the support from China Scholarship Council (CSC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c03793.
High-resolution TEM images, EDX-Mapping of prepared catalysts; IR spectra with propionic acid adsorbed on Au/TiO2 and Pt/TiO2; effect of atmosphere, solvent on the photoactivity; XRD, XPS patterns and TEM images as well as EDX-Mapping of used 1.5Au-0.8Pd/TiO2 catalysts; GC-MS spectra of crude oils and corresponding products by photocatalytic decarboxylation; optimized geometry models and corresponding adsorption energies of RCOOH adsorbed on different surfaces; PDOS of Ti and O orbitals in TiO2 adsorbed sites and PDOS of O, H, and C orbitals in RCOOH and TiO2 orbital in TiO2 adsorbed sites; Optimized structural models and corresponding adsorption energies of H and R• on different surfaces; ICP-AES data; other activity performance (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Taufiqurrahmi N.; Bhatia S. Catalytic cracking of edible and non-edible oils for the production of biofuels. Energy Environ. Sci. 2011, 4 (4), 1087–1112. 10.1039/c0ee00460j. [DOI] [Google Scholar]
- Wang M.; Dewil R.; Maniatis K.; Wheeldon J.; Tan T.; Baeyens J.; Fang Y. Biomass-derived aviation fuels: Challenges and perspective. Prog. Energy Combust. Sci. 2019, 74, 31–49. 10.1016/j.pecs.2019.04.004. [DOI] [Google Scholar]
- Žula M.; Grilc M.; Likozar B. Hydrocracking, hydrogenation and hydro-deoxygenation of fatty acids, esters and glycerides: Mechanisms, kinetics and transport phenomena. Chemical Engineering Journal 2022, 444, 136564–136588. 10.1016/j.cej.2022.136564. [DOI] [Google Scholar]
- Qu L.; Jiang X.; Zhang Z.; Zhang X.-g.; Song G.-y.; Wang H.-l.; Yuan Y.-p.; Chang Y.-l. A review of hydrodeoxygenation of bio-oil: model compounds, catalysts, and equipment. Green Chem. 2021, 23 (23), 9348–9376. 10.1039/D1GC03183J. [DOI] [Google Scholar]
- Pritchard J.; Filonenko G. A.; van Putten R.; Hensen E. J.; Pidko E. A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 2015, 44 (11), 3808–33. 10.1039/C5CS00038F. [DOI] [PubMed] [Google Scholar]
- Wang M.; Zhou H.; Wang F. Photocatalytic Production of Syngas from Biomass. Acc. Chem. Res. 2023, 56 (9), 1057–1069. 10.1021/acs.accounts.3c00039. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Zhao Z.; Zhang C.; Lu J.; Liu H.; Luo N.; Zhang J.; Wang F. Enhanced photocatalytic alkane production from fatty acid decarboxylation via inhibition of radical oligomerization. Nature Catalysis 2020, 3 (2), 170–178. 10.1038/s41929-020-0423-3. [DOI] [Google Scholar]
- Shi J.; Yuan T.; Zheng M.; Wang X. Metal-Free Heterogeneous Semiconductor for Visible-Light Photocatalytic Decarboxylation of Carboxylic Acids. ACS Catal. 2021, 11 (5), 3040–3047. 10.1021/acscatal.0c05211. [DOI] [Google Scholar]
- Long F.; Cao X.; Liu P.; Jiang X.; Jiang J.; Zhang X.; Xu J. Tuning charge transfer of Pt cluster by support defects in Pt/TiO2 for photocatalytic conversion of fatty acid into diesel-like alkane. Journal of Cleaner Production 2022, 375, 133975–133985. 10.1016/j.jclepro.2022.133975. [DOI] [Google Scholar]
- Heciak A.; Morawski A. W.; Grzmil B.; Mozia S. Cu-modified TiO2 photocatalysts for decomposition of acetic acid with simultaneous formation of C1-C3 hydrocarbons and hydrogen. Applied Catalysis B: Environmental 2013, 140–141, 108–114. 10.1016/j.apcatb.2013.03.044. [DOI] [Google Scholar]
- Schwarz J.; König B. Decarboxylative reactions with and without light - a comparison. Green Chem. 2018, 20 (2), 323–361. 10.1039/C7GC02949G. [DOI] [Google Scholar]
