

Abstract
Purpose of Review
Patients with neurofibromatosis type 1 (NF1) are at increased risk for benign and malignant neoplasms. Recently, targeted therapy with the MEK inhibitor class has helped address these needs. We highlight recent successes with selumetinib while acknowledging ongoing challenges for NF1 patients and future directions.
Recent Findings
MEK inhibitors have demonstrated efficacy for NF1-related conditions, including plexiform neurofibromas and low-grade gliomas, two common causes of NF1-related morbidity. Active investigations for NF1-related neoplasms have benefited from advanced understanding of the genomic and cell signaling alterations in these conditions and development of sound preclinical animal models.
Summary
Selumetinib has become the first FDA-approved targeted therapy for NF1 following its demonstrated efficacy for inoperable plexiform neurofibroma. Investigations of combination therapy and the development of a representative NF1 swine model hold promise for translating therapies for other NF1-associated pathology.
Keywords: Neurofibromatosis, MEK inhibitor, Selumetinib, Plexiform neurofibroma, Low-grade glioma, Optic pathway glioma, Malignant peripheral nerve sheath tumor, Combination therapy
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant neurocutaneous and tumor predisposition syndrome with well-defined clinical features developed by a National Institutes of Health (NIH) convened expert panel in 1987 [1]. The underlying genetic alteration is in the neurofibromin 1 gene, NF1, located on chromosome 17 that encodes neurofibromin. NF1 is among the largest genes, and many inactivating, loss of function, and germline mutations have been described. Nonetheless, genetic testing is sensitive, can help differentiate neurocutaneous syndromes, and can confirm a suspected diagnosis, including mosaic disease, before full clinical features develop [2•]. While fully penetrant, there is great variation in disease manifestations, even within families, but morbidity results from multisystem pathology including benign and malignant tumors [3]. A deeper understanding of neurofibromin’s role in the core mitogen-activated protein kinase (MAPK) cell signaling pathway has led to success with disease directed therapy in recent years. This is exemplified by the first FDA-approved therapy for patients with NF1 with selumetinib, for inoperable NF1- associated plexiform neurofibromas in children this year [4•]. The continued development of representative preclinical models and exploration of combination therapy for other neoplastic manifestations of NF1 hold promise to continue improving health outcomes for people with NF1.
Genomic Alterations and Cell Signaling Dysregulation in NF1-Associated Pathology
The Mitogen-Activated Protein Kinase Pathway
Tumorigenic cells develop homeostatic imbalances leading to cell proliferation and to resisting cell death [5]. Ten cell signaling pathways controlling cell growth and cell death are altered in most human cancer [6]. The mitogen-activated protein kinase (MAPK) pathway, first discovered in 1988, is initiated by autophosphorylation of receptor tyrosine kinases (RTKs) leading to sequential downstream activation of RAS GTPases, RAF kinases, MEK kinases, MAPKs ERK1 and ERK2, and finally transcription factors such as ELK1 [7]. The product of the NF1 gene encodes neurofibromin, a protein with a central GTPase activating protein (GAP) domain that interacts with RAS-GTP, dramatically increasing its intrinsic GTPase activity and thereby causing its conversion to RAS-GDP, thus reducing levels of RAS-GTP, the active species of RAS. Thus, neurofibromin functions to reduce RAS-GTP mediated activation of its effector pathways, including the MAPK and phosphoinositide 3-kinase (PI3K) pathways. Dysregulation of these pathways results from biallelic loss of NF1, as occurs in neoplasms in NF1 patients [8]. Congenital alterations of genes involved in the interrelated MAPK and PI3K pathways, collectively called RASopathies, lead to an increased incidence of neoplasms [9, 10].
The discovery of the MAPK pathway and its key role in tumorigenesis spurred the development of targeted therapy for sporadic tumors, in which alterations in genes encoding RTKs, RAS proteins, and RAF and MEK kinases contribute to malignancy. Examples of specific alterations leading to drug development include EGFRT790M, BRAFV600E, and KRASG12C mutations [11]. In these settings, acquired drug resistance is common. Drug resistance can occur due to increased activation of upstream kinases as well as mutations in the targeted receptor or kinase itself. Altered cross-talk to engender resistance can involve alternative MEK activators. Also, cells can use various MEK isoforms, of which at least seven are known, and activate different nuclear transcription factors [7, 12]. The ERK1/2 kinases, in contrast, are specifically activated by MEK1/2. This specific function of MEK1/2 makes them an attractive target given the diverse cellular proliferation and survival responses induced by ERK, especially in NF1 where mutations of upstream kinases and RTKs are not present constitutionally [12].
NF1-Associated Neurofibromas
Neurofibromas are benign tumors that arise from neoplastic Schwann cells [13]. They usually show biallelic loss of NF1, do not express neurofibromin, and demonstrate activated RAS-GTP compared to controls [8]. Cutaneous neurofibromas (cNF) are discrete tumors involving the dermis or epidermis. Virtually all individuals with NF1 develop cNFs during their lifetime, and cNF contribute to disfigurement, dysesthesia, psychosocial distress, and quality of life impairment [14]. Plexiform neurofibromas (pNF) arise internally within virtually any peripheral nerve sheath, grow along nerve tracts, and can invade adjacent tissues. Compression of organs and structures and asymmetric or rapid growth lead to disfigurement and morbidity, with pain and motor morbidities most common [15•]. Children and adolescents with pNF demonstrate significantly worse measures in psychosocial and functional domains [16•].
In contrast to cNF, pNF are less prevalent with 25–50% of NF1 individuals affected. They arise early in life, are possibly congenital, and show peak growth during childhood [13]. Importantly, pNF are at risk of malignant degeneration to malignant peripheral nerve sheath tumors (MPNST), distinct from cNF. While histologically similar, the clinical differences between cNF and pNF suggest underlying biologic differences. In a methylation analysis, differential gene expression was observed between each group, with cNF epigenetically reinforced for RAS/MEK3/p38 and pNF for RAS/ERK [12, 17•].
NF1-Associated Gliomas
Individuals with NF1 are at increased risk for central nervous system tumors, which are mainly indolent low-grade neoplasms but can include aggressive infiltrating disease and develop in an estimated 15–20% of patients. Low-grade gliomas (LGG) are more common in children and most frequently affect the optic pathway. The incidence of high-grade gliomas (HGG) increases with age, with more than 50-fold increased relative risk compared to the general population [18•]. Pediatric LGGs invariably involve upregulation of the MAPK signaling pathway. They are distinct from adult LGG, which are characterized by chromosome 1 and 19 losses and IDH1 mutations and which frequently degenerate into HGG over time [19••]. Of all pediatric LGG, an estimated 14% occur in individuals with NF1, highlighting their shared biology [19••]. NF1-associated LGG have excellent (>95%) 20-year overall survival, but progression-free survival is lower at 41–58%, and morbidity due to tumor location and adverse effects of treatment are significant concerns [19••, 20].
