

Abstract
Background:
Sapanisertib (CB-228/TAK-228) is a potent, selective ATP-competitive, dual inhibitor of mTORC1/2. Metformin is thought to inhibit the mTOR pathway through upstream activation of 5′-AMP-activated protein kinase (AMPK) suggesting combination therapy may enhance antitumor activity of sapanisertib. We report preliminary safety, tolerability, and efficacy from the dose-escalation study of sapanisertib in combination with metformin in patients with advanced solid tumors.
Methods:
Patients with advanced metastatic solid tumors resistant or refractory to standard treatment, with and without mTOR/AKT/PI3K pathway alterations, received sapanisertib 3 or 4 mg daily together with metformin once to three times daily (500–1,500 mg). All patients underwent 14-day titration period for metformin in cycle 1. Tumor measurements were performed following cycle 2 and subsequently every 8 weeks.
Results:
A total of 30 patients were enrolled across four cohorts (3 mg/500 mg; 3 mg/1,000 mg, 4 mg/1,000 mg; 4 mg/1,500 mg). 19 were female (63%), median age was 57 (range: 30–77), all were Eastern Cooperative Oncology Group performance status 1. Tumor types included sarcoma (6), breast (4), ovarian (4), head and neck (3), colorectal (2), lung (2), renal cell (2), endometrial (2), gastroesophageal junction (1), prostate (1), stomach (1), urachus (1), and cervical cancer (1). Median number of prior lines of therapy was 4. Most common genomic alterations included PIK3CA (27%), PTEN (17%), AKT1/2 (10%), mTOR (10%). Of 30 patients evaluable for response, 4 patients achieved partial response (PR); 15 patients achieved stable disease (SD) as best response. Disease control rate (PR+SD) was 63%. Of the responders in PR, 3 of 4 patients had documented PTEN mutations (3/5 patients enrolled with PTEN mutations had PR); 2 of 4 of patients in PR had comutations (patient with leiomyosarcoma had both PTEN and TSC; patient with breast cancer had both PTEN and STK11); 1 of 4 patients in PR had AKT and mTOR mutation; tumor types included leiomyosarcoma (n = 2), breast (n = 1), and endometrial cancer (n = 1). Most common treatment-emergent adverse events included nausea, anorexia, diarrhea, and rash. Grade (G) 3–5 treatment-related adverse events included hyperglycemia (4/30; 13%), fatigue (2/30; 7%), hypertriglyceridemia (1/30; 3%), rash (2/20; 7%), diarrhea (2/30; 7%), creatinine increase (1/30; 3%), acidosis (1/30; 3%). No dose-limiting toxicities (DLT) were reported in the 3 mg/500 mg cohort. One of 6 patient had DLT in the 3 mg/1,000 mg cohort (G3 diarrhea) and 2 of 11 patients had DLTs in the 4 mg/1,500 mg cohort (G3 fatigue, G3 rash). 4 mg/1,000 mg was defined as the MTD.
Conclusions:
The safety profile of mTORC1/2 inhibitor sapanisertib in combination with metformin was generally tolerable, with antitumor activity observed in patients with advanced malignancies harboring PTEN mutations and AKT/mTOR pathway alterations.
Significance:
Sapanisertib (CB-228/TAK-228) is a potent, selective ATP-competitive, next-generation dual inhibitor of mTORC1/2. Metformin is thought to inhibit the mTOR pathway through upstream activation of AMPK suggesting combination therapy may enhance antitumor activity of sapanisertib. This dose-escalation study of sapanisertib and metformin in advanced solid tumors and mTOR/AKT/PI3K pathway alterations, demonstrates safety, tolerability, and early clinical activity in advanced malignancies harboring PTEN mutations and AKT/mTOR pathway alterations.
Clinical trial information: NCT03017833
Introduction
The PI3K/v-Akt Murine Thymoma Viral Oncogene (AKT)/mTOR pathway plays a diverse role in regulation of several cellular functions such as cellular growth, proliferation, and survival (1). This pathway is frequently dysregulated in various cancers, such endometrial, cervical, lung, prostate, skin, and breast cancer (2–4), and mTOR is a key node and central regulator in this pathway.
mTOR exists in two physically and functionally distinct protein signaling complexes, one with raptor, which is sensitive to the mTOR inhibitor rapamycin, and the other with rictor, which is rapamycin insensitive (5). The first, mTOR complex 1 (mTORC1), phosphorylates 4EBP1 (the eukaryotic translation initiation factor 4E-binding protein 1) and p70S6 kinase and results in translation of proteins involved in cell-cycle progression. The second, mTOR complex 2 (mTORC2), has been shown to directly phosphorylate and activate the upstream kinase AKT at serine 473, which promotes proliferation and survival pathways (6). The rapalogs, such as everolimus, exert their inhibitory action predominantly via mTORC1, while their inhibitory effect on mTORC2 is limited or weak (7–10). Consequently, continued signaling through significant pathway feedback loops, results in upregulation of AKT, which has the undesirable effect of accelerating cell proliferation, antagonizing the antiproliferative effect of mTOR inhibition (11). Thus, next-generation dual inhibitors of mTORC1 and mTORC2 have been developed, and preclinical models have supported the potency of this dual inhibition strategy (12–15).
Recently, mTOR inhibitors have been approved by the FDA for the treatment of advanced renal, breast and several other cancers (7–10, 16). Sapanisertib (CB-228, formerly TAK-228 and MLN0128) is a potent, selective ATP-competitive, dual inhibitor of mTORC1/2, regulated by upstream receptor tyrosine kinases, such as insulin-like growth factor-1 receptor (IGF-1R). In early-phase clinical trials, single-agent sapanisertib was well tolerated and demonstrated preliminary antitumor activity in renal cell carcinoma and endometrial cancer (17).
