

Abstract

The role of short strong hydrogen bonds (SSHBs) in ligand–target binding remains largely unexplored, thereby hindering a potentially important avenue in rational drug design. Here we investigate the interaction between the antituberculosis drug bedaquiline (Bq) and the mycobacterial ATP synthase to unravel the role of a specific hydrogen bond to a conserved acidic residue in the target affinity and specificity. Our ab initio molecular dynamics simulations reveal that this bond belongs to the SSHB category and accounts for a substantial fraction of the target binding free energy. We also demonstrate that the presence of an extra acidic residue, i.e., aspartic acid at position 32 (D32), found exclusively in mycobacteria, cooperatively enhances the HB strength, ensuring specificity for the mycobacterial target. Consistently, we show that the removal of D32 markedly weakens the affinity, leading to Bq resistance associated with mutations of D32 to nonacidic residues. By designing simple Bq analogs, we then explore the possibility to overcome the resistance and potentially broaden the Bq antimicrobial spectrum by making the SSHB independent of the presence of the extra acidic residue.
Keywords: ATPase, Tuberculosis, Bedaquiline, Short strong hydrogen bond
Hydrogen bonds (HBs) are ubiquitous intermolecular interactions characterized by directionality and specificity. Due to their versatility and robustness, HBs are essential for many physical phenomena, from protein structure and function to molecular recognition and self-assembly to chemical reactivity.1,2 As hydrogen-bond energies typically vary in the 2–10 kcal/mol range, HBs are generally considered weak non-covalent interactions.3,4 For these weak HBs, the distance between the donor (D) and acceptor (A), dDA, is typically >2.6 Å, and the proton (H) is largely localized on the donor,5,6 which corresponds to a strongly asymmetric double-well potential with a more pronounced minimum at a short D–H distance (purple in Figure 1A).7
Figure 1.

(A) Schematic view of the energy landscape for the displacement of the H-bonded proton between the donor (D) and the acceptor (A) for the weak (purple) and strong (orange) HB. (B) Chemical structure of bedaquiline (Bq). The tertiary amino group involved in forming the hydrogen-bonding interaction with the c ring is indicated by the green circle. (C) Side view of the c ring with bound Bq. (D) Hydrogen bond between the Bq amino group and the c-ring E65. Unique to the mycobacterial c ring is the presence of an additional acidic residue, D32, which due to its location can form an additional HB with E65. For more structural details of the Bq–c-ring interface, see Figure S1.
However, if D and A have similar proton affinities (or pKa’s), the HB energy may greatly exceed the normal range, occasionally reaching 25–40 kcal/mol, while dDA becomes markedly shorter (2.5 Å or even less).7−10 In such cases, the proton experiences a more symmetric low-barrier or single-minimum potential (orange in Figure 1A), and the HB exhibits some covalent character, as evidenced, e.g., by topological analysis of electron density.6,11 If such short strong HBs (SSHBs) form in nonpolar environments, the absence of competing hydrogen bonding to the solvent makes their free energy of formation particularly favorable.4,6,12
So far, the role of unusually strong HBs in ligand–target binding has been largely unexplored, and thus, little attention has been paid to such interactions in medicinal chemistry.14,15 To show that SSHBs can in fact be utilized in structure-based drug design, here we demonstrate that a specific strong HB is critical for the target affinity and specificity of the important antitubercular drug bedaquiline (Bq) and that weakening of this bond due to a mutation is responsible for drug resistance. We also point out potential ways to combat this resistance mechanism or broaden the spectrum of activity of Bq to other bacterial pathogens.