NF1-Associated Malignant Neoplasms
MPNST are the leading cause of mortality in adults with NF1 [21]. They typically arise from prior pNF and have frequently metastasized before diagnosis [13]. Compared to sporadic MPNST, NF1-associated MPNST arise at a younger age and are less responsive to chemotherapy [13]. The progression of pNF to MPNST appears to start with the development of pre-malignant lesions, namely, atypical neurofibroma and atypical neurofibromatous neoplasm of uncertain biological potential (ANNUBP), which commonly acquire loss of CDKN2A/B [13, 22•, 23]. MPNST contain many additional genomic rearrangements; amplification of PDGFR, KIT, and EGFR; and mutations/deletions in SUZ12, EDD, TP53, and/or PTEN along with additional complex RAS-activating mutations [24, 25•].
HGG and breast cancer are additional important causes of neoplasia-related mortality in adults with NF1. Like pNF and MPNST, the biological complexity is distinct between LGG and HGG. LGGs in patients with NF1 are enriched for expression of proteins in the MAPK pathway, and there is a subset (up to 50%) characterized by lymphocyte infiltration, which may contribute to tumor senescence often observed with LGG [26••]. In contrast, when patients with NF1 develop HGG, additional mutations including those in ATRX, TP53, and CDKN2A are acquired, the PI3K pathway is activated, and tumors have low immune infiltration [26••, 27, 28]. The development of breast cancer in females with NF1 occurs earlier than the general population and is linked with poorer outcomes due to an increased proportion of triple negative and HER2 positive subtypes [29]. Somatic acquisition of mutations in TP53, KMT2C, KMT2D, and PIK3CA has been observed [30].
Precision Medicine in NF1—Development of MEK Inhibitors Including Selumetinib
The first MEK inhibitor (MEKi) was discovered in 1995, and subsequent generations of this class saw improved pharmacological properties [12]. These compounds were developed and tested for prevalent cancers such as melanoma and lung cancer, and now they are used for diverse indications [31]. The application of MEKi for NF1, a rare disease, has been accelerated by a remarkable collaboration between the NF community, researchers, funding agencies, and pharmaceutical company stakeholders (Table 1). The Children’s Tumor Foundation and other funding agencies including the NIH, Congressionally Directed Medical Research Programs (CDMRP), NF clinical trials consortium, Gilbert Family Foundation, and the Neurofibromatosis Therapeutics Acceleration Program (NTAP) all support NF research and have contributed to the advancement of research and therapeutics for NF1 patients. Together, these organizations have supported the development of >20 clinical trials for MEKi for NF-related conditions [32–35].
Table 1.
Active clinical trials listed on clinicaltrails.gov (as of Oct 2020) involvingMEK inhibitors, for which NF1-related indications are eligible. This list is organized by NF1 focus. cNF cutaneous neurofibroma, HGG high-grade glioma, JMML juvenile myelomonocytic leukemia, LGG low-grade glioma, MPNST malignant peripheral nerve sheath tumor, OPG optic pathway glioma, pNF plexiform neurofibroma
| Trial identifier | Drug(s) | Indication | Phase | Population | NF specific focus |
|---|---|---|---|---|---|
| NCT03433183 | Selumetinib, sirolimus | MPNST | II | ≥12 years | Yes |
| NCT03962543 | Mirdametinib | Inoperable pNF | II | ≥2 years | Yes |
| NCT03190915 | Trametinib | Relapsed/refractory JMML | II | ≥1 months | Yes |
| NCT02124772 | Trametinib | Refractory pNF | I/II | ≥1 month to ≤17 years | Yes |
| NCT03326388 | Selumetinib | Inoperable pNF, progressive OPG | I/II | ≥3 years to ≤18 years | Yes |
| NCT03231306 | Binimetinib | Symptomatic pNF | II | ≥1 years | Yes |
| NCT01089101 | Selumetinib | Relapsed/refractory LGG | I/II | ≥3 to ≤21 years | Yes |
| NCT02839720 | Selumetinib | Symptomatic cNF | II | ≥18 years | Yes |
| NCT02285439 | Binimetinib | Advanced solid tumors and relapsed/refractory LGG | I/II | ≥1 to ≤18 years | Yes |
| NCT04201457 | Trametinib, dabrafenib, hydroxychloroquine | Recurrent LGG or HGG | I/II | ≥1 to ≤30 years | Yes |
| NCT02407405 | Selumetinib | Inoperable pNF | II | ≥18 years | Yes |
| NCT04435665 | NFX-179 (topical) | cNF | II | ≥18 years | Yes |
| NCT03741101 | Trametinib | pNF | II | ≥1 to <18 years | Yes |
| NCT03871257 | Selumetinib | NF1-associated LGG | III | ≥2 to ≤21 years | Yes |
| NCT03363217 | Trametinib | pNF and relapsed/refractory LGG | I/II | ≥1 month to ≤25 years | Yes |
| NCT04216953 | Cobimetinib, atezolizumab | Advanced soft tissue sarcoma | I/II | ≥6 months | No |
| NCT03516123 | CS3006 | Advanced solid tumors | I | ≥18 years | No |
| NCT03875820 | RO5126766, VS-6063 | Advanced solid tumors | I | ≥18 years | No |
| NCT03905148 | Mirdametinib, BGB-283 | Advanced solid tumors | I | ≥18 years | No |
| NCT02079740 | Trametinib, navitoclax | Advanced solid tumors | I/II | ≥18 years | No |
| NCT01364051 | Selumetinib, cediranib | Advanced solid tumors | I | ≥18 years | No |
| NCT03736850 | CS3006 | Advanced solid tumors | I | ≥18 years | No |
| NCT03976050 | HL-085 | Advanced solid tumors | I | ≥18 years | No |
| NCT01586624 | Selumetinib, vandetanib | Advanced solid tumors | I | ≥18 years | No |
| NCT03839342 | Binimetinib, encorafenib | Advanced solid tumors | II | ≥18 years | No |
| NCT02070549 | Trametinib | Advanced solid tumors | I | ≥18 years | No |
| NCT04485559 | Trametinib, everolimus | Relapsed/refractory LGG | ≥1 to ≤25 years | No | |
| NCT03108131 | Cobimetinib, atezolizumab | Advanced solid tumors | II | ≥18 years | No |
| NCT03434262 | Trametinib, ribociclib (stratum B) | Recurrent/refractory HGG | I | ≥1 to ≤39 years | No |
| NCT01827384 | Trametinib | Advanced solid tumors | II | ≥18 years | No |
Selumetinib (AZD6244; ARRY-142886) is a second-generation allosteric MEKi with an active metabolite that is itself a highly selective kinase inhibitor. Cells treated with the inhibitor show reduced ERK phosphorylation, in vitro and in vivo [36]. It has favorable pharmacokinetics (PK) with a half-life of 12 h and is metabolized by CYP and UGT enzymes [37–39]. The development of genetically engineered mouse NF1 models helped demonstrate the importance of the MAPK pathway in NF1-associated neurofibromas, and the MEKi PD-0325901 was shown to be active in plexiform-like neurofibromas and MPNSTs [40]. Further work demonstrated that even low dose exposure to PD-0325901 reduced tumor volume in neurofibroma-bearing mice [41]. These studies in a murine pNF model were repeated using selumetinib. Neurofibroma-bearing mice showed a sustained decrease in phosphorylated ERK and maximal tumor shrinkage of 30% as determined by volumetric magnetic resonance imaging (MRI) [42, supp appendix]. These preclinical studies provided a rationale for evaluating MEKi in clinical trials for NF1-associated neoplasms.
