

Abstract
The outgrowth and stabilization of nascent dendritic spines are crucial processes underlying learning and memory. Most new spines retract shortly after growth; only a small subset is stabilized and integrated into the new circuit connections that support learning. New spine stabilization has been shown to rely upon activity-dependent molecular mechanisms that also contribute to long-term potentiation (LTP) of synaptic strength. Indeed, disruption of the activity-dependent targeting of the kinase CaMKIIα to the GluN2B subunit of the NMDA-type glutamate receptor disrupts both LTP and activity-dependent stabilization of new spines. Yet it is not known which of CaMKIIα's many enzymatic and structural functions are important for new spine stabilization. Here, we used two-photon imaging and photolysis of caged glutamate to monitor the activity-dependent stabilization of new dendritic spines on hippocampal CA1 neurons from mice of both sexes in conditions where CaMKIIα functional and structural interactions were altered. Surprisingly, we found that inhibiting CaMKIIα kinase activity either genetically or pharmacologically did not impair activity-dependent new spine stabilization. In contrast, shRNA knockdown of CaMKIIα abolished activity-dependent new spine stabilization, which was rescued by co-expressing shRNA-resistant full-length CaMKIIα, but not by a truncated monomeric CaMKIIα. Notably, overexpression of phospho-mimetic CaMKIIα-T286D, which exhibits activity-independent targeting to GluN2B, enhanced basal new spine survivorship in the absence of additional glutamatergic stimulation, even when kinase activity was disrupted. Together, our results support a model in which nascent dendritic spine stabilization requires structural and scaffolding interactions mediated by dodecameric CaMKIIα that are independent of its enzymatic activities.
Keywords: CaMKII, dendritic spine, glutamate uncaging, two-photon imaging
Significance Statement
The stabilization of nascent dendritic spines is thought to support lasting memory of learned experiences. Here, we show that scaffolding and structural interactions, but not the enzymatic activities, of the kinase CaMKIIα are required for activity-dependent new spine stabilization. This study furthers our understanding of the cellular and molecular processes that facilitate learning and memory in the mammalian brain. Understanding the cellular and molecular mechanisms of learning and memory is crucial for our ability to develop therapeutics for memory impairments associated with neurological and neurodegenerative disorders.
Introduction
The dynamic modification of neuronal circuitry underlies learning and memory and is crucial for adaptation and survival. Dendritic spines are the sites of most excitatory synaptic connections in the mammalian cerebral cortex, and the morphological and functional changes that occur at dendritic spines contribute to the neural circuit modifications that support behavior (Kasai et al., 2021). Notably, the stabilization of newly formed spines in the cortex is tightly linked to lasting memory of learned experiences (Xu et al., 2009; Yang et al., 2009; Roberts et al., 2010; Hayashi-Takagi et al., 2015). Interestingly, most new spines are transient (Holtmaat et al., 2005; Berry and Nedivi, 2017) suggesting that stabilization is precisely regulated to favor only a subset of new spines sufficient to support memory. Thus, defining the mechanisms that determine which new spines are stabilized will strengthen our understanding of learning and memory.
Previous studies have shown that synaptic activity enhances the stability of new dendritic spines in the hippocampus and that the enhancement of new dendritic spine stability appears to be specific to patterns of synaptic activity that result in the coordinated long-term enhancement of synaptic strength and spine volume (Matsuzaki et al., 2004) known as long-term potentiation (LTP; De Roo et al., 2008a; Hill and Zito, 2013). NMDA-type glutamate receptor (NMDAR) activation is required for LTP-induced nascent spine stabilization, and disruption of the interaction between the Ca2+/calmodulin-activated kinase CaMKIIα and the GluN2B subunit of the NMDAR prevents activity-dependent new spine stabilization (Hill and Zito, 2013). Notably, CaMKIIα–GluN2B binding facilitates a number of CaMKIIα enzymatic and structural functions that promote LTP induction and maintenance, including binding to densin-180 and α-actinin, activation of signaling molecules, and phosphorylation of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors (Sanhueza and Lisman, 2013; Bayer and Schulman, 2019). Whether these enzymatic and structural activities of CaMKIIα and the downstream cascades they initiate are required for activity-dependent new spine stabilization is not yet known.
Here, we used time-lapse imaging and two-photon glutamate uncaging along with genetic and pharmacological manipulations to elucidate the role of CaMKIIα in activity-dependent new spine stabilization. We found that CaMKIIα is present and enriched at mature levels in new spines shortly after outgrowth on CA1 neurons in hippocampal slice cultures, supporting that CaMKIIα could play an important role in nascent spine stabilization. Surprisingly, high-frequency glutamate uncaging (HFU) enhanced new spine survivorship even when CaMKIIα kinase activity was genetically or pharmacologically inhibited. In contrast, shRNA-mediated knockdown of CaMKIIα blocked activity-dependent new spine stabilization, indicating that CaMKIIα expression is required for new spine stabilization. Finally, we found that phospho-mimetic CaMKIIα-T286D, which generates increased autonomous CaMKIIα interactions, enhanced new spine stabilization even when kinase activity was disrupted with the K42R mutation. Together, our results support a model whereby strong glutamatergic transmission facilitates new spine stabilization through structural and scaffolding functions of CaMKIIα.
Materials and Methods
Preparation and transfection of organotypic slice cultures
Organotypic hippocampal slice cultures were prepared from postnatal day (P) 6–8 C57BL/6J wild-type (WT) mice of both sexes, as described (Stoppini et al., 1991; Opitz-Araya and Barria, 2011). Neurons were transfected 2–3 d prior to imaging using particle-mediated gene transfer, as described in Woods and Zito (2008), except 6–8 µg of DsRed-Express (Clontech), and 6 μg of green fluorescent protein (GFP)-tagged constructs or 5–10 μg of GFP was coated onto 6–7 mg of 1.6 μm gold beads. GFP-tagged constructs included GFP-CaMKIIα, GFP-CaMKIIα-T286D, GFP-CaMKIIα-K42R/T286D (Pi et al., 2010), or GFP-CaMKIIα-K42R (Tullis et al., 2020). CaMKIIα knockdown used 25 µg CaMKIIα-shRNA and rescue also contained 6 µg shRNA-resistant full-length GFP-CaMKIIα* or a truncated (residues 1–325) monomeric GFP-mCaMKIIα* (Lemieux et al., 2012).
Two-photon imaging
Image stacks (512 × 512 pixels, 1 μm z-steps) of 4–6 secondary and tertiary, apical and basal dendritic segments from CA1 pyramidal neurons (6–10 DIV) were acquired on a custom two-photon microscope with a pulsed Ti::Sapphire laser (930 nm, 0.5–3 mW at the sample; Spectra-Physics). Data acquisition was controlled by ScanImage (Pologruto et al., 2003) written in MATLAB (MathWorks). The first time point was acquired in slice culture medium at room temperature after which the slice was maintained in the incubator (35°C). To maximize success rate, two cells were interleaved; therefore, if the first cell had no new spines, the second cell could be pursued instead. After 60 min, new spine identification was performed at the second time point, which was acquired in a bath of recirculating, oxygenated artificial cerebrospinal fluid (ACSF; in mM, 127 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 25 d-glucose, ∼310 mOsm, pH 7.2) with 2 mM Ca2+, 0–0.1 mM Mg2+, and 1 µm tetrodotoxin at 31°C. A total of 2.5–3.5 mm of 4-methoxy-7-nitroindolinyl-caged l-glutamate (MNI-glutamate) was added for uncaging experiments. Staurosporine (1 µM) or an equivalent volume vehicle were added to the bath after new spine identification and 30 min prior to uncaging. Thus, the timing between new spine identification and the HFU stimulation was 30 min on average. All images shown are maximum projections of 3D image stacks after applying a median filter (3 × 3).
