

Abstract
Humans and other primates harbour complex gut bacterial communities that influence health and disease, but the evolutionary histories of these symbioses remain unclear. This is partly due to limited information about the microbiota of ancestral primates. Here, using phylogenetic analyses of metagenome-assembled genomes (MAGs), we show that hundreds of gut bacterial clades diversified in parallel (that is, co-diversified) with primate species over millions of years, but that humans have experienced widespread losses of these ancestral symbionts. Analyses of 9,460 human and non-human primate MAGs, including newly generated MAGs from chimpanzees and bonobos, revealed significant co-diversification within ten gut bacterial phyla, including Firmicutes, Actinobacteriota and Bacteroidota. Strikingly, ~44% of the co-diversifying clades detected in African apes were absent from available metagenomic data from humans and ~54% were absent from industrialized human populations.
In contrast, only ~3% of non-co-diversifying clades detected in African apes were absent from humans. Co-diversifying clades present in both humans and chimpanzees displayed consistent genomic signatures of natural selection between the two host species but differed in functional content from co-diversifying clades lost from humans, consistent with selection against certain functions. This study discovers host-species-specific bacterial symbionts that predate hominid diversification, many of which have undergone accelerated extinctions from human populations.
Humans and other primates evolved in the presence of complex gut microbial communities, motivating efforts to determine the ancestral members of the microbiota1,2. Faithful relationships between microbial lineages and host species over evolutionary timescales lead to congruence between symbiont and host phylogenetic trees (that is, co-diversification)3,4. Thus, in principle, ancestral symbionts in the microbiota can be identified with high confidence despite the lack of a microbiota fossil record. However, previous tests for co-diversification between microbiota and primates have relied on marker-gene approaches, focused on just three bacterial families and yielded conflicting results, such that the extent of ancient, host-species-specific symbioses in the microbiota remains controversial3–6. For example, previous work detected four bacterial clades whose evolutionary trees mirrored the relationships among humans and other African apes5, but additional sampling found conflicting evidence regarding co-diversification for some of these clades6. Moreover, other studies have shown that most gut bacterial lineages are transient within individual hosts’ lifetimes7 and that gut bacterial taxa tend to display low heritability within host populations8,9. Thus, the extent to which constituents of the primate gut microbiota form stable relationships and co-diversify with host species remains contested. Resolving this issue is critical to understanding how the gut microbiota has evolved within humans and non-human primates (NHPs). Identifying the complete set of ancestral, co-diversified symbioses is also pressing for human health, given the importance of endogenous microbiota for the development and function of immune10, metabolic11 and neuroendocrine systems12.
Results
An evolutionary tree of gut bacterial genomes
Previous studies of co-diversification between gut bacteria and primate hosts have focused on marker-gene-based approaches5,6,13, but analyses of whole bacterial genomes would afford greater resolution to detect co-diversification events. To address this issue, we conducted microbiota-wide tests for co-diversification between bacteria and primates using a dataset of gut bacterial metagenome-assembled genomes (MAGs) derived from humans and wild-living NHPs. First, we generated MAGs from the gut microbiota of chimpanzees (Pan troglodytes schweinfurthii and P. t. troglodytes) and bonobos (P. paniscus) residing in the wild throughout equatorial Africa (Extended Data Fig. 1 and Supplementary Table 1). Samples (n = 36) were collected from seven populations of Pan and sequenced deeply on Nanopore and Illumina platforms. Contiguous sequences (contigs) were assembled from both long-read and short-read data, and contigs were binned into genomes using a custom automated workflow (available at https://github.com/CUMoellerLab/sn-mg-pipeline) incorporating multiple previously published genome binning tools, including MetaBat214, MaxBin215, Concoct16 and DASTool17. Overall, our approach yielded 2,614 MAGs with completeness >50%, contamination <5% and strain heterogeneity <0.5%, including 1,449 MAGs with completeness >90% and contamination <5%. Pan MAGs spanned 11 phyla and increased the number of high-quality MAGs available from wild-living Pan by >90-fold relative to previous efforts18. Moreover, the quality of bins generated by our approach, which included both long- and short-read data as well as cross-sample mapping of reads to contigs, was in general higher than previous studies that relied on assembly and binning of MAGs from individual samples. For instance, in previous studies that relied on single-sample MAG assemblies, ~45% of high-quality (>50% complete, <5% contamination) MAGs were >90% complete, whereas ~55% of MAGs generated here were >90% complete. Assembly statistics, taxonomic assignments, completeness and contamination estimates, host IDs and other metadata for Pan MAGs are presented in Supplementary Table 2.
Next, we combined the Pan MAGs with publicly available MAGs from humans and NHPs to construct a phylogeny of bacterial lineages from the primate gut microbiota for downstream analyses. These MAGs were derived from 47 extant human populations representing multiple continents and lifestyles19, archaic 1,000-2,000-year-old human populations sampled from southwestern USA and Mexico20, and 22 NHP species sampled in the wild18. Single-copy core genes from MAGs were identified and aligned using the Genome Taxonomy Database Toolkit21. Alignments were then used to infer a maximum-likelihood phylogenetic tree with IQTree2.022. The final tree contained 9,460 MAGs. Summary statistics regarding the number of host individuals, MAGs per host species, sampling effort per host species and summary statistics for Nanopore sequencing are presented in Supplementary Table 1. Metadata for all MAGs are presented in Supplementary Table 2.
Widespread co-diversification between gut bacteria and primates
We then tested nodes on the phylogeny of primate MAGs for co-diversification with host species. Our method (available at https://github.com/CUMoellerLab/codiv-tools), which is an extension of a method developed for binary host–parasite systems23, traverses the bacterial MAG phylogeny and applies to each node a permutation-based Mantel test for association between bacterial and host subtrees. In these tests, the true association between bacterial and host phylogenetic distances (Mantel’s r) at each node in the symbiont phylogeny is compared against the null distribution of associations generated by permuting the tip labels of both the symbiont and host subtrees. These tests effectively define co-diversification as a significant positive association between symbiont and host divergence estimates. Here, nodes deeper than 1/4th of the root depth of the symbiont phylogeny were not considered for downstream analyses. This root depth was chosen on the basis of the assumption that nodes at these depths represented common ancestors of distantly related bacteria whose divergence probably predated the most-recent common ancestor of primates. Nodes uniting fewer than seven MAGs or MAGs derived from fewer than three host species were not tested. The phylogeny of host species (Fig. 1a) was derived from timetree.org24. Analyses of 1,616 nodes on the symbiont phylogeny identified 206 bacterial clades displaying strong evidence of co-diversification (that is, Mantel’s r > 0.75 and non-parametric P < 0.01) (Fig. 1b). For a subset of these clades, we observed perfect topological congruence between host and symbiont topologies (Fig. 1c,d). The combined use of Mantel’s r and P values to identify co-diversifying clades was supported by post-hoc analyses that assessed the relationships between these values and sampling effort (Supplementary Discussion). Of the 206 clades identified, 168 displayed depths within 1/10th of the root depth of the symbiont phylogeny, indicating that most signal of co-diversification was found towards the tips of the symbiont phylogeny as expected, given the relatively recent timescales of primate evolution.