Clinical Advancements with Selumetinib for NF1
Challenges in Clinical Trial Design for NF
NF1 alterations can be associated with significant neurodevelopmental, neurologic, cardiovascular, and musculoskeletal abnormalities [2•, 43]. Consequently, quality of life can be impacted across psychosocial and health-related domains [43, 44]. Neoplasms, both benign and malignant, may develop at any age and grow with variable kinetics. In some cases, medical therapies must be tolerated for a long duration, be able to reduce tumors of large volume, or treat malignant tumors [45]. Additional challenges for the NF patient population are the relative rarity of the disorder leading to slow clinical trial enrollment, variable disease kinetics among individuals with similar pathology, and variation in disease penetrance and presentation [46•].
The NF community is exemplary for its innovation and collaboration in addressing these challenges. Firstly, the National Cancer Institutes (NCI) natural history study of NF1 has advanced understanding of NF-related morbidities and has been used as a benchmark for clinical study interpretation [46•, 47•]. Secondly, clinical trial design has benefited from standardization of outcome measures by The Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) working groups, created in 2011 [48]. As an illustrative example, historical trials for treatment of pNF were limited by imaging techniques to monitor disease response; REiNS standardized the use of volumetric MRI, which is now incorporated into clinical trials for plexiform neurofibroma [45, 46•, 48–50]. For optic pathway gliomas, visual acuity as an endpoint has surpassed tumor reduction, and other functional outcomes (e.g., motor and respiratory function) in addition to patient-reported outcomes (PRO) of pain inform effectiveness of therapy [48]. While challenges exist in implementation, defining and standardizing clinical trial endpoints precedes clinical trial design that incorporates the reality of NF as a multisystem, lifelong disease state [51].
Efficacy of Selumetinib in Plexiform Neurofibroma
Historically, pNF have been difficult to treat. Agents tested to slow or reverse pNF progression in clinical trials include the farnesyl transferase inhibitor tipifarnib, the antifibrotic agent pirfenidone, the mTOR inhibitor sirolimus, and pegylated interferon alfa-2b. Of these, only pegylated interferon alfa-2b prolonged time to progression with 65% of patients in this trial demonstrating stable disease for 12 months. However, only a minority of patients (5%) achieved tumor volume reduction ≥ 20% [13, 47•, 52]. Imatinib mesylate achieved a 17% rate of ≥ 20% volume reduction primarily in patients with small baseline tumor volumes, and 30% reported symptom improvement [53].
Following promise for MEKi in the treatment of pNF in preclinical models, selumetinib was evaluated in a phase I trial including children with inoperable pNFs. The trial established a maximum tolerated dose of 25 mg/m2/dose that was tolerable for an extended duration (median 30 cycles). In contrast to prior trials, selumetinib showed remarkable success with every patient achieving volume reduction of their pNF after a median of 20 cycles and 71% achieving ≥ 20% volume reduction [42]. Common adverse events of MEKi include dermatologic reactions, gastrointestinal side effects, and cardiotoxicity. Dose-limiting toxicities within the 20 mg/m2 and 25 mg/m2 dosing groups were common at 39%, compared with 25% dose-limiting toxicities in the pegylated interferon trial [42].
A subsequent phase II trial of selumetinib in children with inoperable pNF, SPRINT, evaluated PROs and functional measures (N = 50). Patients were age-matched to 93 patients in the NCI natural history study of NF1 [54••]. After a follow-up of 3 years, 84% of patients had progression-free survival compared to 15% in the controls. Six patients (12%) experienced disease progression, but only two patients failed to have tumor volume reduction as best response. Overall, 70% achieved a tumor volume reduction of ≥ 20%. The median duration to best response was 16 cycles. Seventy-four percent of patients had meaningful improvement of pain, 48% had improvement of health-related quality of life, and improvements in strength and range of motion were reported by 56% and 38% of children, respectively. Adverse events were again common with 38% of patients experiencing dose-limiting toxicity, including 10% who discontinued therapy. All toxicities (Klesse, et al. have reviewed management of MEK inhibitor toxicities) in SPRINT were reversible, but tumor regrowth after discontinuing therapy was common [54••, 55••]. A decreased ratio of circulating proangiogenic hematopoietic stem/progenitor cell populations compared to non-angiogenic circulating cells was identified as a candidate bio-marker to discriminate, early in treatment, those attaining partial response from those with stable/progressive disease [54••, supp appendix].
In comparison to historical trials and untreated patients on the NCI natural history study, the use of selumetinib achieved remarkable success with regard to tumor response and patient-reported outcomes. Selumetinib is generally safe with no irreversible toxicity and can be tolerated for long durations, with 58% of patients remaining on therapy at the time of data cut off in the phase two SPRINT trial. These results led to selumetinib being the first FDA-approved therapy for NF1-associated inoperable pNF.
Other MEKi are currently in clinical trials for pNF including trametinib (GSK-1120212), mirdametinib (PD-0325901), and binimetinib (ARRY-162) [4, 56], but many unanswered questions and challenges remain. For pNFs, adherence to prolonged therapy that commonly causes side effects is required because pNF resume growth if therapy is discontinued. Additionally, there have been no complete responses to therapy. There is a lack of information regarding late toxicities of MEKi therapy. There are no ongoing trials comparing different MEKi head-to-head, nor direct comparisons of MEKi with agents in different classes. Finally, two patients in the SPRINT trial suffered malignant degeneration to MPNST—a reminder of the disease burden carried by patients with NF1 [54••]. Nonetheless, MEK inhibition with selumetinib represents a major advance in the treatment of pNF.