- Du X.; Liu J.; Li D.; Xin H.; Lei X.; Zhang R.; Zhou L.; Yang H.; Zeng Y.; Zhang H.; Zheng W.; Wen X.; Hu C. Structural and electronic effects boosting Ni-doped Mo2C catalyst toward high-efficiency C-O/C-C bonds cleavage. J. Energy Chem. 2022, 75, 109–116. 10.1016/j.jechem.2022.08.024. [DOI] [Google Scholar]
- Yang H.; Zeng Y.; Zhou Y.; Du X.; Li D.; Hu C. One-step synthesis of highly active and stable Ni-ZrO2 catalysts for the conversion of methyl laurate to alkanes. J. Catal. 2022, 413, 297–310. 10.1016/j.jcat.2022.06.035. [DOI] [Google Scholar]
- Zhou Y.; Remón J.; Jiang Z.; Matharu A. S.; Hu C. Tuning the selectivity of natural oils and fatty acids/esters deoxygenation to biofuels and fatty alcohols: A review. Green Energy Environ. 2023, 8, 722–743. 10.1016/j.gee.2022.03.001. [DOI] [Google Scholar]
- Du X.; Peng Y.; Albero J.; Li D.; Hu C.; Garcia H. Synthetic Fuels from Biomass: Photocatalytic Hydrodecarboxylation of Octanoic Acid by Ni Nanoparticles Deposited on TiO2. ChemSusChem 2022, 15 (2), e202102107 10.1002/cssc.202102107. [DOI] [PubMed] [Google Scholar]
- Chen X.; Shen S.; Guo L.; Mao S. S. Semiconductor-based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503–6570. 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Silveri F.; Bayazit M. K.; Ruan Q.; Li Y.; Xie J.; Catlow C. R. A.; Tang J. Bandgap Engineering of Organic Semiconductors for Highly Efficient Photocatalytic Water Splitting. Adv. Energy Mater. 2018, 8 (24), 1801084–1801093. 10.1002/aenm.201801084. [DOI] [Google Scholar]
- Wu X.; Li J.; Xie S.; Duan P.; Zhang H.; Feng J.; Zhang Q.; Cheng J.; Wang Y. Selectivity Control in Photocatalytic Valorization of Biomass-Derived Platform Compounds by Surface Engineering of Titanium Oxide. Chem. 2020, 6 (11), 3038–3053. 10.1016/j.chempr.2020.08.014. [DOI] [Google Scholar]
- Cao S.; Chan T.-S.; Lu Y.-R.; Shi X.; Fu B.; Wu Z.; Li H.; Liu K.; Alzuabi S.; Cheng P.; Liu M.; Li T.; Chen X.; Piao L. Photocatalytic pure water splitting with high efficiency and value by Pt/ porous brookite TiO2 nanoflutes. Nano Energy 2020, 67, 104287. 10.1016/j.nanoen.2019.104287. [DOI] [Google Scholar]
- Wang L.; Xiao Z.; Liu Y.; Cao S.; Ma Z.; Piao L. Mesoporous TiO2 mixed crystals for photocatalytic pure water splitting. Science China Materials 2020, 63 (5), 758–768. 10.1007/s40843-019-1253-8. [DOI] [Google Scholar]
- Ao C. H.; Lee S. C. Indoor air purification by photocatalyst TiO2 immobilized on an activated carbon filter installed in an air cleaner. Chem. Eng. Sci. 2005, 60 (1), 103–109. 10.1016/j.ces.2004.01.073. [DOI] [Google Scholar]
- Cybula A.; Priebe J. B.; Pohl M.-M.; Sobczak J. W.; Schneider M.; Zielińska-Jurek A.; Brückner A.; Zaleska A. The effect of calcination temperature on structure and photocatalytic properties of Au/Pd nanoparticles supported on TiO2. Applied Catalysis B: Environmental 2014, 152–153, 202–211. 10.1016/j.apcatb.2014.01.042. [DOI] [Google Scholar]
- Bowker M.; O’Rourke C.; Mills A. The Role of Metal Nanoparticles in Promoting Photocatalysis by TiO2. Top Curr. Chem. (Cham) 2022, 380 (3), 17. 10.1007/s41061-022-00373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng A.; Zhang L.; Cheng B.; Yu J. Dual Cocatalysts in TiO2 Photocatalysis. Adv. Mater. 2019, 31 (30), e1807660 10.1002/adma.201807660. [DOI] [PubMed] [Google Scholar]
- Caudillo-Flores U.; Muñoz-Batista M. J.; Fernández-García M.; Kubacka A. Bimetallic Pt-Pd co-catalyst Nb-doped TiO2 materials for H2 photo-production under UV and Visible light illumination. Applied Catalysis B: Environmental 2018, 238, 533–545. 10.1016/j.apcatb.2018.07.047. [DOI] [Google Scholar]
