

Abstract
Small molecule toll-like receptor (TLR) 7 agonists have gathered considerable interest as promising therapeutic agents for applications in cancer immunotherapy. Herein, we describe the development and optimization of a series of novel TLR7 agonists through systematic structure–activity relationship studies focusing on modification of the phenylpiperidine side chain. Additional refinement of ADME properties culminated in the discovery of compound 14, which displayed nanomolar reporter assay activity and favorable drug-like properties. Compound 14 demonstrated excellent in vivo pharmacokinetic/pharmacodynamic profiles and synergistic antitumor activity when administered in combination with aPD1 antibody, suggesting opportunities of employing 14 in immuno-oncology therapies with immune checkpoint blockade agents.
Keywords: TLR7 agonist, Tumor, Immuno-oncology, Cancer, PD1
Toll-like receptor 7 (TLR7) is a member of the pattern recognition receptor family and plays a critical role in the innate immune system by detecting pathogens and triggering proinflammatory responses.1 Modulation of TLR7 induces NF-κB and IRF7 activation in a MyD88/IRAK4-dependent manner,2 promoting downstream type I interferon pathways involving plasmacytoid dendritic cells to secrete proinflammatory cytokines such as interferon-α (IFNα).3,4 The potency of TLR7 binders which activate the immune system has garnered interest in utilizing these compounds as therapeutic agents to treat infectious diseases and cancer.1,5−10 Several small molecule TLR7 agonists have been frequently utilized in preclinical and clinical studies, including imiquimod, which obtained FDA approval in 1997 and 2014 for the treatment of genital and perianal warts and superficial basal cell carcinoma, respectively.11−14 Resiquimod is a structurally related analogue of imiquimod and is a potent dual agonist for TLR7 and TLR8, currently being studied in treatments for melanoma and advanced solid tumors.11 More recently, considerable efforts have been made toward combining TLR7/8 agonists with alternative delivery systems15 such as antibody–drug conjugates and prodrug modalities.4,16−24 In addition to being used as a single agent, there is also potential for combining TLR7 agonists with immune checkpoint blockade therapy25−27 to improve response rates in immunologically “cold” tumors.27
In the preceding article, we disclosed a medicinal chemistry campaign which identified compound 1 as a novel and selective TLR7 agonist that showed potent activity in vitro (Table 1).28 On the basis of these initial findings, we embarked on a second round of SAR optimization where our primary objective was to design a compound that (1) is structurally distinct from compound 1, (2) has a favorable ADME profile including good metabolic stability for advancing to in vivo studies, and (3) displays minimal off-target activities including hERG, CYP inhibition, and T-cell toxicity.
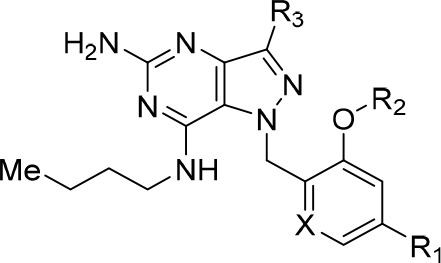
Table 1. Modification at the R1, R2 and R3 Positions and Effects on TLR7 Activity, Metabolic Stability, CYP, and hERG Inhibitiona.

*The CYP inhibition panel included eight isoforms and the most potent IC50 values are shown.
The observed low metabolic stability of compound 1 was attributed to N-dealkylation of the secondary benzylamine substituent, revealed through biotransformation studies. We hypothesized that this liability could be mitigated by replacing the benzylamine moiety with piperazine (Table 1, compound 2). This modification removes the labile benzylamine group while preserving a basic amine in the molecule, allowing for sufficient target engagement in the acidic environment of the endosomal compartment where TLR7 receptors are localized.29,30 Unfortunately, this modification was not tolerated and led to >10-fold reduction in potency in both human and mouse TLR7 reporter assays. While poor mouse metabolic stability was not resolved, significant improvements in human liver microsome stability and CYP inhibition were achieved, where negligible inhibition was observed against eight CYP450 isoforms at concentrations up to 20 μM. We reasoned that the reduction in human and mouse TLR7 activity was due to a decrease in compound basicity, and we were delighted to find that its corresponding piperidine counterpart (compound 3) not only showed improved activity and mouse metabolic stability but also lower PHA blast activity. PHA blast assays were developed to evaluate the cytotoxic potency of TLR agonists toward immune cells. Unfortunately, despite these improvements in potency and metabolic stability, compound 3 now suffered from potent hERG and CYP inhibition.