Promise of Selumetinib in NF1 Low-Grade Glioma
Optic pathway gliomas account for up to 75% of NF1-related LGG and occur at a mean age of 2.7–5.4 years [57, 58]. Due to their variable clinical behavior, treatment is reserved for the 30–50% of individuals experiencing decreased visual acuity or symptoms of hypothalamic dys-function [59, 60•, 61]. Given the potential morbidity of surgery and elevated risk of secondary malignancy from radiation therapy, first-line treatment often entails carboplatin-based chemotherapy [62, 63]. The combination of carboplatin and vincristine (CV) leads to improved vision in one-third of patients, while another third will have deterioration of vision, and the remainder has stable vision [13]. Symptomatic non-optic tract gliomas not amenable to complete resection are treated with similar regimens. Historically, partial response to therapy for LGG has been defined as tumor shrinkage of 50% or more, while progressive disease is defined as a greater than 25% increase.
With advances in the understanding of LGG biology, clinical trials using targeted agents have been performed. Selumetinib was first used in a phase I trial for patients with recurrent or refractory LGG. Selumetinib was tolerable at 25 mg/m2/dose with three dose-limiting toxicities. Across all dosing levels (25 mg/m2, 33 mg/m2, and 43 mg/m2), 57% of patients remained on therapy for 1 year or longer. Four out of five patients with NF1 in this trial completed 20 or more cycles [64•]. Twenty-five patients with NF1 were enrolled in the subsequent phase II trial (NCT01089101) for patients with recurrent or refractory LGG, 13 of which had an optic pathway glioma. Nine patients achieved sustained partial response, 15 had stable disease, and one had progressive disease (Figure 1). Sixty-four percent completed all 26 cycles of treatment, and the 2-year progression-free survival in this subgroup was 96%. No patient with optic glioma had worsening of vision, and 20% had improvement [65••].
Fig. 1.
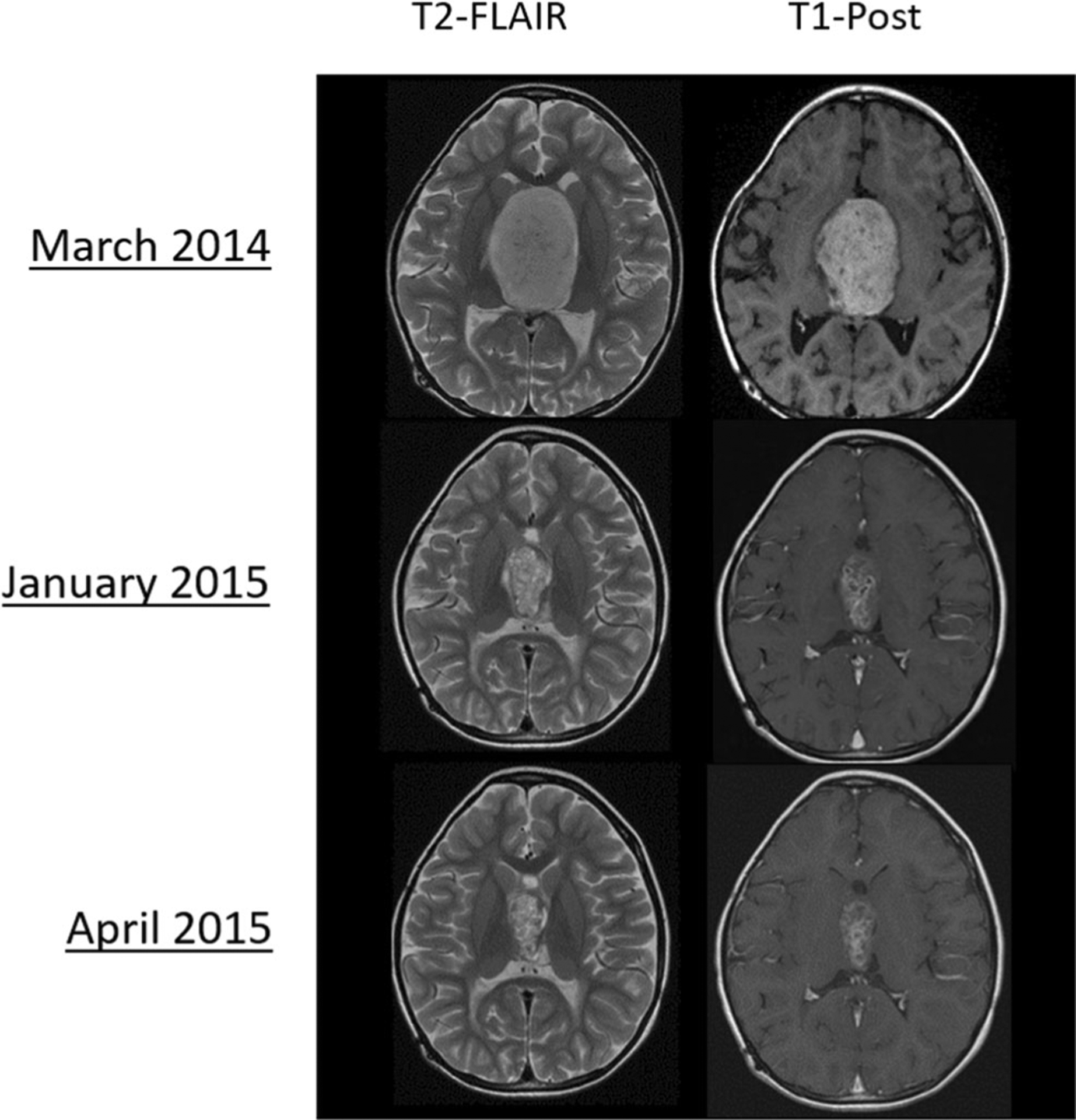
PBTC-029 phase 2, stratum 3 (recurrent NF1-associated low-grade glioma): Example of radiographic response with selumetinib monotherapy. Reproduced with permission by Dr. Jason Fangusaro and the Pediatric Brain Tumor Consortium
These trials of selumetinib demonstrate that LGG in NF1 can be stabilized without excess toxicity. Currently, other targeted agents are being evaluated in clinical trials including trametinib (NCT03363217), binimetinib (NCT02285439, phase I/II and NCT01885195, phase II), everolimus (NCT01158651, phase II), and anti-angiogenesis agents [66•]. To date, no targeted agents have accrued long-term outcome data. The Children’s Oncology Group (COG) Protocol A9952 enrolled 127 patients with NF1-related LGG and assigned them to therapy with CV. Compared to children with sporadic LGG, those with NF1 had superior 5-year event free survival (69% vs 39%) and overall survival (98% vs 87%) [67, 68]. Given that LGG are generally indolent and exhibit progression for up to 10 years from diagnosis, it is unknown whether the favorable responses reported thus far with MEKi therapy will be durable [19••]. Thus, for now, MEKi therapy for NF1-related LGG is promising for treatment of these lesions without the use of radiation or alkylating therapy, which are both associated with second malignant neoplasms in this setting [67]. These results in a refractory population have led to the first phase III trial (NCT03871257) for targeted therapy in NF1 comparing selumetinib vs. CV for previously untreated LGG [69].