- Zielińska-Jurek A. Progress, Challenge, and Perspective of Bimetallic TiO2-Based Photocatalysts. J. Nanomater. 2014, 2014, 1–17. 10.1155/2014/208920. [DOI] [Google Scholar]
- Haldar K. K.; Kundu S.; Patra A. Core-size-dependent catalytic properties of bimetallic Au/Ag core-shell nanoparticles. ACS Appl. Mater. Interfaces 2014, 6 (24), 21946–53. 10.1021/am507391d. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang C.; Yan H.; Yi H.; Lu J. Precisely-controlled synthesis of Au@Pd core-shell bimetallic catalyst via atomic layer deposition for selective oxidation of benzyl alcohol. J. Catal. 2015, 324, 59–68. 10.1016/j.jcat.2015.01.019. [DOI] [Google Scholar]
- Shi D.; Sadier A.; Girardon J.-S.; Mamede A.-S.; Ciotonea C.; Marinova M.; Stievano L.; Sougrati M. T.; La Fontaine C.; Paul S.; Wojcieszak R.; Marceau E. Probing the core and surface composition of nanoalloy to rationalize its selectivity: Study of Ni-Fe/SiO2 catalysts for liquid-phase hydrogenation. Chem. Catalysis 2022, 2 (7), 1686–1708. 10.1016/j.checat.2022.04.009. [DOI] [Google Scholar]
- Fan Y.; Girard A.; Waals M.; Salzemann C.; Courty A. Ag@Pt Core-Shell Nanoparticles for Plasmonic Catalysis. ACS Applied Nano Materials 2023, 6 (2), 1193–1202. 10.1021/acsanm.2c04767. [DOI] [Google Scholar]
- Zong Z.; Xu K.; Li D.; Tang Z.; He W.; Liu Z.; Wang X.; Tian Y. Peptide templated Au@Pd core-shell structures as efficient bi-functional electrocatalysts for both oxygen reduction and hydrogen evolution reactions. J. Catal. 2018, 361, 168–176. 10.1016/j.jcat.2018.02.020. [DOI] [Google Scholar]
- Grirrane A.; Corma A.; Garcia H. Gold-Catalyzed Synthesis of Aromatic Azo Compounds from Anilines and Nitroaromatics. Science 2008, 322, 1661–1664. 10.1126/science.1166401. [DOI] [PubMed] [Google Scholar]
- Segall M D; Lindan P. J D; Probert M J; Pickard C J; Hasnip P J; Clark S J; Payne M C MCPayne, First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter 2002, 14, 2717–2744. 10.1088/0953-8984/14/11/301. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Tkatchenko A.; Scheffler M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102 (7), 073005. 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- Yu H.; Yang X.; Wu Y.; Guo Y.; Li S.; Lin W.; Li X.; Zheng J. Bimetallic Ru-Ni/TiO2 catalysts for hydrogenation of N-ethylcarbazole: Role of TiO2 crystal structure. Journal of Energy Chemistry 2020, 40, 188–195. 10.1016/j.jechem.2019.04.009. [DOI] [Google Scholar]
- Shi D.; Liu J.; Sun R.; Ji S.; Rogers S. M.; Connolly B. M.; Dimitratos N.; Wheatley A. E. H. Preparation of bifunctional Au-Pd/TiO2 catalysts and research on methanol liquid phase one-step oxidation to methyl formate. Catal. Today 2018, 316, 206–213. 10.1016/j.cattod.2018.01.033. [DOI] [Google Scholar]
- Kruse N.; Chenakin S. XPS characterization of Au/TiO2 catalysts: Binding energy assessment and irradiation effects. Applied Catalysis A: General 2011, 391 (1–2), 367–376. 10.1016/j.apcata.2010.05.039. [DOI] [Google Scholar]
- Sun L.; Ren X.; Yin Z.; Jing H.; Zhang M.; Zhou X.; Sun X.; Su H.; Qi C. Promoting effect of trace amount of Pd for p-CNB hydrogenation in Pd-Au/TiO2 bimetallic catalyst. Appl. Surf. Sci. 2023, 623, 157107–157114. 10.1016/j.apsusc.2023.157107. [DOI] [Google Scholar]