Fig. 1 |. Co-diversification of gut microbiota with primate species.
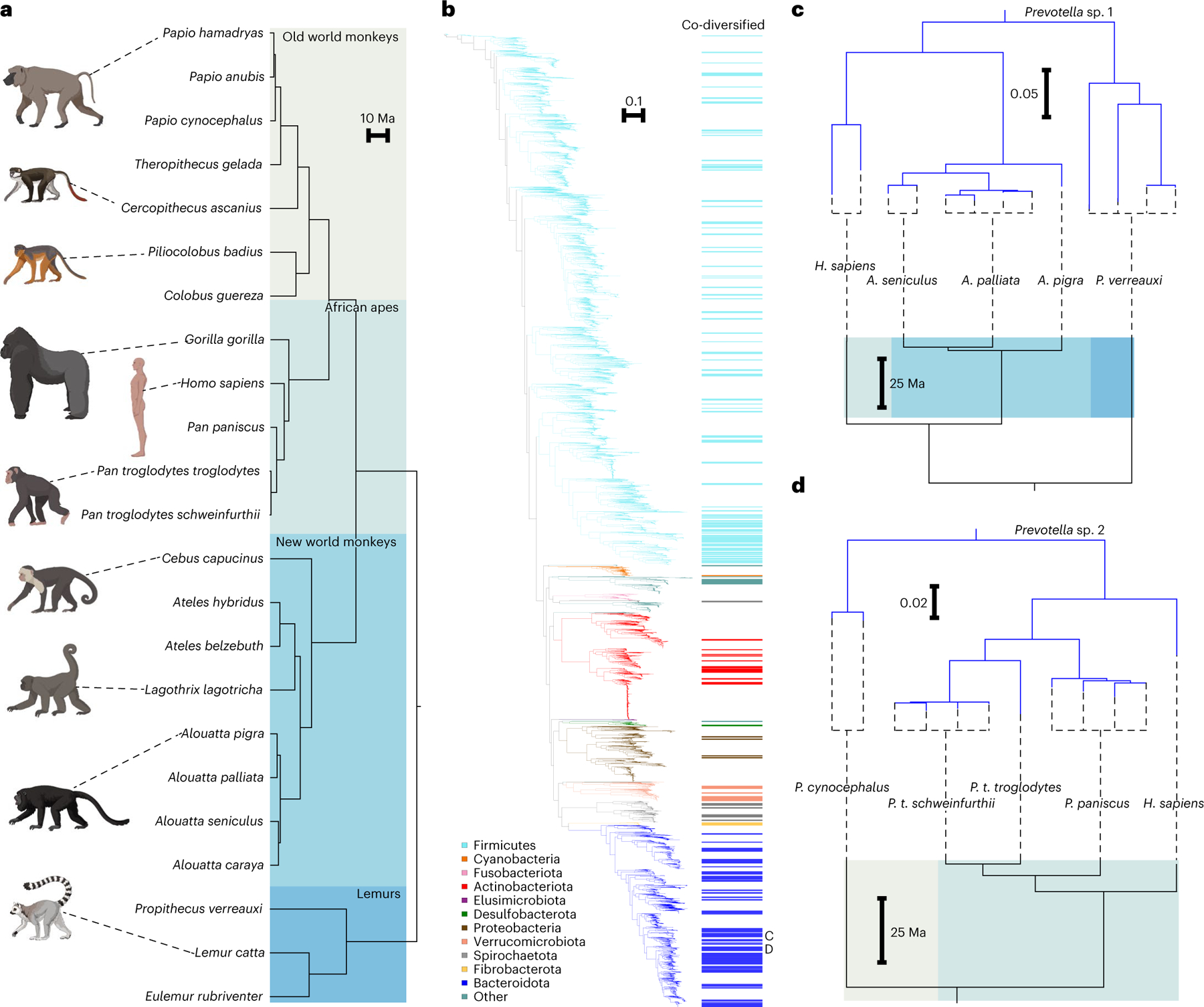
a, Phylogeny shows relationships among primate species from which gut bacterial genomes were derived. Shading delineates Old World monkeys, New World monkeys, African apes and lemurs. b, Phylogeny shows relationships among 9,460 high-quality gut bacterial genomes assembled from the primate gut microbiota. Coloured bars to the right of the phylogeny mark clades showing significant evidence of co-diversification with primate species (Mantel r > 0.75; non-parametric permutation test P < 0.01). Colours of branches and bars correspond to bacterial phyla. Scale indicates amino-acid substitutions per site. Letters C and D indicate clades highlighted in c and d, respectively. c,d, Tanglegrams show examples of correspondence between topologies of bacterial (top of each panel) and host-species (bottom of each panel) phylogenies for two co-diversifying clades of unclassified Prevotella species.
Bootstrap values also provided strong support for the symbiont nodes supporting co-diversification. For 202 of the 206 co-diversifying clades (r > 0.75, non-parametric P < 0.01), all nodes delineating the relationships between MAGs recovered from different host species and supporting co-diversification were supported by >90% of bootstrap replicates, whereas for the other 4 clades these nodes were supported by >75% of bootstrap replicates. Node statistics, subtrees and bootstrap values for clades displaying significant evidence for co-diversification are presented in Supplementary Table 3.
Overall, the clades showing significant evidence of co-diversification represented 10 bacterial phyla and contained 24.084% of the unique branch length of the phylogeny of primate MAGs (Fig. 1b). However, the degree of co-diversification varied across bacterial phyla. A larger fraction of Bacteroidota MAGs than Firmicutes MAGs belonged to clades displaying evidence of co-diversification (734/1,765 compared with 994/5,163). Fibrobacterota MAGs displayed the most consistent evidence of co-diversification with hosts, with 27/27 MAGs belonging to clades that showed significant evidence of co-diversification. The proportions of MAGs belonging to clades displaying evidence of co-diversification for each phylum are presented in Supplementary Table 3.