Challenges and future direction
Through improvements in the understanding of NF1 biology, collaborations between funding agencies, and thoughtful implementation of clinical trial design, therapeutic advancement of MEK inhibition for the most common causes of morbidity for patients with NF1 has been achieved, leading to FDA approval of the MEKi selumetinib for inoperable pNF.
Despite successes, significant challenges in treating patients with NF1 persist. Their life expectancy is 15 years less than the general population, a consequence of malignant neoplasms [21, 70]. Malignant pathology can arise throughout life, with juvenile myelomonocytic leukemia and rhabdomyo-sarcoma prevalent in infancy and MPNST, breast cancer, high-grade glioma, and gastrointestinal stromal tumor prevalent in young adults [44, 56].
Future Treatment Approaches and Paradigms for Preclinical Investigation
In sporadic cancers, MEKi resistance commonly develops by activation of alternative oncogenic pathways (e.g., dysregulation of the RB axis through loss of CDKN2), MEK and ERK reactivation by altered RAS proteins, and activation of receptor tyrosine kinases in response to MEK inhibition [12]. Like observations in sporadic cancer, and in mouse models of MPNST, MEKi monotherapy in malignant NF-associated lesions is not expected to lead to durable treatment response [25•].
Combining targeted agents has the potential to overcome resistance and therapeutic failures for benign and malignant lesions. Agents potentially beneficial for combination therapy in NF1-related malignancy include those targeting other signaling pathways (e.g., mTOR inhibitors) and epigenetic changes in malignant lesions (e.g., CDK4/6 and BET inhibitors) [71••, 72••]. Immunotherapy represents another promising modality for treatment of NF1-associated malignancy. NF1 mutations have broad effects on the immune system and cytokine signaling, driven by hyperactive MAPK signaling [73, 74•]. Programmed death ligand 1 (PDL-1) is expressed in some NF1-related tumors, and tumor infiltrating lymphocytes have been observed in gliomas, neurofibromas, and MPNST [26, 75]. Currently, checkpoint inhibitor therapy is being evaluated in trials for MPNST [72••]. Immunotherapy for NF1 neoplasms may be synergistic with targeted therapy, including MEK inhibitors, or be enhanced by the application of oncolytic viruses or radiation therapy, which can serve to expose neoantigens and enhance immune infiltration in the tumor microenvironment [72••, 74•, 76, 77]. Like targeted therapy, success in treatment of NF1-associated malignant lesions with immunotherapy may be best achieved with combination approaches.
Critical to the design of precision medicine applications for advanced NF1 pathology are the development of accurate preclinical models. As described above, the NF1flox/flox;DhhCre murine model illustrates the potential for pNF MEKi therapy [40, 78]. Induced pluripotent stem cells reprogrammed from tumor cells have been developed and banked, and high-throughput drug screening techniques have been successfully applied to NF1 cell lines [79–81]. Cre-Lox technology has also been used to develop an optic pathway glioma model in a germline NF1+/− mouse model, and MPNST and high-grade gliomas have been modeled by codeletion of NF1 with PTEN or TP53 [18•, 22•, 26••, 58, 82]. However, these models may not capture the full context of NF1-related malignancies, such as epigenetic alterations, so there is interest in expanding patient-derived xenograft (PDX) models [22•]. PDX models permit application of combination agents but lack an immune microenvironment. Investigations of immunotherapy can be assessed in immunocompetent animal models engrafted with syngeneic tumor cell lines.
Genetically engineered mice have additional limitations in the study of cancer, as reviewed by Watson, et al. [83]. In addition to fundamental differences at the molecular and cellular level, limitations include anatomical differences limiting disease modeling, variations in pharmacokinetic and pharmacodynamic (PD) properties making therapeutic translation challenging, and small size limiting investigations into novel imaging or surgical techniques [83]. Swine (Sus scrofa) models provide solutions to many of these issues. Swine have greater genetic homology with humans and are more anatomically representative. Genetically engineered swine have been established to model NF1, and these minipigs phenotypically display clinical features of NF1 present in patients, which is unique compared to other NF1 models. These pigs develop café au lait macules, neurofibromas, and optic pathway gliomas. Importantly, tumor cells undergo spontaneous loss of heterozygosity mimicking the “second-hit” phenomenon that occurs in humans [84••].
Swine models of NF1 present many opportunities for developing therapies to treat NF1. First, the cutaneous manifestations provide an opportunity to address cNF. Active trials include oral selumetinib for adults with cNF (NCT02839720) and the topical MEKi NFX-179 (NCT04435665) to assess PD changes in adults with cNF. Further development of topical therapies will be of great benefit for patients to address this lifelong complication. For systemic therapy, MEKi can be administered orally, as in human patients, and selumetinib has shown PK properties that reflect what is seen in human patients (publication pending). Second, the size of the pig allows for eloquent studies of PK/PD in target tissues. For example, we have been able to successfully characterize the PK/PD of selumetinib in target tissues, including optic nerve and sciatic nerve. Thus, these types of studies can also inform investigators as too the ability of a drug to cross the blood-brain barrier. Additionally, the faithful development of NF1 features and the long lifespan of the pig permits the study of prophylactic interventions for NF1 pathology.
Many significant challenges exist in developing combination therapies, including determining whether drug combinations are safe and effective [85]. Swine models have great potential to address these challenges, as PK, PD, and toxicity can all be measured in a human-sized animal that shows the clinical features of NF1. On a final note, while the focus for NF1 therapy has been on kinase inhibitors, advancements in gene therapy for NF1 are underway, and the NF1 swine model will likely serve as a critical preclinical model, to establish appropriate delivery measures, as well as preclinical safety and efficacy prior to clinical trials [86].
