

Abstract
Experience sculpts brain structure and function. Activity-dependent modulation of the myelinated infrastructure of the nervous system has emerged as a dimension of adaptive change during childhood development and in adulthood. Myelination is a richly dynamic process, with neuronal activity regulating oligodendrocyte precursor cell proliferation, oligodendrogenesis and myelin structural changes in some axonal subtypes and in some regions of the nervous system. This myelin plasticity and consequent changes to conduction velocity and circuit dynamics can powerfully influence neurological functions, including learning and memory. Conversely, disruption of the mechanisms mediating adaptive myelination can contribute to cognitive impairment. The robust effects of neuronal activity on normal oligodendroglial precursor cells, a putative cellular origin for many forms of glioma, indicates that dysregulated or ‘hijacked’ mechanisms of myelin plasticity could similarly promote growth in this devastating group of brain cancers. Indeed, neuronal activity promotes the pathogenesis of many forms of glioma in preclinical models through activity-regulated paracrine factors and direct neuron-to-glioma synapses. This synaptic integration of glioma into neural circuits is central to tumour growth and invasion. Thus, not only do neuron–oligodendroglial interactions modulate neural circuit structure and function in the healthy brain, but neuron–glioma interactions also have important roles in the pathogenesis of glial malignancies.
Introduction
Neuron–glial interactions are central to neural circuit form and function. Astrocytes promote synapse formation during developmental circuit establishment1–3, microglia prune synapses in an activity-dependent manner to refine circuits4,5 and oligodendrocytes form myelin that both provides metabolic support for axons6 and regulates the speed of action potential conduction,7 and thus tunes neural circuit dynamics. Such crosstalk between neurons and glial cells is also central to healthy circuit development, function and adaptive change (see ref. 8 for a review on neuron–glial interactions shaping neural circuit development and function). Thus, disruption or dysregulation of these neuron–glial interactions may contribute importantly to neurological and psychiatric dysfunction in a range of disease states9,10. Similarly, increasing evidence supports the principle that subversion of neuron–glial interactions in glial malignancies are central to brain cancer progression11,12.
In this Review, we discuss neuron–glial interactions that contribute to adaptive brain function, that are disrupted in cognitive disease and that are hijacked in brain cancer. We focus on neuron–oligodendroglial interactions, with consideration of microglia and astrocytes in that context (see refs. 13–16 for dedicated reviews on the rich and complex biology of microglia and astrocytes). Here, we first discuss healthy neuron–oligodendroglial interactions underpinning myelin plasticity, followed by a discussion of how glial malignancies subvert and repurpose these powerful neuron–glial interactions to drive glioma pathophysiology.
Myelin plasticity and adaptive brain function
Myelin structure and opportunities for adaptive tuning
Myelin, concentric wraps of glial membrane that form compact lipid-rich lamellae around axons, is formed by oligodendrocytes in the CNS and by Schwann cells in the peripheral nervous system (for an excellent review on the history of discoveries in myelin biology, see ref. 17). Segments of myelin, called internodes, form between nodes of Ranvier, where voltage-gated sodium channels are concentrated to facilitate action potential propagation7. Ensheathment of axons by myelin internodes serves two major functions. First, myelin provides crucial metabolic support to axons6,18,19. Second, myelin robustly influences the action potential conduction velocity by decreasing transverse capacitance and increasing transverse resistance of the axonal membrane. This insulates the myelinated segments to enable saltatory conduction of action potentials from node to node7. Myelinated axon conduction velocity is 50-fold to 100-fold faster than that of unmyelinated axons20. Parameters of myelin structure that influence conduction velocity include the thickness of the myelin sheath relative to the axon diameter, internode length and internode spacing7,21,22.
In the CNS, myelin development is a remarkably protracted postnatal process, with human CNS myelination extending into the fourth decade of life23–25. Myelination follows a predictable chronological and topographical developmental pattern. In some regions of the nervous system, such as the optic nerves and the spinal cord, myelin development completes relatively early in postnatal development23,24 and myelination is fairly complete after development26,27, with myelination of nearly all axons in the optic nerve and myelinated spinal cord tracts, which have myelin profiles that achieve more ‘optimal’ parameters for maximal conduction velocity7,21,22. In other regions of the nervous system, such as neocortex and intercortical association fibres, myelin development extends over a longer developmental period (into the late 20s to early 30s for humans)23–25. In neocortex and intercortical projections, myelination remains incomplete and axons exhibit heterogeneous myelin profiles after this protracted developmental period. For example, in the corpus callosum, a major white matter tract containing interhemispheric projections, as many as 30% of axons are unmyelinated and many myelinated axons exhibit thinner myelin sheaths than the ‘ideal’ sheath thickness to axon calibre ratio that facilitates maximal conduction velocity28. In the neocortex of both primates and rodents, myelin profiles are variable and frequently do not exhibit geometric parameters predicted to enable maximal conduction velocity29,30. Furthermore, a study applying cutting-edge techniques in electron microscopy in the somatosensory cortex of a young adult mouse revealed that individual axons of neocortical neurons exhibit variable myelination, with regions of intermittent myelination along the axon length30. These observations may reflect delayed myelin development in that neocortical brain region or could underscore the potential to adaptively tune the function of cortical and intercortical circuits through new myelination or myelin remodelling.
Oligodendroglial lineage dynamics over the life span
Oligodendrocytes are generated by oligodendrocyte precursor cells (OPCs) that emerge and expand in the late prenatal and postnatal periods31,32 (see refs. 33,34 for excellent reviews discussing OPC developmental origins in detail). In the adult nervous system, OPCs persist in a regularly arranged, tiled pattern, maintaining a consistent cellular population density35 that accounts for 5–10% of all cells in the CNS36. OPCs are progenitors that can divide asymmetrically to self-renew while producing a daughter oligodendrocyte, divide symmetrically to produce two daughter oligodendrocytes or directly differentiate without proliferation35. In addition to generating myelinating oligodendrocytes, OPCs also have functions unrelated to myelin, including pruning axons37 and synapses38; thus, OPCs join microglia4,5 and astrocytes39 in contributing to synaptic pruning and neural circuit refinement.
OPCs continue to generate new oligodendrocytes throughout the life span of healthy rodents40. Genetic fate-mapping and intravital imaging studies have demonstrated that oligodendrocytes are long-lived, so the ongoing production of new cells cannot be explained by a homeostatic requirement to replace dying oligodendrocytes41–43. In the rodent neocortex and corpus callosum, but not the spinal cord or optic nerve, new oligodendrocytes and new myelin continue to accumulate throughout adulthood41–43. Similarly in non-human primates, oligodendrocytes accumulate in the cortex with age44,45.
In the human brain, OPCs represent the major population of proliferating cells in healthy adult cortex and white matter46. A carbon-dating study that leveraged the carbon-14 by-product of mid-twentieth-century nuclear testing to date human brain cells at autopsy found evidence of substantial myelin turnover in the corpus callosum, but minimal new cell production after childhood47. One interpretation of these carbon-dating findings could be that myelin remodelling by existing oligodendrocytes may occur in the adult human corpus callosum more than new oligodendrocyte production, although this interpretation requires further evidence. This cellular carbon-dating study also demonstrated substantially (10-fold) higher ongoing rates of new oligodendrocyte generation in the adult human prefrontal cortex than in the corpus callosum47. These intriguing human carbon-dating findings have yet to be confirmed using alternative methodologies, but the finding of ongoing human cortical oligodendrogenesis is consistent with histological studies of human neocortex that reveal accumulation of cortical myelin over the life span23.
Activity-regulated myelination
Neuronal activity has a central role in shaping the form and function of neural circuitry throughout the life span (see ref. 8 for a review). Thus, it is not surprising that activity-regulated development and plasticity extend to myelin-forming cells as well. Indeed, a newly appreciated dimension along which neuronal activity modulates structure, and thus function, has emerged in the myelinated infrastructure of the brain. In 1993 Ben Barres first introduced the idea that neuronal activity may influence myelin-forming cell behaviour48, and the well-described but still functionally enigmatic discovery that OPCs receive both glutamatergic and GABAergic synaptic inputs from neurons49–51 stoked interest in the role neurons may have in myelin development and plasticity. Several elegant in vitro studies52,53 and fascinating studies associating experience with changes in myelination in rodents54,55 and white matter structure in non-human primates56 and humans57–59 subsequently supported this concept. However, the extent to which neuronal activity influences myelin-forming cells and modulates myelin structure in the healthy brain during development or adulthood remained a point of controversy, in part because activity-independent modes of myelination clearly also exist60–62.
Robust evidence resulting from the application of newer tools in neuroscience, such as optogenetics and chemogenetics, to this pressing question in glial biology has provided direct evidence that neuronal activity can regulate and modulate myelination, at least in some contexts (Fig. 1). Optogenetic stimulation of premotor cortical projection neuron activity in awake behaving mice resulted in robust proliferation and subsequent differentiation of OPCs and less differentiated pre-OPCs in both juvenile and adult mice, with activity-regulated increase in myelination within the stimulated circuit63. The premotor cortex, a higher-order associative cortical region involved in motor planning, projects down the corticospinal tract (corticofugal projections) and across the corpus callosum (cortico-callosal projections); activity-regulated OPC proliferation was specifically observed in premotor cortico-callosal projections63. This suggests a difference in the oligodendroglial response to activity of intercortical association neurons and neurons projecting to the spinal cord, and underscores potential heterogeneity of neuron–glial interactions in different neuronal subtypes and different regions of the nervous system. Furthermore, activity-regulated, circuit-specific myelination of cortico-callosal projection neurons resulted in positive alterations to motor function that depend on the generation of new oligodendrocytes and the associated activity-regulated changes in myelin structure suited for tuning conduction velocity63. Similar motor circuit OPC proliferation and oligodendrogenesis resulted following a complex wheel motor learning task in mice64, and histological markers of myelination increased following a single-pellet reaching task in rats65. Chemogenetic or whisker stimulation-mediated activation of the somatosensory cortex similarly demonstrated activity-regulated oligodendrogenesis and myelination of the stimulated axons42,66. Activity-regulated myelin changes in the adult rodent brain include both de novo formation of new internodes on previously unmyelinated axons or axon segments30,42,66,67 and remodelling of myelin internodes by existing oligodendrocytes68,69 (Fig. 1).
Fig. 1 |. Myelin plasticity: evidence and implications from rodent models.
