

Abstract
Exercise enhances physical performance and reduces the risk of many disorders such as cardiovascular disease, type 2 diabetes, dementia, and cancer. Exercise characteristically incites an inflammatory response, notably in skeletal muscles. While some effector mechanisms have been identified, regulatory elements activated in response to exercise remain obscure. Here, we have addressed the roles of Foxp3+CD4+ regulatory T cells (Tregs) in the healthful activities of exercise via immunologic, transcriptomic, histologic, metabolic, and biochemical analyses of acute and chronic exercise models. Exercise rapidly induced expansion of the muscle Treg compartment, thereby guarding against over-exuberant production of interferon gamma and consequent metabolic disruptions, particularly mitochondrial aberrancies. Importantly, the performance-enhancing effects of exercise training were dampened in the absence of Tregs. Thus, exercise is a natural Treg booster with therapeutic potential in disease and aging contexts.
One-Sentence Summary:
Regulatory T cells promote muscle adaptation to endurance exercise by limiting inflammation and consequent mitochondrial damage.
Introduction:
Chronic inflammation underpins many modern afflictions, including cardiovascular disease, obesity, type 2 diabetes, neurodegeneration, and cancer (1). Physical activity and exercise (intentional physical activity) improve these conditions. For example, all-cause-mortality risk is reduced by 20% in humans that achieve recommended activity goals, and this amelioration is magnified by greater levels of physical activity up to 10-fold over the minimum (2). Although there is a preponderance of evidence for the benefits of physical activity and exercise in many tissues in both healthy and diseased states, the cellular and molecular mechanisms underpinning exercise-induced adaptations remain ill-defined (3).
Skeletal muscle is the actuator of movement and exercise performance and also plays a critical role in regulation of organismal metabolic homeostasis. Endurance exercise represents a form of transient mechano-metabolic stress to skeletal muscles, which is met with increased expression of genes involved in muscle architecture and intermediary metabolism, in particular oxidative reactions in mitochondria (4, 5). Immunomodulation is also a component of the response to exercise (6). However, since the first description of post-exercise leukocytosis over 100 years ago (7), the study of exercise-induced immunomodulation has been limited to measurements of circulating factors. Little is known about immunological processes operating within muscle, itself, other than the fact that inflammation occurs (8, 9).
Experimental models of muscle injury induce an orchestrated, multi-cellular, inflammatory response followed by a period of tissue repair (10). At steady-state, skeletal muscle harbors a small population of Foxp3+CD4+ regulatory T cells (Tregs) (11); in injured muscle, Tregs are clonally expanded and coordinate a shift from pro-inflammatory to pro-repair processes (11, 12). Induced depletion of Tregs or diminished Treg expansion due to age-related decline in stromal-cell production of IL-33 impairs this transition, leading to reduced myogenesis and increased fibrosis (11, 13). Tregs display a biphasic response to exercise in peripheral blood (14, 15); yet, it is unknown whether exercise promotes mobilization to and proliferation of Tregs in skeletal muscle or whether Tregs are needed to support the benefits of exercise.
Multi-pronged assessment of acute and chronic exercise models allowed us to evidence a role for Tregs in the regulation of exercise-induced muscle inflammation, metabolic reprogramming, and performance enhancement. These findings highlight Treg modulation as a salient component of the cellular and molecular adaptations supporting the benefits of exercise.
Results
Acute endurance exercise induces immunocyte accumulation in specific hindlimb muscles
To investigate the potential of acute endurance exercise (AEX) to modulate immunocyte activities in skeletal muscle, we subjected sedentary C57BL/6J (B6) mice to a 90-minute bout of moderate-intensity running (~55% of maximum speed) on a motorized treadmill after two days of acclimation and performed cytofluorometric analysis on muscle immunocytes on days 1, 3, and 7 post-AEX (Fig. 1A and fig. S1,A to G). Blood lactate measurements taken immediately after exercise were significantly elevated, yet remained below the aerobic-anaerobic threshold previously established for mice of this age, sex, and strain (16) (Fig. 1B). Immunophenotyping of pooled hindlimb muscles – the tibialis anterior (TA), gastrocnemius (Ga), soleus (Sol), and quadriceps (Qd) muscles – from sedentary and acutely exercised mice revealed rapid accumulation of immunocytes (CD45+ cells) in muscles of AEX mice, with peak numbers by the first 24h of recovery (Fig. 1C). The proportion of macrophages macrophages (Ly6G−CD11b+CD64+ cells) was maximum at 24h but stayed elevated for at least one week (Fig. 1D), while the frequency and numbers of pro-inflammatory Ly6Chi macrophages spiked on d1 (Fig. 1E), parallel to what was previously shown in toxin-injured muscles (11, 17). The frequency and numbers of Tregs were also significantly elevated on d1 (Fig. 1F), as was their cell-cycle entry (fig. S1H).
Figure 1. Muscle immunocyte accumulation is induced by acute exercise.
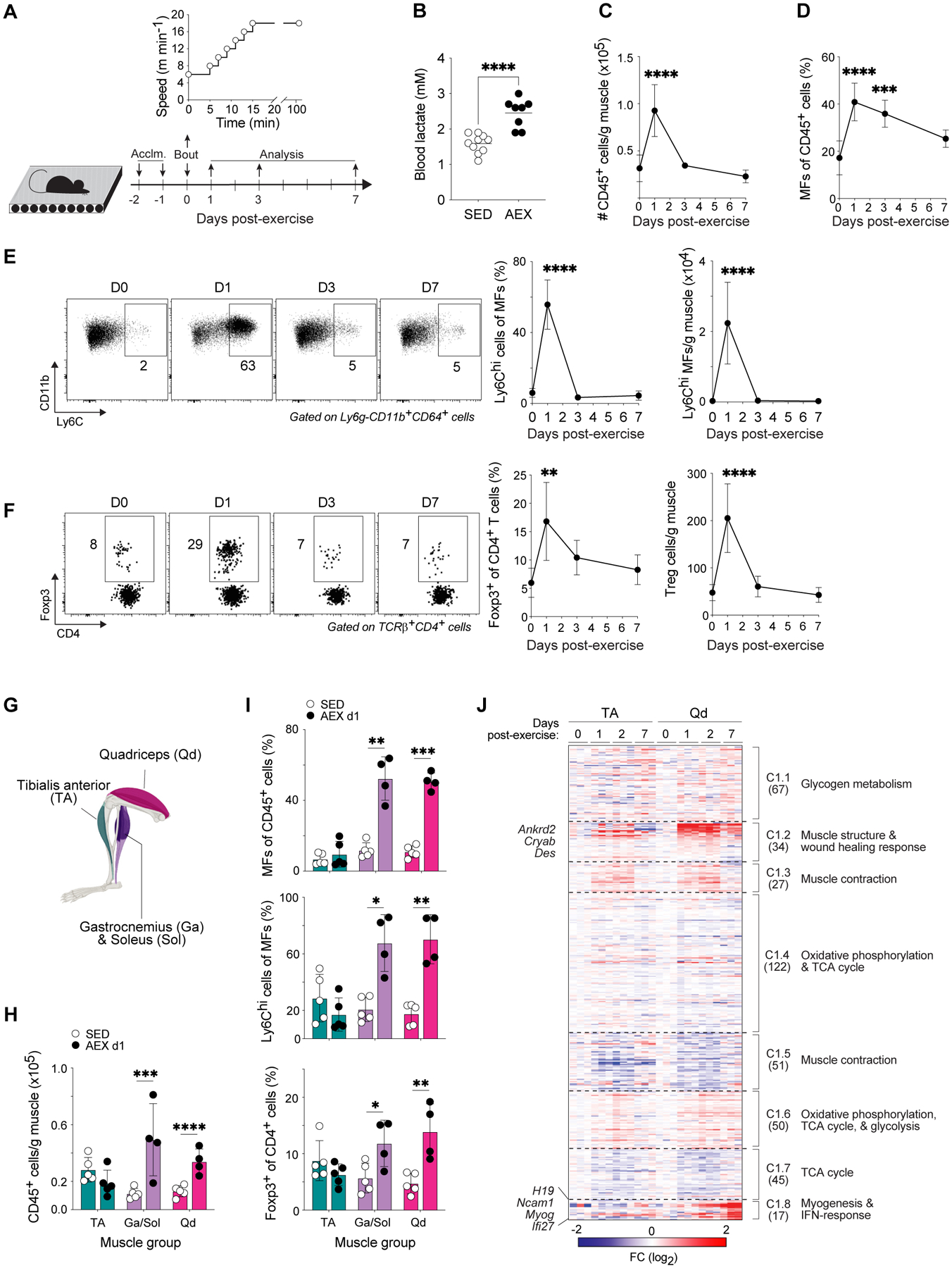
(A-J) 8–10-week-old mice were acclimated to a motorized treadmill for two days and were then made to perform an acute, moderate-intensity bout of endurance exercise. Cytofluorometric (n≥4–5) and transcriptomic (n=3) analyses were performed during recovery.
(A) Schema for AEX experiments.
(B) Blood-lactate concentrations measured at rest and immediately after exercise.
(C) Number of immunocytes (CD45+ cells) per gram of muscle.
(D) Frequency of MFs in muscle.
(E) Representative flow-cytometric plots of pro-inflammatory Ly6Chi MFs in muscle. Numbers beside gates indicate frequencies of gated cells among total MFs. Summary frequencies and numbers per gram of muscle are shown to the right.
(F) Representative flow-cytometric plots of Tregs in muscle. Numbers beside gates indicate frequencies of gated cells among total CD4+ T cells. Summary frequencies and numbers per gram of muscle are shown to the right.
(G) Anatomical map of mouse hindlimb muscles.
(H) Number of immunocytes in specific hindlimb muscles from SED mice or from mice on d1 of recovery after AEX. Each dot represents pooled muscle from 2–3 mice.
(I) Frequencies of MFs, Ly6Chi MFs, and Tregs in muscle from indicated conditions. Each dot represents pooled muscle from 2–3 mice.
(J) k-means clustering of transcripts differentially expressed in TA and Qd before AEX and during recovery (p < 0.05, FDR < 0.05, r2 ≥ 0.65). Numbers in parentheses indicate the number of genes within each cluster. Fold-change as compared to d0 controls from each muscle.
Summary plots show data (means ± SD) pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by an unpaired Student’s t-test (B and H-I) or one-way ANOVA with Dunnett’s multiple comparisons test comparing each post-exercise timepoint to d0 controls (C-F). Acclim., acclimation; AEX, acute exercise; SED, sedentary; MF, macrophage; TCA, tricarboxylic acid.
Although endurance exercise effects changes in virtually all tissues, skeletal muscles experience variable levels of mechanical and metabolic stress, depending on their anatomical location, their fiber-type composition, and the exercise modality employed. Serum creatine kinase, a commonly employed indicator of muscle damage, was not increased at any time after AEX compared with controls (fig. S1I); yet analysis of the membrane permeability of various hindlimb muscles on d1 after AEX showed a greater loss of membrane integrity of combined Ga, Sol, and Qd muscles compared with the TA (fig. S1J). We sought to exploit this natural variation to address the specificity of immunocyte accrual in hindlimb muscles. Analysis of immunocytes from hindlimb muscles grouped by gross anatomy revealed that the Ga/Sol and Qd, but not TA, muscles showed immunocyte accumulation and enrichment of macrophages, Ly6Chi macrophages, Tregs, and neutrophils (Ly6G+CD11b+), concurrent with de-enrichment of Ly6Clo macrophages, CD8+ T cells and conventional CD4+ T cells on d1 post-exercise (Fig. 1, G to I and fig. S1K). To map muscle-group-specific immunomodulation to changes in other processes at the same site, we performed whole-tissue transcriptional profiling of the prototypes of immunologically inactive (TA) and active (Qd) muscles over a timecourse of recovery after AEX. k-means clustering of the transcripts differentially expressed (p < 0.05; FDR < 0.05; r2 ≥ 0.65) by the two muscle groups over the course of recovery highlighted similar shifts in metabolic profiles (clusters C1.1, 1.6, and 1.7); yet two clusters, C1.2 and C1.8, were induced early or late after exercise preferentially in Qd muscles (Fig. 1J). C1.2 included transcripts known to respond to acute exercise, including those encoding factors involved in muscle structure and response to wound healing (Ankrd2, Cryab, Des) (Fig. 1J). The C1.8 transcripts enriched in Qd muscles included: H19, previously highlighted in an analysis of data from the HERITAGE Family Study as part of a signature predictive of the response to exercise training (18); Myog, encoding the myogenic transcription factor Myogenin; and the interferon (IFN)-inducible transcripts Ncam1 and Ifi27 (Fig. 1J).