Metformin, a common antidiabetic medication has been repurposed as antineoplastic agent to enhance the effect of chemotherapy in certain cancers (18). The mechanisms attributed to antihyperglycemic effects of metformin are manifold, including activation of 5′-AMP-activated protein kinase (AMPK), decreased production of cyclic AMP, suppression of mitochondrial complex I of the electron transport chain, glycerophosphate dehydrogenase targeting, and gut microbiome alterations (19). Mechanistically, metformin inhibits the mTOR pathway through upstream activation of AMPK, which results in phosphorylation and activation of the tumor suppressor gene TSC2. This decreases AKT activation, and exerts an inhibitory effect on mTOR (20). Preclinically, metformin-induced activation of AMPK has been shown to disrupt cross-talk between insulin/IGF-1R and G protein–coupled receptors signaling in pancreatic cancer cells, (21) and inhibit cell proliferation, reduce colony formation, and inhibit MAP kinase, AKT, and mTOR in estrogen receptor–positive and –negative, as well as ERBB2-normal and -overexpressing breast cancer cell lines (22). In addition, population studies have reported a decrease in the incidence and mortality rate in patients with cancer taking metformin for management of diabetes (23–26).
Taken together, these observations suggest that combination therapy using metformin and sapanisertib which both target the mTOR pathway could complement and enhance antitumor activity of sapanisertib. Here, we report the preliminary safety, tolerability, and efficacy from the dose-escalation study of sapanisertib in combination with metformin in patients with advanced solid tumors and mTOR/AKT/PI3K pathway alterations.
Materials and Methods
Study Design
This was an open-label, single-center investigator-initiated phase I clinical trial that employed a 3 + 3 dose-escalation design and was conducted at The University of Texas MD Anderson Cancer Center (MDACC; Houston, TX) per Institutional Review Board guidelines. The primary endpoint was to evaluate the safety and tolerability of sapanisertib in combination with metformin, to determine MTD and dose-limiting toxicities (DLT) of the combination in patients with advanced cancers refractory to standard therapy. The secondary objective was to evaluate preliminary antitumor efficacy of this treatment per RECIST version 1.1 (RECIST v1.1; ref. 27). Dose escalation included four dose levels. During dose escalation, patients received one of four dosing schedules: sapanisertib 3 or 4 mg once daily in combination with metformin one to three times daily (500 mg/1,000 mg/1,000 mg/1,500 mg; Fig. 1). The dosing was based on data from previous phase I studies with this agent. Study INK128-001 was a phase I, open-label study of single-agent TAK-228 administered to patients diagnosed with advanced solid malignancies, including, but not limited to, colorectal, renal, endometrial, and urothelial tumors. Four dosing schedules were evaluated (once daily, every week, once daily × 3 days every week, and once daily × 5 days every week). The MTDs for the four schedules investigated in INK128-001 were determined to be 6 mg for once daily dosing, 16 mg for once daily × 3 days every week dosing, 10 mg for once daily × 5 days every week dosing, and 40 mg for every week dosing. On the basis of these data and pharmacokinetic data, it was decided to use the Q Day dosing schema in combination with metformin.
FIGURE 1.

Trial schema.
All patients underwent 14-day titration period for metformin in cycle 1 to limit side effects and commenced sapanisertib on cycle 1 day 15; thereafter, treatment was administered in 4-week cycles. Tumor measurements were performed following cycle 2 and subsequently every 8 weeks. Data cut-off date was March 31, 2021.
DLT was defined if the events occurred within the days 15–42 of the first cycle. Definitions for DLTs, as predetermined in the study protocol, were: any grade ≥3 non-hematologic toxicity, except for the following: grade 3 hyperglycemia lasting ≤14 days (all patients should have received optimal antiglycemic treatment, including insulin, as clinically indicated); grade 3 rash lasting ≤3 days (all patients should have received topical steroid treatment, oral antihistamines, and oral steroids, if indicated); inadequately treated grade 3 nausea and/or vomiting and grade 3 diarrhea (all patients should have received optimal antiemetic and/or antidiarrheal prophylaxis and/or treatment as indicated); grade 4 neutropenia lasting >7 days in the absence of growth factor support; any grade neutropenia of any duration accompanied by fever ≥38.5°C and/or systemic infection; any other ≥ grade 4 hematologic toxicity; inability to administer at least 75% of planned doses of TAK-228 within cycle 1, due to study drug-related toxicity; grade 3 thrombocytopenia with bleeding; any death not clearly due to underlying disease or extraneous causes.
Patients
The study accrual period was from March 12, 2018 to 2020. Eligible patients included patients with metastatic or advanced solid tumors not amendable to standard therapy, an Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0–1 and adequate hematologic, hepatic, and renal function. Patients had to have the ability to swallow oral medications. Diabetic patients with well-controlled diabetes (i.e., normal range HbA1C ≤7%) were permitted and allowed to be on antidiabetic treatment other than metformin. Notable exclusion criteria included poorly controlled diabetes mellitus [defined as glycosylated hemoglobin (HbA1c) >7%], prior treatment with dual PI3K/mTOR inhibitors, TORC1/2 inhibitors or TORC1 inhibitors, significant cardiovascular or pulmonary disease, intercurrent uncontrolled illness, patients receiving corticosteroids (either intravenous or oral steroids, excluding inhalers or low-dose hormone replacement therapy) within 1 week before administration of the first dose of study drug, and malabsorption due to prior gastrointestinal (GI) surgery, GI disease (e.g., patients with enteric stomata were excluded). Patients with treated, stable brain metastases (defined as no evidence of disease progression for ≥3 months before the first dose of study drug, no hemorrhage after treatment, off-treatment with dexamethasone for 4 weeks before administration of the first dose of sapanisertib, no ongoing requirement for dexamethasone or antiepileptic drugs) were permitted.