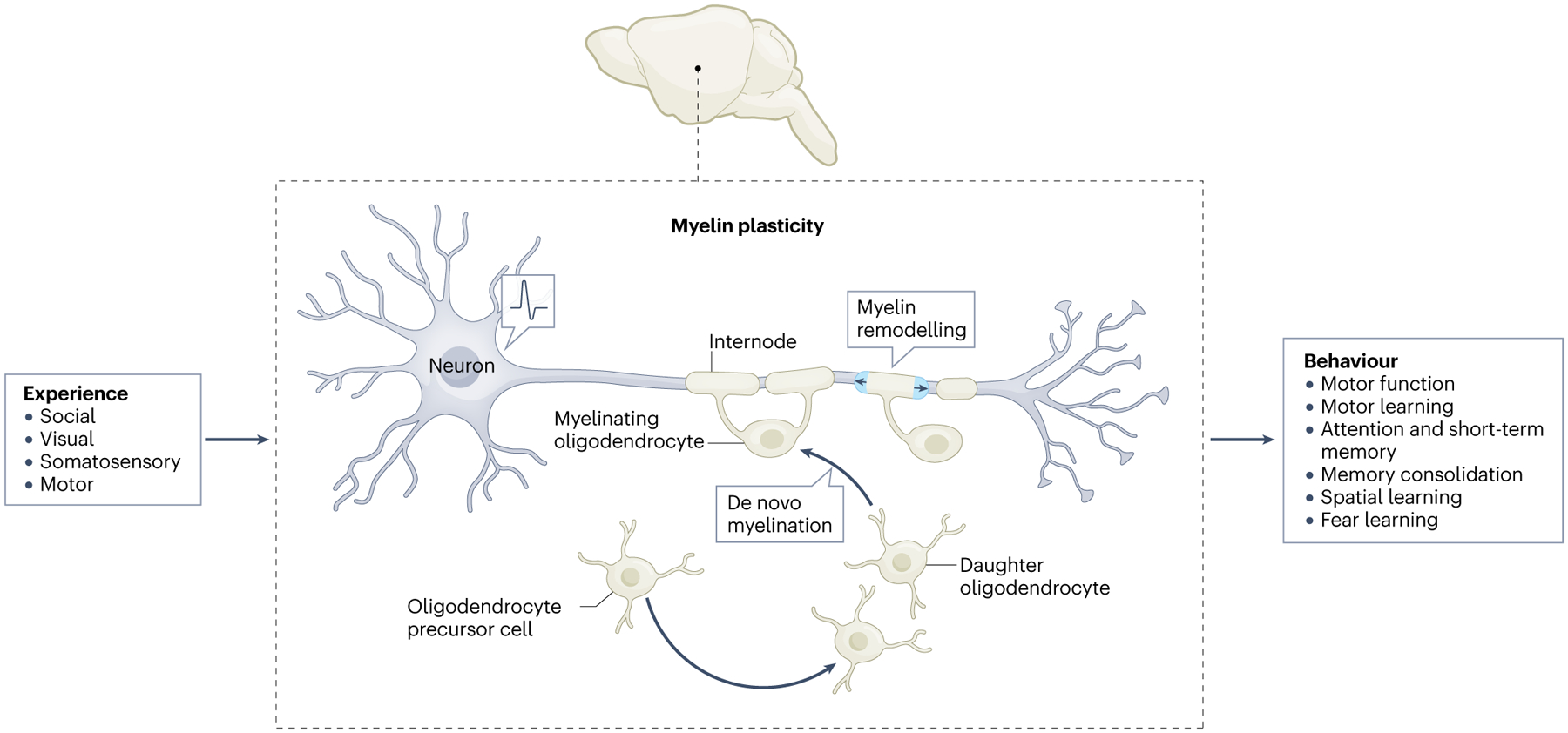
Neuronal activity-regulated changes in myelin are evident in rodent models with direct manipulation of neuronal activity using optogenetics and chemogenetics, and evident with changes in sensory, motor and social experience. Myelin plasticity contributes to a range of neurological functions, including spatial, fear and motor learning.
Mechanisms mediating myelin plasticity
Numerous activity-regulated molecular mechanisms have been hypothesized to mediate the influence of neuronal activity on myelin-forming cells. Moreover, each mechanism may have a role in different contexts defined by neuronal subtype heterogeneity63, OPC heterogeneity70,71, the OPC state72 and the frequency or pattern of neuronal firing52.
One recent study illustrated that activity-regulated brain-derived neurotrophic factor (BDNF) signalling via the tyrosine receptor kinase B (TrkB) receptor on OPCs is a required mechanistic component of myelin plasticity in cortical projection neurons9. Loss of the TrkB receptor specifically in OPCs in the juvenile period or in adulthood completely abrogated OPC proliferation, oligodendrogenesis and myelin changes in response to cortical projection neuronal activity. However, this represents only part of what is likely to be a more complex mechanism that remains to be fully elucidated. Whether BDNF has a central role in myelin plasticity in other circuits and other neuronal types remains to be evaluated; it may be that just as neurons are heterogeneous and oligodendroglial cells are heterogeneous70,71,73, the mechanisms that underlie myelin plasticity are similarly varied.
During development in the zebrafish, axonal activity and vesicular release can influence axon selection for successful myelination74,75; it remains to be determined which molecules packaged in vesicles mediate axon selection. In the juvenile superficial medial prefrontal cortex of mice, social isolation regulates the number of internodes generated by an oligodendrocyte through activity-regulated secretion of endothelin from cerebral vasculature that signals through oligodendrocyte endothelin B receptors68. Elucidating additional aspects of the molecular mechanisms mediating neuron–oligodendroglial interactions — and potential circuit or region-specific expression of such mechanisms — represents an area of intense investigation in the field.
Adaptive myelination and circuit function
Activity-regulated myelin changes and consequent alterations in conduction velocity modulate circuit dynamics to promote coordinated circuit activity67. Increasing evidence suggests that these adaptive myelin changes support healthy cognition. Conditional, inducible genetic mouse models showed that disruption of activity-dependent oligodendrogenesis and/or myelination impairs attention and short-term memory function9, spatial memory consolidation67, fear memory consolidation76 and motor learning64 (Fig. 1). It should be noted that several of these studies probing the functional role of experience and activity-regulated oligodendrogenesis utilized conditional genetic deletion of the transcription factor Myrf, which has roles in cell types other than the oligodendroglial lineage77, and which also could influence myelin-independent functions of OPCs as discussed above.
Computational and experimental evidence supports the principle that myelin plasticity tunes circuit dynamics to promote coordinated circuit function, such as promoting synchronous oscillations between nodes in a neural network67,78,79. For adaptive myelin changes to confer the relatively subtle alterations in conduction velocity that promote coordinated circuit function such as oscillatory synchrony or to enable spike timing-dependent synaptic plasticity, circuit-wide information must be integrated by the oligodendroglial lineage cells. How this occurs with the required precision remains a fascinating mystery. Synapses between neurons and OPCs49,50 have been well characterized, but their function remains incompletely understood. One hypothesis is that these enigmatic neuron–OPC synapses may have an important role for integrating circuit-level information. Mapping of OPC connectivity using monosynaptic rabies-based tracing illustrated brain-wide inputs from the circuits in which the mapped OPCs reside, including both cortical and thalamic projections80. How these inputs to neuron–OPC synapses are integrated and may contribute to adaptive changes in myelin remains to be determined.
Collectively, these insights support the emerging concept that myelin plasticity can contribute to structural changes sculpting adaptive development and ongoing neural plasticity in the adult brain. Given the importance of myelin plasticity to cognitive functions such as attention and learning and memory, it makes sense that loss of myelin plasticity may contribute to disorders of cognition (Box 1). Conversely, activity-regulated myelination may become maladaptive in diseases characterized by abnormally increased circuit activity or abnormal patterns of activity10 (see ref. 81 for a review on maladaptive myelination).
Box 1. Loss of myelin plasticity and cognitive impairment.
Just as myelin plasticity promotes healthy cognitive function, disruption of activity-regulated neuron–oligodendroglial interactions may contribute to cognitive diseases. One illustrative example of a neurocognitive disease in which disruption of myelin plasticity appears to have an important role is the syndrome of cognitive dysfunction that frequently follows cancer chemotherapy, characterized by impaired attention, memory, speed of information processing and multi-tasking. Individuals suffering from chemotherapy-related cognitive impairment, colloquially known as ‘chemobrain’, frequently do not exhibit overt structural damage. However, advanced neuroimaging techniques reveal subtle white matter abnormalities164,165. Histological examination of human post-mortem frontal lobe samples from subjects with previous chemotherapy exposure demonstrates depletion of oligodendroglial lineage cells in subcortical white matter, but not in cortical grey matter. Depletion of oligodendrocyte precursor cells (OPCs) was particularly severe following exposure to high-dose methotrexate, a drug that is especially associated with long-term cognitive impairment166.
A mouse model of high-dose methotrexate exposure demonstrates a similar depletion of white matter OPCs and mature oligodendrocytes166. These changes in oligodendroglial lineage cells are attributable to a perturbation of the gliogenic microenvironment, as chemotherapy-naïve OPCs transplanted into the previously methotrexate-treated brain exhibit similar dysregulation of oligodendroglial lineage populations. This microenvironmental disruption is associated with prominent microglial reactivity, specifically in white matter. Further work demonstrated that methotrexate directly induces microglial reactivity, and that this reactivity persists long after drug exposure. Methotrexate-induced microglial reactivity, in turn, induces neurotoxic astrocyte reactivity, and together these reactive glia cause the observed dysregulation of the oligodendroglial lineage, impaired myelin homeostasis (decreased myelinated axons and thinner myelin sheaths) and myelin plasticity failure9,166,167. The failure of activity-regulated OPC proliferation, oligodendrogenesis and myelination observed in mice previously treated with methotrexate can be understood in the context of a known mechanism required for adaptive myelin change: neuronal brain-derived neurotrophic factor (BDNF) expression is starkly decreased after methotrexate9. Underscoring the central role of microglial reactivity in this multicellular dysregulation, microglial depletion after chemotherapy restores neuronal BDNF expression, normalizes the myelin phenotype and rescues cognitive function in this mouse model of high-dose methotrexate exposure9,166. To establish the relative contribution of the observed myelin plasticity failure to the chemotherapy-induced deficits in cognitive function, the effects of a specific small molecule tyrosine receptor kinase B (TrkB) agonist were tested after methotrexate exposure in a genetic mouse model with or without inducible, OPC-specific deletion of TrkB. The TrkB agonist rescued myelination and cognitive performance even without microglial depletion; importantly, cognitive performance was only rescued in mice with intact OPC expression of TrkB9.
Cognitive symptoms that frequently follow COVID-19 closely mirror those that occur after cancer therapies, and persistent ‘brain fog’ is an important and debilitating component of long COVID-19 (see ref. 168 for a review). Preclinical work has uncovered similar patterns of persistent white matter-selective microglial reactivity, decreased OPC and oligodendrocyte numbers and impaired myelin homeostasis (with decreased myelinated axons in subcortical white matter) after even mild respiratory-restricted COVID-19 (ref. 167). Whether myelin plasticity is similarly impaired after COVID-19 as it is after methotrexate chemotherapy9 and whether similar therapeutic strategies may rescue cognitive function after COVID-19 remain to be determined in future work.