Endurance-exercise training induces muscle inflammation and up-regulates genes involved in oxidative metabolism and interferon signaling
Repeated bouts of endurance exercise produce a drastic increase in muscle oxidative capacity over time (19). Such improvement is partly due to the cumulative effects of transient bursts in expression of mRNAs encoding metabolic enzymes, derived from both nuclear and mitochondrial genomes (20). However, the transcriptomes of chronically exercised muscles show little overlap with their acutely exercised counterparts (21), suggesting that acute and chronic exercise are related yet distinct contexts. Therefore, to investigate whether immunocytes, in particular Tregs, also accumulated in muscles in response to chronic endurance exercise, we individually housed mice with unrestricted access to a running wheel for 2, 3, or 4 weeks, and isolated muscle immunocytes for cytofluorometric analysis after a washout period of 24h (Fig. 2A). Notably, we chose to treat week 1 as an acclimation phase and did not include it in our timecourse as it generally takes mice several days to become accustomed to individual housing and wheels. Similar to acute exercise, exercise training (EXT) resulted in significant accrual of total immunocytes in hindlimb muscles (Fig. 2B). Immunocyte numbers were elevated after all durations of EXT; however, the magnitude of accumulation was greatest at 2 weeks and diminished over additional training times (Fig. 2B). This same pattern of accumulation was observed for the frequency of macrophages, Tregs, and CD8+ T cells, while pro-inflammatory Ly6Chi macrophages and other populations of myeloid-lineage cells were, surprisingly, not enriched after any duration of EXT (Fig. 2C and fig. S2, A to F). Notably, similar to AEX, EXT also failed to induce muscle damage of a magnitude that could be detected by measurement of serum creatine kinase activity (fig. S2G). The differential enrichment of Ly6Chi macrophages in muscles after AEX compared with EXT may reflect the different chemokine profiles of muscles after these interventions. Indeed, comparison of available human and murine muscle transcriptomes after acute and chronic exercise using the MetaMex web app (22) showed acute exercise induced superior enrichment in many chemokines (e.g. Ccl2 and Ccl7) known to attract monocytes.
Figure 2. Muscle immunocyte accumulation, oxidative phosphorylation, and interferon signaling are indued by endurance exercise training.
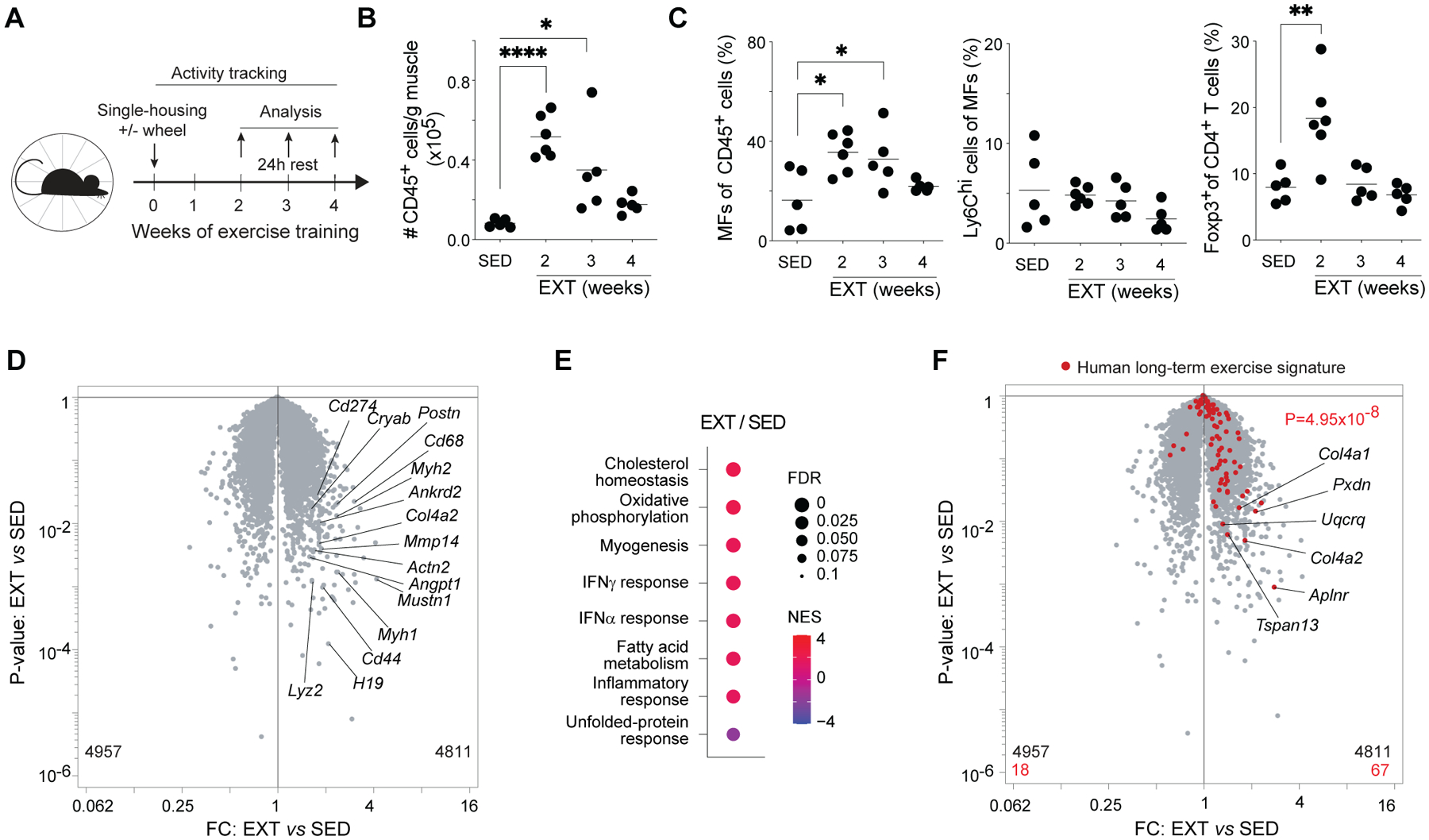
(A-F) 8-week-old mice were individually housed for 2–4 weeks with or without access to running wheels equipped with activity-tracking software. Hindlimb muscles were isolated for cytofluorometric (n≥5) and transcriptomic (n=3) analyses after a 24h washout period.
(A) Schema for EXT experiments.
(B) Number of immunocytes per gram of muscle.
(C) Frequencies of MFs, Ly6Chi MFs, and Tregs.
(D) RNA-seq analysis of Qd muscles from sedentary and 2-week exercise-trained mice.
(E) GSEA showing top Hallmark gene sets enriched or impoverished in Qd muscles after 2wk EXT.
(F) Volcano plot overlain with genes differentially enriched in human muscles after long-term exercise (21).
Summary plots show data pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ****p < 0.0001 by one-way ANOVA with Dunnett’s multiple comparisons test comparing each week of EXT to sedentary controls (B and C) or p value computed by the χ2 test (F). EXT, exercise training; FC, fold-change; FDR, false-discovery rate; NES, normalized enrichment score; all other abbreviations as per prior figures.
The second week of EXT is a phase during which mice have acclimated to using wheels and rapidly accumulate a substantial running distance, which continues to steadily increase for another 1–2 weeks. Although muscle transcriptional and metabolite profiling has typically been performed after 4 or more weeks of EXT, based on the decline in muscle inflammation after 2 weeks, we speculated that 2 weeks might be sufficient for hindlimb muscles to acquire an exercise-adapted phenotype. Transcriptional profiling of Qd muscles from mice exercise-trained for 2 weeks showed up-regulation of transcripts involved in muscle structure and response to wound healing (e.g. Actn2, Ankrd2, Cryab, Myh1, Myh2, Postn), immunological processes (e.g. Cd44, Cd68, Cd274, Lyz2), extracellular matrix remodeling (e.g. Col4a2, Mmp14), and angiogenesis (Angpt1) (Fig. 2D). Gene-set enrichment analysis (GSEA) showed up-regulation of many pathways previously described as hallmarks of adaptation to exercise training, in particular oxidative phosphorylation (Fig. 2E). Increased expression and activity of mitochondrial enzymes involved in oxidative phosphorylation is largely responsible for the enhanced oxidative capacity of muscles after repeated bouts of endurance exercise (4, 5, 19). Enhanced expression of mitochondrial enzymes involved in oxidative phosphorylation is partly regulated by the transcriptional co-activator PPAR-γ co-activator-1 α (PGC1-α) (23). Overlay of a gene-expression signature derived from transgenic mice expressing PGC1-α under control of the muscle creatine kinase promoter (24) showed significant enrichment in muscles after 2 weeks of EXT (fig. S2H). Therefore, the window of training-induced immunomodulation coincides with acquisition of an exercise-adapted muscle transcriptional profile.
Some unexpected pathways were also enriched in exercised-trained relative to sedentary muscles, namely responses to IFNγ and IFNα (Fig. 2E). Although there is a dearth of studies on exercise-induced IFN signaling in skeletal muscle, a recent meta-analysis of human blood and muscle transcriptomes taken before or after various exercise protocols found that many IFN-dependent genes were induced by long-term exercise (21). Overlay of a signature derived from this meta-analysis and comprised of genes differentially up-regulated by long-term exercise in human muscle showed significant enrichment in exercise-trained murine muscles (Fig. 2F).
Tregs control muscle inflammation induced by AEX
As Tregs control muscle inflammation, repair, and regeneration in contexts of acute and chronic injury (11), we wondered whether they also influenced responses to acute exercise. As an initial approach, we performed loss-of-function experiments via diphtheria toxin (DT)-mediated ablation of Foxp3-expressing cells (i.e. Tregs) in Foxp3-IRES-GFP.hDTR mice. DT receptor (DTR)+ and control DTR- littermates were injected with DT during acclimation and immediately before acute exercise (Fig. 3A). This strategy did not affect performance, assessed by measuring post-exercise blood-lactate concentrations (Fig. 3B), and resulted in complete, though transient, depletion of muscle Tregs (Fig. 3C). Immunophenotyping of hindlimb muscles from sedentary and acutely exercised DTR- and DTR+ mice revealed significantly more immunocyte accumulation in DTR+ mice during recovery from AEX (Fig. 3D). The proportions of macrophages were similar in the two genotypes, while the frequency of pro-inflammatory Ly6Chi macrophages was significantly higher in DTR+ muscles after AEX (Fig. 3E). Hematoxylin and eosin (H&E) staining of d7 Qd muscle sections revealed inflammatory lesions characterized by immunocytes encircling one or more myofibers; such lesions were found at a very low frequency in exercised DTR- muscles (Fig. 3Fand fig. S3A). However, muscles of DTR+ mice had a significantly higher frequency of immunocyte congregates and had a significantly larger area involved in each lesion (Fig. 3Fand fig. S3A).
Figure 3. Control of acute exercise-induced muscle inflammation by Tregs.
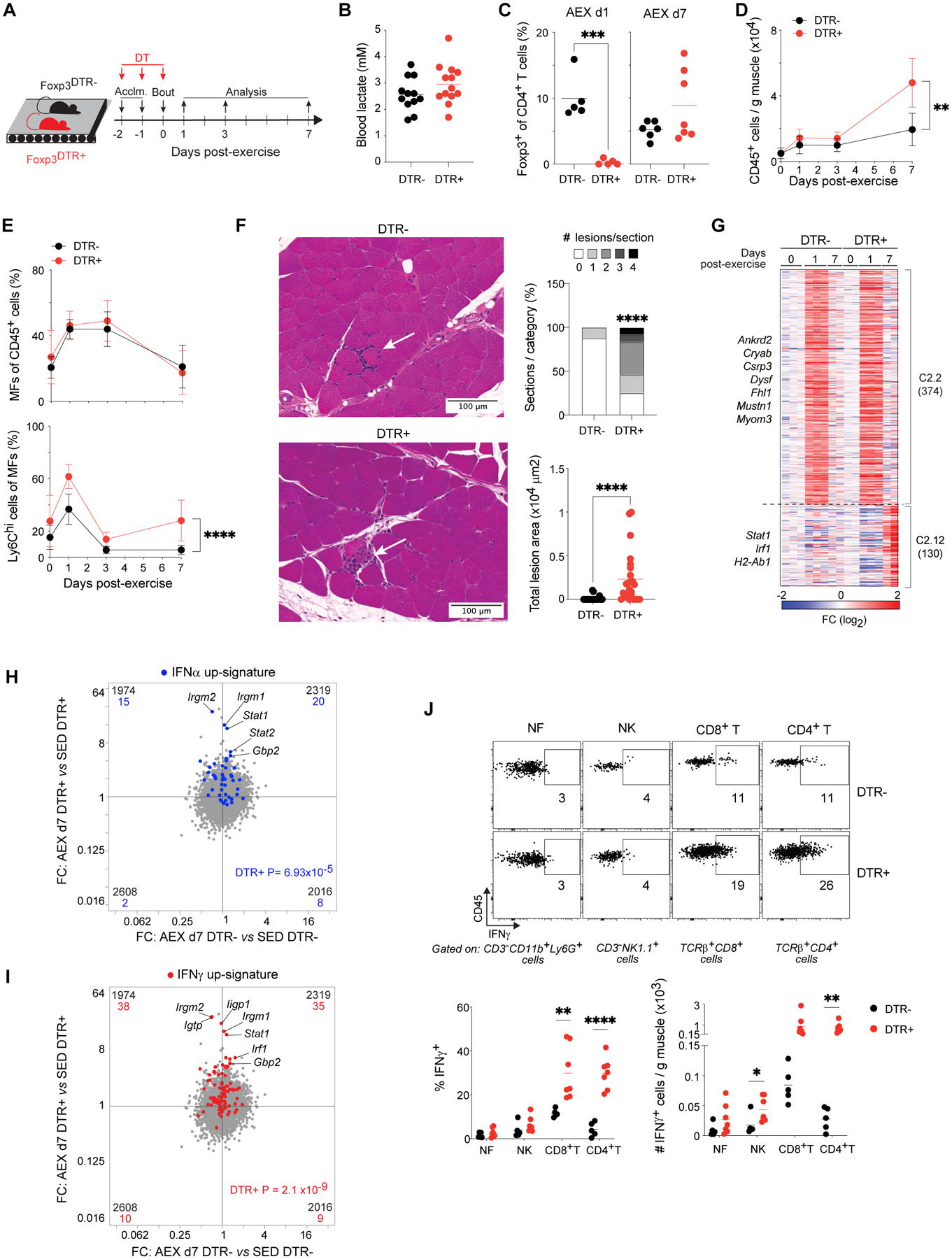
(A) Schema for AEX experiments with DT-induced Treg ablation.