Assessments
Patients who received at least one dose of any study drug were included in the safety population. DLT-evaluable population was defined as all patients who either experienced DLT during cycle 1 or completed treatment with at least 75% of the planned doses of any study drug in cycle 1 and had sufficient follow-up data to allow the investigators to determine whether DLT occurred. The response-evaluable population was defined as all patients who had measurable disease according to RECIST v1.1, who had received at least one dose of any study drug, and who have at least one available postbaseline response assessment as per RECIST v1.1. Response was assessed according to the RECIST v1.1 after every two treatment cycles. Adverse events (AE) were assessed using the NCI Common Terminology Criteria for Adverse Events, version 4.03.
Statistical Analysis
No formal hypotheses were tested, and analyses were descriptive and exploratory. Nonparametric correlations were determined with Spearman rank correlation coefficient.
Ethical Approval and Consent to Participate
The protocol was approved by the Institutional Review Board at The University of Texas MDACC (Houston, TX). The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All patients provided written informed consent before enrollment.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Results
Patients
The patients reported here include all patients with heavily pretreated advanced solid tumors treated as part of a dose-escalation study. A total of 30 patients with advanced or metastatic solid tumors received treatment with sapanisertib and metformin on a range of dose levels. Baseline demographics and characteristics are shown in Table 1. Nineteen patients were female (63%), 11 patients were men (37%), and the overall median age was 57 (range: 30–77 years). The most frequent tumor types enrolled were sarcoma (6), breast (4), ovarian (4), head and neck (3), colorectal (2), lung (2), renal cell (2), endometrial (2), gastroesophageal junction (1), prostate (1), stomach (1), bladder (1), and cervical cancer (1). Patients were enrolled across four cohorts and received sapanisertib in combination with metformin in the following dose combinations (3 mg/500 mg; 3 mg/1,000 mg, 4 mg/1,000 mg; 4 mg/1,500 mg; Fig. 1). Nineteen patients were female (63%), median age was 57 (range: 30–77), all patients were ECOG PS 1. Median number of prior lines of therapy was 4 and 70% of patients (n = 21) and more than three lines of therapy.
TABLE 1.
Baseline patient demographics
| Characteristic | Number (%) |
|---|---|
| Gender | |
| Female | 19 (63) |
| Male | 11 (37) |
| Median age at study enrollment, years (range) | 57 (30–77) |
| Ethnicity | |
| Asian | 3 (10) |
| White | 21 (70) |
| Hispanic | 2 (7) |
| African American | 3 (10) |
| Other | 1 (3) |
| Number of metastatic sites | |
| ≤3 | 27 (90%) |
| >3 | 3 (10%) |
| Disease type | |
| Breast | 4 (13) |
| Colon | 2 (7) |
| Head and neck | 3 (10) |
| Lung | 2 (7) |
| Ovarian | 4 (13) |
| Renal cell carcinoma (RCC) | 2 (7) |
| Sarcoma | 6 (20) |
| Others (endometroid 2, gastroesophageal junction 1, prostate 1, stomach 1, urachus 1, cervix 1) | 7 (23) |
| ECOG PS | |
| 0 | 0 (0) |
| 1 | 30 (100) |
| Number of prior therapies (range) | (0–11) |
| 1–2 | 9 (30) |
| 3–4 | 14 (47) |
| 5 or more | 7 (23) |
DLTs and MTD Determination
Dose escalation, DLTs, and MTDs are summarized in Supplementary Table S1; dose was escalated to level 4. No DLTs were reported in the first cohort treated, dose level 1 (3 mg/500 mg). One patient (1/6) experienced DLT of grade 3 diarrhea in the second cohort, dose level 2 (sapanisertib 3 mg/ metformin 1,000 mg), which was managed supportively with no dose reductions. There were no DLTs in dose level 3. Two further patients (2/11) had DLTs in dose level 4 (sapanisertib 4 mg/metformin 1,500 mg cohort), one patient with grade 3 fatigue, which required two dose-level reductions for sapanisertib; one patient with grade 3 rash, which required reduced dose level on restarting medication. Both these patients were able to continue treatment after dose modification. Of 30 treated patients, 4 patients (13.3%) had dose changes with either drug. Three patients (10%) had dose reduction with sapanisertib. On the basis of these findings, sapanisertib 4 mg in combination with metformin 1,000 mg was defined as the MTD and recommended phase II dose.
Treatment Response
Overall, there were 24 response-evaluable patients per RECIST v1.1. For these 24 patients, 22 had percent change from baseline tumor size measured (Fig. 2). Two patients who had progression due to unequivocal progression in non-target lesions did not have a tumor size measurement. Tumor response across cohorts is summarized in Table 2. Of the 24 evaluable patients, 19 patients (79%) had documented disease control [defined as stable disease (SD) or partial response (PR) per RECIST v1.1 of more than 16 weeks]. Notably, 4 patients (17%) achieved PR as best tumor response; these responses were in patients with leiomyosarcoma (n = 2), breast cancer (n = 1), and endometrial cancer (n = 1; Fig. 2). PRs were achieved at dose level 1 (n = 1), dose level 2 (n = 1), and dose level 3 (n = 2). The most frequent reason for study discontinuation was progressive disease (PD) in 63% of all patients. Overall, the median overall survival was 18 months [95% confidence interval (CI), 7.2–NE] and median progression-free survival was 6.0 months (95% CI, 2.8–14.6); median follow-up was 19 months (Supplementary Fig. S1).
FIGURE 2.