We observed >10-fold more significantly co-diversifying clades than expected under the null hypothesis of the permutation tests assuming independence of clades (16 clades expected at P < 0.01 significance level, 206 observed). However, this null hypothesis is naïve to phylogenetic non-independence of clades introduced by the bacterial tree topology. Pseudoreplication introduced by repeated sampling of host species and subspecies within a symbiont clade may lead to significant results in tests of co-diversification, even if no co-cladogenesis (that is, concordant diversification of symbiont lineages with host speciation events) has occurred6. To address this issue, we conducted additional permutation tests to assess whether there was significantly more evidence in the symbiont phylogeny of co-cladogenesis between host species and bacterial symbiont lineages than expected under the null hypothesis of no co-cladogenesis. In short, these analyses generated 100 host phylogenies with random tip-label assignments (while retaining the repeated sampling per host-tree tip), then performed for each of these host phylogenies the scans of the symbiont MAG phylogeny for co-diversification. In each scan, each node in the symbiont phylogeny less than 1/4th of the total root depth was tested for co-diversification with the host phylogeny by permutation of symbiont and host tip labels, as performed for the real data. Thus, these scans generated a distribution of the number of co-diversifying clades (r > 0.75, P < 0.01) detected by our approach under the null hypothesis due to the given symbiont and host-tree structures and pseudoreplication. These analyses revealed that the scans based on the true host phylogeny detected >3-fold more instances of co-diversifying clades than did the scans based on the host phylogenies with random tip labels. No scan based on host phylogenies with random tip labels detected more instances of co-diversifying clades than the scan based on the true host phylogeny. These results indicate that the symbiont phylogeny contained significantly more instances of co-cladogenesis with primate species than expected under the null hypothesis, even after controlling for tree structures and pseudoreplication (Supplementary Discussion and Extended Data Fig. 2) (z-score = 4.73, P < 0.01). In addition, we performed additional tests for co-diversification within each of the identified co-diversifying clades subsampled to a single MAG per host species per clade. This approach, which ignores information about the monophyly of symbionts derived from the same host species (that is, host-species specificity)—a critical component of strict co-diversification, also supported a history of co-cladogenesis between primate gut bacteria and host species (Supplementary Discussion and Table 3). Moreover, sensitivity analyses, in which scans for co-diversification were performed after MAGs from each host species were removed one host species at a time (Supplementary Discussion), indicated that the approach was not biased to detect a greater number of co-diversifying clades when MAGs from subsets of host species were analysed (Extended Data Fig. 3). Together, these post-hoc analyses provide evidence for widespread co-diversification in the primate gut microbiota beyond what can be explained by spurious detection (that is, false positives) caused by pseudoreplication.
Associations between bacterial and host phylogenies provide evidence for concurrent diversification between bacterial and host lineages. However, these associations could in principle arise due to the successive horizontal colonization of symbionts among closely related host species25. If the bacterial clades identified diversified contemporaneously with their host clades, then the relative depths of the bacterial clades based on genomic divergence should be positively associated with the known ages of the host clades. Across all co-diversifying clades, symbiont clade depths and their corresponding host clade depths were positively associated (R2 = 0.232; P = 4.872 × 10−14) (Extended Data Fig. 4). Moreover, the intercept and slope of this relationship were significant and strikingly consistent across bacterial phyla represented by >10 co-diversifying symbiont clades (that is, Firmicutes, Actinobacteriota and Bacteroidota) (Extended Data Fig. 4). In contrast, a significant positive association was not observed for the clades showing the weakest evidence of co-diversification (r < 0, P > 0.05) (R2 = 0.012, P = 0.393; Extended Data Fig. 5). The associations between depths of co-diversifying clades and known host divergence dates provide an additional line of evidence for concurrent diversification between bacteria and primate species.
Calibration of molecular clocks for co-diversifying gut bacteria
Considering the co-diversifying bacterial clades alongside the known divergence dates of primate species enabled estimation of rates of bacterial molecular evolution—biological parameters that have been difficult to measure due to the paucity of bacterial fossils. To extend the above analysis correlating co-diversifying clade depths with host clade age, we calibrated nucleotide substitution rates within each of the clades that displayed the strongest evidence of co-diversification (that is, Mantel’s r > 0.95). These analyses revealed clock-like rates of evolution of bacterial genomes both within and between primate families (Fig. 2 and Supplementary Table 4). Interestingly, we observed substantial differences in the mean rate of per-clade molecular evolution between bacterial phyla, with Actinobacteriota displaying the fastest rates and Spirochaetota and Melainabacteria/Cyanobacteria the slowest (Extended Data Fig. 6), potentially reflecting systematic differences in doubling times, mutation rates or demographic factors among these taxa. Estimated rates of molecular evolution could also be biased by homologous recombination26, which was not considered here due to a lack of haplotype information. Overall, molecular clock analyses indicated a mean rate of core-gene sequence divergence per clade of 0.0174 substitutions per site per million years, with variation among clades ranging from 0.00153 to 0.063 (Fig. 2 and Supplementary Table 4). These estimates are within the ranges inferred from decadal time series of diverse bacterial pathogens26, millennial time series of Mycobacterium and Yersenia27,28, and time-calibrated phylogenies of Escherichia, Salmonella and Bifidobacterium5,29. Cumulatively, these results further support histories of co-diversification and suggest that host diversification events can inform calibration of molecular clocks for many gut bacteria.
Fig. 2 |. Molecular timescales for bacterial evolution in the primate gut.