- Wu P.; Cao Y.; Zhao L.; Wang Y.; He Z.; Xing W.; Bai P.; Mintova S.; Yan Z. Formation of PdO on Au-Pd bimetallic catalysts and the effect on benzyl alcohol oxidation. J. Catal. 2019, 375, 32–43. 10.1016/j.jcat.2019.05.003. [DOI] [Google Scholar]
- Tabakova T.; Ilieva L.; Petrova P.; Venezia A. M.; Avdeev G.; Zanella R.; Karakirova Y. Complete benzene oxidation over mono and bimetallic Au-Pd catalysts supported on Fe-modified ceria. Chemical Engineering Journal 2015, 260, 133–141. 10.1016/j.cej.2014.08.099. [DOI] [Google Scholar]
- Khder A. S.; Altass H. M.; Jassas R. S.; Al-Rooqi M. M.; Khder M. A.; Morad M.; Gebreil A.; Moussa Z.; Ahmed S. A. Room-Temperature CO Oxidation over Au-Pd Monometallic and Bimetallic Nanoparticle-Supported MgO. ACS Applied Nano Materials 2023, 6 (6), 4243–4252. 10.1021/acsanm.2c05326. [DOI] [Google Scholar]
- Wang P.; Zhan S.; Xia Y.; Ma S.; Zhou Q.; Li Y. The fundamental role and mechanism of reduced graphene oxide in rGO/Pt-TiO2 nanocomposite for high-performance photocatalytic water splitting. Applied Catalysis B: Environmental 2017, 207, 335–346. 10.1016/j.apcatb.2017.02.031. [DOI] [Google Scholar]
- Devaraji P.; Gopinath C. S. Pt - g-C3N4 - (Au/TiO2): Electronically integrated nanocomposite for solar hydrogen generation. Int. J. Hydrogen Energy 2018, 43 (2), 601–613. 10.1016/j.ijhydene.2017.11.057. [DOI] [Google Scholar]
- Tudu B.; Nalajala N.; Prabhakar Reddy K.; Saikia P.; Gopinath C. S. Electronic Integration and Thin Film Aspects of Au-Pd/rGO/TiO2 for Improved Solar Hydrogen Generation. ACS Appl. Mater. Interfaces 2019, 11 (36), 32869–32878. 10.1021/acsami.9b07070. [DOI] [PubMed] [Google Scholar]
- Xing M.; Zhang J.; Chen F.; Tian B. An economic method to prepare vacuum activated photocatalysts with high photo-activities and photosensitivities. Chem. Commun. (Camb) 2011, 47 (17), 4947–9. 10.1039/c1cc10537j. [DOI] [PubMed] [Google Scholar]
- Li G.; Liu C.; Liu Y. Different effects of cerium ions doping on properties of anatase and rutile TiO2. Appl. Surf. Sci. 2006, 253 (5), 2481–2486. 10.1016/j.apsusc.2006.05.002. [DOI] [Google Scholar]
- Wei H.; Liu W.; Chen X.; Yang Q.; Li J.; Chen H. Renewable bio-jet fuel production for aviation: A review. Fuel 2019, 254, 115599–115614. 10.1016/j.fuel.2019.06.007. [DOI] [Google Scholar]
- Wu L.; Li F.; Xu Y.; Zhang J. W.; Zhang D.; Li G.; Li H. Plasmon-induced photoelectrocatalytic activity of Au nanoparticles enhanced TiO2 nanotube arrays electrodes for environmental remediation. Applied Catalysis B: Environmental 2015, 164, 217–224. 10.1016/j.apcatb.2014.09.029. [DOI] [Google Scholar]
- Zhang S.; Zhao W.; Ren A.; Guo B.; Dong Y.; Deng X.-Q. Insight into surface properties of O2 plasma activated Au/TiO2 prepared by DPU in CO oxidation. Catal. Today 2019, 337, 110–116. 10.1016/j.cattod.2019.04.020. [DOI] [Google Scholar]
- Liu Q.; Zhang J.; Xing F.; Cheng C.; Wu Y.; Huang C. Plasmon-enhanced and controllable synthesis of azobenzene and hydrazobenzene using Au/TiO2 composite. Appl. Surf. Sci. 2020, 500, 144214–144219. 10.1016/j.apsusc.2019.144214. [DOI] [Google Scholar]
- Yu H.; Wang X.; Sun H.; Huo M. Photocatalytic degradation of malathion in aqueous solution using an Au-Pd-TiO2 nanotube film. J. Hazard. Mater. 2010, 184 (1–3), 753–758. 10.1016/j.jhazmat.2010.08.103. [DOI] [PubMed] [Google Scholar]
- Nakayama N.; Hayashi T. Preparation of TiO2 nanoparticles surface-modified by both carboxylic acid and amine: Dispersibility and stabilization in organic solvents. Colloids Surf., A 2008, 317 (1–3), 543–550. 10.1016/j.colsurfa.2007.11.036. [DOI] [Google Scholar]