Bq (Figure 1B) is a potent diarylquinoline inhibitor of the mycobacterial ATP synthase that has been used for almost 10 years against multi- and extensively drug-resistant tuberculosis.16,17 X-ray and cryo-EM structures have revealed that Bq binds to the outer surface of the c ring–the rotary part of the membrane-embedded Fo subcomplex of ATP synthase (Figure 1C).18,19 Despite some degree of shape complementarity (Figure S1B), mostly nonpolar interactions between Bq and the shallow binding pocket on the c ring (Figure S1C) cannot alone account for the experimental binding free energy of ∼8 kcal/mol,20,21 especially in the lipid environment where the ligand–protein complex cannot be stabilized by hydrophobic attraction. Apparently, the remaining driver is the only polar contact, i.e., the hydrogen bond between the amino group of Bq (encircled in Figure 1B) and the conserved c-ring glutamate at position 65 (E65 in Figure 1D). Indeed, the Bq/c-ring crystal structure indicates that this bond is unusually short (with the average dDA = 2.4 Å),18 and hence, it likely falls within the SSHB category. The fact that binding mechanism of Bq to the c ring is not fully understood hinders the development of its novel derivatives that would evade the emerging resistance22−24 or target ATP synthases other than the mycobacterial one.25,26
To elucidate this mechanism and, particularly, the role of the short HB in the target affinity and selectivity of Bq, we initially determined the free energy landscape governing the binding of Bq to the membrane-embedded c ring using force-field-based molecular dynamics (MD) simulation (green in Figure S2).
The binding free energy (ΔG) obtained in this way (ca. −1 kcal/mol) turned out to be much smaller than that measured by surface plasmon resonance (−8 kcal/mol),20 even though the simulated structure of the complex closely matches the experimental one (RMSD of 0.22 nm; see Figure S3). In fact, the computed affinity was similarly low even when the HB was prevented by removing the proton from E65 (red in Figure S2). Thus, we concluded that the classical description is not adequate to capture the contribution due to the short HB and that the remaining Bq–c-ring interactions contribute relatively little to the target affinity.
Therefore, to understand the nature of the short HB stabilizing the Bq/c-ring complex, we turned to a hybrid QM/MM ab initio MD approach based on density functional theory as implemented in the NAMD/ORCA interface27 (for the definition of the QM regions and other details, see Figure S4 and SI Methods). Specifically, we determined the effective HB potential, similar to those in Figure 1A, by computing how the free energy changes with the oxygen–proton distance, dOH, using the umbrella sampling (US) method (see SI Methods).
The resulting profile (Figure 2B) shows two free energy wells corresponding to the proton residing on the carboxylate O atom of E65 or the amino N atom of Bq. As can be expected for the low dielectric constant of the lipid bilayer, the “salt bridge” configuration with the proton on Bq is slightly disfavored (by ∼0.8 kcal/mol) compared to the neutral configuration with the proton on E65. Importantly, the barrier for the proton transfer between the two heteroatoms (1 kcal/mol) is markedly lower than the typical zero-point energy in the O–H or N–H stretching modes (5–6 kcal/mol), implying a large degree of proton sharing between O and N which is indicative of the formation of SSHB. Consistently with this conclusion, the O–N distance, dON, is on average 2.54 Å (Figure 2C), and the MD ensemble-averaged energy of the HB in the neutral configuration, approximated at the DFT level and corrected for basis set superposition error, is in the range −(19–21) kcal/mol (Table S1 and Figure S8). As expected, the bond becomes particularly short (2.48 Å) and strong (−32 kcal/mol) when the proton is roughly equidistant from both heteroatoms (Figures 2C, S7, and S8). Also, the average value of electron density at the HB critical point, 0.10 e/A3, is characteristic of quasi-covalent hydrogen bonds, which are usually in the range of 0.08–0.14 e/A3 (Figure S9).28
Figure 2.

(A) Definition of the interatomic distances considered in this work. (B) Free energy profile for the proton transfer between the heteroatoms involved in the Bq–E65 hydrogen bond computed using US and the double-ζ basis set. The bottom panel shows the corresponding probability distribution of the dOH distance. For comparison with the profile obtained using the unbiased MD and triple-ζ basis set, see Figure S5. Figure S6 shows the convergence of the free energy profile. (C) Free energy landscapes describing the correlation between dOH, dON, and dNH. For other correlations, see Figure S7.