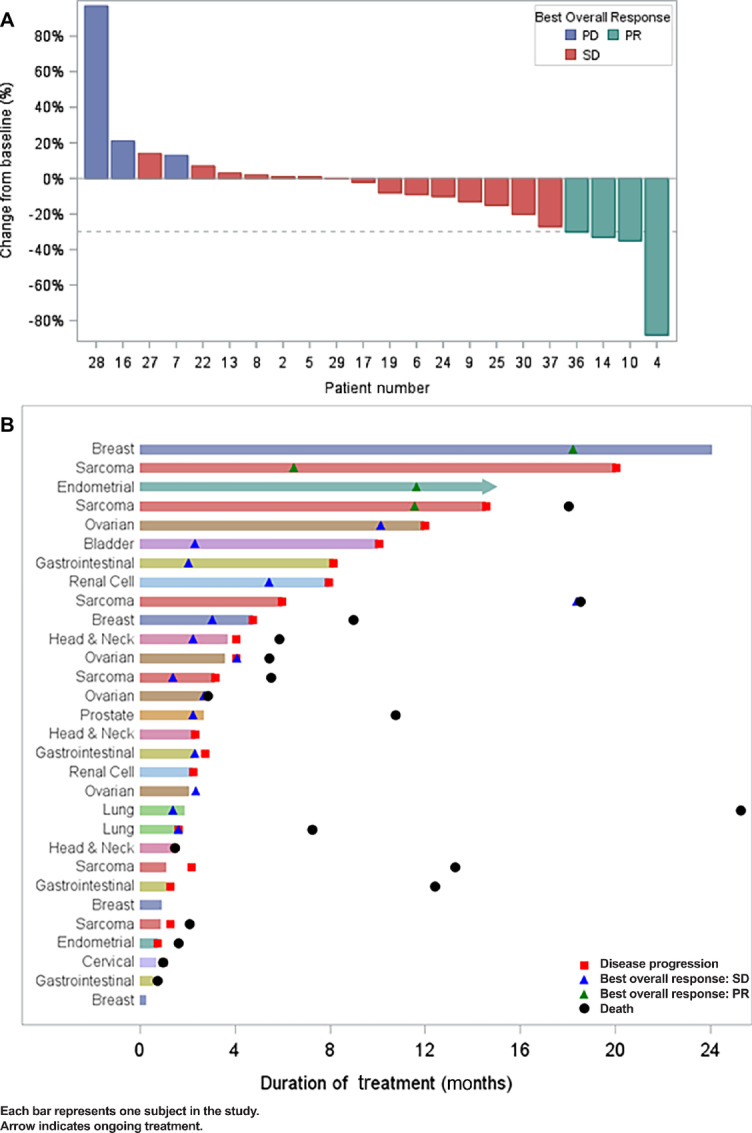
A, Waterfall plot showing best overall response of evaluable patients on trial. Among 30 patients in the dataset, 24 patients were evaluated for best overall response and 6 patients were not evaluated. Among 24 with best overall response, 4 had PR, 15 had SD, and 5 had PD. For these 24 patients, 22 had percent change from baseline tumor size measured. Two patients who had progression due to unequivocal progression in non-target lesions did not have a tumor size measurement. B, Swimmers plot. All 30 patients treated on study are included. PR, SD, PD, death, and ongoing treatment are indicated.
TABLE 2.
Tumor response according to RECIST v1.1 (investigator assessment) in response-evaluable patients
| Best confirmed response | Number of patients |
|---|---|
| Partial response (PR) | 4 (13%) |
| Stable disease (SD) | 15 (50%) |
| Progressive disease (PD) | 5 (17%) |
| Not evaluable | 6 (20%) |
Genomic Alterations
Three out of 4 patients in PR had documented PTEN mutations (3 out of 5 patients with PTEN mutations enrolled on study achieved PR; Supplementary Table S2); 2 of these patients (2/4) had concomitant alterations in the PI3K pathway, that is, these patients had documented lesions in PTEN and STK11 (patient with breast cancer), PTEN and TSC2 (patient with leiomyosarcoma), respectively. The remaining patient in PR had dual mutations in AKT and mTOR; this patient with endometrial carcinoma (and dual AKT E17K and mTOR mutation A1459D) remains on treatment (currently on 26 months of treatment), in ongoing PR. Of all treated patients, the most common genomic alterations included mutations in PIK3CA (27%), PTEN (20%), AKT1/2 (10%), mTOR (10%; Supplementary Fig. S1; Supplementary Table S2). Comutations were frequently observed: 50% of treated patients had ≥2 genomic aberrations, including PIK3CA + NF1, AKT1, TSC2, PTEN; PTEN + STK11, TSC2, PIK3R1, PIK3CA; AKT + mTOR, PIK3CA; mTOR + PTEN, AKT (Fig. 3).
FIGURE 3.

Oncoplot showing mutations and co-occurring alterations in the patients enrolled in this clinical trial.
AEs
The most common treatment-emergent AEs, irrespective of attribution to study drug, are summarized by severity in Table 3 (severity graded per NCI CTCAE criteria v4.03). The most common grade 1–2 treatment-emergent AEs included nausea, vomiting, anorexia, diarrhea, fatigue, rash, hyperglycemia, and weight loss. Grade 3 to 5 treatment-related AEs included hyperglycemia (4/30; 13%), fatigue (2/30; 7%), hypertriglyceridemia (1/30; 3%), rash (2/20; 7%), diarrhea (2/30; 7%), creatinine increase (1/30; 3%), acidosis (1/30; 3%). Grade 5 acidosis was related to a hyperglycemia event. Patient was admitted with diabetic ketoacidosis (blood glucose >350 mg/dL). Hyperglycemia was likely induced by the mTOR inhibition in the setting of concomitant steroids. This event was attributed to the trial. After acute management of diabetic keto acidosis and resolution of hyperglycemia event patient was able to restart at reduced dose.
TABLE 3.