Conclusions
Over the past three decades, advancements in the understanding of NF1 started with the discovery of the NF1 gene and have culminated with the first precision medicine approval for NF1-related pathology, specifically selumetinib for inoperable plexiform neurofibroma. The success of MEKi therapy in NF1 has been spurred by coordinated funding and research for this rare disease and development of relevant endpoints for clinical trial design. NF1 is a multisystem disease and tumor predisposition syndrome, and numerous challenges remain. Malignant neoplasms remain the leading cause of mortality in NF1. To continue improving outcomes, the recent development of sound preclinical models holds promise to translate targeted and combination approaches, taking advantage of advanced understanding of the molecular and immunologic landscape of these diseases.
Declarations
Conflict of Interest Robert Galvin, Nancy Ratner, and Christopher L. Moertel declare no conflict of interest. Adrienne L. Watson is an employee and shareholder of Recombinetics, Inc., and is supported by a grant from the Children’s Tumor Foundation. David A. Largaespada is the co-founder and co-owner of several biotechnology companies including NeoClone Biotechnologies, Inc., Discovery Genomics, Inc. (recently acquired by Immusoft, Inc.), B-MoGen Biotechnologies, Inc. (recently acquired by Bio-Techne Corporation), and Luminary Therapeutics, Inc. He holds equity in, serves as a Senior Scientific Advisor for and Board of Director member for Recombinetics, a genome editing company. The business of all these companies is unrelated to the contents of this article. He consults for Genentech, Inc., which funds some of his research. Sara Osum is supported by a grant from the Children’s Tumor Foundation.
Footnotes
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Friedman JM. Neurofibromatosis 1. 1998. Oct [Updated 2019]. In: Adam MP, Ardinger HH, Pagon RA, et al. , editors. GeneReviews. Seattle: University of Washington, Seattle; 1993–2020. [PubMed] [Google Scholar]
- 2.•.Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR, et al. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019;143(5):e20190660 [DOI] [PubMed] [Google Scholar]; An overview of evidence based recommendations for the evaluation and management of NF1-related pathology.
- 3.Kehrer-Sawatzki H, Mautner V-F, Cooper DN. Emerging genotype–phenotype relationships in patients with large NF1 deletions. Hum Genet. 2017;136(4):349–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.•.Markham A, Keam SJ. Selumetinib: first approval. Drugs. 2020;80(9):931–7 [DOI] [PubMed] [Google Scholar]; An overview of the PK and PD properties of Selumetinib.
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome Atlas. Cell. 2018;173(2):321–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frémin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011;5(6):623–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosser T Neurocutaneous disorders. Continuum (Minneap Minn). 2018;24(1, Child Neurology):96–129. [DOI] [PubMed] [Google Scholar]
- 10.The ICGC/TCGA pan-cancer analysis of whole genomes consortium. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stalnecker CA, Der CJ. RAS, wanted dead or alive: advances in targeting RAS mutant cancers. Sci Signal. 2020;13:eaay6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15(10):577–92. [DOI] [PubMed] [Google Scholar]
- 13.Tadini G, Legius E, Brems H, editors. Multidisciplinary approach to neurofibromatosis type 1. Cham: Springer International Publishing; 2020. [Google Scholar]
- 14.Blakeley JO, Wolkenstein P, Widemann BC, Lee J, Le LQ, Jackson R, et al. Creating a comprehensive research strategy for cutaneous neurofibromas. Neurology. 2018;91(2 Supplement 1): S1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.•.Gross AM, Singh G, Akshintala S, Baldwin A, Dombi E, Ukwuani S, et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro-Oncology. 2018;20(12):1643–51 [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of morbidities resulting from pNF in NF1.
- 16.•.Lai J-S, Jensen SE, Charrow J, Listernick R. Patient reported outcomes measurement information system and quality of life in neurological disorders measurement system to evaluate quality of life for children and adolescents with neurofibromatosis type 1 associated plexiform neurofibroma. J Pediatr. 2019;206:190–0 [DOI] [PubMed] [Google Scholar]; Quality of life effects of NF1-associated pNF.
- 17.•.Grit JL, Johnson BK, Dischinger PS, Essenburg CJ, Campbell S, Pollard K, et al. Distinctive epigenomic alterations in NF1-deficient cutaneous and plexiform neurofibromas drive differential MKK/P38 signaling. Genomics. 2019. 10.1101/833467 [DOI] [PMC free article] [PubMed] [Google Scholar]; Differential epigenomic alterations between NF1-associated cNF and pNF.
- 18.•.Costa ADA, Gutmann DH. Brain tumors in neurofibromatosis type 1. Neuro-Oncol Adv. 2020;2(Supplement_1):i85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]; An overview of NF1-related CNS tumors with a review of preclinical animal model development.
- 19.••.Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. 2020;37(4):569–583.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]; A genomic analysis of a large cohort of pediatric low-grade gliomas that reveals shared underlying biology of pediatric LGG and NF1-related LGG.
- 20.Santoro C, Picariello S, Palladino F, Spennato P, Melis D, Roth J, et al. Retrospective multicentric study on non-optic cns tumors in children and adolescents with neurofibromatosis type 1. Cancers. 2020;12(6):1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duong T, Sbidian E, Valeyrie-Allanore L, Vialette C, Ferkal S, Hadj-Rabia S, et al. Mortality associated with neurofibromatosis 1: a cohort study of 1895 patients in 1980–2006 in France. Orphanet J Rare Dis. 2011;6(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.•.Williams KB, Largaespada DA. New model systems and the development of targeted therapies for the treatment of neurofibromatosis type 1 associated malignant peripheral nerve sheath tumors. Genes. 2020;11(5):477. [DOI] [PMC free article] [PubMed] [Google Scholar]; A detailed overview of the limitations of MEKi monotherapy for NF1-associated malignancies. Additional targets to exploit and preclinical methodologies to translate therapy are explored.
- 23.Carrió M, Gel B, Terribas E, Zucchiatti AC, Moliné T, Rosas I, et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum Mutat. 2018;39(8):1112–25. [DOI] [PubMed] [Google Scholar]
- 24.Martin E, Flucke UE, Coert JH, van Noesel MM. Treatment of malignant peripheral nerve sheath tumors in pediatric NF1 disease. Childs Nerv Syst. 2020;36:2453–62. 10.1007/s00381-020-04687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.•.Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep. 2017;7(1):14992. [DOI] [PMC free article] [PubMed] [Google Scholar]; A molecular analysis of MPNST demonstrating the genomic complexity of this malignancy in comparison with pNF.