The same conclusion may be drawn from the NBO-based analysis of the overlap between the lone-pair orbital of the acceptor, nA, and the antibonding orbital of the D–H bond, σ*DH. The “interaction” energy between these orbitals as a function of the proton position (Figure S10) reaches the value of 130 kcal/mol for the proton equidistant from the heteroatoms, which falls in the range characteristic for the strong HBs.1
In essence, our data strongly indicate that Bq forms a short low-barrier hydrogen bond with E65 on the mycobacterial c ring, which accounts for a significant portion of its affinity for the target. Notably, our QM/MM simulations also revealed that while bound to Bq, E65 also forms a stable H-bond with an additional acidic residue, aspartate D32, which is conserved across mycobacteria (Figure 1D). As indicated by the critical point analysis (Figure S9), this additional interaction itself is a relatively weak HB (dOO = 2.65 Å), but its presence may be crucial for strengthening the Bq–E65 bond. This is because it is known that carboxyl groups are particularly prone to form SSHBs when they participate in cooperative hydrogen-bonding networks.29 To put this hypothesis to the test, we removed the additional carboxylate by replacing D32 with alanine and then used QM/MM umbrella sampling to recompute the free energy profile characterizing the Bq–E65 bond. Figure 3A clearly shows that the profile undergoes a striking change upon D32A mutation, assuming the shape characteristic of a normal-strength HB, with a pronounced proton localization on the donor, which is the carboxylate of E65. In line with these findings, the average energy of the bond is reduced by ∼6 kcal/mol (Figure S8), while the electron density at the bond critical point decreases to 0.06 e/A3, a value typical for a normal-strength HB (Figures 3C and S9).
Figure 3.

(A) Comparison of the free energy profiles characterizing the Bq–E65 HB and the corresponding probability distributions between the wild-type c ring (WT) and its D32A variant. For convergence of the free energy profiles, see Figures S6 and S11. (B) Free energy landscapes describing the correlation between dOH, dON, and dNH for Bq bound to WT and D32A. For other geometric correlations, see Figure S12. (C) Critical point analysis characterizing the strength of the interaction between the Bq amino group and E65 in the WT c ring (left) and its D32A variant (right). For numerical data, see Figure S9.
At the same time, the average dON distance increases from 2.54 to 2.67 Å (Figure 3B), and the HB becomes slightly less colinear and orientationally restrained (Figure S12). The above comparison of WT and the D32A mutant (Figure 3) clearly demonstrates that the extra HB to the aspartate is critical for the formation of the strong low-barrier HB between Bq and E65, which in turn appears to be indispensable for high-affinity binding of Bq to the c ring. More broadly, this is a unique example of a strong cooperativity between two H-bonded carboxylic groups that increases the strength of an intermolecular HB by more than 7.0 kcal/mol.29 Given that the additional conserved aspartate residue (here, D32) occurs exclusively in the mycobacterial c ring, it can be further proposed that it is necessary for the Bq target specificity and hence antitubercular activity. Consistently with this line of reasoning, recent studies have associated the resistance to Bq in clinical isolates of Mycobacterium tuberculosis and Mycobacterium smegmatis with a mutation of the aspartate residue in the c ring to a nonacidic amino acid (valine, glycine, or alanine).16,22,30 Previous attempts to enhance the therapeutic potential of Bq and widen its antibacterial spectrum have mostly focused on modifications to the diarylquinoline scaffold, particularly the naphthyl and phenyl moieties, but with only moderate success.24,26,31,32 However, in light of our current results, much more attention should be paid to the hydrogen bond with the conserved acidic residue (here, E65), which provides most of the affinity. Indeed, ensuring strong character of this bond in a way that is independent of the presence of the additional carboxylate (here, D32) should allow us to evade the emerging resistance and, potentially, extend the application of diarylquinoline agents to other important Gram(+) and Gram(−) pathogens.
As a proof of concept of this approach, we designed two analogs of Bq with the amino group replaced by either 2-hydroxy-1-methylimidazole (I) or formamidic acid (II) substituents that were intended to provide stronger H-bonding interaction with E65. As can be seen in Figure 4, the binding energy for compound I, determined through static quantum calculations as the difference between the energy of the complex and that of the isolated protein and ligand, is indeed considerably larger than the one computed previously in the same way for Bq (by 9.7 and 8.0 kcal/mol for WT and D32A variant of the c ring, respectively). This enhanced affinity, even in the absence of the additional carboxyl group, can be attributed to intramolecular cooperativity where the stronger bond to E65 formed by the imidazole N3 atom (average dON = 2.48 Å) is further strengthened by the weaker bond between the 2-OH group and the second O atom of E65 (dOO = 2.76 Å; see Figure S13). Indeed, when the cooperativity is abolished by removing the 2-OH group, the bond formed by N3 is weakened (dON = 2.58 Å), and consequently, the binding energy is reduced by up to 50% (compound III in Figure S14). In contrast, compound II, although it can also form two H-bonds to E65, is characterized by markedly lower binding energy than the original drug (however, largely independent of the presence of D32). The observed decrease in affinity seems to result from enhanced conformational flexibility of the imide substituent, leading to reduced cooperativity. This emphasizes the importance of fine-tuning the ligand H-bonding properties.