AEs. Most common AEs are summarized by severity, severity based on NCI CTCAE criteria version 4.03
| AE | G1 | G2 | G3 | G4 | G5 | Total grade 1–2 (%) | Total grade 3–5(%) |
|---|---|---|---|---|---|---|---|
| Acidosis | 1 | 1 (3) | |||||
| ALT increased | 1 | 1 (3) | 0 (0) | ||||
| Anemia | 1 | 1 (3) | 0 (0) | ||||
| Anorexia | 11 | 2 | 13 (43) | 0 (0) | |||
| AST increased | 1 | 1 (3) | 0 (0) | ||||
| Constipation | 1 | 1 (3) | 0 (0) | ||||
| Creatinine increased | 1 | 1 | 1 (3) | 1(3) | |||
| Dehydration | 1 | 1 (3) | 0 (0) | ||||
| Diarrhea | 13 | 5 | 2 | 18 (60) | 2(7) | ||
| Dizziness | 2 | 2 (7) | 0 (0) | ||||
| Dry mouth | 2 | 2 (7) | 0 (0) | ||||
| Dyspnea | 1 | 1 (3) | 0 (0) | ||||
| Fatigue | 2 | 4 | 2 | 6 (20) | 2(7) | ||
| Headache | 2 | 2 (7) | 0 (0) | ||||
| Hyperglycemia | 1 | 5 | 4 | 6 (20) | 4(13) | ||
| Hyperlipidemia | 2 | 2 (7) | 0 (0) | ||||
| Hypertriglyceridemia | 2 | 1 | 2 (7) | 1 (3) | |||
| Hyperuricemia | 1 | 1 (3) | 0 (0) | ||||
| Hypomagnesemia | 1 | 1 (3) | 0 (0) | ||||
| Increased HbA1C | 8 | 8 (27) | 0 (0) | ||||
| Mouth sore (mucositis) | 7 | 1 | 8 (27) | 0 (0) | |||
| Nausea | 11 | 3 | 14 (47) | 0 (0) | |||
| Pruritus | 6 | 1 | 7 (23) | 0 (0) | |||
| Rash | 10 | 2 | 2 | 12 (40) | 2(7) | ||
| Taste change | 2 | 2 (7) | 0 (0) | ||||
| Tremor | 1 | 1 | 1 (3) | 0 (0) | |||
| Vomiting | 11 | 1 | 12 (40) | 0 (0) | |||
| Weight loss | 7 | 2 | 9 (30) | 0 (0) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Discussion
This open-label single-institution phase I trial studied the safety and tolerability of the sapanisertib and metformin in combination with patients with advanced refractory cancer and AKT/mTOR/PI3K alterations. The safety profile of mTORC1/2 inhibitor sapanisertib in combination with metformin was generally tolerable, and toxicities observed were attributable to mTOR inhibition consistent with previously published literature. In addition, the combination of sapanisertib 4 mg plus metformin 1,000 mg was defined as the MTD and recommended phase II dose.
Previous groups have demonstrated treatment-related reductions in mTORC biomarkers, including TORC1/2 skin biomarkers (phosphorylated S6, 4EBP1, and PRAS40; ref. 28) which supports the dual TORC1/2 inhibitory activity of sapanisertib. In addition, a recently published phase I study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumors, with an expansion in renal, endometrial, or bladder cancer confirmed the PD effect of sapanisertib on downstream effectors of TORC1 (p4EBP1 and pS6) and TORC2 (pPRAS40 and pNDRG1), with treatment-related decreases in p4EBP1, pS6, pPRAS40, and pNDRG1 using single-agent sapanisertib doses of ≥4 mg (17).
We demonstrate strong preliminary antitumor activity using sapanisertib in combination with metformin in a number of dose levels in molecularly selected patients of a range of tumor types: Disease control was observed in patients of various advanced malignancies harboring PTEN mutations and AKT/mTOR pathway alterations. Notably, 4 out of 24 evaluable patients achieved PR as best tumor response (17%) at a range of dose levels, emphasizing the antitumor activity of this combination. In addition to radiographic responses to this combination in this trial, several patients experienced a durable clinical benefit: out of all evaluable patient per RECIST, 79% (19 patients) had documented disease control (SD + PR). Three out of four PRs achieved were double mutants, that is, these patients had documented lesions in PTEN and STK11, PTEN and TSC2, AKT and mTOR, respectively, suggesting that perhaps hyperactivation of the pathway is important for responses to this combination. No objective responses were seen in patients harboring only PIK3CA mutations (PIK3CA E545K, PIK3CA H1047R, PIK3CA H1047L, other). Best responder was decrease of 15% in target lesions in a patient with liposarcoma harboring PIK3CA p.E545K with co-occuring NF1 p.S1420* alteration.
As P13K/mTOR pathway is concurrently activated in multiple tumors it may be worthwhile to explore combination therapy with agents with non-overlapping toxicity. In a translational study, it was shown that the double mutations hyperactivate PI3K signaling with increased tumor growth in preclinical models and also early clinical trial data showed that breast cancers harboring double mutations were more sensitive and responded better to PI3K inhibitors than those with a single mutation. It is possible that this “double mutation” in the same pathway is functional similarly in other tumors beyond breast cancer.
There are several limitations of this study, including the small number of patients treated, and the inclusion of many patients with comutations in addition to mTOR/AKT/PI3K pathway alterations, which may affect the underlying oncogenic driver pathway. Regardless, we show clinical activity in heavily pretreated patients which suggest that further study of sapanisertib in combination with metformin is warranted, particularly in patients with advanced malignancies harboring PTEN mutations and AKT/mTOR pathway alterations. This regimen could prove to be a highly effective treatment option, and a phase II study is warranted at the recommended phase II dose. In conclusion, sapanisertib had a manageable safety profile and the combination of sapanisertib and metformin was tolerable and exhibited preliminary therapeutic activity.