- 26.••.Packer RJ, Iavarone A, Jones DTW, Blakeley JO, Bouffet E, Fisher MJ, et al. Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro-Oncology. 2020;22(6):773–84 [DOI] [PMC free article] [PubMed] [Google Scholar]; A consensus understanding of the molecular and neuroimmunologic features of NF1-associated gliomas, including high-grade gliomas with recommendations for evaluation and management.
- 27.D’Angelo F, Ceccarelli M, Tala, Garofano L, Zhang J, Frattini V, et al. The molecular landscape of glioma in patients with neurofibromatosis 1. Nat Med. 2019;25(1):176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nix JS, Blakeley J, Rodriguez FJ. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol. 2020;139(4):625–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howell SJ, Hockenhull K, Salih Z, Evans DG. Increased risk of breast cancer in neurofibromatosis type 1: current insights. BCTT. 2017;9:531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap Y-S, Munusamy P, Lim C, Chan CHT, Prawira A, Loke S-Y, et al. Breast cancer in women with neurofibromatosis type 1 (NF1): a comprehensive case series with molecular insights into its aggressive phenotype. Breast Cancer Res Treat. 2018;171(3):719–35. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22(10):1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15(5):290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosa SL, Browder V, Bakker AC, Blakeley JO, Verma SK, Wong LM, et al. Funding community collaboration to develop effective therapies for neurofibromatosis type 1 tumors. EMBO Mol Med. 2020;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stevenson DA, Schill L, Schoyer L, Andresen BS, Bakker A, Bayrak-Toydemir P, et al. The Fourth International Symposium on Genetic Disorders of the Ras/MAPK pathway. Am J Med Genet. 2016;170(8):1959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakker AC, Rosa SL. Rethinking the nonprofit foundation: an emerging niche in the rare disease ecosystem. EMBO Mol Med. 2017;9(9):1179–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–83. [DOI] [PubMed] [Google Scholar]
- 37.Luke JJ, Ott PA, Shapiro GI. The biology and clinical development of MEK inhibitors for cancer. Drugs. 2014;74(18):2111–28. [DOI] [PubMed] [Google Scholar]
- 38.Wu P-K, Park J-I. MEK1/2 inhibitors: molecular activity and resistance mechanisms. Semin Oncol. 2015;42(6):849–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel P, Howgate E, Martin P, Carlile DJ, Aarons L, Zhou D. Population pharmacokinetics of the MEK inhibitor selumetinib and its active N-desmethyl metabolite: data from 10 phase I trials. Br J Clin Pharmacol. 2018;84(1):52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123(1):340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jousma E, Rizvi TA, Wu J, Janhofer D, Dombi E, Dunn RS, et al. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of neurofibromatosis type 1: MEK Inhibition in Neurofibroma. Pediatr Blood Cancer. 2015;62(10):1709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N Engl J Med. 2016;375(26):2550–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3(17004):1–17. [DOI] [PubMed] [Google Scholar]
- 44.Ferner RE, Thomas M, Mercer G, Williams V, Leschziner GD, Afridi SK, et al. Evaluation of quality of life in adults with neurofibromatosis 1 (NF1) using the Impact of NF1 on Quality Of Life (INF1-QOL) questionnaire. Health Qual Life Outcomes. 2017;15(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs. 2013;22(4):443–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.•.Gross AM, Widemann BC. Clinical trial design in neurofibromatosis type 1 as a model for other tumor predisposition syndromes. Neuro-Oncol Adv. 2020;2(Supplement_1):i134–40 [DOI] [PMC free article] [PubMed] [Google Scholar]; Unique considerations for clinical trial design in the NF1 populations.
- 47.•.Gross AM, Dombi E, Widemann BC. Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Childs Nerv Syst. 2020; Available from: 10.1007/s00381-020-04731-2. [DOI] [PubMed] [Google Scholar]; A review of development of MEKi for pNF treatment, including limitations and future directions
- 48.Widemann BC, Plotkin SR. Consensus for NF clinical trials: recommendations of the REiNS collaboration (Supplement II). Neurology. 2016;87(7 Supplement 1):S1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai W, Steinberg SM, Bredella MA, Basinsky G, Somarouthu B, Plotkin SR, et al. Volumetric MRI analysis of plexiform neurofibromas in neurofibromatosis type 1. Acad Radiol. 2018;25(2):144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viskochil D, Linscott LL. Volumetric MRI in neurofibromatosis type 1 (NF1) comes of age to help determine initiation and monitoring of targeted therapies for plexiform neurofibromas. Acad Radiol. 2018. Feb;25(2):141–3. [DOI] [PubMed] [Google Scholar]
- 51.Gripp KW, Schill L, Schoyer L, Stronach B, Bennett AM, Blaser S, et al. The sixth international RASopathies symposium: precision medicine—from promise to practice. Am J Med Genet. 2020;182(3):597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jakacki RI, Dombi E, Steinberg SM, Goldman S, Kieran MW, Ullrich NJ, et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro-Oncology. 2017;19(2):289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robertson KA, Nalepa G, Yang F-C, Bowers DC, Ho CY, Hutchins GD, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol. 2012;13(12):1218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.••.Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430–42 [DOI] [PMC free article] [PubMed] [Google Scholar]; Results of the phase II SPRINT trial leading to FDA approval of selumetinib for inoperable pediatric pNF.
- 55.•.Klesse LJ, Jordan JT, Radtke HB, Rosser T, Schorry E, Ullrich N, et al. The use of mek inhibitors in neurofibromatosis type 1–associated tumors and management of toxicities. Oncologists. 2020; 25(7). [DOI] [PMC free article] [PubMed] [Google Scholar]; MEKi therapy is associated with common toxicities. The management of these is reviewed by Klesse, et al
- 56.Foiadelli T, Naso M, Licari A, Orsini A, Magistrali M, Trabatti C, et al. Advanced pharmacological therapies for neurofibromatosis type 1-related tumors. Acta Bio Medica Atenei Parmensis. 2020;91(7-S):101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shofty B, Ben Sira L, Constantini S. Neurofibromatosis 1–associated optic pathway gliomas. Childs Nerv Syst. 2020;36: 2351–61. 10.1007/s00381-020-04697-1. [DOI] [PubMed] [Google Scholar]
- 58.Khatua S, Gutmann DH, Packer RJ. Neurofibromatosis type 1 and optic pathway glioma: Molecular interplay and therapeutic insights. Pediatr Blood Cancer. 2018;65(3):e26838. [DOI] [PubMed] [Google Scholar]
- 59.Lobbous B, Coffee F, Metrock C, et al. An update on neurofibromatosis type 1-associated gliomas. Cancers. 2020;12(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.•.Fisher MJ, Loguidice M, Gutmann DH, Listernick R, Ferner RE, Ullrich NJ, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro-Oncology. 2012;14(6):790–7 [DOI] [PMC free article] [PubMed] [Google Scholar]; A review of NF1-associated gliomas, including optic pathway gliomas, with an emphasis on the basic science of these tumors and therapeutic development.