(B) Blood-lactate concentrations measured at rest and immediately after exercise (n=12–13).
(C) Frequency of Tregs after AEX with versus without ablation (n≥5).
(D) Timecourse of number of immunocytes in muscles of DTR- and DTR+ mice (n≥4).
(E) Frequencies of MFs (top) and Ly6Chi MFs (bottom) in muscle.
(F) Left: representative H&E-stained sections of Qd muscles of DTR- and DTR+ mice on d7 after AEX. Arrows indicate inflammatory lesions. Right: summary quantification. n=24 sections; 6 mice.
(G) k-means clustering of transcripts differentially expressed in Qd muscles of DTR- and DTR+ mice before AEX and during recovery (p < 0.05, FDR < 0.05, r2 ≥ 0.65). Numbers in parentheses indicate number of genes within each cluster. Fold-change as compared to d0 controls from each genotype.
(H) RNA-seq analysis of Qd muscles from DTR- and DTR+ mice in sedentary and 2-week exercise-trained conditions (n=2). FC/FC plot overlain with a signature of genes differentially enriched (FC ≥ 2) by recombinant IFNα treatment (69).
(I) FC/FC plot from H overlain with a signature of genes differentially enriched (FC ≥ 2) by recombinant IFNγ treatment (32).
(J) Top: representative flow-cytometric plots of immunocyte populations on d7 AEX. Numbers beside gates indicate frequencies of gated cells among all cells of parent subset. Bottom: summary frequencies and numbers (n≥5).
Summary plots show data (means ± SD) pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by an unpaired Student’s t-test (C and J), two-way ANOVA with Šídák’s multiple comparisons test (D and E; p-value indicates genotype, time interaction), a Wilcoxon signed-rank test (F; lesion frequencies), the Mann-Whitney test (F; lesion areas), or p value computed by the χ2 test (H and I). DT, diphtheria toxin; DTR, diphtheria toxin receptor; NK, natural killer cell; all other abbreviations as per prior figures.
To explore the consequences of unleashed inflammation following Treg depletion on the molecular responses to acute exercise, we transcriptionally profiled whole quadriceps muscles of DTR- and DTR+ littermates before exercise and on d1 and d7 after AEX. k-means clustering of the transcripts differentially expressed (p < 0.05; FDR < 0.05; r2 ≥ 0.65) by each genotype over the course of recovery highlighted two clusters, C2.2 and C2.12, containing transcripts that were more strongly induced in Treg-deficient muscles than in control littermates (Fig. 3G). C2.2 included transcripts encoding factors involved in muscle structure (e.g. Ankrd2, Cryab, Dysf) and response to hypoxia (Hif1a) (Fig. 3G). Notably, this cluster shared many transcripts with C1.2 from our comparison of TA and Qd muscles (Fig. 1J), including Csrp3, Mustn1, Ankrd2, Cryab, Fhl1, and Myom3. The C2.12 transcripts enriched in DTR+ muscles belonged to type I and II IFN responses (e.g. Stat1, Irf1, H2-Ab1). Overlay of signatures comprised of IFNα-induced (Fig. 3H) and IFNγ-induced (Fig. 3I) genes showed stronger enrichment in DTR+ than DTR- muscles d7 after AEX. Type I and II IFN signature genes show substantial overlap, so we chose to focus on IFNγ based on the high expression of Ifngr1 in muscle and because punctual Treg ablation is known to result in significantly elevated IFNγ production by NK and T cells in acutely injured muscles (25). Loss of Tregs before acute exercise also increased the number of IFNγ-producing NK cells and CD4+ and CD8+ T cells (Fig. 3J and fig. S3B). All types of non-parenchymal cells residing in muscle were sensitive to IFNγ production, evidenced by increased expression of the IFN-inducible molecules MHCI and PD-L1 on cells from DTR+ compared with DTR- mice (fig. S3C).
Tregs limit muscle inflammation during exercise training and promote performance enhancement
As accrual of Tregs in hindlimb muscles was a shared feature of the responses to acute and chronic endurance exercise (Fig. 1F and Fig. 2C), we questioned whether Tregs influence adaptations to exercise training. Similar to the acute loss-of-function experiments, we ablated Tregs by administering DT to littermate DTR- and DTR+ mice (Fig. 4A). Every-other-day DT injections during the second week of exercise training resulted in complete depletion of muscle Tregs (Fig. 4B) and did not disrupt voluntary wheel-running behavior (Fig. 4C) nor affect body weight (Fig. 4D). Immunophenotyping revealed increased accumulation of total immunocytes after EXT in hindlimb muscles from Treg-deficient mice (Fig. 4E). The proportions of neutrophils, macrophages, and pro-inflammatory Ly6Chi macrophages were significantly higher in muscles of DTR+ mice than in those of DTR- counterparts (Fig. 4F). H&E-stained sections of EXT Qd muscles showed inflammatory lesions reminiscent of those observed in sections of d7 AEX Qd muscles (Fig. 4G and fig. S4A). Sections from trained, Treg-deficient muscles had a significantly higher number of immunocyte congregates as well as a significantly larger area per lesion (Fig. 4G). We measured the exercise capacity of DTR- and DTR+ mice prior to and one day after 2 weeks of EXT (Fig. 4H). As anticipated, training enhanced the exercise capacity of control mice, evidenced by longer running times and more mechanical work performed during the post-test compared with the pre-test (Fig. 4I). However, the performance-enhancing effect of training was significantly stunted in Treg-deficient mice (Fig. 4I). As an alternative loss-of-function approach, we treated mice with an anti-CD25 antibody or control IgG during the second week of exercise training (fig. S4B). Anti-CD25 treatment depleted the fraction and number of muscle Tregs expressing high levels of CD25 (fig. S4C) without affecting the total number of muscle Tregs (fig. S4D). Neither voluntary wheel-running behavior (fig. S4E) nor body weights (fig. S4F) were affected; however, anti-CD25 treatment stunted the performance-enhancing effect of training to a degree similar to that observed after DT-mediated depletion (fig. S4G).
Figure 4. Muscle Tregs control inflammation to support gains in exercise capacity.
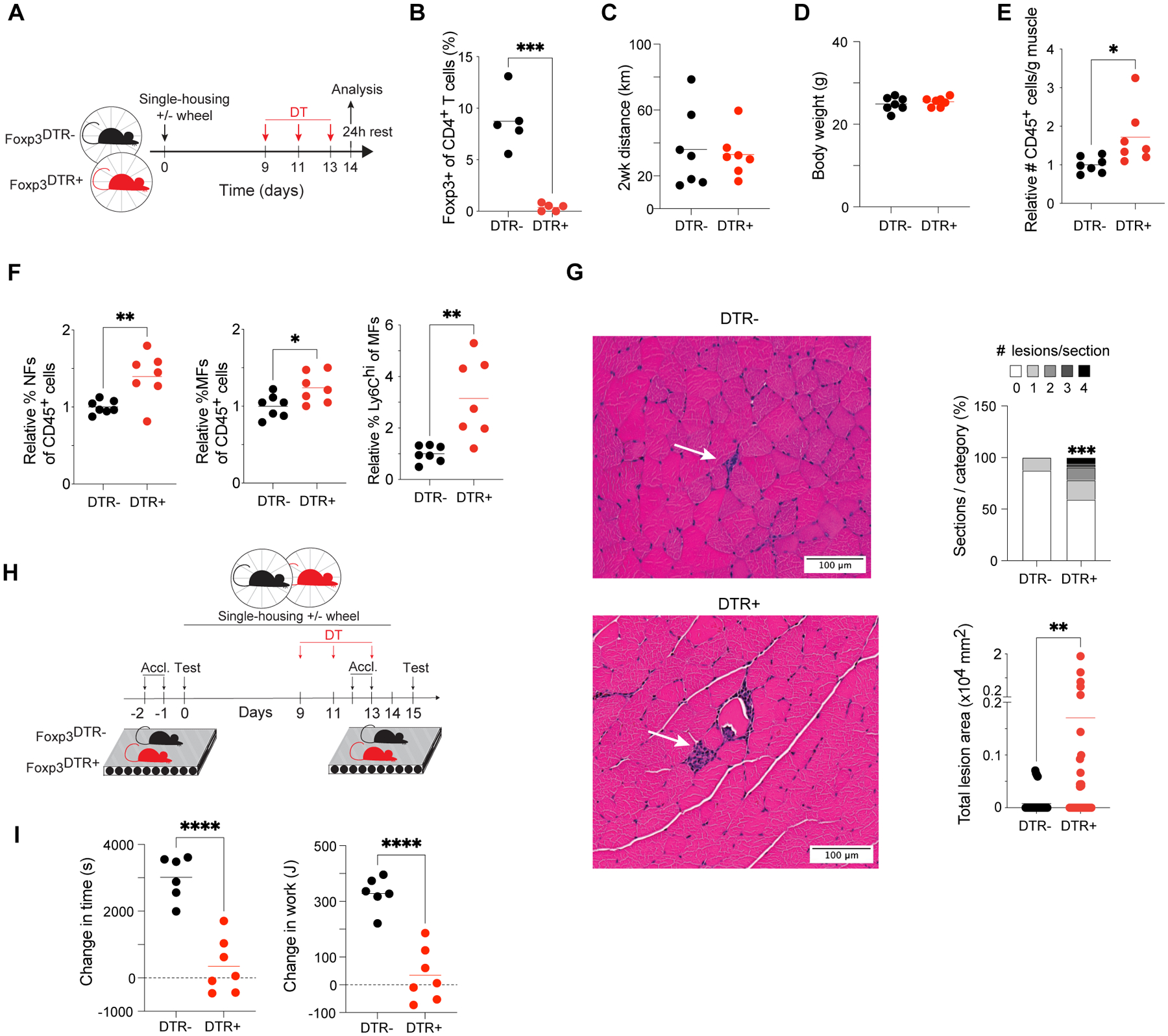
(A) Schema for EXT experiments with DT-induced Treg ablation.
(B) Frequency of Tregs in DTR- and DTR+ mice 48h after completion of DT injections (n=5).
(C) Total distance mice ran voluntarily during EXT (n=7).
(D) Body weights of mice after EXT ± Treg depletion (n=7).
(E) Number of immunocytes in muscle after EXT relative to averages for DTR- mice (n=7).
(F) Muscle immunocyte frequencies after EXT relative to averages for DTR- mice (n=7).
(G) Left: representative H&E-stained sections of Qd muscles from DTR- and DTR+ mice after EXT. Arrows indicate inflammatory lesions. Right: summary plots. n=32 sections; 4 mice.
(H) Schema for exercise-capacity experiments.
(I) Change in time run and work performed by each animal on the post-EXT test compared to the pre-EXT test (n=6–7).
Summary plots show data pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by an unpaired Student’s t-test (B and E-F, I), a Wilcoxon signed-rank test (G; lesion frequencies), or the Mann-Whitney test (G; lesion areas). All abbreviations as per prior figures.
Tregs promote organismal and tissular metabolic adaptations to exercise training
To dissect how ablation of Tregs attenuated the performance-enhancing effect of training, we first assessed muscle contractile function. Ga muscles were stimulated to contract in vivo and the maximum torques during a single contraction cycle (twitch) and during fused contraction cycles (tetanus) were measured. Such measurements were performed prior to and one day after 2 weeks of exercise training, congruent with when our exercise-capacity experiments were performed. Neither DTR- nor DTR+ mice demonstrated significantly altered muscle strength after training (fig. S5A). Transmission electron microscopy (TEM) on EXT Qd muscles supported the finding of unaltered contractile strength in both genotypes, evidenced by the absence of Z-band streaming or other ultrastructural defects in muscle sarcomeres (fig. S5B).
We next measured the whole-body metabolic phenotypes of sedentary and exercise-trained Treg-deficient mice and their control counterparts using commercial metabolic cages. DTR- mice that underwent EXT exhibited an increased respiratory exchange ratio (RER) during the light phase (Zeitgeber time [ZT] 0–12), while Treg-deficient DTR+ mice showed no such change (Fig. 5A). The increase in DTR- RER after training was significant in the portion of the light phase during which the exercise-capacity tests were performed (ZT0–4) (Fig. 5B). Using the updated table of non-protein respiratory quotients (26), we calculated the fractional contribution of glucose and fatty acids to the total energy expenditure of the mice housed in metabolic cages. This analysis of the RER measurements revealed that exercise-trained DTR- mice, but not DTR+ littermates, increased their oxidation of glucose compared with that of sedentary controls (Fig. 5B).