Malignant myelin plasticity: neuron–glioma interactions drive brain tumour progression
Oligodendroglial origins for many gliomas
Malignant gliomas are a leading cause of brain tumour-related death in both children and adults82. Over the past decade, advances in understanding the molecular biology of these brain tumours refined the classification of glioma types83. Gliomas comprise a group of clinically and molecularly distinct entities, and within each tumour exist heterogeneous populations of malignant cells that exhibit transcriptional resemblances to OPCs, oligodendrocytes, astrocytes and — in some tumour types — neural stem or precursor cells84–87 (Table 1). It is thought that the glioma stem-like cell population is responsible for driving tumour initiation, evolution and resistance to therapy87–89. The identity of these stem-like cells can differ between glioma types, with some tumours harbouring stem-like cell populations that resemble neural stem cells and others featuring a more OPC-like population of stem-like cells84–87 (Table 1).
Table 1 |.
Glioma cell state heterogeneity drives distinct mechanisms of network communication which informs potential therapeutic strategies
| Glioma type | Cell state resemblance | Network interactions | Molecular mechanisms and therapeutic targets | Clinically available drugs that impact glioma preclinical models |
|---|---|---|---|---|
| Paediatric low-grade | OPC, astrocyte161 | Paracrineb, neuronal hyperexcitabilityb | NLGN3b, HCNb | ADAM10 inhibitorsb, lamotrigineb |
| IDH and H3 wild-type high-grade glioma | OPC, astrocyte, MES, NPC87 | Neuron–glioma synapse, tumour microtube, neuronal hyperexcitability, paracrine, hub cells (also called pacemaker or oscillatory cells) | AMPAR, potassium channels, CX43, NLGN3, KCa, TSP1, IGF1 | Perampanel, meclofenamate, senicapoc, gabapentin, ADAM10 inhibitors |
| IDH-mutant astrocytoma | NPC and OPC, astrocyte, oligodendrocyte85 | Neuron–glioma synapse, tumour microtube, neuronal hyperexcitability | AMPAR, CX43 | Perampanelc, meclofenamatec |
| IDH-mutant oligodendroglioma | NPC and OPC, astrocyte, oligodendrocyte85,162 | Paracrine, neuronal hyperexcitability | NLGN3 | ADAM10 inhibitorsc |
| H3 G34R/V-mutant DHG | INPC, NPC, astrocyte152 | Unknown | Unknown | Unknown |
| H3 K27M-mutant DMG | OPC, astrocyte, oligodendrocyte, MESa,86,163 | Neuron–glioma synapse, tumour microtube, neuronal hyperexcitability, paracrine, hub cells | AMPAR, potassium channels, CX43, NLGN3 | Perampanel, meclofenamate, ADAM10 inhibitors, senicapocc, gabapentinc |
ADAM10, a disintegrin and metalloproteinase domain-containing protein 10; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CX43, connexin 43; DHG, diffuse hemispheric glioma; DMG, diffuse midline glioma; HCN, hyperpolarization-activated cyclic nucleotide-gated channel; IGF1, insulin-like growth factor 1; INPC, interneuron progenitor cell; MES, mesenchymal cell; NLGN3, neuroligin 3; KCa, calcium-activated potassium channel; NPC, neural precursor cell; OPC, oligodendrocyte precursor cell; TSP1, thrombospondin 1.
Chiefly in older patients.
Studied in neurofibromatosis type 1 (NF1)-associated low-grade optic gliomas.
Drug mechanism has been identified but the drugs have not yet been tested in that specific glioma type.
Gliomas are classified based on their clinical grading and location within the brain. Low-grade gliomas comprise a spectrum of histologic entities, which in adults (but not in children) often progress to higher grade malignancy90,91. High-grade gliomas may arise in the cerebral hemispheres, such as glioblastoma, or in the midline of the CNS, such as diffuse midline gliomas (DMGs) occurring in the thalamus, brainstem — often referred to as diffuse intrinsic pontine glioma (DIPG) — and spinal cord. Although histologically similar, adult and paediatric gliomas have distinct biological profiles and childhood tumours arise with a striking spatiotemporal pattern of incidence and region-specific molecular signatures92. Gliomas arising during childhood tend to occur within midline structures, such as in the brainstem and thalamus, whereas in young and older adults, gliomas chiefly arise in cerebral cortical and subcortical locations.
This striking spatiotemporal pattern of glioma incidence maps well onto the chronological and topographical patterns of developmental myelination. For instance, a discrete wave of pontine myelination in mid-childhood coincides with the peak incidence of pontine DMG at age 6–8 years, and a discrete period of neocortical myelination in adolescence and young adulthood coincides with the timing of young adult hemispheric glioblastomas23,93,94. Concordantly, numerous studies have implicated precursor cells of the oligodendroglial lineage as the cell of origin for many forms of glioma86,95–100. Moreover, the regions in which these tumours typically arise within the adult brain exhibit ongoing oligodendroglial and myelin plasticity (see ref. 101 for an in-depth review).
Neuronal activity promotes glioma initiation, growth and progression
The observation that stem-like cell populations of many forms of glioma transcriptionally resemble OPCs inspired the hypothesis that malignant cells may possess a similar proliferative response to actively firing neurons within the tumour microenvironment. A foundational study applying techniques of modern neuroscience to cancer biology demonstrated that optogenetic stimulation of premotor cortical projection neurons in mice induced a circuit-specific proliferative response of patient-derived paediatric cortical high-grade glioma xenografted to their premotor cortex11. Optogenetically elevating neuronal activity over the course of a week resulted in increased glioma growth and increased tumour burden compared with identically manipulated littermate control mice lacking the light-sensitive opsin11. Further illustrating the effects of neuronal activity on high-grade glioma proliferation, co-culture of glioma cells with neurons exerted a robust, several-fold increase in the glioma proliferation rate12. Concordantly, reducing neuronal activity using a general anaesthetic inhibited patient-derived glioblastoma growth and invasion in mice102.
The effects of neuronal activity on glioma are not restricted to high-grade gliomas such as glioblastoma. A recent study highlighted the effects of optic nerve activity on the initiation, growth and maintenance of optic pathway low-grade glioma103, which frequently arises in young children with neurofibromatosis type 1 (NF1) tumour predisposition syndrome. Genetically engineered mouse models of NF1-associated optic pathway glioma predictably exhibit development of optic nerve tumours at 9 weeks of age104. Optogenetic stimulation of retinal ganglion cell axonal activity in the optic nerve induced an increase in the growth of optic nerve pathway gliomas in NF1-associated optic pathway glioma mice compared with identically manipulated, but not stimulated, littermate controls103. Furthermore, decreasing visual experience and consequent optic nerve activity by rearing these mice in complete darkness prior to tumour development (beginning at 6 weeks of age) abrogated the development of optic pathway gliomas in comparison with littermate control mice, which all developed an optic glioma when raised in normal light cycles103. Reducing optic nerve activity by rearing mice in complete darkness at later ages — at the time of initiation (beginning at 9 weeks of age) or after optic glioma development (beginning at 12 weeks of age) — resulted in fewer and smaller tumours at 16 weeks, demonstrating that optic nerve activity is important for not only initiation and growth but also tumour maintenance103. The reduction in tumour incidence and size when optic nerve activity is reduced after the time of tumour initiation in this model (9 weeks) most likely reflects a role for neuronal activity in glioma cell survival that should be further studied in future work. Subsequent studies also identified a role for peripheral nerve activity in peripheral nerve neurofibroma tumours in the NF1 tumour predisposition syndrome, and identified hyperexcitability in NF1 heterozygous neurons due to NF1-regulated hyperpolarization-activated cyclic nucleotide-gated (HCN) channel function as a mechanism driving both optic nerve glioma and peripheral nerve neurofibromas105. This illustrates that NF1 mutation or loss not only affects the cells that form tumours but also alters the behaviour of other cell types in the tumour microenvironment, such as neurons that contribute to the predisposition for tumour formation105. The role of neuronal activity in the initiation, growth and maintenance of molecularly and clinically distinct subtypes of low-grade gliomas remains to be determined.
Similarly to the findings in the NF1-associated optic pathway low-grade glioma mouse model, olfactory sensory experience and olfactory neuronal activity regulated tumour incidence and growth in a genetically engineered mouse model of olfactory bulb high-grade glioma106. In that mouse model, occluding the nares mechanically or reducing olfactory neuronal activity chemogenetically reduced the growth of high-grade gliomas of the olfactory bulb106.
Paracrine neuron–glioma interactions
Activity-regulated factors secreted from murine cortical slices induce a proliferative effect across multiple distinct subgroups of patient-derived high-grade glioma cultures (including DIPG, adult and paediatric glioblastoma and anaplastic oligodendroglioma)11. Similarly, NF1-associated low-grade glioma proliferate in response to activity-regulated factors secreted from retina–optic nerve explants103. The effects of cortical or retina–optic nerve explants on glioma cell proliferation required explant neuronal activity, as it was lost when blocked using the voltage-gated sodium channel blocker tetrodotoxin11,103. Furthermore, proteomic analyses of the activity-regulated secretome, followed by necessity and sufficiency testing, revealed the presence of activity-regulated glioma mitogens. BDNF was among these mitogens11, concordant with the role that BDNF has in activity-regulated proliferation of healthy OPCs9. In addition, insulin-like growth factor (IGF) mediated activity-regulated neuron–glioma paracrine signalling important for olfactory bulb gliomagenesis in the olfactory bulb glioma study discussed above106. Activity-regulated secreted factors also promote glioma cell migration and invasion, including a cortico-callosal projection neuronal activity-regulated invasion mechanism in adult glioblastoma that requires glioma expression of the axon guidance molecule semaphorin 4F (ref. 107). Together, these studies indicate that different activity-regulated paracrine factors may have distinctively important roles in different brain regions or neural circuits, and future work should seek to identify the region, circuit and neuronal subtype-specific mechanisms regulating brain cancer pathophysiology.
A shed form of the synaptic adhesion protein, neuroligin 3 (NLGN3) was unexpectedly identified in the cortical and retinal explant studies described above as an important activity-regulated paracrine growth factor, with 10 out of 11 diverse glioma models exhibiting a proliferative response to NLGN3 (refs. 11,103). In the healthy brain, the neuroligin protein family has a critical role in synapse form and function as the postsynaptic binding partner for neurexins (see ref. 108 for a review) and NLGN3 contributes to the function of both excitatory and inhibitory synapses109,110. As a synaptic adhesion molecule, it was not previously known that NLGN3 had a pro-proliferative role in any context and even more unexpected was a requirement of NLGN3 for glioma growth, as discussed below.
To test the relative contribution of NLGN3 to glioma progression, patient-derived gliomas were xenografted into the brains of wild-type or Nlgn3 knockout mice. Surprisingly, glioma xenografts failed to grow in brains with an NLGN3-deficient microenvironment111. This unexpected dependency on microenvironmental NLGN3 was identified in patient-derived xenograft models of DIPG, paediatric glioblastoma and adult glioblastoma, but did not extend to a patient-derived model of breast cancer brain metastasis, suggesting specificity for gliomas111. However, over time, a subset of xenografted mice within each experimental cohort began to exhibit tumour growth in the NLGN3-deficient brain111. The mechanisms mediating circumvention of this apparent dependency on microenvironmental NLGN3 in the brain remain to be determined and represent an area of active research.