With increased potency and metabolic stability being achieved, we next focused on optimizing liabilities surrounding CYP and hERG inhibition. Introduction of a polar pyridyl group (compound 4) drastically improved metabolic stability as well as CYP and hERG inhibition profiles at the cost of a 5–10-fold drop in TLR7 reporter assay activity. Attempts to increase lipophilicity through incorporation of a difluoromethoxy group (5) led to a 7-fold drop in TLR7 reporter activity and poor mouse metabolic stability. Installation of small alkyl substituents at the R3 position such as methyl, ethyl, and cyclopropyl groups (6, 7, and 8, respectively) also resulted in significant erosion of potency and worsened mouse metabolic stability.
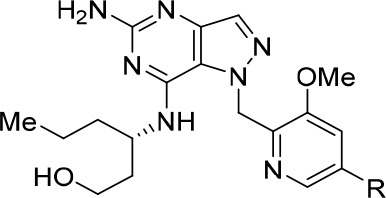
We subsequently shifted our SAR efforts toward modifications at the C-7 position of the core, as well as the piperidine side chain (Table 2) to boost mTLR7 assay potency. We have previously demonstrated that addition of a branched (S)-3-aminohexan-1-ol side chain at the C-7 position provided a favorable combination of potency and physicochemical properties.28 Indeed, incorporation of this aminohexanol side chain in this piperidine series (9) afforded improvements across a range of in vitro parameters without significantly compromising TLR7 activity when compared with compound 4.
Table 2. Effects of Modifications at the R2 Position and R1 Stereochemistry.


Because the piperidine side chain points toward the solvent-exposed region (vide infra), we hypothesized that modifications at the secondary amine could improve potency through tuning of physiochemical properties, namely, membrane permeability. As expected, the compound potency improved substantially via the incorporation of substituents at the R2 position (Table 2). Small hydrophobic groups such as methyl (10, hEC50 = 14 nM) and isopropyl (11, hEC50 = 18 nM) enhanced TLR7 activity, with the methyl analogue providing a 6-fold improvement in potency compared to the unsubstituted piperidine 9, most likely due to greater permeability (PAMPA at pH 7.4 = 9 nm/s (compound 9), 66 (compound 10), and 930 (compound 11)). Capping of the piperidine moiety with an acetonitrile group led to a significant loss of potency, suggesting the importance of the basic piperidine group for TLR7 activity (compound 12). Incorporation of a linear alcohol side chain (13, hEC50 = 49 nM) was found to significantly improve metabolic stability but slightly compromised cellular potency. Pleasingly, installation of a tetrahydropyran (THP) group delivered compound 14 (hTLR7 EC50 = 13 nM), which was equipotent to compound 3. Furthermore, compound 14 demonstrated enhanced stability in the mouse liver microsome stability assay and significantly mitigated hERG and CYP liabilities. Interestingly, tethering the (R) enantiomer of the branched aminohexanol to the C-7 position (15) led to a 21-fold loss of potency, underpinning the importance of stereochemical configuration at the C-7 side chain region. Replacement of the THP group with piperidine (compound 16) attenuated hTLR activity by 24-fold, but potency could be enhanced upon methylation of the piperidine amine (compound 17), underlining the trend between the compound membrane permeability and cellular activity. Overall, the SAR investigations described herein led to the identification of three piperidine capping motifs (methyl, isopropyl, and THP groups), which provided favorable potency, metabolic stability, and minimal inhibition of CYP and hERG.