Figure 5. Tregs support metabolic adaptations to exercise training.
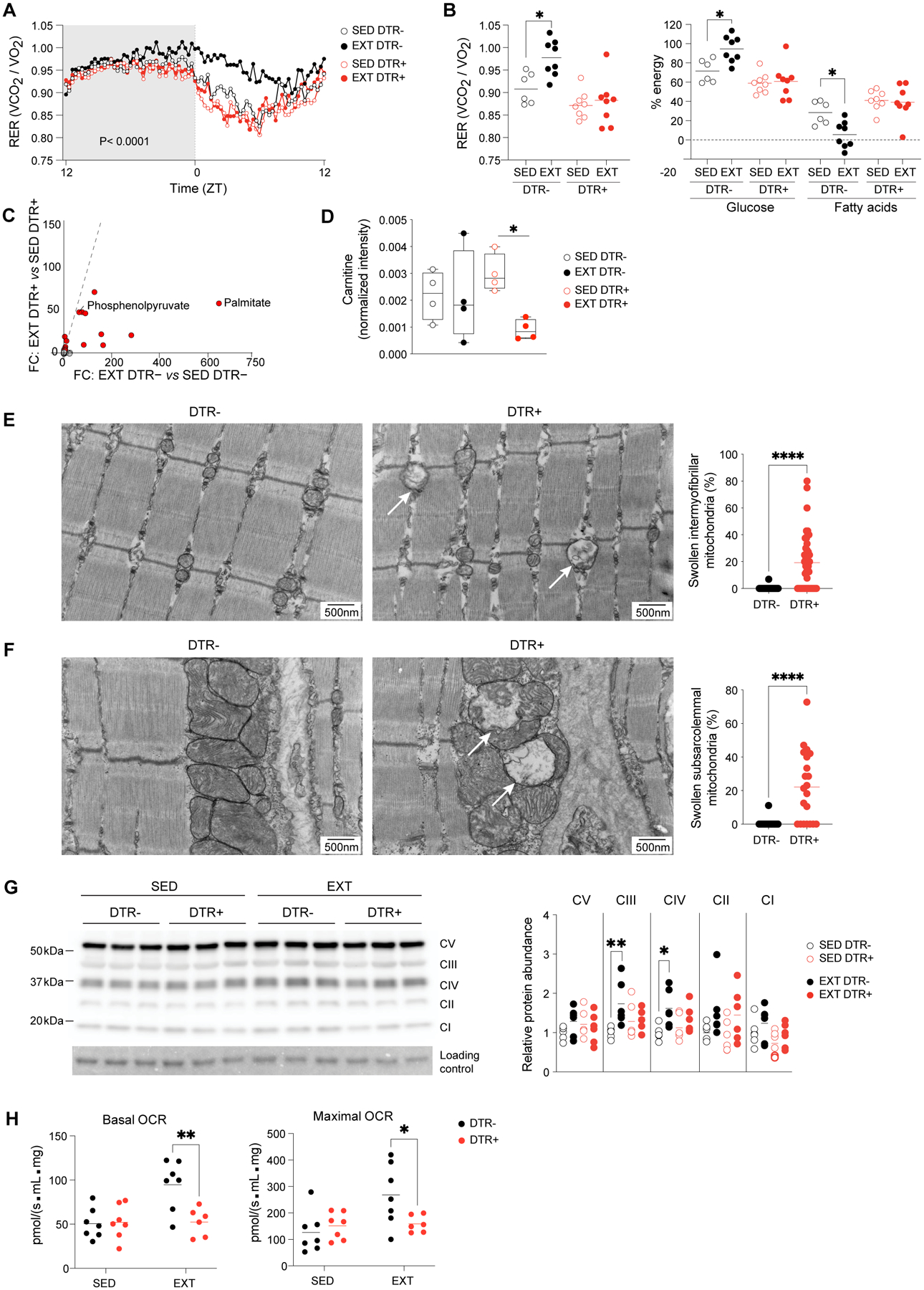
(A) Summary plot of RER of sedentary and exercise-trained DTR- and DTR+ mice housed in metabolic cages (n≥6). Active-phase (ZT12-ZT0) is shaded.
(B) Early rest-phase (ZT0-ZT4) RER and contributions of glucose and fatty acids to total energy expenditure.
(C) Untargeted metabolomic analysis of Qd muscles from DTR- and DTR+ mice in sedentary and 2-week exercise-trained conditions (n=4). FC/FC plot with metabolites differentially enriched (FC ≥ 2) by training highlighted in red.
(D) Normalized intensity of carnitine from metabolomics dataset in panel C.
(E) Left: representative electron micrographs of sarcomeres and intermyofibrillar mitochondria in Qd muscles of DTR- and DTR+ mice after EXT. Arrows indicate swollen mitochondria. Right: frequency of swollen among total intermyofibrillar mitochondria (n≥32; >400 mitochondria).
(F) Left: as per panel E except subsarcolemmal mitochondria. Right: Frequency of swollen among total subsarcolemmal mitochondria (n=21; >170 mitochondria).
(G) Left: representative immunoblot for electron transport chain (ETC) complexes I-V in Qd muscles. Right: summary data (n=6).
(H) Basal and maximal oxygen consumption rates of myofibers from Ga muscles of sedentary and exercise-trained DTR- and DTR+ mice (n=6–7).
Summary plots show data pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ****p < 0.0001 by two-way ANOVA (A; p-value indicates genotype effect), two-way ANOVA with Šídák’s multiple comparisons test (B, D, G, and H), or the Mann-Whitney test (E and F). RER, respiratory exchange ratio; ZT, Zeitgeber time; OCR, oxygen consumption rate; all other abbreviations as per prior figures.
Given the organism-level differences in the metabolic phenotypes of mice that had or did not have a Treg deficiency, we wondered whether there were metabolic deviations in muscle. We first measured the glycogen content in muscles of sedentary and exercise-trained DTR- and DTR+ mice and failed to observe a defect in training-induced supercompensation (fig. S5C). To gain a more comprehensive view of the availability of metabolic substrates and their transformations in muscles after EXT in mice with or without a Treg deficiency, we performed targeted and untargeted metabolomic profiling of quadriceps muscles. Of the ~200 unique metabolites captured by our untargeted analysis, 20 species were induced >2-fold by EXT (Fig. 5C). Interestingly, palmitate was the training-induced metabolite with the greatest differential between DTR- and DTR+ mice (Fig. 5C). Other fatty acid species (e.g. linoleate, oleate, heptadecanoate, and azelate) followed the same pattern: an increase with EXT only in DTR- mice (fig. S5D). L-carnitine was also significantly reduced in muscle from trained DTR+ mice (Fig. 5D), indicating that import of fatty acids into mitochondria for oxidation might have been hindered.
As phosphoenolpyruvate was also among the metabolites that were more enriched in wild-type trained muscles than in those of Treg-deficient mice (Fig. 5C), we looked more broadly at glucose metabolism and found a general de-enrichment in intermediates of glycolysis and the TCA cycle in trained DTR+ muscles compared with DTR- counterparts (fig. S6A), which was consistent with the decrease in glucose oxidation captured by organism-level metabolic profiling (Fig. 5, A and B). Notably, although muscle glycogen content was similar between DTR- and DTR+ mice, glucose 1-phosphate levels were lower in muscles from the latter (fig. S6A), and this decrease corresponded to blunted training-induced cyclic AMP levels in Treg-deficient muscles (fig. S6B), indicating a potential defect in glycogenolysis.
The increased oxidative capacity, and thus fatigue-resistance, of muscles that have undergone repeated bouts of endurance exercise is also a consequence of changes in mitochondrial content and function (4, 5, 19). Therefore, we performed TEM on sections of Qd muscle from EXT mice and focused on mitochondrial morphology. Micrographs from Treg-deficient muscles revealed mitochondria with diminished electron opacity (Fig. 5E), which are reminiscent of “swollen” morphological forms, characterized by loss of cristae profuseness, dilution of matrix contents, and increased diameter (27, 28). Qd muscle sections from DTR+ mice contained a significantly higher fraction of intermyofibrillar mitochondria with a swollen morphology (Fig. 5E). The subsarcolemmal population of mitochondria also showed a greater proportion of swollen forms in Treg-deficient muscles compared with that of control littermates (Fig. 5F). Importantly, we did not observe any differences in mitochondrial swelling in TA muscles (fig. S7, A and B) nor in kidney (fig. S7C), pointing to a local rather than systemic effect of Treg deficiency on mitochondrial morphology. We also quantified the abundance of electron transport chain (ETC) protein complexes in sedentary and trained Qd muscles and found that EXT significantly increased the abundance of ubiquinol-cytochrome c reductase (complex III; CIII) and the rate-limiting cytochrome c oxidase (CIV) only in mice with Tregs (Fig. 5G). To ascertain the effects of Treg depletion on mitochondrial function, we performed high-resolution respirometry on myofibers isolated from fresh Ga muscles of sedentary and trained DTR- and DTR+ mice. EXT increased both basal and maximal respiration of myofibers from wild-type mice; but, by comparison, myofibers from trained mice with a Treg deficiency had significantly lower respiration rates (Fig. 5H).
The performance-enhancing effect of exercise training depends on IFNγ control by Tregs
We then asked whether the stunted gains in performance and diminished metabolic shifts in exercise-trained, Treg-deficient mice could be attributed to particular exercise-induced mediators under Treg control. As EXT up-regulated IFN-response pathways (Fig. 2E), and AEX resulted in exaggerated expression of IFN-responsive molecules and increased IFNγ production by muscle immunocytes in Treg-deficient mice (Fig. 3,H to J and fig. S3C), we looked for signs of increased IFN responses in Qd muscles of DTR+ and DTR- mice after EXT. Overlay of a signature comprised of IFNγ-induced genes showed stronger enrichment in DTR+ than DTR- muscles after training (Fig. 6A). Up-regulation of type I and II IFN-response pathways was confirmed by pathway analysis using GSEA (Fig. 6B). Such up-regulation was at the expense of other pathways known to be induced by exercise training, including cholesterol homeostasis and angiogenesis (Fig. 6B).
Figure 6. Constraint of IFNγ by Tregs is necessary for mitochondrial adaptations and improved performance in response to training.
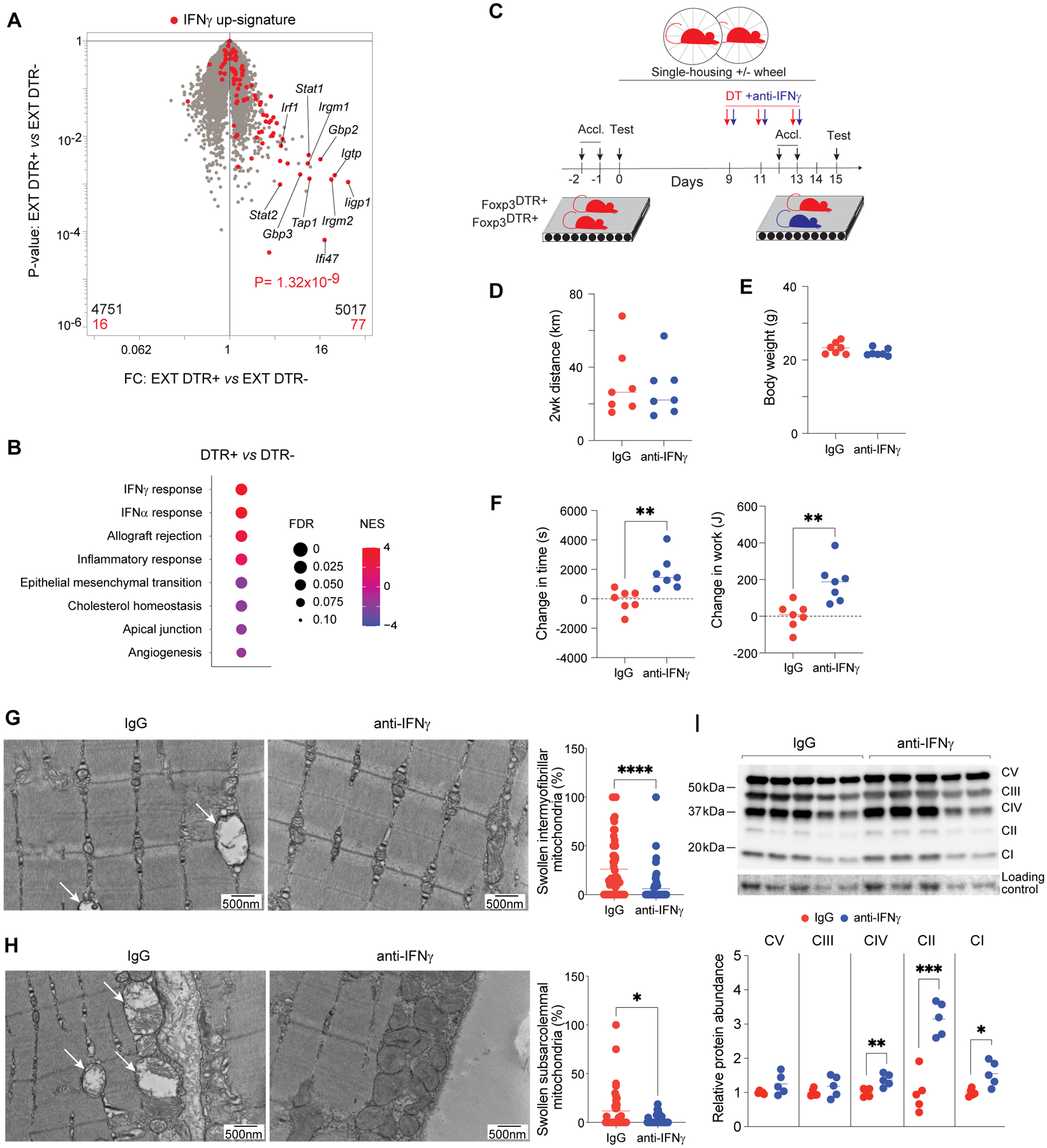
(A) RNA-seq analysis of Qd muscles from 2-week exercise-trained DTR- and DTR+ mice (n=3). Volcano plot overlain with genes differentially enriched by rIFNγ treatment (32).