Supplementary Material
Supplementary figure and tables
Representativeness of Study Participants
Acknowledgments
We thank the patients, and their families who participated in this study. V. Subbiah is an Andrew Sabin Family Foundation fellow at the University of Texas MD Anderson Cancer Center. V. Subbiah acknowledges the support of the Jacquelyn A. Brady Fund. V. Subbiah thanks the team at Draw Impacts for figures. V. Subiah is supported by the US NIH (grants R01CA242845 and R01CA273168); the MD Anderson Cancer Center Department of Investigational Cancer Therapeutics is supported by the Cancer Prevention and Research Institute of Texas (grant RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy (grant 1U01CA180964), NCATS (Center for Clinical and Translational Sciences; grant UL1TR000371) and the MD Anderson Cancer Center Support (grant P30CA016672).
Funding: The trial is an investigator-initiated trial originally funded by Takeda. The current sponsor of the drug is Calithera Biosciences. This work was supported by Takeda and Calithera Bioscience. The University of Texas MD Anderson Cancer Center Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer, the NIH Clinical Translational Science Award (1UL1TR003167), and The University of Texas MD Anderson Cancer Center Support Grant (P30 CA016672). Supported in part by CPRIT PODDS - The Cancer Prevention and Research Institute of Texas (RP150535).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
V. Subbiah reports grants from Takeda and Calithera Biosciences during the conduct of the study; and V. Subbiah reports research funding from Novartis to conduct clinical trials; other grant support for clinical trials from AbbVie, Agensys, Inc, Alfasigma, Altum, Amgen, Bayer, BERG Health, Blueprint Medicines Corporation, Boston Biomedical, Inc, Boston Pharmaceuticals, Celgene Corporation, D3 Bio, Inc, Dragonfly Therapeutics, Inc, Exelixis, Fujifilm, GlaxoSmithKline, Idera Pharmaceuticals, Inc, Incyte Corporation, Inhibrx, Loxo Oncology/Eli Lilly, MedImmune, MultiVir, Inc, NanoCarrier, Co, National Comprehensive Cancer Network, NCI-CTEP, Novartis, PharmaMar, Pfizer, Relay Therapeutics, Roche/Genentech, Takeda, Turning Point Therapeutics, UT MD Anderson Cancer Center, and Vegenics Pty Ltd; travel support from ASCO, ESMO, Helsinn Healthcare, Incyte Corporation, Novartis, and PharmaMar; consultancy or advisory board participation for Helsinn Healthcare, Incyte Corporation, Loxo Oncology/Eli Lilly, MedImmune, Novartis, QED Therapeutics, Relay Therapeutics, Daiichi-Sankyo, and R-Pharm US; and other relationship with Medscape. S.A. Piha-Paul reports other from AbbVie, Inc., ABM Therapeutics, Inc., Acepodia, Inc, Alkermes, Aminex Therapeutics, Amphivena Therapeutics, Inc., BioMarin Pharmaceutical, Inc, Boehringer Ingelheim, Bristol Myers Squib, Cerulean Pharma, Inc., Chugai Pharmaceutical Co., Ltd, Curis, Inc., Cyclacel Pharmaceuticals, Daiichi Sankyo, Eli Lilly, ENB Therapeutics, Epigenetix Inc., Five Prime Therapeutics, F-Star Beta Limited, F-Star Therapeutics, Gene Quantum, Genmab A/S, Gilead Sciences, Inc., GlaxoSmithKline, Helix BioPharma Corp., Hengrui Pharmaceuticals, Co., Ltd., HiberCell, Inc., Immorna Biotherapeutics, Inc., Immunomedics, Inc., Incyte Corp., Jacobio Pharmaceuticals Co., Ltd., Jiangsu Simcere Pharmaceutical Co., Ltd., Lytix Biopharma AS, Medimmune, LLC., Medivation, Inc., Merck Sharp & Dohme Corp., Nectin Therapeutics, Ltd., Novartis Pharmaceuticals, Pieris Pharmaceuticals, Inc., Pfizer, Phanes Therapeutics, Principia Biopharma, Inc., Puma Biotechnology, Inc., Purinomia Biotech, Inc., Rapt Therapeutics, Inc., Replimune, Seattle Genetics, Silverback Therapeutics, Synlogic Therapeutics, Taiho Oncology, Tesaro, Inc., TransThera Bio, ZielBio, Inc. and grants from NCI/NIH, P30CA016672—Core Grant (CCSG Shared Resources) outside the submitted work; and S.A. Piha-Paul has worked as a consultant for CRC Oncology. A.M. Tsimberidou reports grants from OBI Pharmaceuticals, Agenus, IMMATICS, Parker Institute for Cancer Immunotherapy, Tvardi, Tachyon, and Tempus during the conduct of the study; personal fees from Diaccurate, NEX-I, BrYet, VinceRx, and Avstera Therapeutics outside the submitted work. F. Janku reports other from Monte Rosa Therapeutics, Monte Rosa Therapeutics, and Cardiff Oncology outside the submitted work. J. Rodon reports personal fees from Ellipses Pharma, IONCTURA, Clarion Healthcare, Debiopharm, Monte Rosa Therapeutics, Cullgen, Macrogenics, Oncology One, Envision Pharma, Columbus Venture Partners, Sardona Therapeutics, Avoro Capital Advisors, Vall d'Hebron Institute of Oncology/Ministero De Empleo Y Seguridad Social, Chinese University of Hong Kong, Boxer Capital, LLC, Tang Advisors, LLC, Incyte, Alnylam Pharmaceuticals; grants and personal fees from Aadi Bioscience, Pfizer, Merus; grants from Blueprint Medicines, Black Diamond Therapeutics, Merck Sharp & Dohme, Hummingbird, Yingli, Vall d'Hebron Institute of Oncology/Cancer Core Europe, Novartis, Spectrum Pharmaceuticals, Symphogen, BioAtla, GenMab, CytomX, Kelun-Biotech, Takeda-Millennium, GlaxoSmithKline, Taiho, Roche Pharmaceuticals, Bicycle Therapeutics, Curis, Bayer, Nuvation, ForeBio, BioMed Valley Discoveries, Loxo Oncology, Hutchinson Medipharma, Cellestia, Deciphera, Ideaya, Amgen, Tango Therapeutics, Mirati, Linnaeus Therapeutics; other from European Society for Medical Oncology and Vall d'Hebron Institute of Oncology/Ministero De Empleo Y Seguridad Social outside the submitted work. S. Pant reports