Identification of new spines
We defined new spines as any protrusion emanating from the dendrite that was present in the second and/or third images in the time-lapse series (60–90 min later) but not detectable in either the red or green channels in the first image. Persistent neighbor spines were defined as spines present in all images in the time-lapse series. Spines of ambiguous persistence or presence due to fluctuations in dendrite swelling, spine motility, or spine drift in the z-axis were excluded.
High-frequency glutamate uncaging
The HFU stimulus consisted of 60 pulses (720 nm, 8–10 mW at the sample) of 2 ms duration delivered at 2 Hz in the presence of 2.5–3.5 mM MNI-glutamate by parking the beam at a point ∼0.5 μm from the spine head away from the dendrite.
Image analysis
Spine size was estimated from bleed-through-corrected and background-subtracted red (DsRed-Express) fluorescence intensity. Spine brightness measurements give an accurate estimate of relative spine size when compared with electron microscopy (Holtmaat et al., 2005). Spine length was measured from the tip of the spine to the base of the spine neck where it meets the dendrite.
Relative enrichment of GFP-tagged proteins in dendritic spines was calculated using bleed-through-corrected and background-subtracted green (GFP) and red (DsRed-Express) fluorescence intensities from spines and dendrites, as described (Woods et al., 2011). Briefly, the ratio of green fluorescence intensity to red fluorescence intensity (G/R) was calculated for each new spine, size-matched neighboring persistent spines (6–10), and three representative regions on the dendritic shaft (excluding regions dendrite swelling and GFP-puncta, which were indicative of the presence of a z spine). To quantify spine fluorescence intensities, boxes were drawn around whole spines and spine necks using custom software written in MATLAB. Background subtraction was done by drawing a box next to a target spine that was equal on the axis perpendicular to the dendrite as the box drawn around the spine head and neck. The average intensity of that box was multiplied by the number of pixels in the target spine box and subtracted from the integrated intensity from the target spine box. Relative enrichment of spines was calculated by normalizing the G/R ratio of the target spine to the mean G/R ratio of three locations on the adjacent dendrite.
Several criteria were used to ensure that the analyzed data were of high quality. Cells that exhibited lower green fluorescence intensity than the background ROI were excluded. Cells with extremely high levels of GFP-tagged protein expression such that synaptic enrichment was lost were excluded. Cells were also excluded if, after background and bleed-through subtraction, (1) the value of the mean green pixel intensity (G) from neighbor spines was <3.23 a.u., (2) the value of the mean neighbor spine G/R was <0.01, or (3) the ratio of the square of the mean persistent spine G/R to the absolute value of the mean dendrite G/R was <0.05. These criteria allowed unbiased exclusion of cells that returned negative pixel intensity values after background and bleed-through subtraction. Cells that exhibited significant photobleaching (a decline in average integrated fluorescence intensity in the dendrite >20% compared with the first time point) in either the red or green channels were excluded.
Statistical analysis
Survivorship curves were compared using the log-rank test. To compare survivorship at individual time points, we used Fisher's exact test. For comparisons of spine volumes at a given time point to baseline, two-way ANOVAs with Bonferroni’s post hoc tests for multiple comparisons were used. Between-group comparisons of spine baseline volumes, lengths, and rates of new spine outgrowth were performed using a two-tailed unpaired heteroscedastic Student's t test (in the case of two groups), or one-way ANOVA with Tukey’s multiple comparisons (in the case of three groups), unless otherwise noted. Error bars represent standard error of the mean (SEM). Statistical tests were performed using GraphPad Prism 9.2 software.
Results
GFP-CaMKIIα enrichment in new spines is comparable to that in size-matched neighboring spines
To understand the role of CaMKIIα in activity-induced new spine stabilization, we first needed to determine whether CaMKIIα is expressed in new spines and in what time frame. This experiment was an important first step, as we and others have reported that several members of the postsynaptic density-membrane-associated guanylate kinase (PSD-MAGUK) family of postsynaptic scaffolding molecules are present at very low levels in new spines and can take up to 24 h to accumulate to mature enrichment levels (De Roo et al., 2008a; Lambert et al., 2017), indicating that the molecular composition of new spines and their persistent neighbors is distinct, particularly in the earliest stages after new spine outgrowth. We used time-lapse, two-photon imaging to observe spontaneous new spine outgrowth on the dendrites of hippocampal CA1 neurons in slice culture biolistically transfected with mEGFP-tagged CaMKIIα (GFP-CaMKIIα) and a DsRed-Express cell fill (Fig. 1A). We found no difference in the enrichment of GFP-CaMKIIα in new spines as compared with size-matched neighboring control spines (new, 1.5 ± 0.2; neighbor, 1.7 ± 0.1; p = 0.14; Fig. 1B,C). We conclude that CaMKIIα is able to rapidly accumulate at new spines and therefore could play an important role in even the earliest molecular signaling events that support new spine stabilization.
Figure 1.

GFP-CaMKIIα enrichment in new spines is comparable to that in size-matched neighboring spines. A, Images of a dendrite from a hippocampal CA1 neurons in slice culture (DIVs 7–9) expressing DsRed-Express (red) and GFP-CaMKIIα (green) before (open arrowhead) and after (filled arrowhead) spontaneous new spine outgrowth. B, Enrichment (spine:dendrite ratio) of GFP-CaMKIIα in new spines (n = 33 spines/16 cells) was comparable to that in size-matched neighboring spines (n = 21 spines/16 cells). C, Neighboring spines used for enrichment calculations in B were size-matched to new spines (p = 0.62). D, Images of dendrites from CA1 neurons expressing GFP-CaMKIIα-K42R (green) and DsRed-Express (red) before (open arrowhead) and after (filled arrowhead) spontaneous new spine outgrowth. E, Enrichment of GFP-CaMKIIα-K42R in new spines (n = 39 spines/14 cells) was comparable to that in size-matched mature neighboring spines (n = 39 spines/14 cells). Importantly, no difference in relative enrichment was found between new (filled bars) or size-matched neighboring spines (open bars) in the WT (black) and K42R (blue) conditions (new, p = 0.4; neighbors, p = 0.99). Data for GFP-CaMKIIα-WT new spine enrichment is from B. F, Neighboring spines used for enrichment calculations in E were size-matched to new spines (p = 0.99). No difference in new spine size was found between WT (black) and K42R (blue; p = 0.99). Data for GFP-CaMKIIα new spine size is from C. Two-way ANOVA with Bonferroni’s multiple-comparisons test.
Genetic and pharmacological inhibition of CaMKIIα kinase activity does not impair activity-dependent new spine stabilization
To investigate the role of CaMKIIα in activity-dependent new spine stabilization, we tested whether interfering with CaMKIIα kinase activity would disrupt the robust activity-dependent stabilization of new spines induced by HFU of MNI-caged glutamate (MNI-glutamate) at individual new spines (Hill and Zito, 2013). We first chose to use a genetic approach by overexpressing GFP-CaMKIIα containing the K42R point mutation that inhibits CaMKIIα kinase activity (Yamagata et al., 2009; Pi et al., 2010; Tullis et al., 2020). This CaMKIIα-K42R mutant has been shown to act in a dominant-negative manner (Pi et al., 2010; Rossetti et al., 2017). Importantly, enrichment of GFP-CaMKIIα-K42R in new spines was comparable to that in size-matched mature neighboring spines (new, 1.7 ± 0.1; neighbor, 1.8 ± 0.1; p = 0.39), basal spine enrichment levels of GFP-CaMKIIα-K42R in new spines were comparable to those of GFP-CaMKIIα (p = 0.37; Fig. 1D–F), and enrichment levels after HFU did not change compared with baseline for either GFP-CaMKIIα (+1 min, 1.3 ± 0.3; +70 min, 1.0 ± 0.1; p > 0.99) or GFP-CaMKIIα-K42R (+1 min, 0.9 ± 0.1; +70 min, 0.9 ± 0.1; p > 0.99). Furthermore, cells expressing GFP-CaMKIIα-K42R and GFP-CaMKIIα exhibited comparable target new spine sizes, lengths, and rates of new spine outgrowth (Table 1).