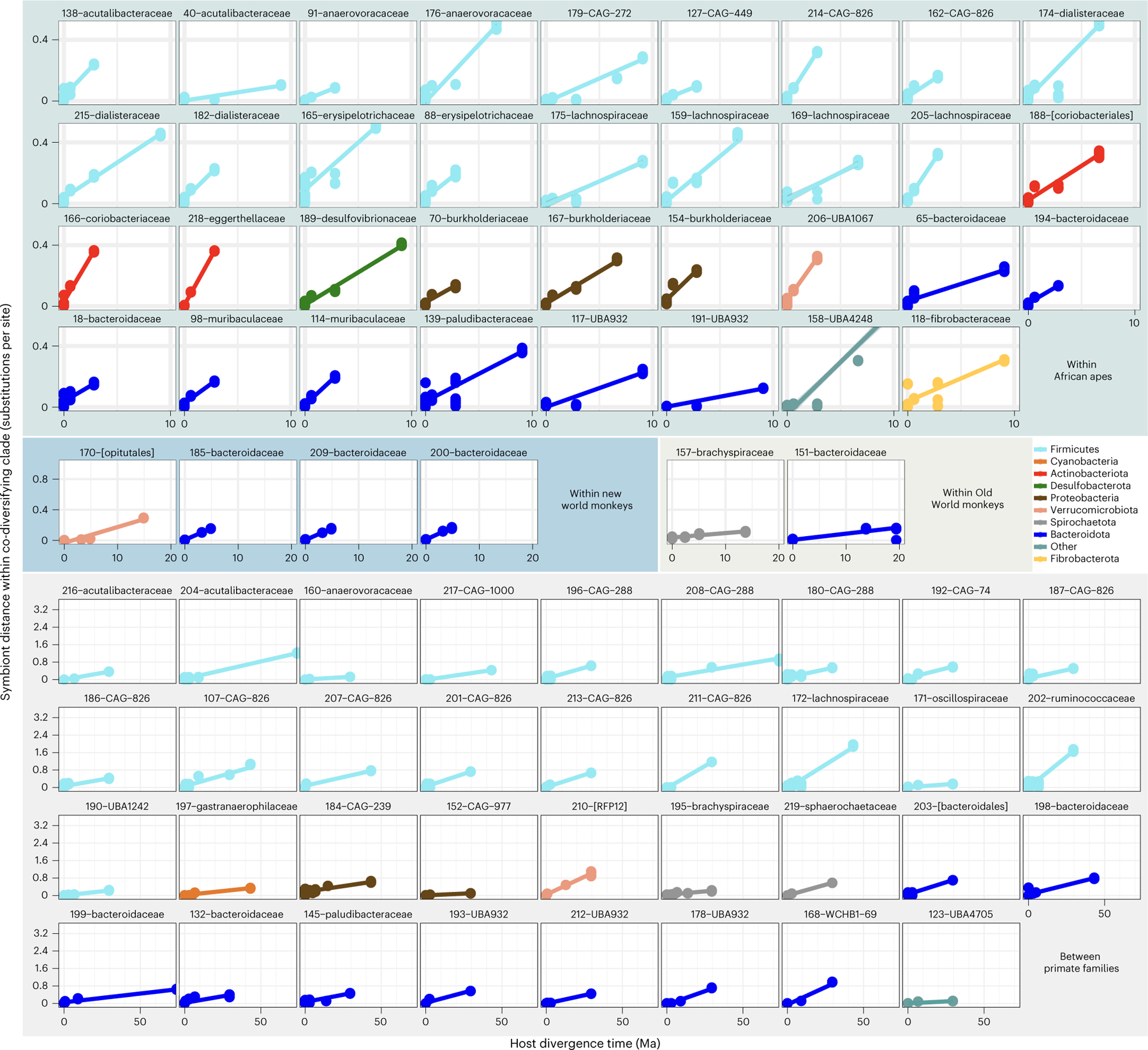
Scatterplots and regression lines show relationships between DNA sequence divergence (nucleotide substitutions per site) of core genes (bac120 marker set) within co-diversifying clades and divergence times of host species from which bacterial genomes were recovered. Each facet corresponds to a single co-diversifying clade. All co-diversifying clades displaying Mantel’s r > 0.95 are shown. Facets are grouped on the basis of the host species from which bacterial genomes were recovered as indicated by backdrop colours corresponding to Fig. 1a. Clade ID numbers and family- or order-level taxonomic assignments (Supplementary Table 4) are presented above each facet.
Genomic content of co-diversifying clades
Given multiple co-diversifying clades distributed throughout the MAG phylogeny, we next tested whether any gene functions or pathways in bacterial genomes were significantly associated with co-diversification independently of bacterial phylogenetic history. These analyses compared co-diversifying bacterial MAGs (that is, those belonging to clades displaying Mantel’s r > 0.75) with MAGs belonging to clades showing the weakest evidence of co-diversification (Mantel’s r < 0). These analyses, which employed phylogenetic methods to account for non-independence on the basis of the structure of the MAG phylogeny, revealed >5-fold more clusters of orthologous genes (COG) functions, categories and pathways displaying significant associations with co-diversification (phylogenetic analysis of variance (ANOVA) P < 0.001) than expected under the null hypothesis (Supplementary Table 3). These results support the idea that multiple gene functions were significantly associated with co-diversification independently of bacterial phylogenetic history. Relative to non-co-diversifying MAGs, co-diversifying MAGs contained fewer functions involved in cell cycle control, cell division, chromosome partitioning and inorganic ion transport and metabolism, but were enriched in multiple uncharacterized proteins, transporters and lipopolysaccharide biosynthesis proteins. In contrast to recent evidence from studies of gut bacteria that co-diversified with human populations30, we found no association independent of bacterial phylogenetic history between bacterial genome size (calculated as observed MAG length multiplied by the inverse of MAG completeness) and co-diversification (phylogenetic ANOVA P = 0.41). Cumulatively, these results identify specific functions significantly overrepresented in co-diversifying gut bacterial genomes independent of phylogenetic history relative to gut bacterial genomes from clades showing the weakest evidence of co-diversification.
Extinction of ancestral gut bacteria from human populations
The discovery of hundreds of gut bacterial clades that co-diversified with primate species over millions of years revealed the striking pattern that many symbiont lineages ancestral to the African apes were not detected in humans. Previous work has shown that individual humans in both industrialized and non-industrialized settings harbour significantly fewer gut bacterial taxa—from species to phyla—than do individual chimpanzees, bonobos or gorillas31–33. This pattern has been attributed to derived aspects of human lifestyles, such as hygiene practices, antibiotic medicines and fibre-poor diets34,35. It has also been proposed that the absence of ancestral bacteria may incur health costs to human populations35,36. Here, specific examples of extinction of ancestral gut bacterial lineages from human populations could be identified as co-diversifying clades containing MAGs derived from Pan and at least one other non-human primate (NHP) species (that is, at least one outgroup to the Hominini), but lacking MAGs derived from humans. Assessing each co-diversifying bacterial clade for this pattern revealed 129 clades ancestral to humans and chimpanzees. Of these, 57 (~44%) lacked human-derived representatives, consistent with extinction from humans. In contrast, 81 clades contained MAGs from humans and at least one non-Pan NHP species, but only 9 (~11%) of these clades did not contain MAGs from Pan. Fisher’s exact tests indicated that a significantly greater proportion of clades ancestral to the Hominini were absent from humans than from Pan (P = 2.829 × 10−7) (Fig. 3a), consistent with an elevated rate of extinction of ancestral clades from humans relative to Pan. In total, bacterial clades lacking human-derived MAGs constituted 52.0% of the branch length of the phylogeny of co-diversifying clades ancestral to the African apes (Fig. 3a). These findings indicate relatively rapid and widespread losses of co-diversifying gut bacterial lineages from humans.
Fig. 3 |. Widespread extinctions of ancestral symbionts from the human microbiota.