(B) GSEA showing top Hallmark gene sets enriched or impoverished in Qd muscles of DTR+ versus DTR- muscles after EXT.
(C) Schema for exercise-capacity experiments with co-administration of IFNγ-neutralizing antibody or IgG with DT.
(D) Total distance mice ran voluntarily during EXT (n=7).
(E) Body weights of mice after EXT (n=7).
(F) Change in time run and work performed by each animal on the post-EXT compared with the pre-EXT test (n=7).
(G) Left: representative electron micrographs of sarcomeres and intermyofibrillar mitochondria in Qd muscles of anti-IFNγ or IgG-treated DTR+ mice after EXT. Arrows indicate swollen mitochondria. Right: frequency of swollen among total intermyofibrillar mitochondria (n≥59; >245 mitochondria).
(H) Left: as per panel G except subsarcolemmal mitochondria. Right: Frequency of swollen among total subsarcolemmal mitochondria (n≥32; >200 mitochondria).
(I) Left: Immunoblot (2 independent experiments: lanes 1–3 and 6–8 versus 4–5 and 9–10) for ETC complexes I-V in Qd muscles from the indicated conditions. Right: summary quantification (n=5).
Summary plots show data pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by an unpaired Student’s t-test (F, I), the Mann-Whitney test (G and H), or p value was computed by the χ2 test (A). IgG, immunoglobulin G; all other abbreviations as per prior figures.
To investigate whether increased IFNγ contributed to the blunted changes in performance and mitochondrial ETC complexes in Treg-ablated mice, we treated DTR+ mice with an IFNγ-neutralizing antibody or control IgG during the window of Treg depletion (Fig. 6C and fig. S8A). This intervention significantly down-regulated IFNγ-responsive transcripts in muscle (fig. S8B) but did not affect voluntary wheel-running behavior (Fig. 6D) or body weights (Fig. 6E). Measuring exercise capacity prior to and one day after 2 weeks of EXT indicated that anti-IFNγ-treated DTR+ mice showed stronger adaptation to training than did their IgG-treated littermates, evidenced by longer running times and more mechanical work performed during the post-test compared with the pre-test (Fig. 6F). The fraction of mitochondria with a swollen morphology was diminished in intermyofibrillar (Fig. 6G) and subsarcolemmal (Fig. 6H) regions of muscles from anti-IFNγ-treated DTR+ mice. Moreover, mitochondrial ETC complexes IV, II (succinate dehydrogenase), and I (NADH:ubiquinone oxidoreductase) were more abundant in anti-IFNγ-treated DTR+ mice compared with their IgG-control littermates (Fig. 6I).
IFNγ limits the beneficial effects of exercise by impairing muscle mitochondrial function
To address whether elevated IFNγ was sufficient for impaired adaptation to exercise training, we treated B6 mice intraperitoneally with recombinant IFNγ (rIFNγ) twice during the second week of voluntary wheel running and performed exercise-capacity tests as above (Fig. 7A). Cytokine treatment did not affect voluntary wheel-running behavior (Fig. 7B) or body weights (Fig. 7C), but mice that received rIFNγ improved their exercise capacity significantly less after EXT than did vehicle-treated littermates (Fig. 7D). To determine whether this anti-ergogenic effect of IFNγ might be related to influences on muscle mitochondrial function, we treated myofibers from fresh Ga muscles with rIFNγ followed by high-resolution respirometry as before. rIFNγ significantly reduced basal and maximal respiration after just 3h (Fig. 7E), and this reduction was sustained during a longer, 24h, treatment (Fig. 7F).
Figure 7. IFNγ directly impairs muscle mitochondrial function to limit the performance-enhancing benefits of exercise.
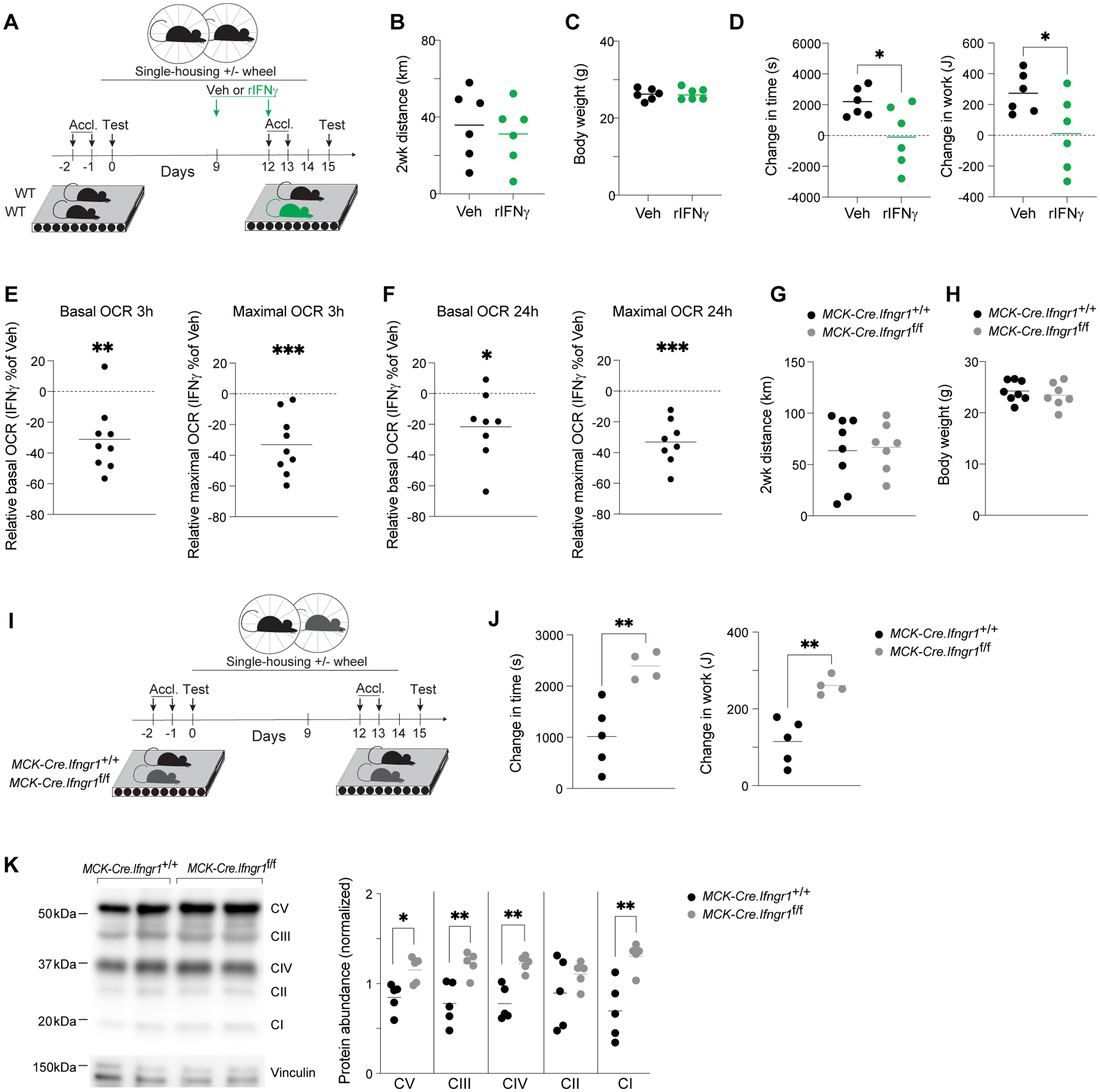
(A) Schema for exercise-capacity experiments with rIFNγ treatment.
(B) Total distance mice ran voluntarily during EXT (n=6).
(C) Body weights of mice after EXT (n=6).
(D) Change in time run and work performed by each animal on the post-EXT versus the pre-EXT test (n=6).
(E) Change in basal and maximal oxygen consumption rates of Ga myofibers treated with rIFNγ for 3h compared with vehicle treatment (n=9).
(F) As per panel E except 24h rIFNγ treatment (n=8).
(G) Total distance wild-type and muscle-specific Ifngr1 KO mice ran voluntarily during EXT (n=7).
(H) Body weights of mice from panel G after EXT (n=7).
(I) Schema for exercise-capacity experiments involving wild-type vs muscle-specific Ifngr1 KO mice.
(J) Change in time run and work performed by each animal on the post-EXT versus the pre-EXT test (n=4–5).
(K) Left: representative immunoblot for electron transport chain (ETC) complexes I-V in Qd muscles. Right: summary data (n=5).
Summary plots show data pooled from ≥2 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001 by an unpaired Student’s t-test (D, J, and K) or a one-sample t-test (E and F). Veh, vehicle; rIFNγ, recombinant IFNγ; all other abbreviations as per prior figures.
Given the pronounced, direct effect of IFNγ on myofiber respiration ex vivo, we questioned whether IFNγ signaling directly to muscle might be limiting the performance-enhancing effect of exercise training. To address this point, we generated a mouse strain harboring conditional deletion of Ifngr1 specifically on myocytes (MCK-Cre.Ifngr1f/f mice). The mutant mice did not run less than their wild-type counterparts during EXT, nor was their body weight significantly different (Fig. 7, G to H). However, the exercise capacity of Ifngr1-mutant mice improved significantly more after training than did that of the controls (Fig. 7, I to J), and this improvement was associated with a differential increase in the abundance of most complexes of the ETC (Fig. 7K). Thus, IFNγ limited the metabolic shifts and performance enhancement typical of exercise training.
Discussion
Given their reported roles in controlling inflammation and potentiating tissue regeneration after injury (11), we addressed the role of Tregs in exercise-induced changes in muscle properties and exercise capacity. Examination of acute and chronic exercise models revealed an early inflammatory response to exercise parallel to that induced by injury, which was controlled by muscle Tregs to permit metabolic reprogramming and improvement in exercise capacity. Loss-of-function experiments uncovered defects in organismal and muscle metabolic adaptations to training in the absence of Tregs, including striking morphological changes to intermyofibrillar and subsarcolemmal mitochondria of load-bearing hindlimb muscles. Treg-deficient mice showed excessive production of IFNγ, which compromised their improvement in endurance after exercise training. A combination of ex vivo cytokine treatments and cell-type-specific deletion of Ifngr1 revealed that IFNγ acts directly on myofibers to reduce mitochondrial function and limit the ergogenic effect of training.
One might ask whether the Treg influences we uncovered were local, mediated by muscle Tregs, or systemic, reflecting systemic influences of Treg loss. To address this question, we first showed that anti-CD25 antibody treatment during exercise training impaired performance to a degree similar to that induced by total Treg ablation without a change in the total number of muscle Tregs. This was consistent with our previous studies of injured muscles, which showed that anti-CD25 antibody treatment impairs Treg function without reducing overall Treg numbers in skeletal muscle or spleen (11). After uncovering mitochondrial defects in quadriceps muscles of Treg-deficient mice after exercise training, we also investigated two tissues distal to exercise-induced inflammation: immunologically inactive TA muscles and the kidneys. Treg deficiency did not affect mitochondrial morphology in these sites. Thus, based on these observations and the obvious parallels with the local impact of Tregs after acute muscle injury (11, 13), we think the Treg influences uncovered by the present study were local.
Exercise studies have primarily been concerned with the function of skeletal muscle as an endocrine organ, with a focus on identifying muscle-secreted factors and their targets (29). But such studies have usually generalized muscle as a conglomeration of fibers, largely ignoring the contributions of non-parenchymal cells. A limited number of recent studies have recognized the roles of stromal cells and immunocytes as important sources of exerkines (22, 30, 31). Our findings demonstrate that modulation of muscle immunocyte activities is not merely a feature of the response to exercise but is also a regulator of exercise adaptations. The role of Tregs in this context was unanticipated, but the increase in IFNγ production and IFN-responsive gene expression in exercised muscles of Treg-deficient mice is highly reminiscent of the “guardian” function of Tregs in other contexts, for example in injured muscle (25) and pancreatic islets of diabetes-prone NOD mice (32, 33).