Table 1.
Target new spine morphological characteristics and new spine outgrowth rates
| Target new spine size | ||||||
|---|---|---|---|---|---|---|
| Groups | n value (spines/cells) | Pixel value/1,000 | Groups compared | p value | Test | |
| Fig. 2 | CaMKIIα | 16/16 | 10 ± 1 | CaMKIIα vs CaMKIIα-K42R | 0.2 | Unpaired two-tailed t test |
| CaMKIIα-K42R | 14/14 | 14 ± 2 | ||||
| Fig. 4 | CaMKIIα-shRNA + CaMKIIα* | 18/18 | 13 ± 2 | CaMKIIα-shRNA + CaMKIIα* vs CaMKIIα-shRNA + mCaMKIIα* | 0.7 | One-way ANOVA with Tukey’s multiple comparisons |
| CaMKIIα-shRNA + mCaMKIIα* | 10/10 | 16 ± 2 | ||||
| CaMKIIα-shRNA | 20/20 | 21 ± 2 | CaMKIIα-shRNA vs CaMKIIα-shRNA + CaMKIIα* | 0.02 | ||
| CaMKIIα-shRNA vs CaMKIIα-shRNA + mCaMKIIα* | 0.3 | |||||
| Fig. 5 | GFP-CaMKIIα-T286D | 60/9 | 14 ± 2 | GFP-CaMKIIα-T286D vs GFP-CaMKIIα-T286D/K42R | 0.5 | |
| GFP-CaMKIIα-T286D/K42R | 29/6 | 17 ± 3 | ||||
| Control (dsRed-Express) | 51/9 | 13 ± 1 | Control vs GFP-CaMKIIα-T286D | 0.9 | ||
| Control vs GFP-CaMKIIα-T286D/K42R | 0.3 | |||||
| Target new spine length | ||||||
| Groups | n value (spines/cells) | µm | Groups compared | p value | Test | |
| Fig. 2 | CaMKIIα | 16/16 | 1.2 ± 0.2 | CaMKIIα vs CaMKIIα-K42R | 0.6 | Unpaired two-tailed t test |
| CaMKIIα-K42R | 14/14 | 1.3 ± 0.2 | ||||
| Fig. 4 | CaMKIIα-shRNA + CaMKIIα* | 18/18 | 1.2 ± 0.1 | CaMKIIα-shRNA + CaMKIIα* vs CaMKIIα-shRNA + mCaMKIIα* | 0.1 | One-way ANOVA with Tukey’s multiple comparisons |
| CaMKIIα-shRNA + mCaMKIIα* | 10/10 | 0.8 ± 0.1 | ||||
| CaMKIIα-shRNA | 20/20 | 1.1 ± 0.1 | CaMKIIα-shRNA vs CaMKIIα-shRNA + CaMKIIα* | 0.9 | ||
| CaMKIIα-shRNA vs CaMKIIα-shRNA + mCaMKIIα* | 0.2 | |||||
| Fig. 5 | GFP-CaMKIIα-T286D | 60/9 | 1.1 ± 0.1 | GFP-CaMKIIα-T286D vs GFP-CaMKIIα-T286D/K42R | 0.1 | |
| GFP-CaMKIIα-T286D/K42R | 29/6 | 1.5 ± 0.2 | ||||
| Control (dsRed-Express) | 51/9 | 1.3 ± 0.1 | Control vs GFP-CaMKIIα-T286D | 0.6 | ||
| Control vs GFP-CaMKIIα-T286D/K42R | 0.3 | |||||
| New spine outgrowth rate | ||||||
| Groups | n value (spines/cells) | # spines/µm /60 min | Groups compared | p value | Test | |
| Fig. 2 | CaMKIIα | 76/16 | 0.07 ± 0.01 | CaMKIIα vs CaMKIIα-K42R | 0.8 | Unpaired two-tailed t test |
| CaMKIIα-K42R | 86/14 | 0.07 ± 0.01 | ||||
| Fig. 4 | CaMKIIα-shRNA + CaMKIIα* | 104/18 | 0.07 ± 0.01 | CaMKIIα-shRNA + CaMKIIα* vs CaMKIIα-shRNA + mCaMKIIα* | 0.3 | One-Way ANOVA with Tukey’s multiple comparisons |
| CaMKIIα-shRNA + mCaMKIIα* | 57/10 | 0.06 ± 0.01 | ||||
| CaMKIIα-shRNA | 148/20 | 0.06 ± 0.01 | CaMKIIα-shRNA vs CaMKIIα-shRNA + CaMKIIα* | 0.8 | ||
| CaMKIIα-shRNA vs CaMKIIα-shRNA + mCaMKIIα* | 0.6 | |||||
| Fig. 5 | GFP-CaMKIIα-T286D | 60/9 | 0.06 ± 0.01 | GFP-CaMKIIα-T286D vs GFP-CaMKIIα-T286D/K42R | 0.5 | |
| GFP-CaMKIIα-T286D/K42R | 29/6 | 0.05 ± 0.01 | ||||
| Control (dsRed-Express) | 51/9 | 0.05 ± 0.01 | Control vs GFP-CaMKIIα-T286D | 0.7 | ||
| Control vs GFP-CaMKIIα-T286D/K42R | 0.9 | |||||
The morphological characteristics of target new spines and outgrowth rates are compared at the time of first appearance (at the end of the first 60 min time-lapse interval).
We proceeded to test whether expression of GFP-CaMKIIα-K42R would disrupt stabilization of nascent dendritic spines. We used time-lapse imaging of dendrites on neurons expressing dsRed-Express and either GFP-CaMKIIα or GFP-CaMKIIα-K42R to identify multiple new spines that spontaneously grew on each cell. One new spine per cell was exposed to HFU stimulation (Fig. 2A). Survivorship of stimulated and unstimulated new spines on the same cell was monitored through time-lapse imaging. Consistent with our observations for cells transfected with GFP alone (Hill and Zito, 2013), our HFU protocol enhanced stimulated new spine survivorship compared with unstimulated new spines on cells expressing GFP-CaMKIIα (Fig. 2B–D; stim, 94%; unstim, 62%; p = 0.03). Surprisingly, we found that HFU also robustly enhanced new spine stabilization on cells expressing GFP-CaMKIIα-K42R (Fig. 2B–D; stim, 100%; unstim, 68%; p = 0.02), suggesting that CaMKIIα kinase activity is not necessary for activity-induced new spine stabilization. Indeed, the rate of stimulated and unstimulated new spine survivorship were not different between the GFP-CaMKIIα or GFP-CaMKIIα-K42R conditions (stim GFP-CaMKIIα vs GFP-CaMKIIα-K42R, p = 0.99; unstim GFP-CaMKIIα or GFP-CaMKIIα-K42R, p = 0.79). Importantly, we confirmed that GFP-CaMKIIα-K42R was acting as a dominant negative, as GFP-CaMKIIα-K42R-transfected neurons exhibited impaired HFU-induced long-term growth of mature spines (K42R, 138 ± 16%; p = 0.11), which is intact in neurons expressing GFP-CaMKIIα (WT, 188 ± 23%; p = 0.01; Fig. 2E,F).
Figure 2.