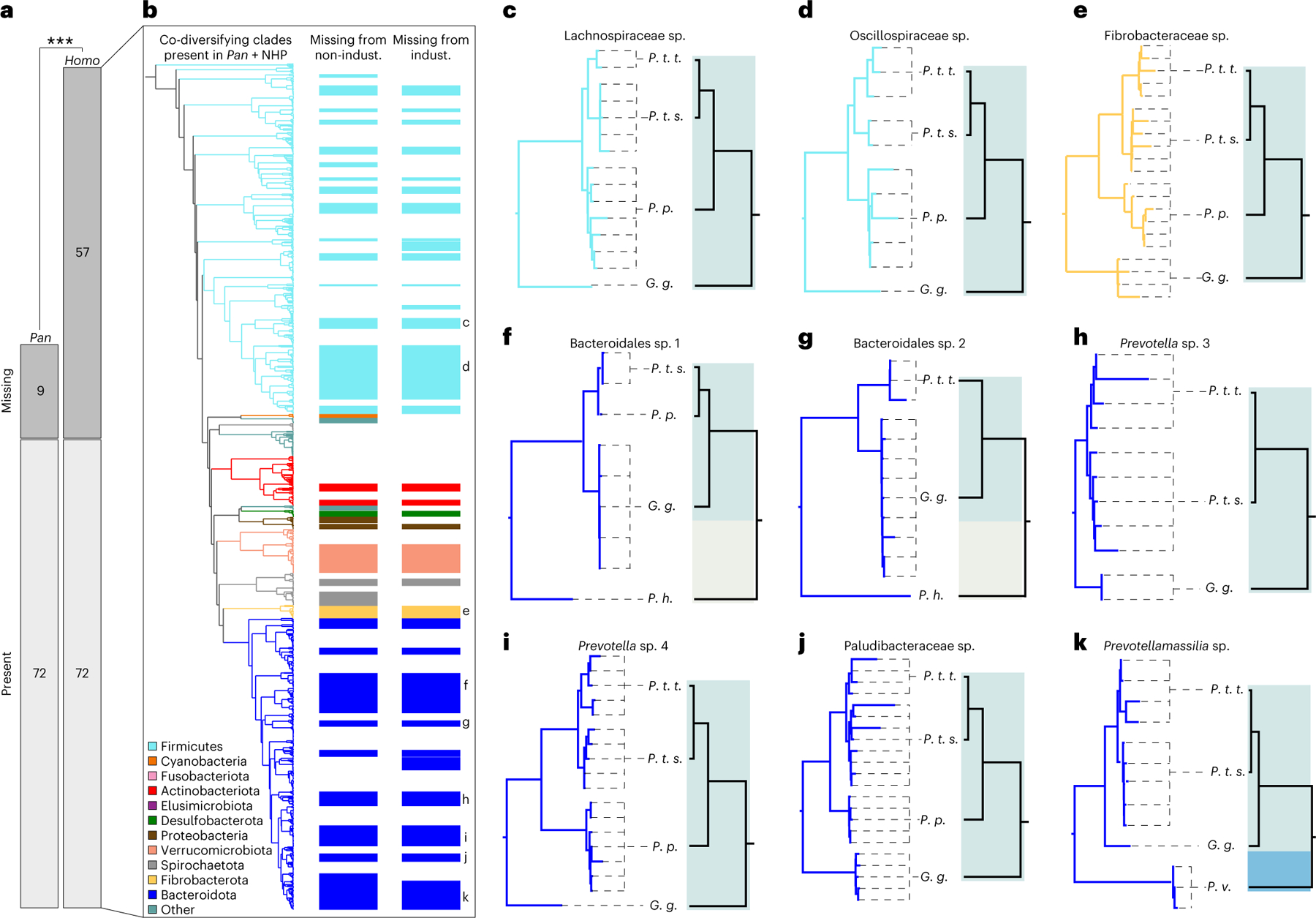
a, Mosaic plot shows that a greater proportion of ancestral co-diversifying clades were absent from humans than from Pan; two-sided Fisher’s exact test ***P = 2.8 × 10−7. Bars indicate the number of clades inferred to be ancestral to humans and chimpanzees. b, Phylogeny shows relationships among co-diversifying clades present in Pan and at least one non-human primate (NHP) outgroup to the Hominini, corresponding to the rightmost bars in a. Colours of branches and bars correspond to bacterial phyla as indicated by the key. Leftmost coloured bars to the right of the phylogeny mark clades not detected in any non-industrialized (non-indust.) human population. Rightmost coloured bars mark clades not detected in any industrialized (indust.) human population. c–k, Tanglegrams show examples of co-diversifying clades ancestral to the Hominini but not detected in humans. Left and right phylogenies in each panel show relationships among bacteria and hosts, respectively. Dashed lines connect bacteria to host species (P. t. t., Pan troglodytes troglodytes; P. t. s., Pan troglodytes schweinfurthii; P. p., Pan paniscus; G. g., Gorilla gorilla; P. h., Papio hamadryas; P. v., Propithecus verreauxi) from which they were recovered. Colours denote bacterial and host taxa as in Fig. 1. All bacterial clades were supported by >75% of bootstrap replicates. Note the absence of human-derived bacterial lineages from each clade.
In addition, we observed lifestyle-specific absence of ancestral gut bacterial symbionts from human populations. Several of the clades ancestral to the African apes contained MAGs from non-industrialized human populations but not from industrialized populations (that is, the human-derived species genome bin (SGB) present in these clades lacked representatives from industrialized human populations) (Fig. 3b). Conversely, some clades contained MAGs from industrialized human populations but not non-industrialized populations. However, these clades numbered fewer than those lacking MAGs from industrialized human populations, despite a bias in sampling effort towards industrialized human populations relative to non-industrialized human populations. This observation suggests an accelerated rate of loss of co-diversifying clades from industrialized populations relative to non-industrialized populations.
Examples of co-diversifying bacterial clades displaying evidence of extinction from all human populations are shown in Fig. 3c–k. The proportion of co-diversifying clades ancestral to the Hominini but absent from humans did not differ among bacterial phyla (Fisher’s exact test P > 0.05 in each pair of comparisons). However, multiple COGs, COG categories and COG pathways were enriched in these clades relative to ancestral co-diversifying clades retained in humans (Extended Data Fig. 7 and Supplementary Table 5). Functions enriched in clades absent from humans included pathways contributing to the urea cycle and the biosynthesis of lysine, serine, biotin and aromatic amino acids. Moreover, certain functions were enriched in co-diversifying clades missing from industrialized humans compared with co-diversifying clades retained in non-industrialized humans, including pathways for gluconeogenesis and lipid biosynthesis (Supplementary Table 5). These results identify specific functions enriched in co-diversifying clades displaying evidence of extinction from human populations.