We next shifted our efforts toward expanding the SAR at the piperidine side chain, focusing on isosteric replacements of the piperidine side chain which has been previously reported to provide lipophilicity-lowering effects compared to the corresponding six-membered heterocycle (Table 3).31 As expected, loss in potency was observed for spirocyclic amine analogues that lacked capping motifs (compounds 18, 19, and 20) most likely due to reduced cellular permeability, and potency was generally restored upon installation of THP (compounds 21, 22, and 23) and isopropyl (compounds 24, 25, and 26) capping groups. Although these investigations did not improve the overall properties of our TLR7 agonists, these observations further validated our design strategy of introducing capping groups at the basic piperidine group to fine-tune compound physicochemical properties. On the basis of TLR7 potency, metabolic stability, and liability profiles, the piperidine series (compounds 11, 13, and 14) were advanced to in vivo studies.
Table 3. Replacement of the Piperidine Group with Spirocyclic Bioisosteres.

We used nontumor bearing mice to establish proof-of-concept of promoting cytokine production with the TLR7 agonists. First, compounds 11, 13, and 14 were evaluated in a single-dose (1 mg/kg) PK/PD study in Balb/C female mice and type I interferon levels were measured at 2 h postadministration (see Supporting Information). Compound 14 was found to be the most effective at promoting IFNα secretion and was progressed to PK/PD evaluation with compound 27, which is disclosed in the preceding manuscript28 (Figure 1a).
Figure 1.
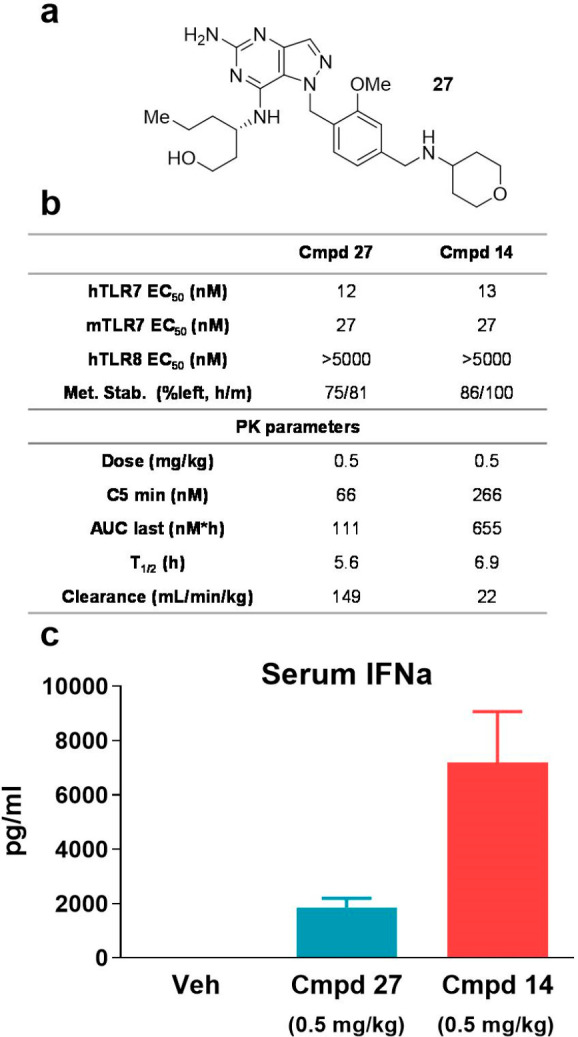
PK/PD studies for compounds 14 and 27 in nontumor bearing female Balb/C mice. (a) Chemical structure of compound 27.28 (b) Comparison of TLR7 potency, metabolic stability and PK profiles for compounds 14 and 27. (c) Production of cytokine IFNα following treatment with TLR7 agonists 14 and 27 2 h postadministration. N = 9 mice per group.
While compounds 14 and 27 showed equipotent TLR7 activity in both human and mouse in vitro reporter assays, compound 14 demonstrated superior metabolic stability, which is also reflected in its in vivo PK profile such as lower clearance rate and higher AUC (area under the curve) compared to 27 (Figure 1b). Pharmacodynamic effects were also assessed by evaluating serum IFNα concentration levels at the 2 h postadministration time point, where compound 14 achieved higher levels of cytokine production compared to 27 (Figure 1c).