While control of IFNγ production is a well-documented function of Tregs, the muscle-specific effects of excessive IFNγ signaling on local cellular and molecular processes had not been parsed. Our data showed that elevated IFNγ production as a consequence of Treg ablation had a negative impact on the abundance of several mitochondrial ETC complexes in muscle. These effects are consistent with previous in vitro experiments showing that IFN treatment reduces expression of genes important for mitochondrial biogenesis and oxidative phosphorylation in C2C12 myoblasts (34) and brown adipocytes (35). We also showed that excessive IFNγ due to Treg ablation or exogenous supplementation was associated with mitochondrial swelling and reduced respiratory capacity. Although elucidation of a precise mechanism will require further investigation, there is a small number of previous studies that provide insight. Treatment of C2C12 myoblasts with IFNγ revealed that repression of mitochondrial gene expression was dependent on the upregulation of the class II transactivator (CIITA) and its repression of Sirtuin 1 (SIRT1) transcription by deacetylation of histones H3 and H4 in the SIRT1 proximal promoter (34). This mechanism would therefore act directly on the AMP-activated protein kinase (AMPK)/SIRT1/PGC-1α signaling axis, which serves as one of the main sensors of exercise-induced energetic stress and activates mitochondrial and other metabolic adaptations (36, 37). However, we and others have observed that, under conditions of chronic IFN signaling, transcripts for mitochondrial factors actually increase without a corresponding increase in the proteins they encode. Type II IFN also regulates translation via suppression of mTORC1 signaling and eIF4E activity (38). Thus, regulation of the abundance of mitochondrial ETC complexes by IFNγ could be dependent on stalled translation. Finally, it is possible that the rapid reduction in respiration we observed in our ex vivo experiments with freshly isolated myofibers could be related to VDAC1 oligomerization, opening of the mitochondrial permeability transition pore, or another post-translational mechanism.
The roles of Tregs and IFNγ in regulating muscle oxidative phenotype and exercise capacity have several important implications for human physiology and disease. There is an inverse dose-response relationship between levels of physical activity and all-cause-mortality risk in humans (2), but the mechanisms underpinning this relationship are ill-defined. Many pro-inflammatory cytokines become chronically elevated during late life (e.g. IL-1β, IL-12, IFNγ, and TNFα), and the anti-inflammatory effects of chronic exercise are a proposed basis for the lower disease burden of active versus sedentary individuals (39). Of these pro-inflammatory“gerokines,” IFNγ has been highlighted as an attractive therapeutic target because transcriptional and proteomic profiling showed strong age-dependent enrichment of IFN responses across tissues, including hindlimb skeletal muscle (40, 41). Life-long exercise in mice prevented the age-dependent increase in numerous pro-inflammatory gerokines, including IFNγ; reduced the incidence of sarcopenia, dynapenia, and organ pathology; and completely protected against the occurrence of malignant tumors that afflicted sedentary animals (42). Strategies aimed at reducing IFNγ production or IFNγ signaling may be an approach to prevent or reverse age-dependent decline in muscle function. Inflammatory myopathies such as dermatomyositis (DM) are also associated with unleashed IFN production, particularly from muscle T cells (43–45). Deltoid muscles of DM patients showed abnormal mitochondrial morphology, including swollen forms reminiscent of those observed in Treg-deficient mice, and IFN-responsive transcript expression inversely correlated with mitochondrial gene expression, oxidative phosphorylation, and aerobic exercise capacity (46). Indeed, there is currently a Phase 2 clinical trial investigating the efficacy of anti-IFNβ antibody infusions in DM (NCT05192200). IFNγ also contributes to muscle pathology in the mdx mouse model of muscular dystrophy (47), and Tregs were shown to limit type I inflammation and mitigate muscle damage in mdx mice (11, 12). As low-intensity exercise improves muscle function in mdx mice and in patients with muscular dystrophy, it will be useful to know whether this beneficial effect is mediated by modulation of Tregs (48).
As aerobic exercise capacity is a strong, independent predictor of all-cause and disease-specific mortality in humans (49), the development of strategies to enhance muscle mitochondrial function are of clear interest. In addition to directly targeting IFNs, we propose that modulation of Tregs might be an effective alternative approach to controlling excessive IFNγ production in contexts of aging and inflammatory myopathies. Additionally, our findings add exercise to the collection of Treg-modulating interventions.
Materials and Methods
Study design
The major aims of this study were threefold: 1) to map the dynamics of exercise-induced inflammation in skeletal muscle to its transcriptional, metabolic, and functional responses; 2) to identify key regulatory elements involved in these processes; 3) to elucidate the impact of loss of regulation on the benefits of exercise. To these ends, we performed immunologic, transcriptomic, histologic, metabolic, and biochemical analyses of acute and chronic exercise models coupled with Treg ablation and modulation IFNγ signaling.
Mice
All mice were maintained under SPF conditions at Harvard Medical School (HMS) facilities in accordance with Institutional Animal Care and Use Committee guidelines (IS00001257). C57BL/6J, Foxp3-DTR, D2.B10-Dmdmdx/J, B6.FVB(129S4)-Tg(Ckmm-cre)5Khn/J (MCK-Cre), and C57BL/6N-Ifngr1tm1.1Rds/J (Ifngrf) mice were originally purchased from Jackson Laboratory, and mice were bred and maintained in-house before males between 8–10 weeks of age were used in experiments. All experiments using these strains were conducted with littermate controls.
Exercise
For acute exercise and exercise-capacity testing, mice were acclimatized to a motorized treadmill (Columbus Instruments, Exer 3/6 Treadmill) for two consecutive days, followed by a full bout at least 24h later. Treadmill running began between Zeitgeber time (ZT) 0 and 1, and concluded before ZT4 to minimize circadian disruption. For acute exercise, a 5 min warmup at 6 m/min was followed by a step-wise acceleration of 2 m/min every 2 min; the treadmill belt speed was capped at 18 m/min and sustained for 90 min. For exercise capacity testing, mice went through a similar warmup, and the treadmill belt speed was sustained between 26–32 m/min (segmented into 40 min stages with 2 m/min jumps) until mice reached exhaustion, which was defined as inability to remain on the treadmill belt for more than 5 s. Change in work was calculated as done previously (50). For exercise training, mice were housed individually in cages equipped with running-wheels (Columbus Instruments, Home Cage Running Wheel). Voluntary wheel running was recorded at intervals of 10 s using software from the running-wheels manufacturer.
Cell isolation
Isolation of immunocytes from skeletal muscle was performed as previously described (11). Briefly, hindlimb muscles were dissected, minced and incubated for 30 min at 37°C in digestion buffer (Dulbecco’s Modified Eagle Medium [DMEM], 2% fetal calf serum [FCS], collagenase II [2 mg/mL; Gibco] and DNase I [100 μg/mL; Sigma-Aldrich]). Immunocytes were enriched by density gradient centrifugation. The interphase between 40% and 80% Percoll (GE Healthcare) layers was recovered, washed, and stained for cytofluorometric analysis. For Foxp3-DTR experiments, a harsher isolation protocol was implemented (51) to increase recovery of myeloid cells, as done previously (52). Briefly, hindlimb muscles were minced and incubated in digestion buffer (Ham’s F10, 10% horse serum, collagenase II [800 U/mL; Gibco]) for 90 min at 37°C. Partially digested tissue and cells were sedimented by centrifugation, followed by further digestion using collagenase II (80 U/mL) and dispase (1 U/mL) for 30 min at 37°C. Fully digested tissue was mechanically disrupted by passage through a syringe and then filtered through a cell strainer. Isolated cells were washed and stained for cytofluorometric analysis.
Cytofluorometric analysis
To identify and analyze the cell subsets examined in this study, we stained immunocytes with combinations of the following antibodies: anti-CD45 (30-F11), -TCRβ (H57–597), -CD4 (GK1.5), -CD8 (53–6.7), -CD11b (M1/70), -CD11c (N418), -CD64 (X54–5/7.1), -Ly6G (1A8), -Ly6C (HK1.4), -CD3 (145–2C11), -NK1.1 (PK136), -TCRγδ (GL3), -CD19 (6D5), -CD31 (MEC13.3), -Sca1 (D7), -CD140a (APA5), -PD-L1 (10F.9G2), -MHCI (M1/42) (all from BioLegend), and anti-Ki67 (B56) (BD Pharmingen). Viability was assessed using the Zombie UV Fixable Viability Kit (BioLegend). For identification of Tregs, cells were fixed, permeabilized and intracellularly stained for Foxp3 (FJK-16s) using the Foxp3/Transcription Factor Staining Buffer Set according to the manufacturer’s instructions (eBioscience). For assessment of cytokine production, cells were stimulated for 3.5 h at 37°C with 50 ng/mL phorbol myristate acetate (PMA) and 1 μM ionomycin (both Sigma-Aldrich) in the presence of a protein transport inhibitor cocktail (eBioscience) followed by surface staining and fixation/permeabilization using a Cytofix/Cytoperm Kit (BD Biosciences). Intracellular IFNγ was detected by staining with an anti-IFNγ antibody (XMG1.2). Samples were acquired on a FACSymphony A5 flow cytometer (BD Biosciences) or Aurora spectral flow cytometer (Cytek). Data were analyzed using FlowJo software (Treestar).
Quantification of exercise-induced muscle damage
Creatine kinase activity was measured in serum using a commercially available kit (Sekisui Diagnostics) as described previously (11). Muscle Evans Blue Dye (EBD) uptake was measured as previously described (52). Briefly, 24h before endpoint, mice were injected i.p. with a 1% w/v solution of EBD at 1% of animal bodyweight. At the designated endpoints, individual muscles were dissected, weighed, and incubated in formamide at 55 °C overnight. The next day, extracts were spun down, and absorbance of supernatants was measured at 620 nm along with EBD standards. Absorbance values were converted to ng of EBD using the standard curve, and values were normalized to starting tissue weights to derive ng EBD per mg muscle.
RNA sequencing and analysis
For whole-muscle RNA-seq, RNA was isolated from Qd muscles using TRIzol (Invitrogen) according to the manufacturer’s instructions. To generate libraries, we resuspended 2 ng RNA in 5 μL of TCL buffer (Qiagen) containing 1% 2-mercaptoethanol (Sigma-Aldrich). Library construction and sequencing and data processing were performed according to the standard Immunological Genome Project protocols (https://www.immgen.org/img/Protocols/ImmGenULI_RNAseq_methods.pdf). Smart-seq2 libraries were prepared and sequenced as previously described (53). Briefly, RNA was captured and purified using RNAClean XP beads (Beckman Coulter), and polyadenylated (poly-A) mRNA was selected using an anchored oligo(dT) primer (5´-AAGCAGTGGTATCAACGCAGAGTACT30VN-3´). Poly-A mRNA was converted to cDNA by the reverse-transcription reaction followed by limited PCR amplification of first-strand cDNA. Tn5 transposon-based fragmentation was performed using the Nextera XT DNA Library Preparation Kit (Illumina). To enable pooling before sequencing, samples were PCR-amplified for an additional 12 cycles using barcoded primers such that each sample carried a specific combination of Illumina P5 and P7 barcodes. Paired-end sequencing was performed on an Illumina NextSeq500 (two full NextSeq runs per plate for an average of 10M raw reads per sample) using two 38-bp reads with no further trimming. Reads were aligned to the mouse genome (GENCODE GRCm38/mm10 primary assembly and gene annotations vM16) using STAR 2.7.3a. Transcripts annotated as ribosomal RNA were removed, and gene-level quantification was calculated using the Subread 2.0 command featureCounts. The DESeq2 package from Bioconductor was used to normalize raw read counts according to the median of ratios method (samples with <1 million uniquely mapped reads or with fewer than 8000 genes with over 10 reads were excluded from normalization). Normalized data were converted to GCT and CLS files, which were used in downstream analyses. Additional quality control after normalization included removal of biological replicates with a poor Pearson’s correlation (<0.9) and/or poor congregation by principal component analysis of the top 1000 variable genes. Genes with fewer than 15 reads across samples or a high coefficient of variation (>0.30) were removed in Multiplot Studio (GenePattern; Broad Institute).
To highlight gene-expression dynamics across muscle groups and between DTR- and DTR+ mice, we applied a two-step regression-modeling strategy using maSigPro (54). An alpha of 0.05 was used to account for multiple-hypothesis testing, and a false discovery rate of 5% was used to identify differentially expressed transcripts. To abstract modules of genes that were co-expressed within each timecourse, we implemented silhouette analysis to partition differential genes into clusters (cluster correlation, r2 ≥0.65). Heatmaps were generated using Morpheus (Broad Institute). Pathway analysis on gene lists from each cluster was performed using Metascape (55). Volcano plots and FC/FC plots were generated using Multiplot Studio. Pathway analysis was performed using Metascape or GSEA Hallmark gene sets (56).
Histology
Quadriceps muscles were fixed in 4% paraformaldehyde and processed by the Rodent Histopathology Core at HMS. Four or eight sections, 200 μm apart, were produced from each sample and stained with H&E. Sections were imaged using a Nikon Ti inverted microscope and analyzed using QuPath (57) and Fiji (58). Analysis was performed by two individuals, independently and blinded.