NLGN3 is also key to optic nerve activity-regulated optic glioma pathophysiology, and deletion of Nlgn3 prevented optic glioma development in NF1-associated optic pathway glioma mice with normal visual experience103. Supporting an important role for NLGN3 in glioblastoma pathobiology, a retrospective analysis of bulk RNA sequencing data in The Cancer Genome Atlas database revealed that tumour Nlgn3 expression levels inversely correlate with patient overall survival11. Stratifying tumours by molecular subtype (proneural, neural, classical and mesenchymal) revealed the lowest Nlgn3 expression levels in tumours whose transcriptional profile is dominated by mesenchymal tumour cell states11. This point frames the question about which glioma cellular states are enriched for particular neuron–glioma interaction mechanisms (Box 2).
Box 2. Heterogeneous cellular substates in glioma are enriched for neuron–glioma interaction mechanisms.
The introduction of next-generation sequencing technology has elucidated important principles of intertumoural and intratumoural cellular heterogeneity in gliomas. A landmark paper in 2010 examined bulk RNA sequencing data from glioblastoma cases in The Cancer Genome Atlas and found that tumours generally fit into one of four transcriptional phenotypes — ‘neural’, ‘proneural’, ‘classical’ and ‘mesenchymal’ — each associated with specific oncogenic mutations169. Later, single-cell RNA sequencing studies elucidated that each glioblastoma is composed of cells in these four states, and the bulk sequencing results reflected the dominant signature of that tumour at the time it was sampled87. Each of these four transcriptional signatures — neural, proneural, classical and mesenchymal — represents a cellular state that can also be described by the normal cell type it most resembles: neural precursor cell (NPC)-like (neural), oligodendrocyte precursor cell (OPC)-like (proneural), astrocyte-like (classical) and mesenchymal cell (MES)-like (mesenchymal). Specific oncogene aberrations and microenvironmental factors favour expression of each cellular state87. Glioblastoma cells are protean, so can change cellular states from one of these transcriptional phenotypes to another in response to therapeutic pressure, microenvironmental influences and tumour evolution over the disease course. As the various glioma subtypes have been examined using single-cell techniques, characteristic cellular compositions and hierarchies have emerged. For example, diffuse intrinsic pontine glioma (DIPG) and other diffuse midline gliomas (DMGs) — tumours that typically occur in childhood and are characterized by mutations in genes encoding histone H3 — are chiefly composed of OPC-like/oligodendrocyte-like and astrocyte-like tumour cells (Table 1), with OPC-like cells representing the tumour-initiating cancer stem cell population86. Unlike in IDH-wild-type glioblastoma, NPC-like cells are not present86,163 and MES-like cells are chiefly found in older patients163.
Each of these glioma cellular states is enriched for particular types of interactions within malignant neuron–glioma networks. Neuron-to-glioma synapses are enriched in the OPC-like and NPC-like populations of glioma cells12,123, whereas gap junction-coupled glioma-to-glioma tumour microtubes are enriched in astrocyte-like and MES-like tumour cells123. K+-evoked currents are prominent in glioma cells exhibiting gap junction-coupled tumour microtubes12, which implies that K+-evoked currents are prominent in, but likely not limited to, astrocyte-like tumour cells. Glioma ‘hub cells’ expressing the calcium-activated potassium channel KCa3.1 largely fall into the MES-like category in adult IDH-wild-type glioblastoma but can also have the other cellular states141. Astrocyte-like tumour cells secrete synaptogenic paracrine factors such as glypican 3 (ref. 150) and thrombospondin 1 (TSP1) (ref. 140), contributing to neuronal hyperexcitability and network remodelling140,145,150. Cellular substates, associated malignant neuron–glioma network interactions and relevant therapeutic targets and drugs that hit those targets have been found in most forms of glioma studied to date (Table 1). Not all aspects of neuron–glioma interactions have been tested in all glioma subtypes; for example, neuronal activity-regulated paracrine factors are known to contribute to optic glioma pathophysiology, but neuron-to-glioma synapses remain to be studied in this tumour subtype.
The receptor or interaction partner(s) of NLGN3 in glioma are presently unknown, so targeting NLGN3 for glioma therapy has to date focused on mechanisms of NLGN3 shedding. NLGN3 is proteolytically cleaved at the membrane in an activity-regulated manner by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) enzyme, releasing the large amino-terminal ectodomain (shed NLGN3) into the tumour microenvironment111. Pharmacological inhibition of ADAM10 demonstrates significant preclinical efficacy in a range of glioma types, from adult glioblastoma to DIPG111 and NF1-associated optic pathway glioma103. A phase I clinical trial using an ADAM10 inhibitor to block NLGN3-mediated neuron–glioma interactions is presently ongoing112.
Mechanistically, shed NLGN3 binding to glioma cells stimulates several oncogenic signalling cascades, with the early activation of focal adhesion kinase and the initiation of its downstream PI3K–mTOR, Ras and Src effector pathways111. NLGN3 induces its own feedforward expression in glioma cells and increases GJA1 and TTYH1 expression111 (Fig. 2). Connexin 43 (CX43) and tweety homologue 1 have roles in the formation of tumour microtubes, long processes connected to each other by CX43-mediated gap junctions that establish a tumour to tumour cell network113,114. These microtubes connecting glioma cancer cells contribute to glioma therapeutic resistance and progression12,113,114. In addition, NLGN3 exposure increased expression of NTRK2 (encoding the BDNF receptor TrkB), several synapse-related genes (including those encoding glutamate receptor subunits), THBS1 (encoding the synaptogenic factor thrombospondin 1 (TSP1)), axon guidance genes and numerous genes involved in ion transport (encoding several potassium (K+) channels, calcium (Ca2+) channels and excitatory amino acid transporter 1)111. Thus, NLGN3 promotes transcriptomic enrichment for multiple aspects of glioma network integration (Fig. 2). As discussed below, such network integration is central to glioma pathophysiology, and these transcriptomic effects of NLGN3 may help explain the unexpected dependency of glioma on microenvironmental NLGN3 (ref. 111). An open question is whether redundant mechanisms regulate the expression of these processes that might explain the evolution of glioma resistance to NLGN3 loss111 described above.
Fig. 2 |. Neuronal activity-regulated mechanisms of glioma growth.
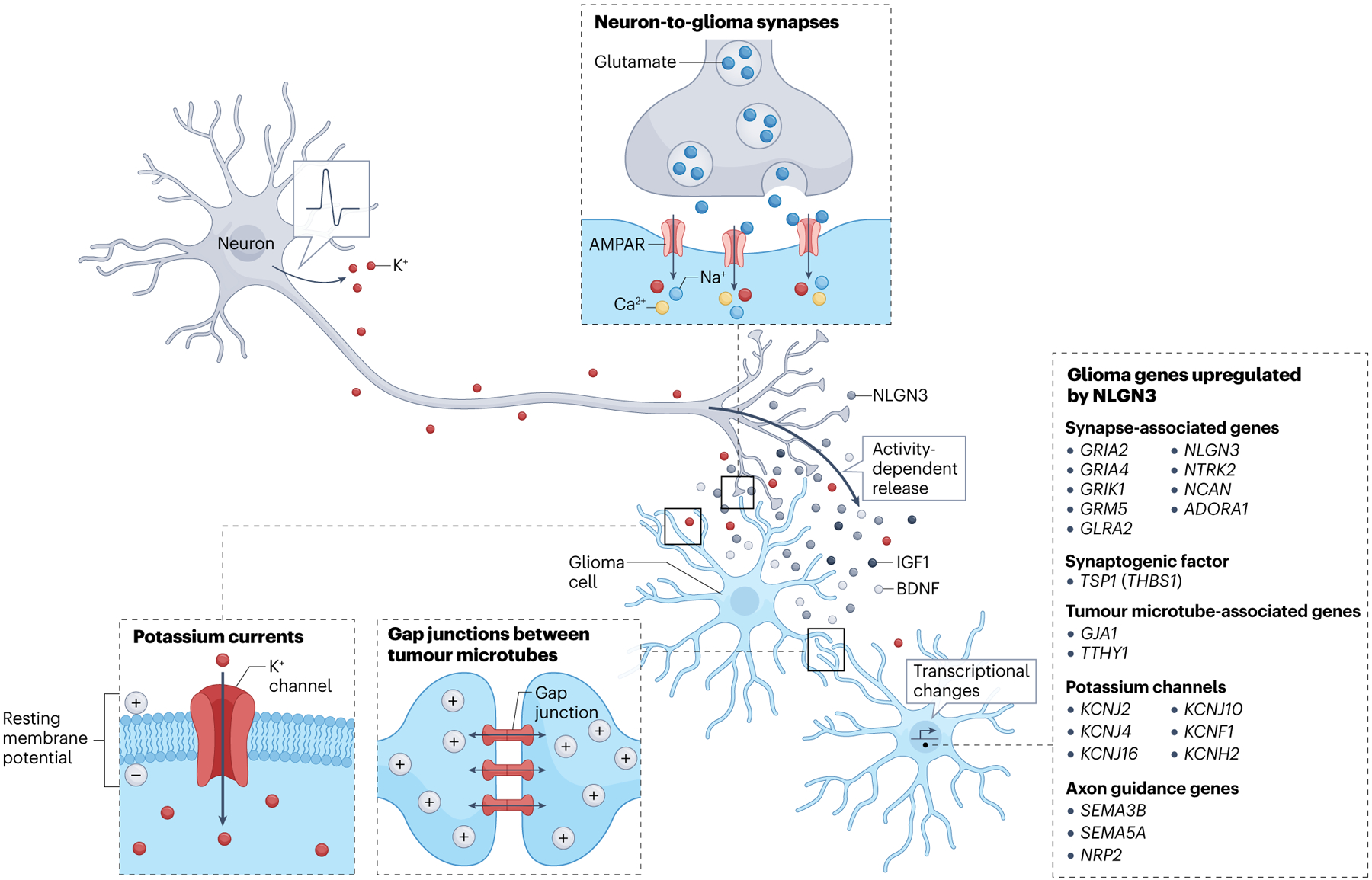
Neuronal activity drives glioma proliferation, growth and progression through activity-regulated paracrine factors including neuroligin 3 (NLGN3)11,103,111, brain-derived neurotrophic factor (BDNF)11,120, insulin-like growth factor 1 (IGF1)106, activity-regulated increases in potassium (K+) that evoke K+ currents in glioma cells12,102 and bona fide neuron-to-glioma synapses12,102,120,123. K+-evoked currents are amplified through gap junction coupling between glioma cells via tumour microtubes12,102. Activity-regulated interactions also induce gene expression changes in glioma cells relevant to multiple aspects of glioma network integration11,111. In particular, NLGN3 signalling induces glioma gene expression changes that underpin other neuron–glioma interactions, including upregulating synapse-associated genes, the BDNF receptor tyrosine receptor kinase B (TrkB) (NTRK2), the synaptogenic factor thrombospondin 1 (TSP1), genes encoding K+ channels, tumour microtube-associated genes including connexin 43 (CX43) (GJA1) and axon guidance genes including semaphorins (implicated in glioma invasion107)111. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor.