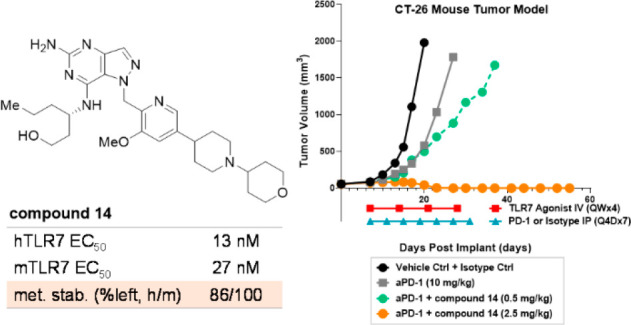
The outcomes of the experiments described above suggested that compound 14 can elicit strong immunostimulatory responses, and to further evaluate its effects on tumor growth, an in vivo efficacy study was performed in a syngeneic CT-26 tumor mouse model in combination with aPD1 antibodies. As shown in Figure 2, aPD1 alone (gray, 10 mg/kg, IP route, Q4Dx7) did not show significant tumor regression, but dose-dependent tumor growth delay was observed when administration of aPD1 was combined with compound 14 at 0.5 (green) and 2.5 (orange) mg/kg (QWx4). Notably, complete tumor regression was observed at the 2.5 mg/kg dose, exhibiting no relapse for the remaining study period (27 days) after halting compound treatment. Overall, this study demonstrated dose-dependent synergistic antitumor activity of TLR7 agonist 14 when coadministered with aPD1 antibodies in vivo.
Figure 2.
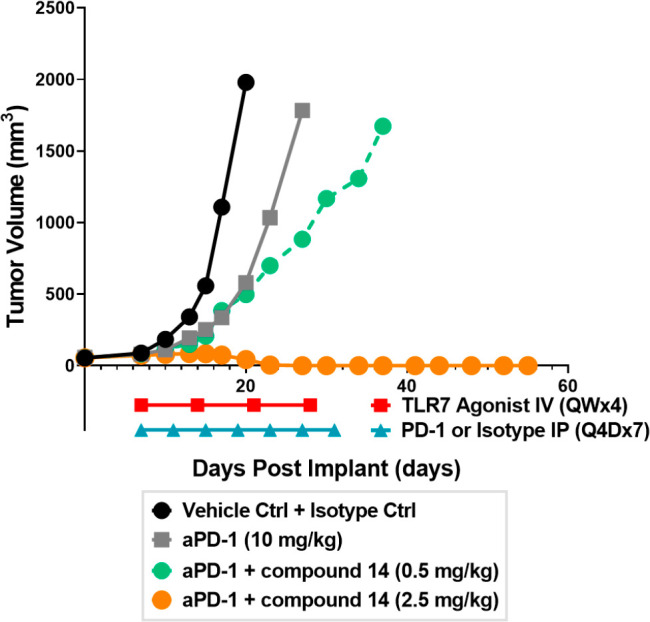
Efficacy of compound 14 in a CT-26 mouse tumor model.
A 2.93 Å resolution cocrystal structure of compound 14 bound to the monkey TLR7 ectodomain highlights key interactions with the target protein. As shown in Figure 3, the small molecule sits at the dimer interface, and the 2-aminopyrimidine group makes a critical bipartite salt bridge interaction with the acidic side chain of Asp555 as well as backbone carbonyl of Thr586. Further stabilization of the complex is achieved through two π–π interactions between the (i) pyrazolopyrimidine core and Phe408 (see Supporting Information) and (ii) methoxy pyridyl side chain and Tyr356. The nitrogen atom of this pyridyl group also engages in a water-mediated hydrogen bond with N-2 of the pyrazolo core. As anticipated, the N–H group at the C-7 position and the methoxy group forms an intramolecular hydrogen bond, where a similar interaction was found in the cocrystal structure of compound 27 and monkey TLR7 protein.28 Binding of the basic piperidine amine to Gln354 is also conserved between the two crystal structures. Furthermore, the aliphatic hydroxyl group of the C-7 side chain engages in extensive electrostatic interactions with the side chains of Thr586 and Tyr264. This cocrystal structure thus provides further evidence of target engagement between compound 14 with the TLR7 receptor.