advisory/consultancy (personal): Zymeworks, Ipsen,Novartis, Janssen, Boehringer Ingelheim; research grant/funding (Institution): Mirati Therapeutics, Merck, Pfizer, Lilly, Xencor, Novartis, Bristol Myers Squibb, Ipsen, Astellas, Purple Biotech, 4D Pharma, Boehringer Ingelheim, NGM Biopharmaceuticals, Janssen, Arcus Biosciences, Elicio Therapeutics, Zymeworks, BioNtech. E.E. Ileana Dumbrava reports grants from Bayer HealthCare Pharmaceuticals Inc, Immunocore LTD, Amgen, Aileron Therapeutics, Compugen Ltd, TRACON Pharmaceuticals Inc, Gilead/Immunomedics, BOLT Therapeutics, Aprea Therapeutics, Bellicum Pharmaceuticals, PMV Pharma, Triumvira, Segan Inc, Mereo BioPharma 5 Inc, Sanofi, Astex Therapeutics, Rain Oncology, Poseida, Genentech, Boehringer Ingelheim, Mersana Therapeutics; other from BOLT Therapeutics, Mersana Therapeutics, Orum Therapeutics, Summit Therapeutics, PMV Pharma, ASCO, LFSA Association, and Rain Oncology; non-financial support from Catamaran Bio during the conduct of the study. D.S. Hong reports research(Inst)/grant funding (Inst): AbbVie, Adaptimmune, Adlai-Nortye, Amgen, AstraZeneca, Bayer, Biomea, Bristol Myers Squibb, Daiichi-Sankyo, Deciphera, Eisai, Eli Lilly, Endeavor, Erasca, F. Hoffmann-LaRoche, Fate Therapeutics, Genentech, Genmab, Immunogenesis, Infinity, Kyowa Kirin, Merck, Mirati, Navier, NCI-CTEP, Novartis, Numab, Pfizer, Pyramid Bio, Revolution Medicine, SeaGen, STCube, Takeda, TCR2, Turning Point Therapeutics, VM Oncology; travel, accommodations, expenses: AACR, ASCO, CLCC, Bayer, Genmab, SITC, Telperian; consulting, speaker, or advisory role: 28Bio, Abbvie, Acuta, Adaptimmune, Alkermes, Alpha Insights, Amgen, Affini-T, Astellas, Aumbiosciences, Axiom, Baxter, Bayer, Boxer Capital, BridgeBio, CARSgen, CLCC, COG, COR2ed, Cowen, Ecor1, Erasca, Fate Therapeutics, F.Hoffmann-La Roche, Genentech, Gennao Bio, Gilead, GLG, Group H, Guidepoint, HCW Precision Oncology, Immunogenesis, InduPro, Janssen, Liberium, MedaCorp, Medscape, Numab, Oncologia Brasil, ORI Capital, Pfizer, Pharma Intelligence, POET Congress, Prime Oncology, Projects in Knowledge, Quanta, RAIN, Ridgeline, SeaGen, Stanford, STCube, Takeda, Tavistock, Trieza Therapeutics, Turning Point Therapeutics, WebMD, YingLing Pharma, Ziopharm; other ownership interests: Molecular Match (advisor), OncoResponse (founder, advisor), Telperian (founder, advisor). Y. Yuan reports personal fees from AbbVie, Amgen, Bexion Pharmaceuticals, BeyondSpring Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Century Therapeutics, Enliven Therapeutics, GT Medical Technologies, NGM Biopharmaceuticals, Repare Therapeutics, Servier Pharmaceuticals, Starpax Pharmaceuticals, Vertex Pharmaceuticals, A2 Bio, and Xinthera outside the submitted work. F. Meric-Bernstam reports grants from Calithera Biosciences, Takeda Pharmaceutical during the conduct of the study; personal fees from AbbVie, Aduro BioTech Inc., Alkermes, AstraZeneca, Black Diamond, Biovica, Chugai Biopharmaceuticals, Daiichi Sankyo Co. Ltd., DebioPharm, Ecor1 Capital, eFFECTOR Therapeutics, Eisai, F. Hoffman-La Roche Ltd., FogPharma, GT Apeiron, Genentech Inc., Harbinger Health, IBM Watson, Infinity Pharmaceuticals, Immunomedics, Inflection Biosciences, Jackson Laboratory, Karyopharm Therapeutics, Kolon Life Science, Lengo Therapeutics, Loxo Oncology, Menarini Group, Mersana Therapeutics, OnCusp Therapeutics, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Protai Bio Ltd, Puma Biotechnology Inc., Samsung Bioepis, Sanofi, Seattle Genetics Inc., Silverback Therapeutics, Spectrum Pharmaceuticals, Tallac Therapeutics, Tyra Biosciences, Xencor, Zymeworks, Theratechnologies, Zentalis; grants from Aileron Therapeutics, Inc., AstraZeneca, Bayer Healthcare Pharmaceutical, Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., Debiopharm International, eFFECTOR Therapeutics, Genentech Inc., Guardant Health Inc., Klus Pharma, Novartis, Puma Biotechnology Inc., Taiho Pharmaceutical Co.; and other from European Organisation for Research and Treatment of Cancer (EORTC), European Society for Medical Oncology (ESMO) outside the submitted work. A. Naing reports grants from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor/Millendo, Amplimmune, ARMO BioSciences, Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, Bristol Myers Squibb, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Eli Lilly, Kymab, PsiOxus, Arcus Biosciences, NeoImmuneTech, Immune-Onc Therapeutics, Surface Oncology, Monopteros Therapeutics, BioNTech SE, Seven & Eight Biopharma, and SOTIO Biotech AG; personal fees from Deka Biosciences, NGM Bio, PsiOxus Therapeutics, Immune-Onc Therapeutics, STCube Pharmaceuticals, OncoSec KEYNOTE-695, Genome & Company, CytomX Therapeutics, Nouscom, Merck Sharp & Dohme Corp, OncoNano, Servier, Lynx Health, AbbVie, PsiOxus, and AKH Inc, The Lynx Group, Society for Immunotherapy of Cancer (SITC), Korean Society of Medical Oncology (KSMO), Scripps Cancer Care Symposium, ASCO Direct Oncology Highlights, European Society for Medical Oncology (ESMO), CME Outfitters outside the submitted work; non-financial support from ARMO BioSciences, NeoImmuneTech; and spouse research funding: The Texas Medical Center Digestive Diseases Center, Jeffery Modell Foundation, Immune Deficiency Foundation, Baxalta US Inc, Chao Physician-Scientist Foundation; consultant/advisory board: Takeda, Pharming Healthcare Inc, and Horizon Therapeutics USA, Inc.; ad hoc consultancy speaker: Alfaisal University. No disclosures were reported by the other authors.