Underscoring the importance of these transcriptional programmes, pathway enrichment analysis of a large cohort of DIPG and other paediatric high-grade glioma tumours (>1,000) identified an enrichment for dysregulation of gene sets involved in neuronal communication across the diversity of the tumours92. More granular analysis of high-grade glioma single-cell transcriptomes identified robust expression of synapse-related genes enriched in the OPC-like population of these tumours12. These transcriptomic profiles highlighted the potential that, similar to the formation of neuron-to-OPC synapses in the normal brain49, gliomas may retain postsynaptic capabilities, and that activity-regulated paracrine factors such as NLGN3 may promote neuron-to-glioma synaptogenesis.
Neuron-to-glioma synapses
Back to back publications in 2019 using electrophysiological and ultra-structural approaches in primary tumour tissue and patient-derived xenograft models demonstrated the presence of bona fide synapses between presynaptic neuronal axons and postsynaptic glioma cells12,102. Co-culture of glioma cells with Nlgn3 knockout or wild-type mouse neurons supported the hypothesis that NLGN3 promotes synapse formation between neurons and glioma cells12. Whole-cell patch clamp electrophysiological recordings in both paediatric and adult glioblastomas revealed subpopulations of tumour cells that exhibit excitatory postsynaptic currents (ranging from 10 pA to 100 pA in magnitude) in response to evoked neuronal activity12,102. These functional neuron-to-glioma synapses are mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type ionotropic glutamate receptors on the glioma cells. In normal neuronal cells, the vast majority of AMPA receptors (AMPARs) are impermeable to calcium, either not containing a GluA2 subunit or containing GluA2 subunits that have undergone RNA editing at the glutamine or arginine (Q or R) site to alter ion permeability. Gliomas harboured under-edited GluA2 mRNA12,102, which rendered the AMPARs Ca2+ permeable115,116. Calcium permeability of AMPARs can contribute to synaptic plasticity in neurons (for review, please see ref. 117); however, the functional consequences of AMPAR-mediated calcium influx in glioma remains to be fully elucidated.
The properties and function of AMPARs in OPCs, the putative cell of origin for many forms of high-grade glioma, remain somewhat enigmatic. However, OPCs exhibit neuron-to-OPC synapses also mediated by Ca2+-permeable AMPARs49, which may influence OPC proliferation and survival118,119. In both paediatric and adult high-grade glioma models, blocking signalling through AMPAR-mediated neuron-to-glioma synapses either pharmacologically using AMPAR-targeting anti-seizure medication (perampanel) or genetically by expressing a dominant-negative form of the GluA2 AMPAR subunit starkly reduced glioma proliferation, growth and progression12,102.
Subsequent work has found that malignant neuron-to-glioma synapses hijack mechanisms of adaptive plasticity normally operant in synapses between neurons. For instance, activity-regulated secretion of BDNF promoted increased numbers of neuron-to-glioma synapses, as well as increased synaptic strength through increased trafficking of AMPARs to the postsynaptic membrane120. It is presently unknown whether other types of synapses, mediated by different neurotransmitter receptors, exist between additional neuronal subtypes and glioma cells. However, there is early evidence for GABAergic currents that are depolarizing (owing to high intracellular chloride concentration) and tumour growth-promoting in some types of gliomas121,122.
A recent study in adult glioblastoma illustrates an important role for AMPAR-mediated neuron-to-glioma synapses in glioma invasion and expansion of the tumour throughout the brain123. Neuron-to-glioma synapses involving cells at the invasive edge of the tumour that resemble NPC-like cells and OPC-like cells promoted the extension of invasive tumour microtubes and increased the speed of tumour cell invasion123. Over time, these dynamic, invasive cells become stationary and transition from an NPC-like or OPC-like phenotype to a more astrocyte-like and mesenchymal-like cellular phenotype (Box 2), and interconnect with other glioma cells and with astrocytes via gap junctional coupling. Meanwhile, the NPC-like or OPC-like glioma cells at the tumour margins form new synapses within neural circuits as they invade and propagate the tumour radially to colonize increasing areas of the brain123. Concordantly, single-cell genomic studies of gliomas at initial diagnosis and at recurrence illustrate increased synaptic gene expression at later stages of the disease course124. Taken together, the studies discussed above elucidate an emerging picture of dynamic, evolving neural–glioma networks regulated, in part, by the activity within tumour-infiltrated neural circuits.
Other electrochemical neuron–glioma interactions
In addition to an excitatory postsynaptic current, neuronal activity can trigger a non-synaptic inward current that exhibits long duration kinetics (timescale of seconds) in a subset of glioma cells. These prolonged currents are evoked by extracellular K+ released by actively firing neurons12,102, with extracellular K+ levels further increased owing to impaired K+ clearance in the glioma microenvironment125. Such K+-evoked currents are amplified across a network of glioma cells via glioma-to-glioma gap junction connections12 and are reminiscent of the activity-dependent currents observed in normal astrocytes126. Seminal work by Frank Winkler’s group established that a glioma-to-glioma network is formed through tumour microtubes that promote tumour resistance to therapy114. Such cellular coupling between glioma cells propagated synchronous Ca2+ wave formation via CX43 gap junction-mediated communication114. Pharmacological gap junction blockade not only de-synchronized the neuronal activity-dependent Ca2+ transients throughout the glioma network but also decreased the amplitude of neuronal activity-dependent, K+-evoked currents12,102. Blocking gap junctions, for example with the pain medication meclofenamate, decreased glioma proliferation in patient-derived orthotopic xenograft mouse models of glioma12.
Voltage-dependent mechanisms of glioma progression
Membrane depolarization can trigger the opening of voltage-gated ion channels and downstream intracellular signalling cascades (Fig. 3) that regulate numerous cellular functions and gene expression (see refs. 127,128 for reviews). The importance of membrane depolarization in the developing brain to NPC proliferation, migration, differentiation and survival129–134 raises the possibility that gliomas may be hijacking similar voltage-dependent mechanisms. In the prenatal brain, depolarization-induced Ca2+ waves occur in neural stem cells135,136 and changes in membrane potential instruct precursor cell fate decisions during corticogenesis137.
Fig. 3 |. Glioma membrane depolarization promotes tumour cell proliferation.
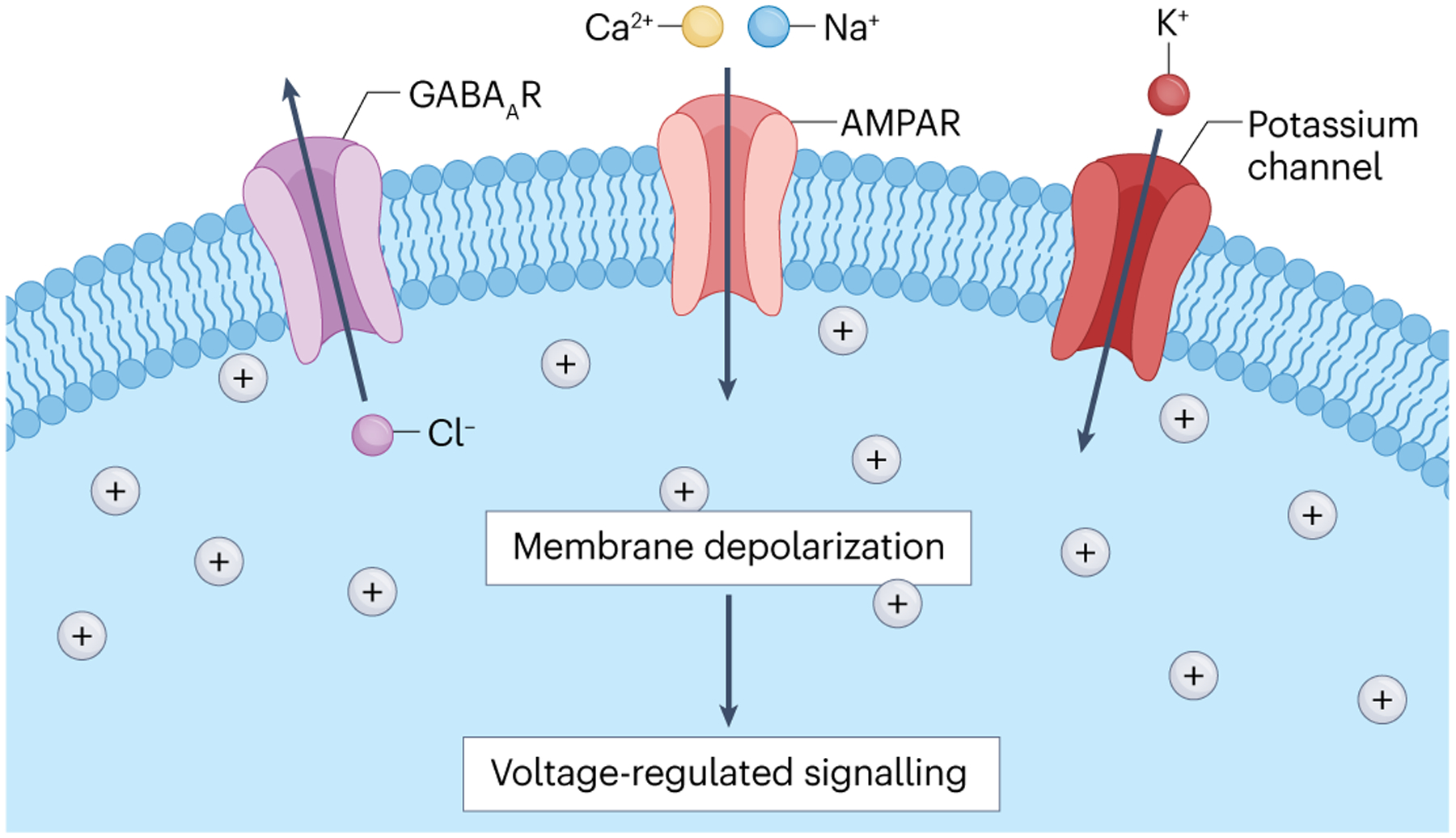
Gliomas exhibit multiple mechanisms of membrane depolarization, including calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated neuron-to-glioma synapses12,102, activity-dependent potassium-evoked currents12,102 and, in some glioma types, depolarizing GABAergic currents mediated by outward flux of chloride through GABAA receptors122. Membrane depolarization alone is sufficient to drive glioma proliferation12. Membrane depolarization can trigger opening of voltage-gated ion channels and consequent intracellular signalling events, but the details of voltage-sensitive mechanisms in glioma remain to be elucidated.