The more frequent absence of ancestral bacterial lineages from humans than from Pan could not be explained by biases in sampling effort, as metagenomic data from humans exceeded those from Pan and other NHPs by >10-fold. Similarly, the more frequent absence of ancestral symbionts from humans was not the result of excluding replicate human-derived MAGs within 95% average nucleotide identity (ANI bins; Supplementary Discussion) because each co-diversifying bacterial clade from which human MAGs were absent spanned >10% ANI (Supplementary Table 3). In addition, reperforming phylogenetic analyses with additional human-derived gut bacterial genomes from the Unified Human Gastrointestinal Genome (UHGG) collection37 indicated that the co-diversifying clades identified as extinct were also not represented in this genome database, further supporting the extinction of co-diversified lineages from humans (Supplementary Discussion). Moreover, the median mappability of human metagenomic samples to the human-derived MAGs included here exceeded 95%16, suggesting that the failure to detect ancestral gut bacterial lineages in humans was not due to lack of representative MAG diversity from humans. Additional mapping analyses of human metagenomic data also failed to detect the ancestral co-diversifying clades that were missing human-derived MAGs (Supplementary Discussion), indicating that the lineages identified as extinct from humans were not present in humans at abundances above the detection threshold afforded by available metagenomic datasets.
In contrast to the significantly co-diversifying clades (r > 0.75, P < 0.01), which showed marked evidence of extinction from humans, the clades displaying the weakest evidence of co-diversification (r < 0, P > 0.05) showed no evidence of widespread extinctions from humans. Of the clades displaying the weakest evidence of co-diversification, 168 clades contained MAGs from Pan and at least one other NHP. Of these 168 clades, only five lacked human-derived representatives (Supplementary Table 6). These results indicate that extinctions from humans of symbiont clades found in Pan and other NHPs have been largely restricted to clades displaying significant evidence of co-diversification.
Signatures of selection in co-diversifying bacterial genomes
The observation that nearly half of the co-diversified symbionts ancestral to the African apes were absent from humans (Fig. 3) suggests altered pressures of natural selection acting on the microbiota in humans relative to Pan. To explore whether symbiont genomes displayed evidence of divergent selection between host species, we next tested within the co-diversifying clades detected in humans whether human-derived MAGs contained signatures of positive and purifying selection different from those found in closely related Pan-derived MAGs. For each co-diversifying clade detected in both humans and Pan, we identified the core gene (that is, open reading frame) families shared by all MAGs from the clade. We then aligned each core gene family, constructed a maximum-likelihood phylogeny and calculated the ratio of non-synonymous to synonymous substitutions per site (that is, dN/dS) along each branch. For each core gene, we compared dN/dS ratios between the branch leading to human-derived MAGs and the branch leading to Pan-derived MAGs. To minimize the effects of sequencing or binning errors present in MAGs38, dN/dS was calculated on the basis of synapomorphies that differentiated clades of human- and Pan-derived MAGs; that is, these analyses of dN/dS were based on MAGs assembled from multiple individuals per host group. Thus, these analyses revealed bacterial genes that displayed phylogenetically independent signatures of positive selection (dN/dS > 1) in either humans or Pan, or in both humans and Pan.
Results showed that most core open reading frames (CORFs) have evolved primarily under purifying selection (dN/dS < 1), with a minority displaying evidence of positive selection (dN/dS > 1) (Extended Data Fig. 8 and Supplementary Table 7). Interestingly, patterns of purifying and positive selection in bacterial genomes were generally consistent between human and Pan. Per-CORF dN/dS values in humans and Pan were positively associated (regression P = 3.581 × 10−21; R2 = 0.674), and multiple CORFs displayed evidence of positive selection in both host groups (Extended Data Fig. 8 and Supplementary Table 7). In contrast, a subset of genes displayed evidence of positive selection in humans or Pan but evidence of purifying selection in the other host (upper-left and lower-right quadrants of Extended Data Fig. 8). For example, elongation factor P of a Prevotella lineage displayed significant evidence of positive selection in humans (dN/dS = 5.51) but significant evidence of purifying selection in Pan (dN/dS = 0.033) (Supplementary Table 5). This gene has been previously shown to influence virulence and antibiotic drug resistance in Salmonella enterica39. Although the specific selective agents responsible for divergent dN/dS between humans and Pan cannot be determined from our analyses, results identify co-diversifying bacterial genes that display significant evidence of adaptive evolution in primate host species.
Discussion
The tight evolutionary relationships between primates and many of their gut bacteria provide windows into the deep history of the human microbiota. Considering these host–microbe relationships in a comparative context using high-quality genomic data allowed us to identify signatures of co-diversification and natural selection across bacterial phyla. Many of the ancestral members of the hominid microbiota were absent from human populations. Some co-diversifying lineages appear to have been lost exclusively from industrialized human populations, but most co-diversifying lineages absent from industrialized human populations were also absent from non-industrialized human populations, suggesting that extinctions of these bacteria occurred in humans’ more distant evolutionary past.
The extinction of ancestral gut bacterial lineages from Homo sapiens regardless of lifestyle may have resulted from immune, physiological or behavioural changes that occurred along the human lineage. For instance, changes in diet that occurred after humans diverged from Pan, such as the transition away from eating raw leaves and fruit towards cooking food and higher consumption of animal fat and proteins40, may have altered the selective environment within the gut in a manner that selects against the assembly of microbiota as diverse as those found in other apes. In contrast, co-diversifying symbionts absent specifically from industrialized human populations were probably driven to local extinction by recent lifestyle changes that differentiate these populations from non-industrialized populations. The observation that co-diversifying clades (r > 0.75) appeared to show elevated rates of extinction from humans not observed for non-co-diversifying clades (r < 0) (57/129 versus 5/167) suggests that host-species-specific symbionts may be particularly susceptible to extinction from humans. By identifying bacterial symbioses that predate the divergence of humans from other primates, this study generates high-priority targets for efforts to preserve humans’ endogenous microbiota diversity.
Methods
Ethics approval
All research described in this manuscript was compliant with ethical regulations as approved by the Institutional Biosafety Committee at Cornell University.
Sample collection
Collection of faecal samples from wild chimpanzee and bonobo populations and their genetic analysis have been previously reported41–46. Briefly, bonobo (Pan paniscus) samples were collected at four field sites in the Democratic Republic of the Congo (LK, KR, IK, TL2). Samples from central chimpanzees (Pan troglodytes troglodytes) were collected at field sites in Cameroon (DP) and the Republic of the Congo (GT), while samples from eastern chimpanzees (Pan troglodytes schweinfurthii) were collected in Gombe National Park in Tanzania (GM) (Supplementary Table 1). When possible, samples were collected from nest sites in the morning to minimize the possibilities of degradation and contamination from the external environment. All samples were stored in RNAlater immediately upon collection and for shipping to the University of Pennsylvania (UPenn) in Philadelphia, Pennsylvania, where they were placed in −80 °C freezers for long-term storage. Faecal DNAs were used to determine host mitochondrial haplotypes (D loop) and to identify individuals using short tandem repeat analyses of nuclear DNA by capillary electrophoresis as previously described42–46 (Supplementary Table 1). Aliquots of samples were shipped on dry ice from UPenn to Cornell University in Ithaca, New York, where they were processed and analysed. All samples were obtained with permission from local authorities as previously reported41–46. Samples were shipped in compliance with the regulations of the Convention on International Trade in Endangered Species of Wild Fauna and Flora, and with governmental export and import permits.