Authors’ Contributions
V. Subbiah: Conceptualization, resources, formal analysis, supervision, funding acquisition, validation, investigation, visualization, writing-original draft, writing-review and editing. N. Coleman: Writing-original draft, writing-review and editing. S.A. Piha-Paul: Investigation, writing-review and editing. A.M. Tsimberidou: Investigation, writing-review and editing. F. Janku: Investigation, writing-review and editing. J. Rodon: Writing-original draft, writing-review and editing. S. Pant: Investigation, writing-review and editing. E.E. Ileana Dumbrava: Investigation, writing-review and editing. S. Fu: Investigation, writing-review and editing. D.S. Hong: Investigation, writing-review and editing. S. Zhang: Investigation, project administration. M. Sun: Investigation, project administration. Y. Jiang: Supervision, project administration. J. Roszik: Software, investigation, writing-review and editing. J. Song: Data curation, formal analysis. Y. Yuan: Data curation, software, formal analysis. F. Meric-Bernstam: Resources, supervision, investigation, project administration, writing-review and editing. A. Naing: Conceptualization, investigation, writing-review and editing.
References
- 1. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 2014;4:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther 2002;1:707–17. [PubMed] [Google Scholar]
- 3. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol 2016;82:943–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hinz N, Jucker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun Signal 2019;17:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48. [DOI] [PubMed] [Google Scholar]
- 6. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006;6:729–34. [DOI] [PubMed] [Google Scholar]
- 7. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andre F, O'Regan R, Ozguroglu M, Toi M, Xu BH, Jerusalem G, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 2014;15:580–91. [DOI] [PubMed] [Google Scholar]
- 10. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma:: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56. [DOI] [PubMed] [Google Scholar]
- 11. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66:1500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zheng B, Mao JH, Qian L, Zhu H, Gu DH, Pan XD, et al. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett 2015;357:468–75. [DOI] [PubMed] [Google Scholar]
- 13. Janes MR, Vu C, Mallya S, Shieh MP, Limon JJ, Li LS, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia 2013;27:586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C, et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther 2011;10:1394–406. [DOI] [PubMed] [Google Scholar]
- 15. Korets SB, Musa F, Curtin J, Blank SV, Schneider RJ. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol Oncol 2014;132:468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016;387:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Voss MH, Gordon MS, Mita M, Rini B, Makker V, Macarulla T, et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br J Cancer 2020;123:1590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajjar J, Habra MA, Naing A. Metformin: an old drug with new potential. Expert Opin Investig Drugs 2013;22:1511–7. [DOI] [PubMed] [Google Scholar]
- 19. Li M, Li X, Zhang H, Lu Y. Molecular mechanisms of metformin for diabetes and cancer treatment. Front Physiol 2018;9:1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 2010;123:271–9. [DOI] [PubMed] [Google Scholar]
- 21. Zakikhani M, Dowling RJO, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res 2008;1:369–75. [DOI] [PubMed] [Google Scholar]
- 22. Alimova IN, Liu BL, Fan ZY, Edgerton SM, Dillon T, Lind SE, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009;8:909–15. [DOI] [PubMed] [Google Scholar]
- 23. Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang ZJ, Bi YY, Li SY, Zhang QJ, Zhao GM, Guo Y, et al. Reduced risk of lung cancer with metformin therapy in diabetic patients: a systematic review and meta-analysis. Am J Epidemiol 2014;180:11–4. [DOI] [PubMed] [Google Scholar]
- 25. Kim YS, Choi EA, Lee JW, Kim Y, You HS, Han YE, et al. Metformin use reduced the overall risk of cancer in diabetic patients: a study based on the Korean NHIS-HEALS cohort. Nutr Metab Cardiovas 2020;30:1714–22. [DOI] [PubMed] [Google Scholar]
- 26. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006;29:254–8. [DOI] [PubMed] [Google Scholar]
- 27. Kim JH. Comparison of the RECIST 1.0 and RECIST 1.1 in patients treated with targeted agents: a pooled analysis and review. Oncotarget 2016;7:13680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Infante JR, Tabernero J, Cervantes A, Jalal S, Burris HA, Macarulla T, et al. A phase 1, dose-escalation study of MLN0128, an investigational oral mammalian target of rapamycin complex 1/2 (mTORC1/2) catalytic inhibitor, in patients (pts) with advanced non-hematologic malignancies. Mol Cancer Ther 2013;12:C252. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure and tables
Representativeness of Study Participants
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.