Whereas many details about the voltage-sensitive mechanisms regulating normal neurodevelopment remain to be fully elucidated, the importance of the precise control of ionic flux during prenatal development has been highlighted by the identification of mutations in ion channels and neurotransmitter receptor genes in individuals exhibiting prominent cortical malformations138,139. To test the functional consequences of membrane depolarization in glioma, patient-derived glioma cells were engineered to express the light-sensitive cation channel channelrhodopsin 2, xenografted to the mouse cortex and then optogenetically depolarized12. This demonstrated that glioma membrane depolarization alone is sufficient to promote malignant cell proliferation12. The intracellular events triggered by changes to membrane potential that regulate glioma cell proliferation remain an open question, as does the possibility that membrane depolarization influences additional cancer cellular functions beyond proliferation. Elucidating the voltage-sensitive signalling pathway(s) that mediate the functional effects of glioma membrane depolarization represents an area of intense ongoing investigation.
Thus, glioma is an electrically active tissue in which neuron-to-glioma synapses and glioma-to-glioma gap junctional coupling promote synchronous membrane depolarizations and consequent Ca2+ transients throughout networks of cancer cells. Glioma cells form a network that integrates structurally and electrically into the neural circuitry of the brain. In adult patients with hemispheric glioblastoma, intraoperative electrophysiology demonstrates that the degree of functional connectivity of the tumour with the rest of the brain is strongly inversely correlated with survival140.
Autonomous currents in the glioma network
Underscoring the importance of activity-evoked Ca2+ transients in glioma, a small population of tumour cells exhibit autonomous oscillatory Ca2+ transients in both paediatric12 and adult141 high-grade gliomas. These cells with oscillatory Ca2+ transients (representing ~4% of total glioma cells in glioblastoma) are highly connected to other glioma cells within the gap junction-connected glioma-to-glioma network, forming a ‘hub’ within the network141. Autonomous, rhythmic oscillations of Ca2+ transients in these ‘hub cells’ are regulated by the Ca2+-activated K+ channel KCa3.1 — also expressed by pacemaker sinoatrial node cardiac myocytes142 — and propagated to the connected network of gap junction-coupled tumour cells141. Analyses of signalling pathways regulated by these periodic Ca2+ transients highlighted tumour cell mitogen-activated protein kinase 2 and nuclear factor-κB pathways, both known to contribute to cancer cell behaviours141. Genetically or pharmacologically targeting KCa3.1, which abrogates these autonomous Ca2+ transients, reduced glioma cell viability, mitigated glioma growth and prolonged mouse survival in preclinical models of adult glioblastoma141.
Glioma-induced neuronal excitability
Gliomas are an electrically active tissue, responding to neuronal input in both a synaptic manner and a non-synaptic manner12,102. These malignant cells are, in turn, able to promote excitability of neurons in both adult and paediatric glioma types12,143–145 (Fig. 4). Glioma-associated seizures are a common symptom for patients with both low-grade and high-grade glioma59,146,147. Intraoperative electrocorticography in awake patients with hemispheric high-grade gliomas has demonstrated glioma-induced hyperexcitability in tumour-infiltrated cortex at rest12 and during cognitive language tasks such as visual and auditory naming140.
Fig. 4 |. Neuron–glioma interactions drive glioma pathobiology.
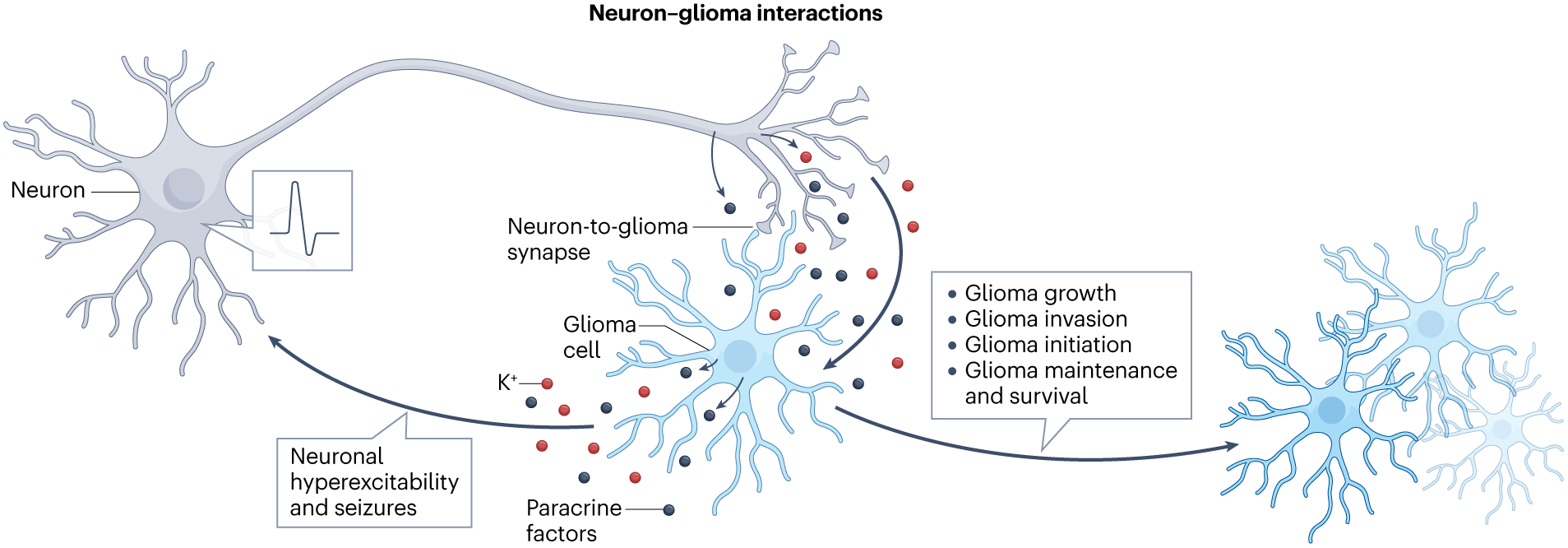
Paracrine and synaptic interactions between neurons and glioma cells promote cancer cell proliferation, invasion and survival, driving tumour initiation and growth. Reciprocally, glioma cells remodel neural circuits and promote neuronal hyperexcitability, resulting in glioma-associated seizures and increased neuronal input to the tumour.
Several mechanisms of glioma-induced neuronal hyperexcitability have been identified, including tumour cell release of glutamate via cysteine–glutamate transport systems143 and alterations in the neuronal network owing to loss of perineuronal nets148. Glioma-associated loss of inhibitory interneurons and glioma-associated switch from a hyperpolarizing to a depolarizing effect of GABA in excitatory neurons148,149, elevated extracellular K+ levels owing to impaired K+ clearance in the glioma microenvironment125 and promotion of neuronal synaptogenesis through glioma secretion of synaptogenic factors (such as glypican 3 (refs. 145,150) and TSP1 (ref. 140)) can also promote neuronal hyperexcitability. The glioma-associated loss of inhibitory interneurons and perineuronal nets occurred early in the course of disease in an adult glioblastoma mouse model148 and seizure severity increased with tumour progression, correlated to an increasing proportion of tumour cells secreting synaptogenic factors145.
The full consequences of glioma-induced neural circuit integration and remodelling on neural circuit function are beginning to come into sharp relief. Recent evidence from a landmark intraoperative electrophysiology study illustrates that high-grade gliomas functionally remodel neural circuitry: in patients with glioblastoma in the left hemisphere, involving but not limited to language areas, expressive language tasks recruited activity not only in the expected frontotemporal language areas but also in all cortical areas infiltrated by the tumour140. Tumour cell secretion of the synaptogenic factor TSP1 correlated with the degree of functional connectivity in patients and TSP1 was shown experimentally to promote neuron–glioma interactions and contribute to glioma pathophysiology140, including both synaptogenesis140 and tumour microtube formation140,151. The tumour-infiltrated language cortex remains functional, albeit at reduced efficiency140, explaining some of the cognitive effects of glioma and also underscoring the well-established clinical observation that — unlike more other neurological disease processes — brain areas affected by gliomas retain a surprising degree of functionality. It is now increasingly clear how that preservation of activity drives brain cancer progression (Fig. 4).
Therapeutic targeting of neuron–glioma interactions
Identifying the mechanisms involved in neuron–glioma interactions will enable clinically approved brain-penetrant drugs to be repurposed for modulation of tumour cell dependency on the active microenvironment. Neuronal activity-evoked tumour excitatory postsynaptic currents are targetable using the AMPAR-blocking anti-epileptic drug perampanel and such treatment has demonstrated preclinical efficacy in glioma patient-derived xenografts12,102. Glioma growth and invasion is decreased through targeting the gap junction-mediated Ca2+ transients using the anti-inflammatory drug meclofenamate in preclinical models12,102. Targeting glioma-induced synaptogenesis by blocking TSP1 signalling with the anti-epileptic drug gabapentin reduced glioma growth in glioma patient-derived xenografts140. As variable responses of NPCs to neuronal activity are observed depending on the neurodevelopmental context129–134, it is difficult to predict the clinical relevance of targeting these neuron–glioma interactions in tumours that arise from different progenitor populations within the brain, such as those that arise from neuronal precursor cells152–154. The mechanistic effects of electrophysiological responses in other primary brain cancers will likely vary from those of gliomas, for example in those tumours which arise from neuronal-lineage precursor cells, such as medulloblastoma. The neuroscience of each individual brain tumour type must be studied to elucidate tumour-specific neural mechanisms regulating pathogenesis.
Whereas the role of neuronal activity in other primary brain tumours is yet to be fully appreciated, new insights into secondary brain cancers have confirmed an interaction with neurons that drives malignant progression. Brain metastatic breast cancer cells acquire an increased expression of a NMDA receptor subtype within the brain microenvironment which enables them to successfully colonize the brain155. Tumour progression is facilitated by the integration of the metastatic cells amongst glutamatergic neurons and subsequent stimulation of glutamate-activated NMDA receptor signalling155, along with the formation of astrocyte–tumour gap junction coupling156. Investigations into neuron–tumour communication across all brain cancer subtypes will uncover specific signalling interactions, either by synaptic, perisynaptic or paracrine mechanisms, that are susceptible to therapeutic targeting.
Conclusions and outlook
Although the importance of neuron–glial interactions in brain development, adaptive plasticity and disease has become increasingly evident, numerous pressing questions remain. Elucidating the molecular and cellular mechanisms mediating adaptive oligodendroglial cell responses — which likely exhibit developmental stage, brain region, neural circuit and neuron subtype-specific heterogeneity — will be important for fundamental understanding of nervous system development, plasticity and function. Oligodendroglial plasticity is implicated in a growing list of brain functions, and maladaptive or deficient oligodendroglial plasticity may have important pathophysiological roles in an expanding number of neurological and neuropsychiatric diseases. Developing appropriate therapeutic interventions to target dysfunction of neuron–oligodendroglial interactions will require such fundamental mechanistic understanding. A stark example of this are glial malignancies.
The mechanistic parallels evident in the neuron–oligodendroglial interactions that contribute to myelin plasticity and the neuron–glioma interactions that drive the pathogenesis of glial malignancies highlight the extent to which brain cancers hijack mechanisms of normal neural development and plasticity and underscore the importance of understanding the neuroscience of brain cancers. Elucidating key interactions between neurons and brain cancer cells and neuronal mechanisms hijacked by malignant cells reveals an avenue of potential therapeutic interventions that may improve outcomes for these lethal cancers. Major challenges to achieving the potential of this therapeutic approach include the need to adapt therapies to malignant neuron–glioma networks that will likely change and evolve in complex ways over the course of the disease, and to develop therapeutic interventions that selectively disrupt mechanisms in the cancer cells rather than in healthy neural cells.