Muscle physiology
Muscle isometric torque was assessed in vivo on an Aurora 1300A 3-in-1 Whole Animal System for mice according to the standard Treat-NMD Neuromuscular Network protocol (https://www.treat-nmd.org/wp-content/uploads/2023/07/cmd-MDC1A_M.2.2.002-73.pdf). Briefly, mice were anesthetized by inhalation of isoflurane (4% initial, followed by 2% for maintenance) with 1–1.5 L/min 100% O2. Mice were placed on a warming pad (37 °C), ophthalmic ointment was applied to their eyes, and hindlimbs were depilated and cleaned with 70% ethyl-alcohol. To induce ankle plantar flexion, monophasic square pulses (0.1 ms) were delivered to the tibial nerve via subcutaneous electrodes. Pulse amplitude was adjusted to optimize twitch tension. At resting length, the frequency of pulses in a 300 ms pulse-train were progressively increased until maximal fused tetany was obtained. Three separate twitches and tetanic contractions were recorded for both right and left legs, and measurements were normalized to body-weight. Pre- and post-exercise assessments were performed blinded.
Electron microscopy
Mice were euthanized and immediately perfusion-fixed using PBS followed by 2% formaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences, #15949–1L). Quadriceps muscles were dissected, and superficial longitudinal tissue sections were produced using a scalpel. Sections were processed by the Electron Microscopy Core at HMS as follows: small pieces (1–2 mm cubes) of fixed tissue were washed in 0.1 M sodium cacodylate buffer and post-fixed for 1h in a buffer containing 1% osmiumtetroxide (OsO4) and 1.5% potassiumferrocyanide (KFeCN6). Cubes were then washed twice in water followed by one wash in 50 mM maleate buffer, pH 5.15 (MB) and incubated for 1h in 1% uranyl acetate in MB. Cubes were washed once in MB and twice more in water followed by dehydration in grades of alcohol (10min each: 50%, 70%, 90%, 100%, 100%). Samples were then put in propyleneoxide for 1h and infiltrated overnight in a 1:1 mixture of propyleneoxide and TAAB Epon (TAAB Laboratories Equipment Ltd, https://taab.co.uk). The following day, samples were embedded in TAAB Epon and polymerized at 60°C for 48h.
Ultra-thin sections (~80 nm) were cut on a Reichert Ultracut-S microtome, picked up onto copper grids, stained with lead citrate, and examined in a JEOL 1200EX transmission electron microscope or a TecnaiG2 Spirit BioTWIN. Images were recorded with an AMT 2k CCD camera at 2,000x and 12,000x for analysis of muscle sarcomere structure and subsarcolemmal and intermyofibrillar mitochondrial structures, and images were saved as TIFF files. Micrographs from two to three mice per group were analyzed. Image analysis was performed blinded using Fiji (58).
Metabolic cages
Mice were transferred from their existing cages into a Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments) in the early rest phase. VO2 and VCO2 were measured and used to calculate respiratory exchange ratio (RER; VCO2/VO2) over a 48h period. Only measurements taken after the first 6–8h of housing in CLAMS were used for analysis (beginning of first active phase). CLAMS measurements were analyzed using CalR (59). The percentage of energy provided by oxidation of glucose or fatty acids was calculated from RER values as described previously (26).
Metabolite profiling
Quadriceps muscles were dissected and snap frozen in liquid N2. Samples were weighed and homogenized in 0.5 mL of extraction solvent (70% ethanol spiked with 1 μM of U-13C6 glucose [Cambridge Isotope Laboratories, #CLM-1396-PK] and unlabeled caffeine [Sigma-Aldrich, #PHR1009] for internal standards) in screw-cap microcentrifuge tubes at 10°C in a Precellys Evolution homogenizer (Bertin technologies) using metallic beads. Metabolites were extracted from homogenates by addition of 7 mL of extraction solvent at 70°C for 1 minute with occasional vortexing to ensure complete extraction.
Targeted metabolite analysis was conducted on a QExactive HF-X mass spectrometer equipped with a heated electrospray ionization probe (HESI II probe). The mass spectrometer was coupled to a Vanquish binary UPLC system (Thermo Fisher Scientific, San Jose, CA). For chromatographic separation prior to mass analysis, 5 μL of the sample was injected onto an Ethylene Bridged Hybrid zwitterionic hydrophilic interaction chromatography (BEH Z-HILIC) column (100 mm, 1.7 μM particle size, 2.1 mm internal diameter, Waters). Samples were diluted 1:5 in acetonitrile for the analysis. Mobile phase A was 15 mM ammonium bicarbonate in 90% water and 10% acetonitrile, and mobile phase B was 15 mM ammonium bicarbonate in 95% acetonitrile and 5% water. The column oven was held at 45°C and autosampler at 4°C. The chromatographic gradient was carried out at a flow rate of 0.5 mL/min as follows: 0.75 min initial hold at 95% B; 0.75–3.00 min linear gradient from 95% to 30% B, 1.00 min isocratic hold at 30% B. B was brought back to 95% over 0.50 minutes, after which the column was re-equilibrated under initial conditions. The mass spectrometer was operated in full-scan negative mode, with the spray voltage set to 3 kV, the capillary temperature to 320°C, and the HESIII probe to 300°C. The sheath gas flow was set to 50 units, the auxiliary gas flow was set to 10 units, and the sweep gas flow was set to 1 unit. Mass acquisition was performed in a range of m/z = 70–900, with the resolution set at 120,000. Retention times were determined using authentic standards. Raw data were converted to .mzML and processed using emzed (60). Raw peak areas were produced by integrating a retention time window determined by m/z, chemical standards, fragmentation patterns, and isotopologue patterns. The peak areas were normalized to baseline, internal standard, and biomass. Water and acetonitrile were purchased from Fisher and were Optima LC/MS grade. Ammonium bicarbonate powder was purchased from Merck, and ethanol from Decon laboratories. To build a library of metabolites for untargeted metabolomics, top-15 data-dependent acquisitions were performed on a pooled study sample. The instrumentation was as described above. MS1 resolution was set to 60,000 and MS2 to 30,000. The sample was injected twice, excluding ions selected for fragmentation on the first run from the second run. Proteowizard MSConvert (61) was used to convert the MS2 libraries to mgf format, and MS2 features were compared to Human Metabolome Database (62), Global Natural Products Social Molecular Networking (63), Massbank (64), and MassBank of North America (https://mona.fiehnlab.ucdavis.edu) databases using ModifiedCosine similarity from matchms (65) Python package. The similarity threshold was set to 0.5 and the number of matching m/z threshold was set to 3. In addition, retention times and m/z values of a full-scan acquisition on a pooled study sample was compared to an in-house database. All features identified in the pooled study sample were evaluated and false peak identifications were removed. The final library entailed 204 compounds. Data are available in the Metabolights (66) repository.
Muscle glycogen quantification
Glycogen was extracted from gastrocnemius muscles and quantified as described previously (67). Briefly, muscles were snap frozen in liquid N2 after dissection and were later pulverized using a frozen mortar and pestle. Powdered gastrocnemius muscles were weighed, and metabolites were extracted using 1 M HCl at 95°C for 2h, followed by neutralization using 2 M NaOH. Extracts were clarified by centrifugation, and supernatants were diluted for subsequent quantification of glucose using a glucose oxidase activity assay according to the manufacturer’s instructions (Sigma-Aldrich, #GAGO20). Absorbance was measured at 340 nm, and glucose generated from hydrolyzed glycogen was quantified based on a standard curve and normalized to starting tissue weights.
Immunoblot assay
Tissue lysates were generated from fresh-frozen quadriceps muscles homogenized in ice-cold buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM dithiothreitol, and 1x protease and phosphatase inhibitor cocktail (ThermoFisher, #78440). Protein concentration in each lysate was determined by bicinchoninic acid (BCA) assay (ThermoFisher, #23235), and 15 μg were separated by SDS-PAGE, followed by transfer to PVDF membrane by wet transfer. Blocking was performed for 30 minutes at room temperature in 5% skim milk in tris-buffered saline (pH 7.4) with 0.1% Tween 20 (TBST), and membranes were incubated with primary antibodies overnight at 4°C in 5% bovine serum albumin in TBST. The Total OXPHOS Rodent WB Antibody Cocktail (Abcam, #ab110413) was used for measurement of protein complexes in the electron transport chain. Loading controls were based on Ponceau S staining (Sigma-Aldrich, #P7170) or Vinculin antibody labeling (Sigma-Aldrich, #V9264). Chemiluminescence was imaged using a BioRad ChemiDoc, and densitometry was performed on blot images using Fiji (58).
High-resolution respirometry
Myofibers were isolated from gastrocnemius muscles by incubation in digestion buffer (Ham’s F10, 10% horse serum, collagenase II [800 U/mL; Gibco]) for 75 min at 37°C. To avoid injuring gastrocnemius fibers, all neighboring muscles were removed, and the entire hindlimb, was subjected to digestion. After digestion, hindlimbs were transferred to warm wash medium (digestion buffer without collagenase, with 10 mM HEPES) in a serum-coated 10 cm petri dish. The Achilles tendon was cut at its most distal point and used as a handle to lift the gastrocnemius to allow removal of the soleus muscle. Myofibers were released by flushing gastrocnemius muscles with wash medium using a serum-coated, wide-bore pipet. Healthy myofibers, or fibers with a straight, non-contracted morphology, were either washed in assay medium (DMEM, 5 mM glucose, 2 mM L-glutamine, 1 mM sodium pyruvate, no phenol red, no sodium bicarbonate) and loaded into an Oroboros O2K high-resolution respirometer or washed in culture medium (digestion buffer without collagenase; with 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate) and incubated in culture medium for the duration of recombinant IFNγ treatment (100 ng/mL). For cytokine treatment experiments, myofibers from one mouse were divided equally into vehicle and IFNγ groups to create paired samples. At the time of respirometry, myofibers were resuspended in 400 μL of assay medium and loaded into an Oroboros O2K high-resolution respirometer for measurement of oxygen flux during basal respiration. Maximal oxygen consumption rates were obtained by addition of FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone)) to a final concentration of 5 μM. At the end of respirometry, myofibers were retrieved from the assay chamber and lysed in ice-cold buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM dithiothreitol, and 1x protease and phosphatase inhibitor cocktail (ThermoFisher, #78440). Protein concentration in each lysate was determined by BCA assay (ThermoFisher, #23235) and used for normalization of oxygen flux values (~500 μg per sample).
In vivo antibody and cytokine treatments
Depletion of CD25+ cells was accomplished by i.p. injection of 100 μg of anti-CD25 mAb (clone PC61) or IgG. Effects on muscle immunophenotype and exercise capacity were determined one day after the final injection. For IFNγ-neutralization experiments, mice were injected i.p. with 250 μg anti-mouse IFNγ mAb (BioLegend, clone XMG1.2) or mouse IgG1 isotype control (BioXCell, clone IgG1) every-other-day for one week. Effects of neutralization on muscle transcriptomes, protein levels of mitochondrial ETC complexes, and exercise capacity were determined two days after the final injection. For cytokine treatments, mice were injected i.p. with 10 μg recombinant IFNγ (Biolegend) twice a week for one week as done previously (68). Effects of recombinant IFNγ on protein levels of mitochondrial ETC complexes and exercise capacity were determined at the end of treatment.
Statistical analyses
Data are presented as mean ± SD. Unless stated otherwise, GraphPad Prism software was used to perform statistical testing: Student’s t-test for two-group comparisons or one-way ANOVA or two-way ANOVA for multiple-group comparisons. p-values for gene signature enrichment in data presented in volcano plots were determined using a χ2 test. p = *, <0.05; **, <0.01; ***, <0.001; ****, <0.0001. Mice with the same genotypes were randomly assigned to interventions in all cases except for exercise-capacity experiments. In experiments involving exercise capacity testing before and after neutralizing antibody or recombinant cytokine treatments, mice were allocated to treatment groups based on starting exercise capacity to ensure equal distribution of latent performance capacities between groups. Image analyses and exercise capacity tests were performed blinded.
Supplementary Material
Acknowledgements:
We thank K. Hattori, A. Ortiz-Lopez, L. Yang, K. Seddu, A. Baysoy, J. Lee, R. Ramirez, O. Yaghi, and B. Hana for experimental assistance; C. Laplace for assistance with graphics; R. Stephansky, P. Anekal, and P. Montero Llopis for transmitted light microscopy assistance; the Dana-Farber Cancer Institute Metabolomics Core; the Harvard Medical School (HMS) Rodent Histopathology Core; the HMS Electron Microscopy Facility; and the HMS Immunology Department Flow Cytometry Core. The anatomical map of mouse hindlimb muscles in Figure 1 was created with BioRender.com.
Funding:
This work was supported by the National Institutes of Health (grants R01 AR070334 to DM, F32 AG072874 to PKL, and F32 AG069363 to ALM), and The JPB Foundation (to DM).
Footnotes
Competing interests: The authors declare that they have no competing interests. ALM is currently an editor for Cell Metabolism.
Data and materials availability:
Transcriptomes from RNA-seq of whole skeletal muscles have been deposited in the Gene Expression Omnibus (GSE220181). Metabolomes from metabolite profiling of whole skeletal muscles have been deposited in the Metabolights repository (MTBLS8391). All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.