Hybrid metagenomic sequencing of chimpanzee and bonobo gut microbiota
We employed a hybrid metagenome sequencing approach to enable the assembly of genomes from the gut microbiota of chimpanzees and bonobos. For Illumina sequencing, we extracted DNAs from all samples using a bead beating approach as implemented in the PowerLyzer Qiagen kit. Libraries for Illumina sequencing were prepared using a TruSeq–equivalent approach at the Cornell Biotechnology Resource Center as previously described47 and pooled in equimolar amounts for sequencing. Pooled libraries were sequenced on a NovaSeq S4 flow cell at the University of California, Davis Genome Centre.
For Nanopore sequencing, we extracted DNAs using a three-step approach consisting of (1) enzymatic lysis, (2) osmotic lysis and (3) bead beating following previously described methods48 with some modifications. Briefly, 200 µl DNA/RNA Shield (Zymo) was added to 40–50 mg faecal material, homogenized with a pipette tip and rotated for 10 min at 20 r.p.m. after a ~2 s vortexing. Supernatants were transferred to clean 2 ml tubes after centrifuging at 5,000 × g for 5 min. Pellets were then washed with 100 µl PBS once and supernatants were transferred to the previous 2 ml tube after centrifuging at 5,000 × g for 5 min. Pellets were then washed again with 1,000 µl PBS, lysed by adding 100 µl PBS and 5 µl MetaPolyzyme (Sigma-Aldrich), and incubated at 35 °C for 2 h. DNA/RNA shield (100 µl), 10 µl 10% SDS and 10 µl 20 mg ml−1 Proteinase K were then added to the mixture and the mixture further incubated at 55 °C for 30 min at 300 r.p.m. After centrifuging at 5,000 × g for 5 min, supernatants were transferred to the previous 2 ml tube. Pellets were resuspended in 750 µl genomic lysis solution (Zymo), transferred to a ZR BashingBead lysis tube (Zymo) and bead-beaten on an Omni Bead Ruptor Elite (OMNI) for 1 cycle of 40 s at 6 m s−1. Supernatants were combined with previous steps after centrifuging at 10,000 × g for 1 min. Pooled supernatants from each step were then mixed with one volume of MagBinding buffer (Zymo) and 50 µl MagBinding beads (Zymo), and bound on a tube rotator for 10 min at 20 r.p.m. Tubes were placed on magnetic racks and incubated for 2 min until clear, after which supernatants were discarded. DNA elution buffer (100 µl) was added to the tube and mixed 10 times before adding another 500 µl Quick-DNA MagBinding buffer. After binding on tube rotators for 10 min at 20 r.p.m., samples were put on a magnetic rack and incubated for 2 min until clear, after which the supernatant was discarded. Beads were then washed with 900 µl DNA pre-wash buffer and 900 µl gDNA wash buffer (Zymo). DNA elution buffer (900 µl; Zymo) was then added and removed immediately. Final DNA was eluted with 50 µl of DNA elution buffer and stored at 4 °C for later use. Libraries were prepared using the Nanopore Ligation Sequencing kit (SQK-LSK110) and sequenced on the MinION platform, with one flow cell dedicated to each sample.
Illumina-based assemblies and binning
We developed and employed a snakemake49 workflow to assemble and bin contigs from Illumina shotgun metagenomic reads. MAGmaker is a flexible and modular bioinformatic pipeline for quality filtering, taxonomic profiling, assembly, binning and optimal bin-selection of short-read metagenomic sequencing data from an arbitrarily large number of samples. In short, raw reads were quality-filtered using cutadapt v4.150 and then optionally mapped against a reference Pan genome (GCF_002880755.1) using Bowtie2 v2.351. Read quality profiles were generated using fastqc. High-quality non-host reads were then assembled using metaSPAdes v3.1552. Assemblies were evaluated for quality with Quast v5.0.253. Finally, a Jaccard distance matrix of samples was generated using SourMash v4.054, a fast and efficient MinHash algorithm. For binning, fastqs from metagenome samples were mapped in an all-by-all manner against the assembled contigs of samples from the same host population using Minimap2 v2.2455. Coverage results from abundance mapper were then input into binning algorithms CONCOCT v0.4.216, MetaBAT2 v2.1514 and MaxBin v2.215. After binning, DASTool v1.1.317 was used to select the optimal set of bins from the three binning algorithms on the basis of default thresholds for genome completion and contamination. All code used for assembly and binning is available at https://github.com/CUMoellerLab/sn-mg-pipeline.
Nanopore base calling and hybrid metagenomic assembly
Base calling was performed on a Lambda Labs workstation containing two NVIDIA RTX 3090 graphical processing units (GPUs) with Guppy v6.1.2 using the following settings: –chunk_size 3000 –chunks_per_runner 768 –qscore_filtering –min_qscore 7 –config dna_r9.4.1_450bps_hac.cfg –calib_detect –compress_fastq.
To assemble MAGs from Illumina and Nanopore data from chimpanzees, we employed the reticulatus snakemake workflow (https://github.com/SamStudio8/reticulatus). All GPU-accelerated assembly and polishing was conducted on a Lambda Labs workstation containing two NVIDIA RTX 3090 GPUs. Pan reads were removed from fastq files using dehumanizer against Pan reference genome GCF_002880755.1. Nanopore reads for each sample were assembled into contigs with Flye v2.956 and contigs were polished with racon v1.4.357 and medaka v1.4.0 (https://github.com/nanoporetech/medaka) using Illumina reads derived from each faecal sample sequenced on a MinION.
Strain heterogeneity
Strain heterogeneity of all MAGs generated for this study was estimated using CMSeq as previously described19, employing an approach in which Illumina reads from each sample were mapped to every MAG assembled from the sample (https://github.com/SegataLab/cmseq). All MAGs displaying strain heterogeneity >0.5% were excluded from downstream analyses. Strain heterogeneity was calculated only for MAGs for which >100 positions in the genome were covered by at least 10 reads with base quality >30. The strain-heterogeneity threshold and mapping criteria were chosen to enable direct comparisons with existing genome databases analysed in this study.
Phylogenomic analyses
We combined all 2,614 chimpanzee MAGs with previously assembled MAGs from NHPs18 and humans19,37. For these analyses, whose goal was to determine the ancestral composition of the primate gut microbiota, we focused only on MAGs from NHPs sampled in the wild because captive NHPs have previously been shown to acquire certain gut bacterial lineages from humans6,58,59, potentially obscuring ancient signals of co-diversification events. For phylogenetic inference, we used the representative genome from each human gut-derived 95% ANI SGB identified in ref. 19 or the species-level representative genomes from the UHGG catalogue37. These MAG databases were chosen for our analyses due to the broad representation of publicly available metagenomic datasets, and >90% of human gut metagenomic reads were recruitable by mapping to the MAG databases19.