Beyond brain cancer, interactions between the nervous system and cancers throughout the body are emerging as crucially important pathogenic mechanisms and promising therapeutic targets (for reviews on the emerging field of cancer neuroscience, see refs. 157–159). In every example studied to date, cancer cells subvert normal mechanisms by which the nervous system maintains tissue and organ homeostasis or promotes development and regeneration (see ref. 160 for a review on the neural regulation of ontogeny and oncology). Thus, fundamental discoveries in normal development and physiology are robustly synergistic with the study of diseases such as cancers. In the case of seemingly intractable brain cancers such as high-grade gliomas, progress towards effective therapy must take a multidisciplinary approach, incorporating crucially important lessons from neuroscience. In turn, mechanisms discovered in brain cancer may help inform principles of healthy brain development and plasticity.
Acknowledgements
The authors thank M. Filbin for helpful input on Table 1. This work was supported by grants from Cancer Research UK (to M.M.), ChadTough Defeat DIPG (to M.M.), National Institute of Neurological Disorders and Stroke (R01NS092597 to M.M.), National Institutes of Health (NIH) Director’s Pioneer Award (DP1NS111132 to M.M.), National Cancer Institute (P50CA165962, R01CA258384, U19CA264504 to M.M.), Damon Runyon Cancer Research Foundation (to K.R.T.), Stanford Maternal and Child Health Research Institute (to K.R.T.), Gatsby Charitable Foundation (Gatsby Initiative in Brain Development and Psychiatry to M.M.), HHMI Emerging Pathogens Initiative (EPI), Oscar’s Kids Foundation (to M.M.), McKenna Claire Foundation (to M.M.), Virginia and D. K. Ludwig Fund for Cancer Research (to M.M.), Oligo Nation (to M.M.), Waxman Family Research Fund (to M.M.) and Will Irwin Research Fund (to M.M.).
Footnotes
Competing interests
M.M. is on the SAB for TippingPoint Biosciences, and her family holds equity in MapLight Therapeutics.
References
- 1.Christopherson KS et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433 (2005). [DOI] [PubMed] [Google Scholar]; This work establishes that astrocytes secrete synaptogenic factors including thrombospondins.
- 2.Ullian EM, Harris BT, Wu A, Chan JR & Barres BA Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol. Cell Neurosci 25, 241–251 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Eroglu C et al. Gabapentin receptor α2δ−1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens B et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Schafer DP et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funfschilling U et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that myelinating oligodendrocytes not only modulate conduction velocity but also provide metabolic support to axons.
- 7.Huxley AF & Stämpeli R Evidence for saltatory conduction in peripheral myelinated nerve fibres. J. Physiol 108, 315–339 (1949). [PubMed] [Google Scholar]
- 8.Pan Y & Monje M Activity shapes neural circuit form and function: a historical perspective. J. Neurosci 40, 944–954 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geraghty AC et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron 103, 250–265.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knowles JK et al. Maladaptive myelination promotes generalized epilepsy progression. Nat. Neurosci 25, 596–606 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatesh HS et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that neuronal activity can promote the growth of high-grade gliomas.
- 12.Venkatesh HS et al. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated the existence of bona fide neuron-to-glioma synapses that robustly promote tumor growth; published back-to-back with ref. 102.
- 13.Lyon KA & Allen NJ From synapses to circuits, astrocytes regulate behavior. Front. Neural Circuits 15, 786293 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilton DK, Dissing-Olesen L & Stevens B Neuron–glia signaling in synapse elimination. Annu. Rev. Neurosci 42, 107–127 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Khakh BS & Deneen B The emerging nature of astrocyte diversity. Annu. Rev. Neurosci 42, 187–207 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Nagai J et al. Behaviorally consequential astrocytic regulation of neural circuits. Neuron 109, 576–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boullerne AI The history of myelin. Exp. Neurol 283, 431–445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown AM, Evans RD, Black J & Ransom BR Schwann cell glycogen selectively supports myelinated axon function. Ann. Neurol 72, 406–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saab AS, Tzvetanova ID & Nave KA The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol 23, 1065–1072 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Zalc B & Colman DR Origins of vertebrate success. Science 288, 271–272 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Smith RS & Koles ZJ Myelinated nerve fibers: computed effect of myelin thickness on conduction velocity. Am. J. Physiol 219, 1256–1258 (1970). [DOI] [PubMed] [Google Scholar]
- 22.Wu LMN, Williams A, Delaney A, Sherman DL & Brophy PJ Increasing internodal distance in myelinated nerves accelerates nerve conduction to a flat maximum. Curr. Biol 22, 1957–1961 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yakovlev PI in Regional Development of the Brain in Early Life Ch. 1 (ed. Minkowski A) 3–70 (Blackwell Scientific, 1967). [Google Scholar]
- 24.Lebel C et al. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage 60, 340–352 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Miller D et al. Prolonged myelination in human neocortical evolution. Proc. Natl Acad. Sci. USA 109, 16480–16485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donaldson HAH & Hoke GW On the areas of the axis cylinder and medullary sheath as seen in cross sections of the spinal nerves of vertebrates. J. Comp. Neurol 15, 1–15 (1905). [Google Scholar]
- 27.Honjin R, Sakato S & Yamashita T Electron microscopy of the mouse optic nerve: a quantitative study of the total optic nerve fibers. Arch. Histol. Jpn 40, 321–332 (1977). [DOI] [PubMed] [Google Scholar]
- 28.Olivares R, Montiel J & Aboitiz F Species differences and similarities in the fine structure of the mammalian corpus callosum. Brain Behav. Evol 57, 98–105 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Peters A & Sethares C Myelinated axons and the pyramidal cell modules in monkey primary visual cortex. J. Comp. Neurol 365, 232–255 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Tomassy GS et al. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344, 319–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu QR et al. Common developmental requirement for olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Kessaris N et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci 9, 173–179 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergles D & Richardson W Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol 8, a020453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitew S et al. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 276, 29–47 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Hughes E, Kang S, Fukaya M & Bergles D Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci 16, 668–676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson MR, Polito A, Levine JM & Reynolds R NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell Neurosci 24, 476–488 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Buchanan J et al. Oligodendrocyte precursor cells ingest axons in the mouse neocortex. Proc. Natl Acad. Sci. USA 119, e2202580119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work introduces the concept that OPCs can prune axons, illustrating a myelin-independent function for oligodendroglial lineage cells.
- 38.Auguste YSS et al. Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat. Neurosci 25, 1273–1278 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work introduces the concept that — similar to microglia and astrocytes — OPCs can prune synapses, illustrating a myelin-independent function for oligodendroglial lineage cells.
- 39.Chung WS et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young K et al. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi RB et al. Remarkable stability of myelinating oligodendrocytes in mice. Cell Rep. 21, 316–323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hughes EG, Orthmann-Murphy JL, Langseth AJ & Bergles DE Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci 21, 696–706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill RA, Li AM & Grutzendler J Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci 21, 683–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters A & Sethares C Oligodendrocytes, their progenitors and other neuroglial cells in the aging primate cerebral cortex. Cereb. Cortex 14, 995–1007 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Peters A, Verderosa A & Sethares C The neuroglial population in the primary visual cortex of the aging rhesus monkey. Glia 56, 1151–1161 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Geha S et al. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol. 20, 399–411 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yeung M et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 159, 766–774 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Barres BA & Raff MC Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260 (1993). [DOI] [PubMed] [Google Scholar]
- 49.Bergles DE, Roberts JD, Somogyi P & Jahr CE Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191 (2000). [DOI] [PubMed] [Google Scholar]; This work demonstrates the existence of bona fide neuron-to-OPC synapses.
- 50.Lin SC & Bergles DE Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat. Neurosci 7, 24–32 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Karadottir R, Cavelier P, Bergersen LH & Attwell D NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stevens B, Tanner S & Fields RD Control of myelination by specific patterns of neural impulses. J. Neurosci 18, 9303–9311 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishibashi T et al. Astrocytes promote myelination in response to electrical impulses. Neuron 49, 823–832 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Makinodan M, Rosen K, Ito S & Corfas G A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J et al. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat. Neurosci 15, 1621–1623 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez MM, Hearn EF, Do D, Rilling JK & Herndon JG Differential rearing affects corpus callosum size and cognitive function of rhesus monkeys. Brain Res. 812, 38–49 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Scholz J, Klein MC, Behrens TE & Johansen-Berg H Training induces changes in white-matter architecture. Nat. Neurosci 12, 1370–1371 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that humans who learn a complex motor task (juggling) exhibit white matter changes consistent with myelination in relevant brain regions following training.
- 58.Schlegel A, Rudelson J & Tse P White matter structure changes as adults learn a second language. J. Cogn. Neurosci 24, 1664–1670 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Berendsen S et al. Prognostic relevance of epilepsy at presentation in glioblastoma patients. Neuro Oncol. 18, 700–706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee S, Chong SYC, Tuck SJ, Corey JM & Chan JR A rapid and reproducible assay for modeling myelination by oligodendrocytes using engineered nanofibers. Nat. protoc 8, 771–782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Redmond SA et al. Somatodendritic expression of JAM2 inhibits oligodendrocyte myelination. Neuron 91, 824–836 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bechler ME, Byrne L & Ffrench-Constant C CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr. Biol 25, 2411–2416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson EM et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that premotor cortical projection neuron activity promotes OPC proliferation, oligodendrogenesis and myelin changes that alter motor function in an oligodendrogenesis and myelin-dependent manner.
- 64.McKenzie I et al. Motor skill learning requires active central myelination. Science 346, 318–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that oligodendrogenesis is induced by motor learning and that motor learning in a complex wheel task depends on oligodendrogenesis.
- 65.Sampaio-Baptista C et al. Motor skill learning induces changes in white matter microstructure and myelination. J. Neurosci 33, 19499–19503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitew S et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. commun 9, 306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steadman PE et al. Disruption of oligodendrogenesis impairs memory consolidation in adult mice. Neuron 105, 150–164 e156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that learning-induced oligodendrogenesis and de novo myelination is required for memory consolidation in spatial and fear learning tests, and shows that these adaptive oligodendroglial changes promote oscillatory synchrony between the hippocampus and frontal cortex.
- 68.Swire M, Kotelevtsev Y, Webb DJ, Lyons DA & Ffrench-Constant C Endothelin signalling mediates experience-dependent myelination in the CNS. eLife 8, e49493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang SM, Michel K, Jokhi V, Nedivi E & Arlotta P Neuron class-specific responses govern adaptive myelin remodeling in the neocortex. Science 370, eabd2109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marques S et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vigano F, Mobius W, Gotz M & Dimou L Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci 16, 1370–1372 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Lundgaard I et al. Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS Biol. 11, e1001743 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dimou L, Simon C, Kirchhoff F, Takebayashi H & Gotz M Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci 28, 10434–10442 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mensch S et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci 18, 628–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that synaptic vesicle release by axons regulates the number of myelin internodes that an oligodendrocyte forms and promotes myelination during development in zebrafish.