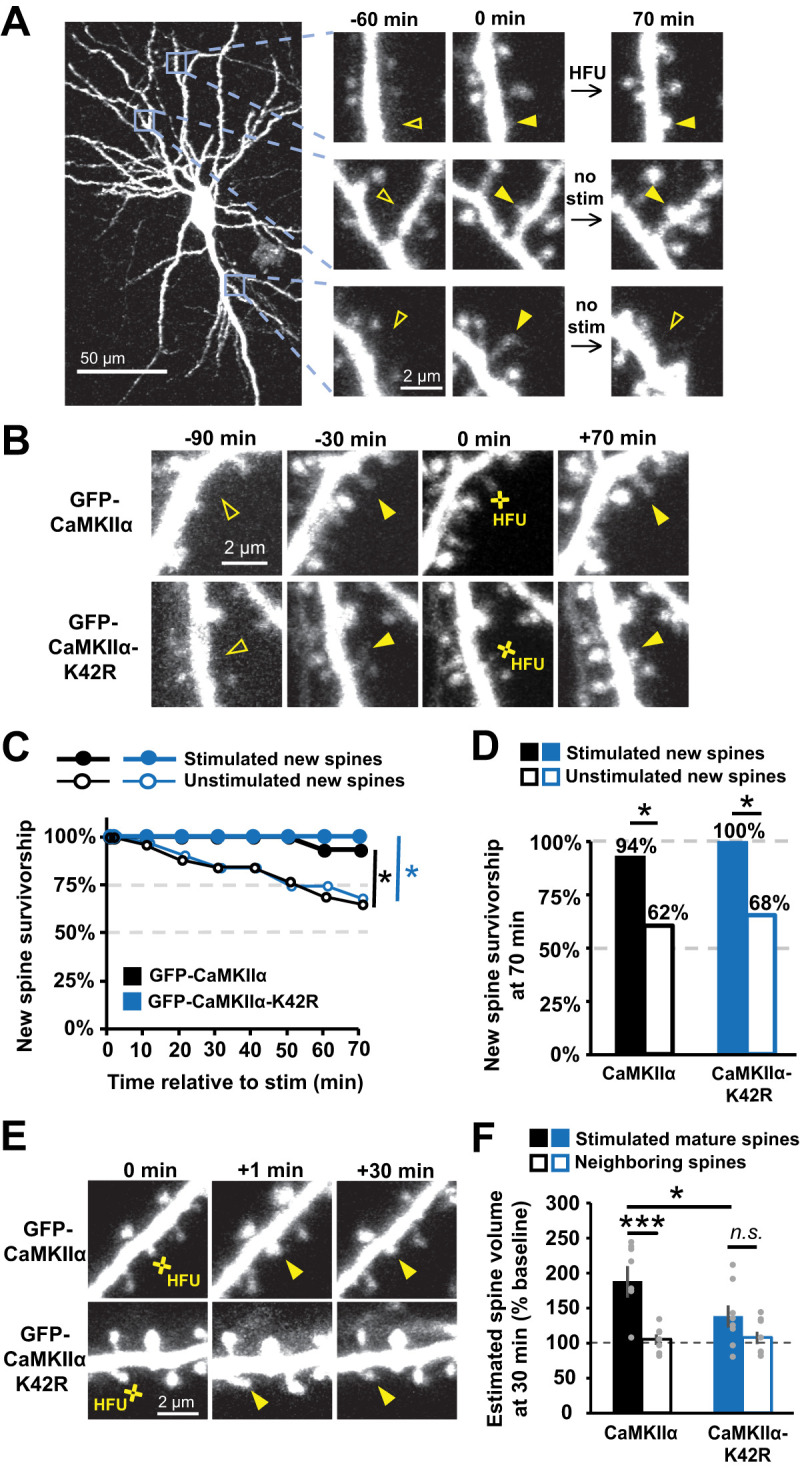
GFP-CaMKIIα-K42R does not impair activity-dependent new spine stabilization. A, Images of red fluorescence showing dendrites from CA1 neurons in slice culture (DIVs 7–9) expressing DsRed-Express and GFP-CaMKIIα. Three new spines appeared (solid arrowheads), one of which was stimulated with HFU. One unstimulated spine was eliminated (open arrowhead at +70 min). B, Images (red channel) of dendrites from CA1 neurons expressing DsRed-Express and either GFP-CaMKIIα (top row) or GFP-CaMKIIα-K42R (bottom row) showing new dendritic spines stimulated with HFU at 0 min. C, HFU stimulation enhanced new spine survivorship (filled circles; WT, n = 16 spines/16 cells; K42R, n = 14 spines/14 cells) relative to unstimulated new spines (open circles; WT, n = 31 spines/16 cells; K42R, n = 32 spines/14 cells) on the same cells for both GFP-CaMKIIα-WT (black) and GFP-CaMKIIα-K42R (blue). D, Survivorship of HFU-stimulated new spines (filled bars) at 70 min was increased compared with unstimulated new spines (open bars) on the same cells for both GFP-CaMKIIα-WT (black) and GFP-CaMKIIα-K42R (blue). E, Images of red fluorescence showing dendrites before and after HFU at mature spines at 0 min. F, GFP-CaMKIIα-K42R expression impaired HFU-induced long-term growth of mature spines (filled blue; n = 8 spines/8 cells) that is retained in cells expressing GFP-CaMKIIα (filled black; n = 8 spines/8 cells). Log-rank task in C, Barnard's exact test in D, and two-way ANOVA with Bonferroni’s multiple-comparisons test in F. *p < 0.05, **p < 0.01, ***p < 0.001.
While the K42R mutation acts in a dominant-negative manner, it retains residual kinase activity in response to glutamatergic stimulation (Rossetti et al., 2017; Tullis et al., 2020), and we were also concerned that transfected cells might contain fully endogenous CaMKII holoenzymes lacking the mutant subunit. Residual levels of CaMKII activity could be sufficient to promote the enzymatic interactions and signaling cascades necessary to stabilize new spines. As an independent means to test the role of CaMKIIα enzymatic activity in activity-induced new spine stabilization, we pharmacologically inhibited CaMKIIα using staurosporine, a potent, broad-spectrum kinase inhibitor that competitively binds the ATP-binding pocket of CaMKIIα. Unlike many of the more widely used CaMKIIα kinase inhibitors with higher specificity, staurosporine does not interfere with the interaction between activated CaMKIIα and the GluN2B subunit (Barcomb et al., 2013). Using staurosporine to inhibit CaMKIIα thus allowed us to distinguish between the requirement for GluN2B binding (Hill and Zito, 2013) and the potential requirement for kinase activity in activity-induced new spine stabilization.
Using time-lapse two-photon imaging of dendrites on hippocampal CA1 neurons expressing GFP, we identified multiple new spines that spontaneously grew on each cell (Fig. 3A). We then added staurosporine (final concentration of 1 µM) or an equivalent volume of vehicle for the remainder of the experiment. After a 30 min incubation in either staurosporine or vehicle, one new spine per cell was exposed to HFU stimulation, and survivorship was monitored for stimulated and unstimulated new spines on the same cell. We found that stimulated new spines were more stable than unstimulated new spines on the same cells after incubation in either vehicle (Veh; stim, 100%; unstim, 65%; p = 0.04) or staurosporine (Sta; stim, 100%; unstim, 70%; p = 0.04; Fig. 3B,C). Furthermore, the rate of stimulated and unstimulated new spine survivorship was not different between the vehicle and staurosporine conditions (stim Veh vs Sta, p = 0.99; unstim Veh vs Sta, p = 0.80). Importantly, we confirmed that HFU-induced long-term growth of mature spines was blocked by staurosporine (101 ± 9%; p = 0.03) but intact in vehicle (140 ± 11%; p = 0.99), indicating the effectiveness of staurosporine as a kinase inhibitor (Fig. 3D,E). Our results with staurosporine are consistent with our finding that genetic inhibition of CaMKIIα kinase activity did not impair activity-induced new spine stabilization. Together, these results strongly support that CaMKIIα kinase activity is not necessary for activity-dependent new spine stabilization.
Figure 3.