Figure 4.
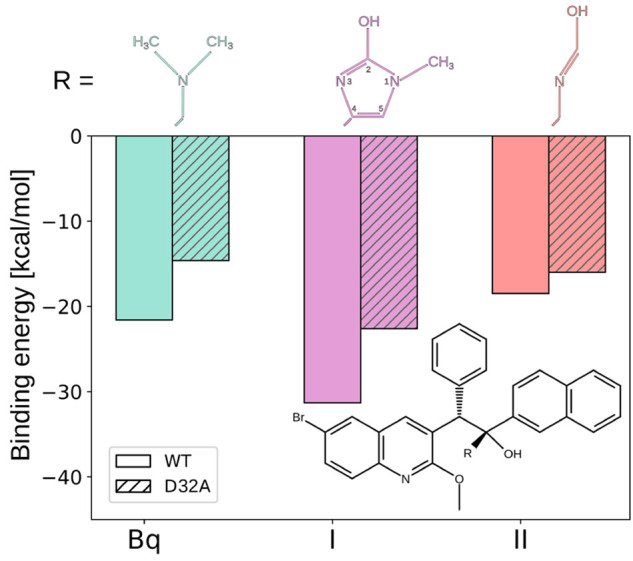
Hydrogen-bond strength between Bq or its two analogs (I and II) and E65 in the WT c ring and its D32A variant, quantified as the binding energy between the ligand’s HB-forming group (R) and either E65/D32 pair (WT) or E65 alone (D32A).
To sum up, in this work we used ab initio and force-field-based MD simulation to show that bedaquiline (Bq), an important antitubercular drug, forms a short strong (low-barrier) hydrogen bond with the conserved carboxylate residue in the c ring of mycobacterial ATP synthase (E65 in Mycobacterium phlei) and that this bond accounts for a significant portion of Bq affinity for the target. We further demonstrated that the unique additional carboxylate residue in the mycobacterial c ring (D32) cooperatively strengthens this HB, providing a mechanistic explanation of the target specificity. Indeed, when this additional carboxylate is removed, the Bq–E65 HB is weakened by ∼32%, which possibly is responsible for the emergence of the Bq-resistant isolates with D32 mutated to nonacidic residues. Building upon these findings, we proposed a novel strategy for overcoming the resistance of mycobacteria to Bq and potentially for broadening its spectrum to other bacterial pathogens. Specifically, we show that the simple imidazole analog of Bq, which regardless of the presence of D32 preserves the strong nature of the HB to E65, has markedly higher affinity for both the WT c ring and its D32A variant. More broadly, our results underscore the importance of considering short strong HBs in drug design and resistance mechanisms, pointing to exciting prospects in the development of novel therapeutics.
Acknowledgments
This work was funded by the Polish National Science Centre under Sonata Bis Grant 2017/26/E/NZ2/00472. S.S. acknowledges the funding provided by the Gdansk University of Technology through the Nobelium grant (16/2021/IDUB/I.1) under “The Excellence Initiative - Research University” (IDUB) Program. This research was supported in part by PL-Grid Infrastructure. Computational resources were also provided by the TASK, WCSS, and ICM Centers.
Glossary
Abbreviations
- SSHB
short strong hydrogen bond
- Bq
bedaquiline
- MD
molecular dynamics
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00509.