Figure 3.

X-ray cocrystal structure of compound 14 with monkey TLR7 ectodomain (RSCB PDB 8S9H).
The synthesis of compound 14 commenced with 1,3-bis(methoxycarbonyl)-2-methyl-2-thiopseudourea (28) (Scheme 1). Condensation with methyl 4-amino-1H-pyrazole-5-carboxylate (29) followed by treatment with sodium methoxide afforded the 4-hydroxypyrazolopyrimidine core (30), which was subsequently iodinated using N-iodosuccinimide to afford compound 31. Alkylation of 31 with benzyl chloride 32 provided N1-functionalized product 33 as the major regioisomer. Amination of hydroxypyrazolopyrimidine 33 with secondary amine 34 was achieved with BOP in the presence of DBU to afford compound 35. This material was then reduced with zinc in acetic acid to furnish the pyrazolopyrimidine core 36. Installation of the piperidine side chain was accomplished by Suzuki cross-coupling with vinyl boronate 37 to give compound 38. Hydrogenation, Boc deprotection with trifluoroacetic acid, and reductive amination using tetrahydro-4H-pyran-4-one led to the formation of compound 39. Sequential deprotection of the TBS and methyl carbamate protecting groups successfully delivered compound 14. The synthetic route to access compound 14 was adapted accordingly to generate other analogues discussed in this manuscript (see Supporting Information).
Scheme 1. Synthesis of Compound 14.

Regents and reaction conditions: (a) acetic acid; (b) sodium methoxide, 92% over two steps; (c) N-iodosuccinimide, 95%; (d) 32, Cs2CO3, 76%; (e) 34, BOP, DBU, 71%; (f) zinc, acetic acid, 62%; (g) 37, Pd(dppf)Cl2, K2CO3, 70 °C, 78%; (h) Pd–C/H2; (i) TFA; (j) tetrahydro-4H-pyran-4-one, NaBH(OAc)3, acetic acid; (k) 4 M HCl(aq); (l) NaOH(aq), 80 °C, 55% over five steps
In summary, we report the discovery of a novel series of TLR7 agonists that feature a pyridyl-piperidine side chain tethered to a pyrazolopyrimidine core. Our SAR studies began with compound 1 which was outlined in the preceding manuscript. Despite its decent potency in the reporter assay, compound 1 displayed poor metabolic stability, as well as an unfavorable CYP and hERG inhibition profile. Starting from compound 1, our systematic SAR efforts led to the identification of a new class of pyrazolopyrimidines which contain a piperidine side chain, addressing poor metabolic stability issues associated with N-dealkylation. Subsequent design iterations including incorporation of a pyridyl group and piperidine capping motifs resulted in the discovery of TLR7 agonist 14 that demonstrated nanomolar potency and excellent metabolic stability and was clean in CYP and hERG panels. Importantly, compound 14 demonstrated superior in vivo PK/PD properties compared to lead compound 27, which was disclosed in the preceding manuscript.28In vivo efficacy studies with compound 14 showed synergistic antitumor activity with aPD1 in a CT-26 tumor model, suggesting the potential of combining these TLR7 agonists with immune checkpoint blockade agents in the clinic.
Glossary
Abbreviations
- BOP
(benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate)
- DBU
1,8-diazabicyclo(5.4.0)undec-7-ene
- TFA
trifluoroacetic acid.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00456.