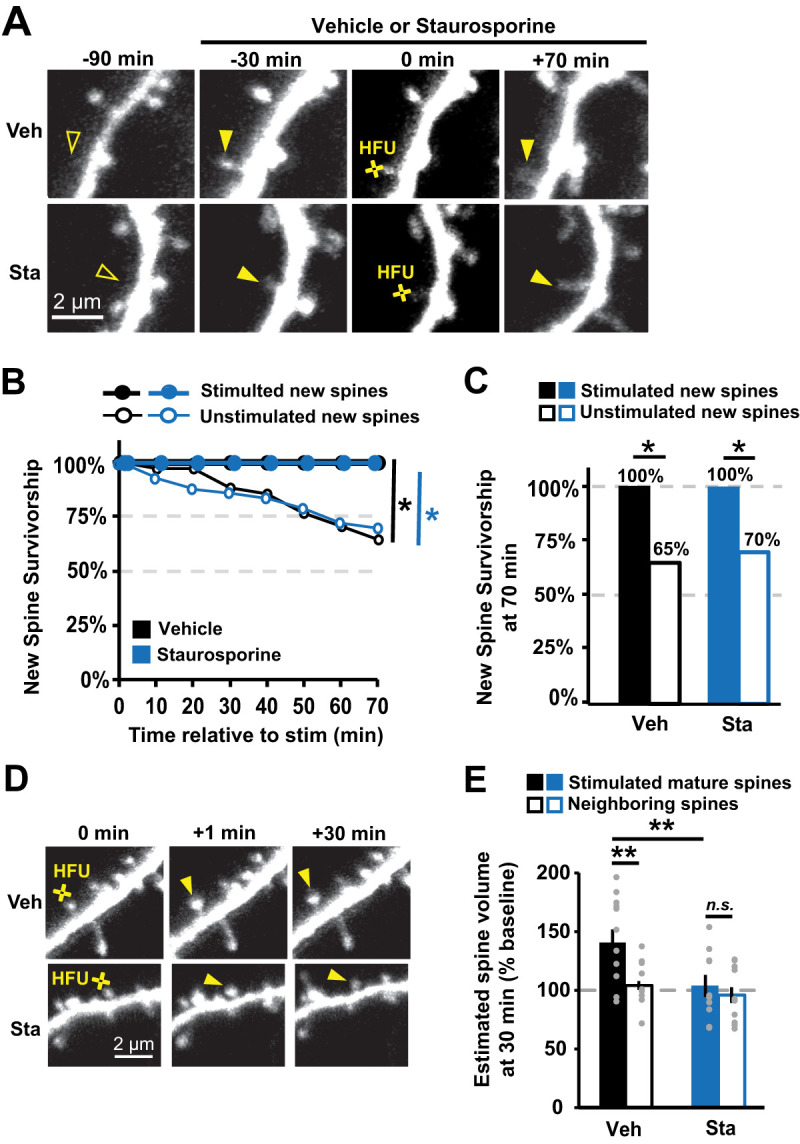
Inhibition of CaMKIIα kinase activity with staurosporine does not impair activity-dependent new spine stabilization. A, Images of green fluorescence showing dendrites from GFP-transfected CA1 neurons in slice culture (DIVs 7–9). One new spine (filled arrowhead at 0 min) per neuron was stimulated with HFU at 0 min, following 30 min pre-incubation in either vehicle (Veh, top row) or 1 µm staurosporine (Sta, bottom row). B, Survivorship of stimulated new spines (filled circles; Veh, 9 spines/9 cells; Sta, 11 spines/11 cells) was enhanced relative to unstimulated new spines on the same cells (open circles; Veh, 33 spines/9 cells; Sta, 43 spines/11 cells) in both vehicle (black) and staurosporine (blue) conditions. C, Survivorship of HFU-stimulated new spines (filled bars) at 70 min was increased compared with unstimulated new spines (open bars) on the same cells in both vehicle (black) and staurosporine (blue) conditions. D, Images of green fluorescence showing dendrites on GFP-transfected CA1 neurons before and after HFU (yellow circle). E, Incubation with 1 µM staurosporine (Sta; filled blue; n = 10 spines/10 cells) blocked HFU-induced long-term growth of mature spines (101 ± 9%; p = 0.03), which was intact in vehicle conditions (Veh; filled black; n = 12 spines/12 cells; 140 ± 11%; p = 0.99). Volume of unstimulated neighbors was unchanged (open bars; Veh, 104 ± 3%; p = 0.99; K42R, 96 ± 7%; p = 0.99). Log-rank task in B, Barnard's exact test in C, and two-way ANOVA with Bonferroni’s multiple-comparisons test in E. *p < 0.05, **p < 0.01, ***p < 0.001.
Knockdown of CaMKIIα blocks activity-dependent new spine stabilization
We next set out to test whether kinase-independent functions of CaMKIIα are required for nascent spine stabilization. Beyond its enzymatic activities, CaMKIIα plays a number of structural and scaffolding roles, independent of those performed by CaMKIIβ, most of which are facilitated by interactions with other synaptic proteins such as α-actinin, densin-180, the GluN2B subunit of the NMDAR, the proteasome, and PSD-MAGUKs (Walikonis et al., 2001; Krapivinsky et al., 2004; Bingol et al., 2010). Some of these scaffolding and structural roles of CaMKIIα are distinct from its enzymatic roles and do not require CaMKIIα kinase activity (Krapivinsky et al., 2004; Bingol et al., 2010; Pi et al., 2010; Barcomb et al., 2013). These interactions would require precise regulation of the amounts of available CaMKIIα and its physical interactions with potential binding partners. Thus, decreased levels of endogenous CaMKIIα would likely interfere with these structural and scaffolding activities, some of which may be necessary for activity-dependent new spine stabilization.
We tested whether structural and/or scaffolding activities of CaMKIIα are needed to support activity-dependent nascent spine stabilization using an shRNA-mediated knockdown of endogenous CaMKIIα with an shRNA that was designed and validated in previous work (Lemieux et al., 2012). We validated this CaMKIIα-shRNA in our preparation by demonstrating that HFU-induced long-term growth of mature spines was blocked by knockdown of CaMKIIα (98 ± 10%; p = 0.99) and rescued by co-expression of shRNA-resistant GFP-CaMKIIα* (200 ± 26%; p = 0.04; Fig. 4A,B). We next examined the effect of CaMKIIα-shRNA on HFU-induced new spine stabilization. We found that knockdown of CaMKIIα disrupted HFU-induced new spine stabilization (Fig. 4C–E; stim, 60%; unstim, 61%; p = 0.98). To rule out possible effects of nonspecific shRNA activity, we rescued the knockdown by co-expressing an shRNA-resistant form of GFP-CaMKIIα (GFP-CaMKIIα*). Rescuing CaMKIIα levels restored activity-dependent new spine stabilization, as new spines that received the HFU stimulus were again more stable than unstimulated control new spines (Fig. 4C–E; stim, 100%; unstim, 67%; p = 0.03). Target new spine sizes, lengths, and outgrowth rates were comparable in cells expressing CaMKIIα-shRNA alone and those expressing both the shRNA and the shRNA-resistant GFP-CaMKIIα*, with the exception of new spine size being significantly larger in the CaMKIIα-shRNA alone condition relative to the rescue with shRNA-resistant GFP-CaMKIIα* (Table 1), which would be expected to stabilize CaMKIIα-shRNA new spines, instead of destabilize them, as we observed. These results confirm a role for CaMKIIα in activity-induced new spine stabilization. Together with our previous results, we conclude that nonenzymatic CaMKIIα function is required to enhance new spine stabilization downstream of strong glutamatergic stimulation.
Figure 4.