- 61.Farazdaghi MK, Katowitz WR, Avery RA. Current treatment of optic nerve gliomas. Curr Opin Ophthalmol. 2019;30(5):356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bandopadhayay P, Bergthold G, London WB, Goumnerova LC. Morales La Madrid A, Marcus KJ, et al. Long-term outcome of 4, 040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database: PLGG SEER Long-Term Outcome. Pediatr Blood Cancer. 2014;61(7):1173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Blank P, Bandopadhayay P, Haas-Kogan D, Fouladi M, Fangusaro J. Management of pediatric low-grade glioma. Curr Opin Pediatr. 2019;31(1):21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.•.Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro-Oncology. 2017;19(8):1135–44 [DOI] [PMC free article] [PubMed] [Google Scholar]; Results of the phase I trial using selumetinib for recurrent/refractory pediatric LGG, including a cohort of NF1 patients.
- 65.••.Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011–22 [DOI] [PMC free article] [PubMed] [Google Scholar]; Results of the phase II trial evaluating selumetinib for recurrent/refractory pediatric LGG, including a cohort of NF1 patients.
- 66.•.Lobbous M, Korf BR. Therapeutic development in neurofibromatosis. In: Signorelli F, Messina R, editors. Neurofibromatosis - Current Trends and Future Directions. IntechOpen; 2020. Available from: https://www.intechopen.com/books/neurofibromatosis-current-trends-and-future-directions/therapeutic-development-in-neurofibromatosis. [Google Scholar]; A review of current therapeutic developments for NF1, including lists of clinical trials using diverse strategies for NF1-associated pathology
- 67.Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the children’s oncology group. J Clin Oncol. 2012;30(21):2641–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ater JL, Xia C, Mazewski CM, Booth TN, Freyer DR, Packer RJ, et al. Non-randomized comparison between neurofibromatosis type 1 (NF1) and non-NF1 children who received carboplatin and vincristine (CV) for progressive low grade glioma (LGG): a report from the children’s oncology group (COG). Cancer. 2016;122(12):1928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fangusaro J, Witt O, Hernáiz Driever P, Bag AK, de Blank P, Kadom N, et al. Response assessment in paediatric low-grade glioma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020;21(6):e305–16. [DOI] [PubMed] [Google Scholar]
- 70.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.••.Brosseau J-P, Liao C-P, Le LQ. Translating current basic research into future therapies for neurofibromatosis type 1. Br J Cancer. 2020;123(2):178–86 [DOI] [PMC free article] [PubMed] [Google Scholar]; A review of current therapeutic targets under investigation, including MEKi and immunotherapy, and an additional focus on potential avenues for novel therapeutics in NF1 based on advances in the basic science understanding of NF1-associated tumors.
- 72.••.Martin E, Lamba N, Flucke UE, Verhoef C, Coert JH, Versleijen-Jonkers YMH, et al. Non-cytotoxic systemic treatment in malignant peripheral nerve sheath tumors (MPNST): a systematic review from bench to bedside. Crit Rev Oncol Hematol. 2019;138:223–32 [DOI] [PubMed] [Google Scholar]; An extensive review of preclinical in vivo studies and current clinical trials utilizing non-cytotoxic monotherapy and combination therapy approaches to address MPNST.
- 73.Karmakar S, Reilly KM. The role of the immune system in neurofibromatosis type 1-associated nervous system tumors. CNS Oncol. 2017;6(1):45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.•.Kumar S, Principe DR, Singh SK, Viswakarma N, Sondarva G, Rana B, et al. Mitogen-activated protein kinase inhibitors and t-cell-dependent immunotherapy in cancer. Pharmaceuticals. 2020;13(1):–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; An overview of the MAPK signaling network and its interaction with the immune system when altered, with implications for immunotherapy.
- 75.Wang S, Liechty B, Patel S, Weber JS, Hollmann TJ, Snuderl M, et al. Programmed death ligand 1 expression and tumor infiltrating lymphocytes in neurofibromatosis type 1 and 2 associated tumors. J Neuro-Oncol. 2018;138(1):183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Antoszczyk S, Rabkin SD. Prospects and progress of oncolytic viruses for treating peripheral nerve sheath tumors. Expert Opin Orphan Drugs. 2016;4(2):129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. [DOI] [PubMed] [Google Scholar]
- 78.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, et al. Plexiform and Dermal Neurofibromas and Pigmentation Are Caused by Nf1 Loss in Desert Hedgehog-Expressing Cells. Cancer Cell. 2008;13(2):105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carrió M, Mazuelas H, Richaud-Patin Y, Gel B, Terribas E, Rosas I, et al. Reprogramming captures the genetic and tumorigenic properties of neurofibromatosis type 1 plexiform neurofibromas. Stem Cell Rep. 2019;12(2):411–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo J, Grovola MR, Xie H, Coggins GE, Duggan P, Hasan R, et al. Comprehensive pharmacological profiling of neurofibromatosis cell lines. Am J Cancer Res. 2017;7(4):923–34. [PMC free article] [PubMed] [Google Scholar]
- 81.Kraniak JM, Chalasani A, Wallace MR, Mattingly RR. Development of 3D culture models of plexiform neurofibroma and initial application for phenotypic characterization and drug screening. Exp Neurol. 2018;299:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ricker CA, Pan Y, Gutmann DH, Keller C. Challenges in drug discovery for neurofibromatosis type 1-associated low-grade glioma. Front Oncol. 2016;6. 10.3389/fonc.2016.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watson AL, Carlson DF, Largaespada DA, Hackett PB, Fahrenkrug SC. Engineered swine models of cancer. Front Genet. 2016;7(78). 10.3389/fgene.2016.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.••.Isakson SH, Rizzardi AE, Coutts AW, Carlson DF, Kirstein MN, Fisher J, et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun Biol. 2018;1(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]; Report on the development of a genetically engineered swine model that recapitulates key molecular and phenotypic features of NF1.
- 85.Lopez JS, Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat Rev Clin Oncol. 2017;14(1):57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leier A, Bedwell DM, Chen AT, Dickson G, Keeling KM, Kesterson RA, et al. Mutation-directed therapeutics for neurofibromatosis type I. Mol Ther–Nucleic Acids. 2020;20:739–53. [DOI] [PMC free article] [PubMed] [Google Scholar]