REFERENCES
- 1.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW, Miller AH, Mantovani A, Weyand CM, Barzilai N, Goronzy JJ, Rando TA, Effros RB, Lucia A, Kleinstreuer N, Slavich GM, Chronic inflammation in the etiology of disease across the life span. Nat Med. 25, 1822–1832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arem H, Moore SC, Patel A, Hartge P, Berrington de GA, Visvanathan K, Campbell PT, Freedman M, Weiderpass E, Adami HO, Linet MS, Lee IM, Matthews CE, Leisure time physical activity and mortality: a detailed pooled analysis of the dose-response relationship. JAMA Intern. Med 175, 959–967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanford JA, Nogiec CD, Lindholm ME, Adkins JN, Amar D, Dasari S, Drugan JK, Fernandez FM, Radom-Aizik S, Schenk S, Snyder MP, Tracy RP, Vanderboom P, Trappe S, Walsh MJ, Molecular Transducers of Physical Activity Consortium (MoTrPAC): mapping the dynamic responses to exercise. Cell. 181, 1464–1474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffey VG, Hawley JA, The molecular bases of training adaptation. Sports Med. 37, 737–763 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Egan B, Zierath JR, Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 17, 162–184 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Pedersen BK, Toft AD, Effects of exercise on lymphocytes and cytokines. Br. J Sports Med 34, 246–251 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulz G, in Deutshes Archiv fur Klinische Medicin, von Ziemssen H, von Zenker FA, Eds. (Verlag von F.C.W. Vogel, Leipzig, 1893), chap. 10, pp. 234–281. [Google Scholar]
- 8.Armstrong RB, Ogilvie RW, Schwane JA, Eccentric exercise-induced injury to rat skeletal muscle. J Appl. Physiol Respir. Environ. Exerc. Physiol 54, 80–93 (1983). [DOI] [PubMed] [Google Scholar]
- 9.Marklund P, Mattsson CM, Wahlin-Larsson B, Ponsot E, Lindvall B, Lindvall L, Ekblom B, Kadi F, Extensive inflammatory cell infiltration in human skeletal muscle in response to an ultraendurance exercise bout in experienced athletes. J Appl. Physiol 114, 66–72 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Tidball JG, Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol. 17, 165–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D, A special population of regulatory T cells potentiates muscle repair. Cell. 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA, Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl. Med 6, 258ra142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, Mathis D, Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity. 44, 355–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clifford T, Wood MJ, Stocks P, Howatson G, Stevenson EJ, Hilkens CMU, T-regulatory cells exhibit a biphasic response to prolonged endurance exercise in humans. Eur. J Appl. Physiol 117, 1727–1737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proschinger S, Winker M, Joisten N, Bloch W, Palmowski J, Zimmer P, The effect of exercise on regulatory T cells: A systematic review of human and animal studies with future perspectives and methodological recommendations. Exerc Immunol Rev. 27, 142–166 (2021). [PubMed] [Google Scholar]
- 16.Billat VL, Mouisel E, Roblot N, Melki J, Inter- and intrastrain variation in mouse critical running speed. J Appl. Physiol 98, 1258–1263 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Arnold L, Henry A, Poron F, Baba-Amer Y, van RN, Plonquet A, Gherardi RK, Chazaud B, Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp. Med 204, 1057–1069 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Timmons JA, Knudsen S, Rankinen T, Koch LG, Sarzynski M, Jensen T, Keller P, Scheele C, Vollaard NB, Nielsen S, Akerstrom T, MacDougald OA, Jansson E, Greenhaff PL, Tarnopolsky MA, van Loon LJ, Pedersen BK, Sundberg CJ, Wahlestedt C, Britton SL, Bouchard C, Using molecular classification to predict gains in maximal aerobic capacity following endurance exercise training in humans. J Appl. Physiol (1985.) 108, 1487–1496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holloszy JO, Booth FW, Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 38, 273–291 (1976). [DOI] [PubMed] [Google Scholar]
- 20.Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL, Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol. 588, 4795–4810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amar D, Lindholm ME, Norrbom J, Wheeler MT, Rivas MA, Ashley EA, Time trajectories in the transcriptomic response to exercise - a meta-analysis. Nat Commun. 12, 3471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pillon NJ, Gabriel BM, Dollet L, Smith JAB, Sardon PL, Botella J, Bishop DJ, Krook A, Zierath JR, Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat Commun. 11, 470 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puigserver P, Spiegelman BM, Peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr. Rev 24, 78–90 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM, Transcriptional co-activator PGC-1 α drives the formation of slow-twitch muscle fibres. Nature. 418, 797–801 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Panduro M, Benoist C, Mathis D, Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc Natl Acad Sci U S A. 115, E2585–E2593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Péronnet F, Massicotte D, Table of nonprotein respiratory quotient: an update. Can. J Sport Sci 16, 23–29 (1991). [PubMed] [Google Scholar]
- 27.Hackenbrock CR, Ultrastructural bases for metabolically linked mechanical activity in mitochondria: I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 30, 269–297 (1966). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LEHNINGER AL, Water uptake and extrusion by mitochondria in relation to oxidative phosphorylation. Physiol Rev. 42, 467–517 (1962). [DOI] [PubMed] [Google Scholar]
- 29.Bay ML, Pedersen BK, Muscle-organ crosstalk: focus on immunometabolism. Front Physiol. 11, 567881 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knudsen NH, Stanya KJ, Hyde AL, Chalom MM, Alexander RK, Liou YH, Starost KA, Gangl MR, Jacobi D, Liu S, Sopariwala DH, Fonseca-Pereira D, Li J, Hu FB, Garrett WS, Narkar VA, Ortlund EA, Kim JH, Paton CM, Cooper JA, Lee CH, Interleukin-13 drives metabolic conditioning of muscle to endurance exercise. Science. 368, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Vamvini M, Nigro P, Ho LL, Galani K, Alvarez M, Tanigawa Y, Renfro A, Carbone NP, Laakso M, Agudelo LZ, Pajukanta P, Hirshman MF, Middelbeek RJW, Grove K, Goodyear LJ, Kellis M, Single-cell dissection of the obesity-exercise axis in adipose-muscle tissues implies a critical role for mesenchymal stem cells. Cell Metab. 34, 1578–1593 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D, How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 31, 654–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sitrin J, Ring A, Garcia KC, Benoist C, Mathis D, Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J Exp. Med 210, 1153–1165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li P, Zhao Y, Wu X, Xia M, Fang M, Iwasaki Y, Sha J, Chen Q, Xu Y, Shen A, Interferon gamma (IFN-γ) disrupts energy expenditure and metabolic homeostasis by suppressing SIRT1 transcription. Nucleic Acids Res. 40, 1609–1620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kissig M, Ishibashi J, Harms MJ, Lim HW, Stine RR, Won KJ, Seale P, PRDM16 represses the type I interferon response in adipocytes to promote mitochondrial and thermogenic programing. EMBO J. 36, 1528–1542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J, AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 458, 1056–1060 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J, Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, Cross JR, Ratsch G, Rice CM, Ivashkiv LB, Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 16, 838–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duggal NA, Niemiro G, Harridge SDR, Simpson RJ, Lord JM, Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat Rev Immunol. 19, 563–572 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Cao W, IFN-aging: coupling aging with interferon response. Front Aging. 3, 870489 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shavlakadze T, Morris M, Fang J, Wang SX, Zhu J, Zhou W, Tse HW, Mondragon-Gonzalez R, Roma G, Glass DJ, Age-related gene expression signature in rats demonstrate early, late, and linear transcriptional changes from multiple tissues. Cell Rep. 28, 3263–3273 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Nilsson MI, Bourgeois JM, Nederveen JP, Leite MR, Hettinga BP, Bujak AL, May L, Lin E, Crozier M, Rusiecki DR, Moffatt C, Azzopardi P, Young J, Yang Y, Nguyen J, Adler E, Lan L, Tarnopolsky MA, Lifelong aerobic exercise protects against inflammaging and cancer. PLoS One. 14, e0210863 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moneta GM, Pires MD, Marasco E, Rosina S, Verardo M, Fiorillo C, Minetti C, Bracci-Laudiero L, Ravelli A, De BF, Nicolai R, Muscle expression of type I and type II interferons is increased in juvenile dermatomyositis and related to clinical and histologic features. Arthritis Rheumatol. 71, 1011–1021 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, Milisenda JC, Grau-Junyent JM, Selva-O’Callaghan A, Paik J, Albayda J, Christopher-Stine L, Lloyd TE, Corse AM, Mammen AL, Identification of distinctive interferon gene signatures in different types of myositis. Neurology. 93, e1193–e1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigolet M, Hou C, Baba AY, Aouizerate J, Periou B, Gherardi RK, Lafuste P, Authier FJ, Distinct interferon signatures stratify inflammatory and dysimmune myopathies. RMD. Open 5, e000811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer A, Laverny G, Allenbach Y, Grelet E, Ueberschlag V, Echaniz-Laguna A, Lannes B, Alsaleh G, Charles AL, Singh F, Zoll J, Lonsdorfer E, Maurier F, Boyer O, Gottenberg JE, Nicot AS, Laporte J, Benveniste O, Metzger D, Sibilia J, Geny B, IFN-β-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 134, 655–666 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, Tidball JG, IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J Immunol. 187, 5419–5428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanna BS, Yaghi OK, Langston PK, Mathis D, The potential for Treg-enhancing therapies in tissue, in particular skeletal muscle, regeneration. Clin Exp Immunol. 211, 138–148 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harber MP, Kaminsky LA, Arena R, Blair SN, Franklin BA, Myers J, Ross R, Impact of cardiorespiratory fitness on all-cause and disease-specific mortality: advances since 2009. Prog. Cardiovasc. Dis 60, 11–20 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM, Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J Biol Chem. 282, 30014–30021 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Liu L, Cheung TH, Charville GW, Rando TA, Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc 10, 1612–1624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mann AO, Hanna BS, Munoz-Rojas AR, Sandrock I, Prinz I, Benoist C, Mathis D, IL-17A-producing γδT cells promote muscle regeneration in a microbiota-dependent manner. J Exp Med. 219, e20211504 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R, Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Nueda MJ, Tarazona S, Conesa A, Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics. 30, 2598–2602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK, Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl. Acad Sci U S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW, QuPath: Open source software for digital pathology image analysis. Sci Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis. Nat Methods. 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mina AI, LeClair RA, LeClair KB, Cohen DE, Lantier L, Banks AS, CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiefer P, Schmitt U, Vorholt JA, eMZed: an open source framework in Python for rapid and interactive development of LC/MS data analysis workflows. Bioinformatics. 29, 963–964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P, A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 30, 918–920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wishart DS, Guo A, Oler E, Wang F, Anjum A, Peters H, Dizon R, Sayeeda Z, Tian S, Lee BL, Berjanskii M, Mah R, Yamamoto M, Jovel J, Torres-Calzada C, Hiebert-Giesbrecht M, Lui VW, Varshavi D, Varshavi D, Allen D, Arndt D, Khetarpal N, Sivakumaran A, Harford K, Sanford S, Yee K, Cao X, Budinski Z, Liigand J, Zhang L, Zheng J, Mandal R, Karu N, Dambrova M, Schioth HB, Greiner R, Gautam V, HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res. 50, D622–D631 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu WT, Crusemann M, Boudreau PD, Esquenazi E, Sandoval-Calderon M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu CC, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw CC, Yang YL, Humpf HU, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, CAB P, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodriguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard PM, Phapale P, Nothias LF, Alexandrov T, Litaudon M, Wolfender JL, Kyle JE, Metz TO, Peryea T, Nguyen DT, VanLeer D, Shinn P, Jadhav A, Muller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BO, Pogliano K, Linington RG, Gutierrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N, Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 34, 828–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T, MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom. 45, 703–714 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Huber F, Verhoeven S,C Meijer H Spreeuw EM Villanueva Castilla C Geng JJJ van der Hooft S Rogers ADF Belloun JH Sparks, matchms - processing and similarity evaluation of mass spectrometry data. Journal of Open Source Software. 52, 2411 (2020). [Google Scholar]
- 66.Haug K, Cochrane K, Nainala VC, Williams M, Chang J, Jayaseelan KV, O’Donovan C, MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sato S, Basse AL, Schonke M, Chen S, Samad M, Altintas A, Laker RC, Dalbram E, Barres R, Baldi P, Treebak JT, Zierath JR, Sassone-Corsi P, Time of exercise specifies the impact on muscle metabolic pathways and systemic energy homeostasis. Cell Metab. 30, 92–110 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Li C, Wang G, Sivasami P, Ramirez RN, Zhang Y, Benoist C, Mathis D, Interferon-α-producing plasmacytoid dendritic cells drive the loss of adipose tissue regulatory T cells during obesity. Cell Metab. S1550–4131(21)00277–1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, Lee M, Casanova JL, Clark JD, Hegen M, Telliez JB, Hacohen N, De Jager PL, Regev A, Mathis D, Benoist C, Parsing the interferon transcriptional network and its disease associations. Cell. 164, 564–578 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptomes from RNA-seq of whole skeletal muscles have been deposited in the Gene Expression Omnibus (GSE220181). Metabolomes from metabolite profiling of whole skeletal muscles have been deposited in the Metabolights repository (MTBLS8391). All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.