CaMKIIα knockdown blocks activity-dependent new spine stabilization. A, Images of red fluorescence showing mature spines on dendrites from hippocampal CA1 neurons in slice culture (DIVs 7–9) transfected with dsRed-Express and CaMKIIα shRNA alone (top row) or co-expressed with shRNA-resistant CaMKIIα* variants (middle and bottom rows), showing mature dendritic spines stimulated with HFU at 0 min. B, shRNA-mediated knockdown of CaMKIIα (filled blue; n = 8 spines/8 cells) impaired HFU-induced long-term growth of mature spines that was restored with co-expression of shRNA-resistant GFP-CaMKIIα* (black; n = 14 spines/14 cells) but not with co-expression of monomeric mGFP-CaMKIIα* (gray; n = 10 spines/10 cells). C, Images of red fluorescence showing dendrites on CA1 neurons (DIVs 7–9) expressing a DsRed-Express cell fill and either CaMKIIα shRNA (top row) or CaMKIIα shRNA + shRNA-resistant GFP-CaMKIIα* variants (bottom two rows). HFU-induced new spine (filled arrowheads) stabilization failed following knockdown of CaMKIIα (open arrowhead at 70 min), which was rescued with expression of shRNA-resistant GFP-CaMKIIα*, but not with expression of monomeric shRNA-resistant mGFP-CaMKIIα*. D, Knockdown of CaMKIIα disrupted the stabilization of new spines (filled circles) as compared with unstimulated new spines (open circles) at times beyond 30–40 min (blue; stimulated, 20 spines/20 cells; unstimulated, 103 spines/20 cells). Rescuing with full-length shRNA-resistant CaMKIIα* restored activity-dependent new spine stabilization (black; stim, 18 spines/18 cells; unstim, 98 spines/18 cells), but not shRNA-resistant monomeric mCaMKIIα* (gray; stim, 10 spines/10 cells; unstim, 47 spines/10 cells). E, Activity-dependent new spine stabilization at 70 min (filled bars) was not different from that of unstimulated new spines (open bars) following knockdown of CaMKIIα (black), which was restored when CaMKIIα is rescued with shRNA-resistant CaMKIIα* (blue), but not with shRNA-resistant monomeric mCaMKIIα*. Two-way ANOVA with Bonferroni’s multiple-comparisons test in B, log-rank task in D, and Barnard's exact test in E. *p < 0.05, **p < 0.01, ***p < 0.001.
In order to test whether the dodecameric structure of CaMKIIα is critical for activity-dependent nascent spine stabilization, we utilized a truncated monomeric version of CaMKIIα (amino acids 1–325) that removes the association domain but retains the regulatory domain (Lemieux et al., 2012). Notably, in conjunction with shRNA-CaMKIIα knockdown, rescuing CaMKIIα levels with shRNA-resistant monomeric GFP-mCaMKIIα* did not restore activity-dependent new spine stabilization, as new spines that received the HFU stimulus were not more stable than unstimulated control new spines on the same cell (Fig. 4C–E; stim, 70%; unstim, 64%; p = 0.7). Importantly, enrichment levels in new spines of GFP-CaMKIIα* (1.4± 0.1) and monomeric GFP-mCaMKIIα* (1.3 ± 0.1) were comparable (p = 0.8). Furthermore, target new spine sizes, lengths, and outgrowth rates were comparable in cells expressing CaMKIIα-shRNA alone and those expressing both the shRNA and the shRNA-resistant monomeric GFP-mCaMKIIα* (Table 1). Thus, we conclude that the dodecameric structure of CaMKIIα is critical for activity-dependent nascent spine stabilization.
Overexpression of pseudo-autophosphorylated CaMKIIα enhances basal spine survivorship independent of kinase activity
We next probed whether CaMKIIα's nonenzymatic structural and/or scaffolding activities are not only necessary but sufficient to enhance activity-dependent new spine stabilization. We took advantage of phospho-mimetic CaMKIIα mutants that increase basal levels of CaMKIIα–GluN2B binding (Barcomb et al., 2014), specifically the replacement of threonine 286 with an aspartic acid, or CaMKIIα-T286D (Pi et al., 2010). The T286D mutation renders CaMKIIα constitutively active, allowing interactors and substrates access to the kinase and regulatory domains. Pairing this mutation with the K42R point mutation generates increased autonomous CaMKIIα interactions, while blocking CaMKIIα enzymatic activities.
To determine the effect of autonomous CaMKIIα on new spine survivorship with and without kinase activity, we expressed the GFP-CaMKIIα-T286D or GFP-CaMKIIα-K42R/T286D with a dsRed-Express cell fill in organotypic hippocampal slice cultures. For these experiments where we wanted to determine whether constitutively autonomous CaMKIIα was sufficient to enhance new spine survivorship, we did not expose new spines to our HFU protocol; instead, we monitored basal new spine stability over a period of 70 min using time-lapse imaging. We found that new spines were more stable on cells expressing either GFP-CaMKIIα-T286D or GFP-CaMKIIα-K42R/T286D compared with new spines on cells expressing only DsRed-Express (Fig. 5A–C; DsRed, 63%; T286D, 83%; p = 0.02; K42R/T286D, 85%; p = 0.01). Importantly, expression of WT GFP-CaMKIIα did not alter new spine survivorship as compared with dsRed-Express alone (Fig. 5C,D; DsRed, 67%; WT, 65%; p = 0.84), so increased survivorship was due to pseudo-autophosphorylated CaMKIIα, independent of kinase activity. Furthermore, new spine size, length, and outgrowth rate were comparable between control cells and those expressing GFP-CaMKIIα-T286D or GFP-CaMKIIα-K42R/T286D (Table 1). When combined with our previous published results (Hill and Zito, 2013), our findings support a model in which CaMKIIα–GluN2B binding facilitates nonenzymatic CaMKIIα functions that are both necessary and sufficient for enhancing new spine stabilization.
Figure 5.

Overexpression of constitutively autonomous CaMKIIα enhances basal spine survivorship independent of kinase activity. A, Images of red fluorescence showing new spine outgrowth (filled arrowhead at 0 min) and stabilization (filled arrowhead at 70 min) on dendrites from hippocampal neurons (DIVs 7–9) expressing DsRed-Express alone (top row) or co-expressed with GFP-CaMKIIα-T286D (middle row) or GFP-CaMKIIα-K42R/T286D (bottom row). B, Basal spine survivorship rates were higher on cells expressing GFP-CaMKIIα-T286D (dark blue; 63 spines/10 cells) and GFP-CaMKIIα-K42R/T286D (light blue; 53 spines/7 cells) compared with survivorship rates on cells expressing only DsRed-Express (black; 51 spines/10 cells). Barnard's exact test with Bonferroni’s multiple-comparisons correction. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Molecular composition of nascent dendritic spines
There is substantial evidence indicating that the formation of new spines and their ability to persist and integrate into functional synaptic circuits is crucial to learning (Xu et al., 2009; Yang et al., 2009; Roberts et al., 2010; Hayashi-Takagi et al., 2015; Albarran et al., 2021). Despite this vital role, the molecular composition and signaling pathways at play in new spines remain largely unexplored. New spines do share some molecular and functional properties with mature spines; new spine AMPAR currents are comparable to those recorded from mature spines of similar size (Zito et al., 2009; Kwon and Sabatini, 2011) and ultrastructural evidence shows that a subset of new spines are found directly apposed to presynaptic boutons (Trachtenberg et al., 2002; Knott et al., 2006; Zito et al., 2009), suggesting that new spines are rapidly equipped to respond to glutamatergic stimulation and incorporated into neural circuits.
Still, new spines differ from mature spines in several key ways. Most notably, new spines exhibit very low expression levels of the PSD-family MAGUKs (De Roo et al., 2008a; Lambert et al., 2017), key scaffolding molecules that regulate synaptic strength, maturation, and stability (Ehrlich and Malinow, 2004; Boehm et al., 2006; Elias et al., 2008; Cane et al., 2014; Taft and Turrigiano, 2014). NMDAR currents are also smaller in new spines (Zito et al., 2009; Kwon and Sabatini, 2011), where they demonstrate greater diffusional coupling to the dendrite (Zito et al., 2009). PSD-family MAGUKS, NMDAR-mediated signaling, and spine morphologies associated with a high degree of compartmentalization are all thought to regulate synaptic stability (De Roo et al., 2008a,b; Cane et al., 2014; Taft and Turrigiano, 2014; Lambert et al., 2017), suggesting that the low basal survivorship rates of new spines may be due to their distinct molecular composition and signaling. Identifying the molecular signaling pathways at play in new spines is therefore crucial to understand the mechanisms involved in their stabilization.