- 75.Hines JH, Ravanelli AM, Schwindt R, Scott EK & Appel B Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci 18, 683–689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that myelination preferentially forms on active axons during development in zebrafish.
- 76.Pan S, Mayoral SR, Choi HS, Chan JR & Kheirbek MA Preservation of a remote fear memory requires new myelin formation. Nat. Neurosci 23, 487–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang H, Zhou F, Zhou S & Qiu M MYRF: a mysterious membrane-bound transcription factor involved in myelin development and human diseases. Neurosci. Bull 37, 881–884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pajevic S, Basser P & Fields R Role of myelin plasticity in oscillations and synchrony of neuronal activity. Neuroscience 276, 135–147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noori R et al. Activity-dependent myelination: a glial mechanism of oscillatory self-organization in large-scale brain networks. Proc. Natl Acad. Sci. USA 117, 13227–13237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mount CW, Yalcin B, Cunliffe-Koehler K, Sundaresh S & Monje M Monosynaptic tracing maps brain-wide afferent oligodendrocyte precursor cell connectivity. eLife 8, e49291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Knowles JK, Batra A, Xu H & Monje M Adaptive and maladaptive myelination in health and disease. Nat. Rev. Neurol 18, 735–746 (2022). [DOI] [PubMed] [Google Scholar]
- 82.Ostrom QT et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol. 24, v1–v95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Louis DN et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen J et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh SK et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Ryall S et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37, 569–583.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Claus EB et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg. Focus 38, E6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mackay A et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dvorak AV et al. An atlas for human brain myelin content throughout the adult life span. Sci. Rep 11, 269 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kinney HC, Brody BA, Kloman AS & Gilles FH Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J. Neuropathol. Exp. Neurol 47, 217–234 (1988). [DOI] [PubMed] [Google Scholar]
- 95.Galvao RP et al. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl Acad. Sci. USA 10.1073/pnas.1414389111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alcantara Llaguno SR et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell 28, 429–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu C et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper implicates OPCs as the cell of origin for glioblastoma.
- 98.Monje M et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl Acad. Sci. USA 108, 4453–4458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nagaraja S et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Z et al. Cell lineage-based stratification for glioblastoma. Cancer Cell 38, 366–379.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Filbin M & Monje M Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat. Med 25, 367–376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Venkataramani V et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538 (2019). [DOI] [PubMed] [Google Scholar]; Published back to back together with ref. 12, this work demonstrates the existence of bona fide neuron-to-glioma synapses that robustly promote tumour growth.
- 103.Pan Y et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 594, 277–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that visual experience regulates the initiation, growth and maintenance of NF1-associated optic pathway glioma in a genetically engineered mouse model.
- 104.Toonen JA, Ma Y & Gutmann DH Defining the temporal course of murine neurofibromatosis-1 optic gliomagenesis reveals a therapeutic window to attenuate retinal dysfunction. Neuro Oncol. 19, 808–819 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anastasaki C et al. Neuronal hyperexcitability drives central and peripheral nervous system tumor progression in models of neurofibromatosis-1. Nat. commun 13, 2785 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen P et al. Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature 606, 550–556 (2022). [DOI] [PubMed] [Google Scholar]
- 107.Huang-Hobbs E et al. Remote neuronal activity drives glioma progression through SEMA4F. Nature 619, 844–850 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bemben MA, Shipman SL, Nicoll RA & Roche KW The cellular and molecular landscape of neuroligins. Trends Neurosci. 38, 496–505 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sudhof TC Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Budreck EC & Scheiffele P Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur. J. Neurosci 26, 1738–1748 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Venkatesh HS et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.US National Library of Medicine. ClinicalTrials.gov, h. c. g. c. s. N. [DOI] [PubMed]
- 113.Jung E et al. Tweety-homolog 1 drives brain colonization of gliomas. J. Neurosci 37, 6837–6850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Osswald M et al. Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98 (2015). [DOI] [PubMed] [Google Scholar]; This work demonstrates that glioma cells connect to each other through long processes called tumour microtubes to form a gap junction-coupled network.
- 115.Sommer B, Kohler M, Sprengel R & Seeburg PH RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19 (1991). [DOI] [PubMed] [Google Scholar]
- 116.Hollmann M, Hartley M & Heinemann S Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science 252, 851–853 (1991). [DOI] [PubMed] [Google Scholar]
- 117.Cull-Candy SG & Farrant M Ca2+-permeable AMPA receptors and their auxiliary subunits in synaptic plasticity and disease. J. Physiol 599, 2655–2671 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen TJ et al. In vivo regulation of oligodendrocyte precursor cell proliferation and differentiation by the AMPA-receptor subunit GluA2. Cell Rep. 25, 852–861.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Kougioumtzidou E et al. Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. eLife 6, e28080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taylor KR et al. Glioma synapses recruit mechanisms of adaptive plasticity. Preprint at bioRxiv 10.1101/2021.11.04.467325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Labrakakis C, Patt S, Hartmann J & Kettenmann H Functional GABAA receptors on human glioma cells. Eur. J. Neurosci 10, 231–238 (1998). [DOI] [PubMed] [Google Scholar]
- 122.Barron T et al. GABAergic neuron-to-glioma synapses in diffuse midline gliomas. Preprint at bioRxiv 10.1101/2022.11.08.515720 (2022). [DOI] [Google Scholar]
- 123.Venkataramani V et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917.e31 (2022). [DOI] [PubMed] [Google Scholar]; This work shows that neuron-to-glioma synapses promote glioblastoma invasion.
- 124.Varn FS et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199.e16 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Curry RN et al. Glioma epileptiform activity and progression are driven by IGSF3-mediated potassium dysregulation. Neuron 111, 682–695.e9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sibille J, Pannasch U & Rouach N Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J. Physiol 592, 87–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abdul Kadir L, Stacey M & Barrett-Jolley R Emerging roles of the membrane potential: action beyond the action potential. Front. Physiol 9, 1661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang M & Brackenbury WJ Membrane potential and cancer progression. Front. Physiol 4, 185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.LoTurco JJ, Owens DF, Heath MJ, Davis MB & Kriegstein AR GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298 (1995). [DOI] [PubMed] [Google Scholar]
- 130.Canudas AM et al. PHCCC, a specific enhancer of type 4 metabotropic glutamate receptors, reduces proliferation and promotes differentiation of cerebellar granule cell neuroprecursors. J. Neurosci 24, 10343–10352 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Luk KC, Kennedy TE & Sadikot AF Glutamate promotes proliferation of striatal neuronal progenitors by an NMDA receptor-mediated mechanism. J. Neurosci 23, 2239–2250 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Luk KC & Sadikot AF Glutamate and regulation of proliferation in the developing mammalian telencephalon. Dev. Neurosci 26, 218–228 (2004). [DOI] [PubMed] [Google Scholar]
- 133.Paez-Gonzalez P, Asrican B, Rodriguez E & Kuo CT Identification of distinct ChAT+ neurons and activity-dependent control of postnatal SVZ neurogenesis. Nat. Neurosci 17, 934–942 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang Z et al. Activation of type 5 metabotropic glutamate receptor promotes the proliferation of rat retinal progenitor cell via activation of the PI-3-K and MAPK signaling pathways. Neuroscience 322, 138–151 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Gu X, Olson EC & Spitzer NC Spontaneous neuronal calcium spikes and waves during early differentiation. J. Neurosci 14, 6325–6335 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weissman TA, Riquelme PA, Ivic L, Flint AC & Kriegstein AR Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661 (2004). [DOI] [PubMed] [Google Scholar]
- 137.Vitali I et al. Progenitor hyperpolarization regulates the sequential generation of neuronal subtypes in the developing neocortex. Cell 174, 1264–1276.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smith RS et al. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 99, 905–913.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith RS & Walsh CA Ion channel functions in early brain development. Trends Neurosci. 43, 103–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krishna S et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 617, 599–607 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that glioblastoma remodels functional neural circuitry in the human brain to augment functional connectivity with the tumour, that the synaptogenic factor TSP1 has an important role in this and that the degree of functional connectivity between glioma and the rest of the brain is strongly inversely correlated with survival.
- 141.Hausmann D et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 613, 179–186 (2023). [DOI] [PubMed] [Google Scholar]
- 142.Weisbrod D et al. SK4 Ca2+ activated K+ channel is a critical player in cardiac pacemaker derived from human embryonic stem cells. Proc. Natl Acad. Sci. USA 110, E1685–E1694 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Buckingham SC et al. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med 17, 1269–1274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that glioma cells promote neuronal hyperexcitability through glutamate secretion, contributing to the common clinical phenomenon of glioma-associated seizures.
- 144.Campbell SL, Buckingham SC & Sontheimer H Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 53, 1360–1370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.John Lin CC et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci 20, 396–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.van Breemen MS, Wilms EB & Vecht CJ Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol. 6, 421–430 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Drumm MR et al. Postoperative risk of IDH-mutant glioma-associated seizures and their potential management with IDH-mutant inhibitors. J. Clin. Invest 133, e168035 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hatcher A et al. Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Invest 130, 2286–2300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Campbell SL et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yu K et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 578, 166–171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that glioma cells secrete synaptogenic factors such as glypican 3 to promote neuronal hyperexcitability and seizures.
- 151.Joseph JV et al. TGF-β promotes microtube formation in glioblastoma through thrombospondin 1. Neuro Oncol. 24, 541–553 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen CCL et al. Histone H3.3G34-mutant interneuron progenitors co-opt PDGFRA for gliomagenesis. Cell 183, 1617–1633.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gibson P et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 468, 1095–1099 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Smith KS et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature 609, 1012–1020 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zeng Q et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen Q et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Monje M et al. Roadmap for the emerging field of cancer neuroscience. Cell 181, 219–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Winkler F et al. Cancer neuroscience: state of the field, emerging directions. Cell 186, 1689–1707 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mancusi R & Monje M The neuroscience of cancer. Nature 618, 467–479 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Venkatesh H & Monje M Neuronal activity in ontogeny and oncology. Trends Cancer 3, 89–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Reitman ZJ et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat. commun 10, 3731 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu I et al. The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location. Nat. Genet 54, 1881–1894 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Beera KG et al. Altered brain morphology after focal radiation reveals impact of off-target effects: implications for white matter development and neurogenesis. Neuro Oncol. 20, 788–798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Menning S et al. Changes in brain white matter integrity after systemic treatment for breast cancer: a prospective longitudinal study. Brain Imaging Behav. 12, 324–334 (2018). [DOI] [PubMed] [Google Scholar]
- 166.Gibson EM et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 176, 43–55.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fernandez-Castaneda A et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 185, 2452–2468.e16 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Monje M & Iwasaki A The neurobiology of long COVID. Neuron 110, 3484–3496 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Verhaak RG et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]