Full experimental details for key compounds, biological assay protocols, and crystallography details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kobold S.; Wiedemann G.; Rothenfußer S.; Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunotherapy 2014, 6 (10), 1085–95. 10.2217/imt.14.75. [DOI] [PubMed] [Google Scholar]
- Chi H.; Li C.; Zhao F. S.; Zhang L.; Ng T. B.; Jin G.; Sha O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Frontiers in Pharmacology 2017, 8, 304. 10.3389/fphar.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy E. J.; Parker A. E.; O’Neill L. A. J. Targeting Toll-like receptors: emerging therapeutics?. Nat. Rev. Drug Discovery 2010, 9 (4), 293–307. 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- Bhagchandani S.; Johnson J. A.; Irvine D. J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Delivery Rev. 2021, 175, 113803. 10.1016/j.addr.2021.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön M. P.; Schön M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008, 27 (2), 190–199. 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- Mackman R. L.; Mish M.; Chin G.; Perry J. K.; Appleby T.; Aktoudianakis V.; Metobo S.; Pyun P.; Niu C.; Daffis S.; Yu H.; Zheng J.; Villasenor A. G.; Zablocki J.; Chamberlain J.; Jin H.; Lee G.; Suekawa-Pirrone K.; Santos R.; Delaney W. E. I. V.; Fletcher S. P. Discovery of GS-9688 (Selgantolimod) as a Potent and Selective Oral Toll-Like Receptor 8 Agonist for the Treatment of Chronic Hepatitis B. J. Med. Chem. 2020, 63 (18), 10188–10203. 10.1021/acs.jmedchem.0c00100. [DOI] [PubMed] [Google Scholar]
- Embrechts W.; Herschke F.; Pauwels F.; Stoops B.; Last S.; Pieters S.; Pande V.; Pille G.; Amssoms K.; Smyej I.; Dhuyvetter D.; Scholliers A.; Mostmans W.; Van Dijck K.; Van Schoubroeck B.; Thone T.; De Pooter D.; Fanning G.; Jonckers T. H. M.; Horton H.; Raboisson P.; McGowan D. 2,4-Diaminoquinazolines as Dual Toll-like Receptor (TLR) 7/8 Modulators for the Treatment of Hepatitis B Virus. J. Med. Chem. 2018, 61 (14), 6236–6246. 10.1021/acs.jmedchem.8b00643. [DOI] [PubMed] [Google Scholar]
- McGowan D. C.; Herschke F.; Pauwels F.; Stoops B.; Smyej I.; Last S.; Pieters S.; Embrechts W.; Khamlichi M. D.; Thoné T.; Van Schoubroeck B.; Mostmans W.; Wuyts D.; Verstappen D.; Scholliers A.; De Pooter D.; Dhuyvetter D.; Borghys H.; Tuefferd M.; Arnoult E.; Hong J.; Fanning G.; Bollekens J.; Urmaliya V.; Teisman A.; Horton H.; Jonckers T. H. M.; Raboisson P. Identification and Optimization of Pyrrolo[3,2-d]pyrimidine Toll-like Receptor 7 (TLR7) Selective Agonists for the Treatment of Hepatitis B. J. Med. Chem. 2017, 60 (14), 6137–6151. 10.1021/acs.jmedchem.7b00365. [DOI] [PubMed] [Google Scholar]
- Schiaffo C. E.; Shi C.; Xiong Z.; Olin M.; Ohlfest J. R.; Aldrich C. C.; Ferguson D. M. Structure–Activity Relationship Analysis of Imidazoquinolines with Toll-like Receptors 7 and 8 Selectivity and Enhanced Cytokine Induction.. J. Med. Chem. 2014, 57 (2), 339–347. 10.1021/jm4004957. [DOI] [PubMed] [Google Scholar]
- Roethle P. A.; McFadden R. M.; Yang H.; Hrvatin P.; Hui H.; Graupe M.; Gallagher B.; Chao J.; Hesselgesser J.; Duatschek P.; Zheng J.; Lu B.; Tumas D. B.; Perry J.; Halcomb R. L. Identification and Optimization of Pteridinone Toll-like Receptor 7 (TLR7) Agonists for the Oral Treatment of Viral Hepatitis. J. Med. Chem. 2013, 56 (18), 7324–7333. 10.1021/jm400815m. [DOI] [PubMed] [Google Scholar]