Here, we show that, unlike GFP-tagged PSD-family MAGUKs, new spines express GFP-CaMKIIα at levels comparable to those in mature spine levels, independent of CaMKIIα kinase activity. CaMKIIα's presence in new spines supports that CaMKIIα signaling could play a critical role in new spine function. Indeed, evidence supports a requirement for the CaMKIIα–GluN2B interaction not only in activity-dependent new spine stabilization (Hill and Zito, 2013) but also in spontaneous and activity-dependent new spine outgrowth (Hamilton et al., 2012), suggesting that CaMKIIα's functions at new spines may precede any form of synaptic stimulation.
Role of CaMKIIα kinase activity in new spine stabilization
Despite our finding that CaMKIIα is present at mature levels in new spines, we were surprised to find that CaMKIIα kinase activity is not required for enhanced new spine stabilization induced either by strong glutamatergic stimulation at single spines or by overexpression of the CaMKIIα-K42R/T286 phospho-mutant. Our results in new spines are in contrast with what is known regarding the important role of CaMKIIα kinase activity in stabilization of the long-term growth of mature spines (Araki et al., 2015; Cornelia Koeberle et al., 2017). However, major changes to the molecular composition of new spines occur during the maturation process, including the recruitment of PSD-family MAGUKs (De Roo et al., 2008a; Lambert et al., 2017), no doubt creating a vastly different biochemical signaling environment in the new spine as it develops. Indeed, it is possible that, while CaMKIIα kinase activity is not required to enhance new spine stabilization on the time scale of 70–130 min after new spine growth, as we observed in our experiments, it may be necessary at later times, for example, following the delayed recruitment of other synaptic proteins, such as PSD-family MAGUKS. Overall, our data demonstrate that CaMKIIα kinase function is not required for the early steps of new spine stabilization, within the first few hours following new spine outgrowth.
Role of GluN2B–CaMKIIα binding in new spine stabilization
Our finding that CaMKIIα kinase activity is not required for activity-dependent new spine stabilization leaves an undefined role for the required CaMKIIα–GluN2B interaction (Hill and Zito, 2013). This interaction has long been known to be important in the regulation of basal synaptic transmission and LTP maintenance (Barria and Malinow, 2002; Halt et al., 2012; Barcomb et al., 2014; Incontro et al., 2018), where it is thought to play a role in bringing Ca2+/CaM-activated CaMKII closer to its targeted substrates to alter synaptic transmission and synaptic strengthening in a kinase-dependent manner. In new spines, our results instead support a nonenzymatic role for CaMKIIα in new spine stabilization. Indeed, we show that, although CaMKIIα kinase activity is not required, knockdown of CaMKIIα disrupts activity-dependent new spine stabilization. Altogether our results suggest that the interaction between GluN2B and CaMKIIα is required to support a primarily structural or scaffolding role for CaMKIIα in new spine stabilization.
While we found that CaMKIIα-T286D, which enhanced the interaction between GluN2B and CaMKIIα, also increased basal spine stabilization, survivorship rates for CaMKIIα-T286D were lower than observed for new spines that received HFU stimulation. At mature spines, glutamatergic stimulation initiates a number of concurrent signaling cascades and molecular changes, such as NMDAR and mGluR activation and downstream signaling mechanisms (Lee et al., 2003; Malinow, 2003; Murakoshi et al., 2011; Bosch et al., 2014; Stein et al., 2021) that are not replicated by overexpression of the CaMKIIα-T286D phospho-mutant. It is likely that at least a subset of these mechanisms acts in conjunction with GluN2B–CaMKIIα binding to enhance activity-dependent new spine stabilization. In addition, the GluN2B–CaMKIIα interaction may serve to bring CaMKIIβ, which complexes with CaMKIIα at a 3:9 ratio in the hippocampus, within optimal proximity to its binding partners in order to regulate cytoskeletal stability (Okamoto et al., 2007; Kim et al., 2015; Kim et al., 2019).
Nonenzymatic CaMKIIα function in new spine stabilization
Although CaMKIIβ is perhaps more well recognized than CaMKIIα for its nonenzymatic functions in regulating the spine actin cytoskeleton, CaMKIIα has also been shown to participate in several functionally important scaffolding and structural interactions that are distinct from those made by CaMKIIβ (Hell, 2014; Bayer and Schulman, 2019). Some of these interactions are less likely to be relevant for the earliest stages of new spine stabilization, such as roles with stargazin, TARP γ-8, or the Rac-1 activating RAKEC (Opazo et al., 2010, 2012; Park et al., 2016; Saneyoshi et al., 2019), as they require either binding to PSD-family MAGUKs or CaMKIIα kinase activity. However, other known CaMKIIα interactions are independent of these requirements and therefore would make attractive candidates for roles in new spine stabilization, such as the activity-dependent binding of CaMKIIα directly to the 26S proteasome, and indirect interactions of CaMKIIα with SynGAP-1α via the multi-PDZ domain protein MUPP-1 (Krapivinsky et al., 2004; Bingol et al., 2010).
Indeed, CaMKIIα's nonenzymatic interactions with the proteasome and the MUPP1–SynGAP-1α complex appear particularly promising in the context of understanding new spine stabilization. SynGAP-1α is a negative regulator of synapse maturation, and its exclusion from synapses contributes to synaptic strengthening, precocious PSD-95 accumulation, and increased spine volume (Vazquez et al., 2004; Clement et al., 2012; Aceti et al., 2015; Araki et al., 2015). Interestingly, activity-dependent dissociation of the MUPP1–SynGAP-1α complex from CaMKIIα does not require CaMKIIα kinase activity (Krapivinsky et al., 2004) and thus may provide a mechanism for SynGAP-1α dispersion (Araki et al., 2015) from new spines, independent of PSD-family MAGUKs and kinase activity. Furthermore, activity-dependent new spine formation requires the proteasome (Hamilton et al., 2012), which may remain accumulated at sites of new spine formation, where it could play a role in the activity-dependent degradation of negative regulators of synapse stability and maturation. Indeed, there is evidence that the proteasome mediates degradation of SynGAP (Zhang et al., 2020) and Ephexin5 (Hamilton et al., 2017), which both have roles in regulating dendritic spine stability. The elucidation of the role of these two proteins and of other nonenzymatic functions of CaMKIIα downstream of GluN2B–CaMKIIα to promote new spine survivorship is an intriguing and compelling avenue for future study.