Detailed description of computational methods, additional analyses of unbiased and free energy simulations, and results obtained with different functionals and basis sets (PDF)
Author Contributions
∥ J.S. and S.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Grabowski S. J. What Is the Covalency of Hydrogen Bonding?. Chem. Rev. 2011, 111, 2597–2625. 10.1021/cr800346f. [DOI] [PubMed] [Google Scholar]
- Scheiner S. The Hydrogen Bond: A Hundred Years and Counting. J. Indian Inst. Sci. 2020, 100, 61–76. 10.1007/s41745-019-00142-8. [DOI] [Google Scholar]
- Sheu S. Y.; Yang D. Y.; Selzle H. L.; Schlag E. W. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12683–12687. 10.1073/pnas.2133366100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D.; Pinney M. M. Hydrogen Bonds: Simple after All?. Biochemistry 2018, 57, 3338–3352. 10.1021/acs.biochem.8b00217. [DOI] [PubMed] [Google Scholar]
- Saunders L. K.; Nowell H.; Hatcher L. E.; Shepherd H. J.; Teat S. J.; Allan D. R.; Raithby P. R.; Wilson C. C. Exploring short strong hydrogen bonds engineered in organic acid molecular crystals for temperature dependent proton migration behaviour using single crystal synchrotron X-ray diffraction (SCSXRD). CrystEngComm 2019, 21, 5249–5260. 10.1039/C9CE00925F. [DOI] [Google Scholar]
- Zhou S.; Wang L. Unraveling the structural and chemical features of biological short hydrogen bonds. Chem. Sci. 2019, 10, 7734–7745. 10.1039/C9SC01496A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerz I.; Gutmann M. J. Intermolecular OHN hydrogen bond with a proton moving in 3-methylpyridinium 2,6-dichloro-4-nitrophenolate. RSC Adv. 2015, 5, 95576–95584. 10.1039/C5RA06733B. [DOI] [Google Scholar]
- Cleland W. W.; Kreevoy M. M. Low-Barrier Hydrogen Bonds and Enzymic Catalysis. Science 1994, 264, 1887–1890. 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- Perrin C. L. Are short, low-barrier hydrogen bonds unusually strong?. Acc. Chem. Res. 2010, 43, 1550–1557. 10.1021/ar100097j. [DOI] [PubMed] [Google Scholar]
- Kemp M. T.; Lewandowski E. M.; Chen Y. Low barrier hydrogen bonds in protein structure and function. Biochim. Biophys. Acta, Proteins Proteomics 2021, 1869, 140557. 10.1016/j.bbapap.2020.140557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikita H.; Saito K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. R. Soc., Interface 2014, 11, 20130518. 10.1098/rsif.2013.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala P. A.; Ruben E. A.; Liu C. W.; Piccoli P. M. B.; Hohenstein E. G.; Martínez T. J.; Schultz A. J.; Herschlag D. Determination of Hydrogen Bond Structure in Water versus Aprotic Environments To Test the Relationship Between Length and Stability. J. Am. Chem. Soc. 2015, 137, 5730–5740. 10.1021/ja512980h. [DOI] [PubMed] [Google Scholar]
- Chen D.; Oezguen N.; Urvil P.; Ferguson C.; Dann S. M.; Savidge T. C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. 10.1126/sciadv.1501240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurczab R.; Śliwa P.; Rataj K.; Kafel R.; Bojarski A. J. Salt Bridge in Ligand–Protein Complexes—Systematic Theoretical and Statistical Investigations. J. Chem. Inf. Model. 2018, 58, 2224–2238. 10.1021/acs.jcim.8b00266. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Göhlmann H. W. H.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. A Diarylquinoline Drug Active on the ATP Synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Diacon A. H.; Pym A.; Grobusch M. P.; de los Rios E.; Gotuzzo J. M.; Vasilyeva I.; Leimane V.; Andries K.; Bakare T.; De Marez N.; Haxaire-Theeuwes M.; Lounis N.; Meyvisch P.; De Paepe E.; van Heeswijk R. P.; Dannemann B. Multidrug-Resistant Tuberculosis and Culture Conversion with Bedaquiline. N. Engl. J. Med. 2014, 371, 723–732. 10.1056/NEJMoa1313865. [DOI] [PubMed] [Google Scholar]
- Preiss L.; Langer J. D.; Yildiz Ö.; Eckhardt-Strelau L.; Guillemont J. E. G.; Koul A.; Meier T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Courbon G. M.; Bueler S. A.; Mai J.; Liu J.; Rubinstein J. L. Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature 2021, 589, 143–147. 10.1038/s41586-020-3004-3. [DOI] [PubMed] [Google Scholar]