- Cao X.; Cordova A. F.; Li L. Therapeutic Interventions Targeting Innate Immune Receptors: A Balancing Act. Chem. Rev. 2022, 122 (3), 3414–3458. 10.1021/acs.chemrev.1c00716. [DOI] [PubMed] [Google Scholar]
- Keshavarz A.; Pourbagheri-Sigaroodi A.; Zafari P.; Bagheri N.; Ghaffari S. H.; Bashash D. Toll-like receptors (TLRs) in cancer; with an extensive focus on TLR agonists and antagonists. IUBMB Life 2021, 73 (1), 10–25. 10.1002/iub.2412. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang S.; Li H.; Wang H.; Zhang T.; Hutchinson M. R.; Yin H.; Wang X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53 (5), 1046–1055. 10.1021/acs.accounts.9b00631. [DOI] [PubMed] [Google Scholar]
- Kaushik D.; Kaur A.; Petrovsky N.; Salunke D. B. Structural evolution of toll-like receptor 7/8 agonists from imidazoquinolines to imidazoles. RSC Medicinal Chemistry 2021, 12 (7), 1065–1120. 10.1039/D1MD00031D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodell C. B.; Ahmed M. S.; Garris C. S.; Pittet M. J.; Weissleder R. Development of Adamantane-Conjugated TLR7/8 Agonists for Supramolecular Delivery and Cancer Immunotherapy. Theranostics 2019, 9 (26), 8426–8436. 10.7150/thno.35434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A. T.; Pulukuri A. J.; Davaritouchaee M.; Abbasi A.; Hendricksen A. T.; Opp L. K.; Burt A. J.; Nielsen A. E.; Mancini R. J. Comparing the immunogenicity of glycosidase-directed resiquimod prodrugs mediated by cancer cell metabolism. Acta Pharmacologica Sinica 2020, 41 (7), 995–1004. 10.1038/s41401-020-0432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.-H.; Li Y.-T.; Zhang R.-Y.; Liu Y.-L.; You Z.-W.; Bian M.-M.; Wen Y.; Wang J.; Du J.-J.; Guo J. Alum Adjuvant and Built-in TLR7 Agonist Synergistically Enhance Anti-MUC1 Immune Responses for Cancer Vaccine. Frontiers in Immunology 2022, 13, 857779. 10.3389/fimmu.2022.857779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignacio B. J.; Albin T. J.; Esser-Kahn A. P.; Verdoes M. Toll-like Receptor Agonist Conjugation: A Chemical Perspective. Bioconjugate Chem. 2018, 29 (3), 587–603. 10.1021/acs.bioconjchem.7b00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Wang L.; Wang Z.; Li T.; Chen H.; Zhang Y.; Hu Z.; Dimitrov D. S.; Du J.; Liao X. Immune Modulating Antibody–Drug Conjugate (IM-ADC) for Cancer Immunotherapy.. J. Med. Chem. 2021, 64 (21), 15716–15726. 10.1021/acs.jmedchem.1c00961. [DOI] [PubMed] [Google Scholar]
- Debnath S.; Hao G.; Guan B.; Thapa P.; Hao J.; Hammers H.; Sun X. Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists. International Journal of Molecular Sciences 2022, 23 (13), 7160. 10.3390/ijms23137160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S.; Brems B. M.; Olawode E. O.; Miller J. T.; Brooks T. A.; Tumey L. N. Design and Characterization of Immune-Stimulating Imidazo[4,5-c]quinoline Antibody-Drug Conjugates. Mol. Pharmaceutics 2022, 19 (9), 3228–3241. 10.1021/acs.molpharmaceut.2c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F.; Han S.-W.; Doi T.; Amatu A.; Ajani J. A.; Kuboki Y.; Cortez A.; Cellitti S. E.; Mahling P. C.; Subramanian K.; Schoenfeld H. A.; Choi S. M.; Iaconis L. A.; Lee L. H.; Pelletier M. R.; Dranoff G.; Askoxylakis V.; Siena S. Preclinical Characterization and Phase I Study of an Anti–HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunology Research 2022, 10 (12), 1441–1461. 10.1158/2326-6066.CIR-21-0722. [DOI] [PubMed] [Google Scholar]