- Li J.; Liu T.; Sui G.; Zhen D. Photocatalytic Performance of a Nd-SiO2-TiO2 Nanocomposite for Degradation of Rhodamine B Dye Wastewater. J. Nanosci. Nanotechnol. 2015, 15 (2), 1408–15. 10.1166/jnn.2015.9611. [DOI] [PubMed] [Google Scholar]
- CAPECCHI G.; FAGA M. G.; MARTRA G.; COLUCCIA S.; IOZZI M. F.; COSSI M. Adsorption of CH3COOH on TiO2:IR and theoretical investigations. Res. Chem. Intermed. 2007, 33, 269–284. 10.1163/156856707779238748. [DOI] [Google Scholar]
- McEntee M.; Tang W.; Neurock M.; Yates J. T. Mechanistic Insights into the Catalytic Oxidation of Carboxylic Acids on Au/TiO2: Partial Oxidation of Propionic and Butyric Acid to Gold Ketenylidene through Unsaturated Acids. ACS Catal. 2015, 5 (2), 744–753. 10.1021/cs5014255. [DOI] [Google Scholar]
- Ojamae L.; Aulin C.; Pedersen H.; Kall P.-O. IR and quantum-chemical studies of carboxylic acid and glycine adsorption on rutile TiO2 nanoparticles. J. Colloid Interface Sci. 2006, 296 (1), 71–78. 10.1016/j.jcis.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Woll C. IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: bridging the materials gap. Chem. Soc. Rev. 2017, 46 (7), 1875–1932. 10.1039/C6CS00914J. [DOI] [PubMed] [Google Scholar]
- Qian R.; Zong H.; Schneider J.; Zhou G.; Zhao T.; Li Y.; Yang J.; Bahnemann D. W.; Pan J. H. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: An overview. Catal. Today 2019, 335, 78–90. 10.1016/j.cattod.2018.10.053. [DOI] [Google Scholar]
- Maity P.; Mohammed O. F.; Katsiev K.; Idriss H. Study of the Bulk Charge Carrier Dynamics in Anatase and Rutile TiO2 Single Crystals by Femtosecond Time-Resolved Spectroscopy. J. Phys. Chem. C 2018, 122 (16), 8925–8932. 10.1021/acs.jpcc.8b00256. [DOI] [Google Scholar]
- Kumaravel V.; Mathew S.; Bartlett J.; Pillai S. C. Photocatalytic hydrogen production using metal doped TiO2: A review of recent advances. Applied Catalysis B: Environmental 2019, 244, 1021–1064. 10.1016/j.apcatb.2018.11.080. [DOI] [Google Scholar]
- Wang A.; Wu S.; Dong J.; Wang R.; Wang J.; Zhang J.; Zhong S.; Bai S. Interfacial facet engineering on the Schottky barrier between plasmonic Au and TiO2 in boosting the photocatalytic CO2 reduction under ultraviolet and visible light irradiation. Chemical Engineering Journal 2021, 404, 127145–127152. 10.1016/j.cej.2020.127145. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jonsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Zhang H.; Zhou P.; Ji H.; Ma W.; Chen C.; Zhao J. Enhancement of photocatalytic decarboxylation on TiO2 by water-induced change in adsorption-mode. Applied Catalysis B: Environmental 2018, 224, 376–382. 10.1016/j.apcatb.2017.10.020. [DOI] [Google Scholar]
- van Santen R. A.; Neurock M.; Shetty S. G. Reactivity Theory of Transition-Metal Surfaces: A Brønsted-Evans-Polanyi Linear Activation Energy-Free-Energy Analysis. Chem. Rev. 2010, 110, 2005–2048. 10.1021/cr9001808. [DOI] [PubMed] [Google Scholar]
- Sutton J. E.; Vlachos D. G. A Theoretical and Computational Analysis of Linear Free Energy Relations for the Estimation of Activation Energies. ACS Catal. 2012, 2 (8), 1624–1634. 10.1021/cs3003269. [DOI] [Google Scholar]
- Kumar A.; Choudhary P.; Kumar A.; Camargo P. H. C.; Krishnan V. Recent Advances in Plasmonic Photocatalysis Based on TiO2 and Noble Metal Nanoparticles for Energy Conversion, Environmental Remediation, and Organic Synthesis. Small 2022, 18 (1), e2101638 10.1002/smll.202101638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.