- Haagsma A. C.; Podasca I.; Koul A.; Andries K.; Guillemont J.; Lill H.; Bald D. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS One 2011, 6, e23575. 10.1371/journal.pone.0023575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy J. P.; Ragunathan P.; Shin J.; Cooper C. B.; Upton A. M.; Grüber G.; Dick T. TBAJ-876 Retains Bedaquiline’s Activity against Subunits c and ε of Mycobacterium tuberculosis F-ATP Synthase. Antimicrob. Agents Chemother. 2019, 63, e01191-19. 10.1128/AAC.01191-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segala E.; Sougakoff W.; Nevejans-Chauffour A.; Jarlier V.; Petrella S. New Mutations in the Mycobacterial ATP Synthase: New Insights into the Binding of the Diarylquinoline TMC207 to the ATP Synthase C-Ring Structure. Antimicrob. Agents Chemother. 2012, 56, 2326–2334. 10.1128/AAC.06154-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard M.; Nøhr-Meldgaard K.; Bojer M. S.; Krogsgård Nielsen C.; Meyer R. L.; Slavetinsky C.; Peschel A.; Ingmer H. Inhibition of the ATP Synthase Eliminates the Intrinsic Resistance of Staphylococcus aureus towards Polymyxins. mBio 2017, 8, e01114-17. 10.1128/mBio.01114-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarathy J. P.; Ragunathan P.; Cooper C. B.; Upton A. M.; Grüber G.; Dick T. TBAJ-876 Displays Bedaquiline-Like Mycobactericidal Potency without Retaining the Parental Drug’s Uncoupler Activity. Antimicrob. Agents Chemother. 2020, 64, e01540-19. 10.1128/AAC.01540-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagsma A. C.; Abdillahi-Ibrahim R.; Wagner M. J.; Krab K.; Vergauwen K.; Guillemont J.; Andries K.; Lill H.; Koul A.; Bald D. Selectivity of TMC207 towards Mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. 10.1128/AAC.01393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans W.; Vranckx L.; Lounis N.; Pop O.; Guillemont J.; Vergauwen K.; Mol S.; Gilissen R.; Motte M.; Lançois D.; De Bolle M.; Bonroy K.; Lill H.; Andries K.; Bald D.; Koul A. Novel Antibiotics Targeting Respiratory ATP Synthesis in Gram-Positive Pathogenic Bacteria. Antimicrob. Agents Chemother. 2012, 56, 4131–4139. 10.1128/AAC.00273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo M. C.; Bernardi R. C.; Rudack T.; Scheurer M.; Riplinger C.; Phillips J. C.; Maia J. D.; Rocha G. B.; Ribeiro J. V.; Stone J. E.; Neese F.; Schulten K.; Luthey-Schulten Z. NAMD goes quantum: An integrative suite for hybrid simulations. Nat. Methods 2018, 15, 351–354. 10.1038/nmeth.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata I.; Molins E.; Alkorta I.; Espinosa E. Topological Properties of the Electrostatic Potential in Weak and Moderate N···H Hydrogen Bonds. J. Phys. Chem. A 2007, 111, 6425–6433. 10.1021/jp071924c. [DOI] [PubMed] [Google Scholar]
- Trevisan L.; Bond A. D.; Hunter C. A. Quantitative Measurement of Cooperativity in H-Bonded Networks. J. Am. Chem. Soc. 2022, 144, 19499–19507. 10.1021/jacs.2c08120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrella S.; Cambau E.; Chauffour A.; Andries K.; Jarlier V.; Sougakoff W. Genetic Basis for Natural and Acquired Resistance to the Diarylquinoline R207910 in Mycobacteria. Antimicrob. Agents Chemother. 2006, 50, 2853–2856. 10.1128/AAC.00244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C.; Preiss L.; Wang B.; Fu L.; Wen H.; Zhang X.; Cui H.; Meier T.; Yin D. Structural Simplification of Bedaquiline: the Discovery of 3-(4-(N,N-Dimethylaminomethyl)phenyl)quinoline-Derived Antitubercular Lead Compounds. ChemMedChem 2017, 12, 106–119. 10.1002/cmdc.201600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong A. S.; Choi P. J.; Blaser A.; Sutherland H. S.; Tsang S. K.; Guillemont J.; Motte M.; Cooper C. B.; Andries K.; Van Den Broeck W.; Franzblau S. G.; Upton A. M.; Denny W. A.; Palmer B. D.; Conole D. 6-Cyano Analogues of Bedaquiline as Less Lipophilic and Potentially Safer Diarylquinolines for Tuberculosis. ACS Med. Chem. Lett. 2017, 8, 1019–1024. 10.1021/acsmedchemlett.7b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.