- Cortez A.; Li Y.; Miller A. T.; Zhang X.; Yue K.; Maginnis J.; Hampton J.; Hall D. S.; Shapiro M.; Nayak B.; D’Oro U.; Li C.; Skibinski D.; Mbow M. L.; Singh M.; O’Hagan D. T.; Cooke M. P.; Valiante N. M.; Wu T. Y. H. Incorporation of Phosphonate into Benzonaphthyridine Toll-like Receptor 7 Agonists for Adsorption to Aluminum Hydroxide. J. Med. Chem. 2016, 59 (12), 5868–5878. 10.1021/acs.jmedchem.6b00489. [DOI] [PubMed] [Google Scholar]
- Brant M. G.; Garnett G. A. E.; Guedia J.; Lasalle M.; Lawn S.; Petersen M. E.; Duan R.; Mendez-Campos J.; Hirkala-Schaefer T.; Winters G. C.; Barnscher S. D. Generation and structure-activity relationships of novel imidazo-thienopyridine based TLR7 agonists: application as payloads for immunostimulatory antibody drug-conjugates. Bioorg. Med. Chem. Lett. 2023, 91, 129348. 10.1016/j.bmcl.2023.129348. [DOI] [PubMed] [Google Scholar]
- Sato-Kaneko F.; Yao S.; Ahmadi A.; Zhang S. S.; Hosoya T.; Kaneda M. M.; Varner J. A.; Pu M.; Messer K. S.; Guiducci C.; Coffman R. L.; Kitaura K.; Matsutani T.; Suzuki R.; Carson D. A.; Hayashi T.; Cohen E. E. W. , Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397. 10.1172/jci.insight.93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Gao Y.; He L.; Sun S.; Xia T.; Hu L.; Yao L.; Wang L.; Li D.; Shi H.; Liao X. Structure-Based Design of Highly Potent Toll-like Receptor 7/8 Dual Agonists for Cancer Immunotherapy. J. Med. Chem. 2021, 64 (11), 7507–7532. 10.1021/acs.jmedchem.1c00179. [DOI] [PubMed] [Google Scholar]
- Yu X.; Long Y.; Chen B.; Tong Y.; Shan M.; Jia X.; Hu C.; Liu M.; Zhou J.; Tang F.; Lu H.; Chen R.; Xu P.; Huang W.; Ren J.; Wan Y.; Sun J.; Li J.; Jin G.; Gong L. PD-L1/TLR7 dual-targeting nanobody-drug conjugate mediates potent tumor regression via elevating tumor immunogenicity in a host-expressed PD-L1 bias-dependent way. Journal for ImmunoTherapy of Cancer 2022, 10 (10), e004590 10.1136/jitc-2022-004590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudel Y. B.; He L.; Cox M.; Zhang Q.; Johnson W.; Cong Q.; Cheng H.; Chowdari N.; Tarby C.; Donnell A.; Broekema M.; O’Malley D.; Andappan M.; Wang B.; Li Y.-X.; Sivaprakasam P.; Critton D.; Mulligan D.; Sandhu B.; Xie C.; Ramakrishnan R.; Murtaza A.; Schieven G.; Mathur A.; Gavai A.; Vite G.; Gangwar S., Discovery of Novel TLR7 Agonists as Systemic Agent for Combination with aPD1 for Use in Immuno-oncology. ACS Med. Chem. Lett. 2024, 10.1021/acsmedchemlett.3c00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. T.; Bess L. S.; Mwakwari S. C.; Livesay M. T.; Li Y.; Cybulski V.; Johnson D. A.; Bazin H. G. Synthetic Toll-like Receptors 7 and 8 Agonists: Structure–Activity Relationship in the Oxoadenine Series. ACS Omega 2019, 4 (13), 15665–15677. 10.1021/acsomega.9b02138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taar A.; Ganguly D.; Roy S.; Das N.; Sarkar D. Structural Evolution and Translational Potential for Agonists and Antagonists of Endosomal Toll-like Receptors. J. Med. Chem. 2021, 64 (12), 8010–8041. 10.1021/acs.jmedchem.1c00300. [DOI] [PubMed] [Google Scholar]
- Degorce S. L.; Bodnarchuk M. S.; Scott J.S. Lowering Lipophilicity by Adding Carbon: AzaSpiroHeptanes, a log D Lowering Twist. ACS Med. Chem. Lett. 2019, 10, 1198. 10.1021/acsmedchemlett.9b00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.