References
- Aceti M, Creson TK, Vaissiere T, Rojas C, Huang WC, Wang YX, Petralia RS, Page DT, Miller CA, Rumbaugh G (2015) Syngap1 haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol Psychiatry 77:805–815. 10.1016/j.biopsych.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarran E, Raissi A, Jaidar O, Shatz CJ, Ding JB (2021) Enhancing motor learning by increasing the stability of newly formed dendritic spines in the motor cortex. Neuron 109:3298–3311 e3294. 10.1016/j.neuron.2021.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL (2015) Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85:173–189. 10.1016/j.neuron.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Buard I, Coultrap SJ, Kulbe JR, O’Leary H, Benke TA, Bayer KU (2014) Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J 28:3810–3819. 10.1096/fj.14-250407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcomb K, Coultrap SJ, Bayer KU (2013) Enzymatic activity of CaMKII is not required for its interaction with the glutamate receptor subunit GluN2B. Mol Pharmacol 84:834–843. 10.1124/mol.113.089045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35:345–353. 10.1016/S0896-6273(02)00776-6 [DOI] [PubMed] [Google Scholar]
- Bayer KU, Schulman H (2019) CaM kinase: still inspiring at 40. Neuron 103:380–394. 10.1016/j.neuron.2019.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96:43–55. 10.1016/j.neuron.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M (2010) Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140:567–578. 10.1016/j.cell.2010.01.024 [DOI] [PubMed] [Google Scholar]
- Boehm J, Ehrlich I, Hsieh H, Malinow R (2006) Two mutations preventing PDZ–protein interactions of GluR1 have opposite effects on synaptic plasticity. Learn Mem 13:562–565. 10.1101/lm.253506 [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82:444–459. 10.1016/j.neuron.2014.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane M, Maco B, Knott G, Holtmaat A (2014) The relationship between PSD-95 clustering and spine stability in vivo. J Neurosci 34:2075–2086. 10.1523/JNEUROSCI.3353-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, et al. (2012) Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151:709–723. 10.1016/j.cell.2012.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelia Koeberle S, Tanaka S, Kuriu T, Iwasaki H, Koeberle A, Schulz A, Helbing DL, Yamagata Y, Morrison H, Okabe S (2017) Developmental stage-dependent regulation of spine formation by calcium-calmodulin-dependent protein kinase IIalpha and Rap1. Sci Rep 7:13409. 10.1038/s41598-017-13728-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Mendez P, Poglia L, Muller D (2008a) Activity-dependent PSD formation and stabilization of newly formed spines in hippocampal slice cultures. Cereb Cortex 18:151–161. 10.1093/cercor/bhm041 [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Muller D (2008b) LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol 6:e219. 10.1371/journal.pbio.0060219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R (2004) Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci 24:916–927. 10.1523/JNEUROSCI.4733-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA (2008) Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A 105:20953–20958. 10.1073/pnas.0811025106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31:1203–1216. 10.1038/emboj.2011.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Lambert JT, Parajuli LK, Vivas O, Park DK, Stein IS, Jahncke JN, Greenberg ME, Margolis SS, Zito K (2017) A dual role for the RhoGEF Ephexin5 in regulation of dendritic spine outgrowth. Mol Cell Neurosci 80:66–74. 10.1016/j.mcn.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K (2012) Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74:1023–1030. 10.1016/j.neuron.2012.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525:333–338. 10.1038/nature15257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TC, Zito K (2013) LTP-induced long-term stabilization of individual nascent dendritic spines. J Neurosci 33:678–686. 10.1523/JNEUROSCI.1404-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K (2005) Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45:279–291. 10.1016/j.neuron.2005.01.003 [DOI] [PubMed] [Google Scholar]
- Incontro S, Diaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, Nicoll RA (2018) The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Commun 9:2069. 10.1038/s41467-018-04439-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Ziv NE, Okazaki H, Yagishita S, Toyoizumi T (2021) Spine dynamics in the brain, mental disorders and artificial neural networks. Nat Rev Neurosci 22:407–422. 10.1038/s41583-021-00467-3 [DOI] [PubMed] [Google Scholar]
- Kim K, et al. (2015) A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87:813–826. 10.1016/j.neuron.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, et al. (2019) Autophosphorylation of F-actin binding domain of CaMKIIbeta is required for fear learning. Neurobiol Learn Mem 157:86–95. 10.1016/j.nlm.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K (2006) Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci 9:1117–1124. 10.1038/nn1747 [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE (2004) SynGAP–MUPP1–CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron 43:563–574. 10.1016/j.neuron.2004.08.003 [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL (2011) Glutamate induces de novo growth of functional spines in developing cortex. Nature 474:100–104. 10.1038/nature09986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JT, Hill TC, Park DK, Culp JH, Zito K (2017) Protracted and asynchronous accumulation of PSD95-family MAGUKs during maturation of nascent dendritic spines. Dev Neurobiol 77:1161–1174. 10.1002/dneu.22503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, et al. (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112:631–643. 10.1016/S0092-8674(03)00122-3 [DOI] [PubMed] [Google Scholar]
- Lemieux M, Labrecque S, Tardif C, Labrie-Dion E, Lebel E, De Koninck P (2012) Translocation of CaMKII to dendritic microtubules supports the plasticity of local synapses. J Cell Biol 198:1055–1073. 10.1083/jcb.201202058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R (2003) AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci 358:707–714. 10.1098/rstb.2002.1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472:100–104. 10.1038/nature09823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A 104:6418–6423. 10.1073/pnas.0701656104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D (2010) CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67:239–252. 10.1016/j.neuron.2010.06.007 [DOI] [PubMed] [Google Scholar]
- Opazo P, Sainlos M, Choquet D (2012) Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol 22:453–460. 10.1016/j.conb.2011.10.010 [DOI] [PubMed] [Google Scholar]
- Opitz-Araya X, Barria A (2011) Organotypic hippocampal slice cultures. J Vis Exp:2462. 10.3791/2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chavez AE, Mineur YS, Morimoto-Tomita M, Lutzu S, Kim KS, Picciotto MR, Castillo PE, Tomita S (2016) CaMKII phosphorylation of TARPgamma-8 is a mediator of LTP and learning and memory. Neuron 92:75–83. 10.1016/j.neuron.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Otmakhov N, El Gaamouch F, Lemelin D, De Koninck P, Lisman J (2010) CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc Natl Acad Sci U S A 107:14437–14442. 10.1073/pnas.1009268107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2:13. 10.1186/1475-925X-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R (2010) Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature 463:948–952. 10.1038/nature08759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti T, Banerjee S, Kim C, Leubner M, Lamar C, Gupta P, Lee B, Neve R, Lisman J (2017) Memory erasure experiments indicate a critical role of CaMKII in memory storage. Neuron 96:207–216 e202. 10.1016/j.neuron.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T, Matsuno H, Suzuki A, Murakoshi H, Hedrick NG, Agnello E, O’Connell R, Stratton MM, Yasuda R, Hayashi Y (2019) Reciprocal activation within a kinase–effector complex underlying persistence of structural LTP. Neuron 102:1199–1210 e1196. 10.1016/j.neuron.2019.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Lisman J (2013) The CaMKII/NMDAR complex as a molecular memory. Mol Brain 6:10. 10.1186/1756-6606-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IS, Park DK, Claiborne N, Zito K (2021) Non-ionotropic NMDA receptor signaling gates bidirectional structural plasticity of dendritic spines. Cell Rep 34:108664. 10.1016/j.celrep.2020.108664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. 10.1016/0165-0270(91)90128-M [DOI] [PubMed] [Google Scholar]
- Taft CE, Turrigiano GG (2014) PSD-95 promotes the stabilization of young synaptic contacts. Philos Trans R Soc Lond B Biol Sci 369:20130134. 10.1098/rstb.2013.0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- Tullis JE, Rumian NL, Brown CN, Bayer KU (2020) The CaMKII K42M and K42R mutations are equivalent in suppressing kinase activity and targeting. PLoS One 15:e0236478. 10.1371/journal.pone.0236478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB (2004) SynGAP regulates spine formation. J Neurosci 24:8862–8872. 10.1523/JNEUROSCI.3213-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, Kennedy MB (2001) Densin-180 forms a ternary complex with the (alpha)-subunit of Ca2+/calmodulin-dependent protein kinase II and (alpha)-actinin. J Neurosci 21:423–433. 10.1523/JNEUROSCI.21-02-00423.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, Zito K (2011) Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J Neurosci 31:12129–12138. 10.1523/JNEUROSCI.6662-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods G, Zito K (2008) Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J Vis Exp:675. 10.3791/675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y (2009) Rapid formation and selective stabilization of synapses for enduring motor memories. Nature 462:915–919. 10.1038/nature08389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, et al. (2009) Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci 29:7607–7618. 10.1523/JNEUROSCI.0707-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB (2009) Stably maintained dendritic spines are associated with lifelong memories. Nature 462:920–924. 10.1038/nature08577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, et al. (2020) PSD-93 interacts with SynGAP and promotes SynGAP ubiquitination and ischemic brain injury in mice. Transl Stroke Res 11:1137–1147. 10.1007/s12975-020-00795-z [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K (2009) Rapid functional maturation of nascent dendritic spines. Neuron 61:247–258. 10.1016/j.neuron.2008.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]