Single-copy core genes from each genome were extracted, concatenated and aligned using the Genome Taxonomy Database Toolkit (GTDB-Tk) R06-RS202 bac120 collection21. A phylogeny was inferred from the amino-acid alignment with IQTree2 v2.1.2 using WAG+G4 substitution model and 1,000 bootstrap replicates. The substitution model was selected by ModelFinder as implemented in IQTree222.
Identifying co-diversified and host-species-specific clades
To identify co-diversified and host-species-specific gut bacterial lineages from the MAG phylogeny, we developed a workflow on the basis of an existing approach for detecting co-diversification in simpler host–parasite systems23. Ref. 23 utilized permutation-based Mantel tests, in which the topology of the parasite phylogeny is permuted to generate a null distribution ofconcordance between parasite and host phylogenies. Here we extended this method to allow tests for co-diversification across a phylogeny of lineages derived from complex microbiota from a clade of host species. Our approach, which is available at https://github.com/CUMoellerLab/codiv-tools, takes as input an incidence table indicating from which host species each symbiont lineage was recovered, a symbiont phylogeny and a host phylogeny. It then applies a permutation test for co-diversification for each node of the symbiont phylogeny. Here we tested each node present in the most distal 1/4 of the symbiont phylogeny because deeper nodes (for example, those representing the common ancestors of different phyla) are expected to predate the diversification ofprimates. In addition, the rooting ofeach bacterialclade was obtained by the nearest outgroup for the clade within the symbiont phylogeny (that is, for permutation tests, each clade was extracted, with its root, from the MAG phylogeny). This workflow outputs a table of P values and r correlation coefficients for each node indicating results of Mantel tests between host phylogenetic distances and symbiont phylogenetic distances. Low P values and high r coefficients indicate high concordance between host and symbiont phylogenies—a pattern indicative of ancient associations and co-diversification.
In addition, we conducted additional permutation-based analyses to assess the degree to which pseudoreplication introduced by sampling multiple individuals per host species and subspecies may have affected the detection of bacterial clades showing significant evidence of co-diversification. For these analyses, we randomly permuted the host-tree tip labels and reran the scan of the symbiont phylogeny for co-diversifying clades described above 100 times. Within each of these 100 scans, each node in the distal 1/4th of the symbiont tree was tested for co-diversification with the host tree using the permutation-based Mantel test introduced in ref. 23 in which both host and symbiont tips were permuted 999 times. Thus, the 100 scans generated a null distribution of the number of significantly co-diversifying clades (r > 0.75, P < 0.01) expected on the basis of the pseudoreplication present within and the structure of the symbiont MAG phylogeny (Extended Data Fig. 2).
We also conducted analyses to determine the sensitivity of our results to the inclusion of MAGs individual host species. In these analyses, we removed all MAGs from individual host species one host species at a time and performed scans for co-diversification on the reduced dataset. Full details of these analyses and their results are described in Supplementary Information.
Phylogenetically independent associations of gene functions with co-diversification
We tested for gene functions in bacterial genomes that were significantly overrepresented in co-diversifying clades relative to non-co-diversifying clades while accounting for bacterial phylogenetic history and non-independence. Genes from MAGs were annotated against the COG database in Anvi’o v7.060. For these analyses, we employed phylogenetic ANOVA using 1,000 permutations and default settings as implemented in phytools v1.561 to test for COG functions, categories and pathways significantly enriched in MAGs from bacterial clades showing Mantel’s r > 0.75 compared to MAGs from clades showing Mantel’s r < 0. These r-value thresholds were chosen to contrast MAGs showing the strongest evidence of co-diversification with those showing the weakest evidence of co-diversification. These analyses focused on only >90% complete MAGs to avoid false inferences regarding the absence of genes from individual MAGs. Results from these tests are presented in Supplementary Table 3.
In addition to testing copy number of COG functions, categories and pathways, we tested whether estimated genome size differed between co-diversifying and non-co-diversifying MAGs independently of host bacterial phylogenetic history. Estimated genome size was calculated as observed genome length multiplied by the inverse of the MAG’s completeness.
Calibration of molecular clocks in the primate gut microbiota
To estimate genome-wide rates of evolution in co-diversifying clades, phylogenies were estimated for each clade from the 120 bacterial marker genes identified by CheckM v1.1.662. For each clade, we extracted the unaligned single-copy marker gene nucleotide sequences from each constituent MAG and generated a multiple sequence alignment for each marker gene using MACSE V263, a codon-aware sequence aligner. Then, the aligned sequences for each marker gene within a clade were concatenated and a phylogeny estimated for the clade using RAxML v864. Rates of sequence evolution were estimated using a linear regression of the genetic distance calculated between each pair of MAGs and the evolutionary divergence time of their respective hosts. Code used for molecular clock analyses is available at https://github.com/CUMoellerLab/Sanders-etal-2022-analysis.
Identification of functions enriched in clades absent from humans
The absence from humans of co-diversifying bacterial clades that were detected in Pan and other NHP species raised questions about which metagenomic functions have been lost from humans. To address this issue, we tested for differentially abundant genes between Pan-derived MAGs from co-diversifying clades missing from humans and Pan-derived MAGs from co-diversifying clades present in humans. These tests asked whether any gene functional groups differentiated the ancestral bacterial clades extinct from humans from those present in humans. Genes from MAGs were annotated against the COG database in Anvi’o60. Genome fasta files were imported as contigs databases using anvi-gen-contigs-database, and genes were called using anvi-run-hmms and annotated against the COG20 database using anvi-run-ncbi-cogs. Contigs databases from Pan MAGs from co-diversifying clades present in Pan and at least one outgroup NHP to the Hominini were imported into a genomes storage database using anvi-gen-genomes-storage, and pangenome analyses were run using anvi-pan-genome. Pan MAGs in the pangenome were annotated using anvi-import-misc-data on the basis of whether the MAGs were from clades that lacked or contained human-derived representatives. Functional enrichment of COG categories, pathways and functions between these groups was then calculated using anvi-compute-functional-enrichment.
Mapping analyses and identification of extinction events
Raw metagenomic reads from the Human Microbiome Project Healthy Human Subjects cohort were downloaded from https://hmpdacc.org/. Raw metagenomic reads from Hadza hunter gatherers were downloaded from NCBI SRA (SRP056480, Bioproject ID PRJNA278393). Mapping of metagenomic reads to reference genome databases was conducted with Minimap255 using default settings. SAM and BAM files were converted and analysed with Samtools65.
Extinction of co-diversifying clades from human populations was also validated by incorporating all MAGs from each SGB reported in ref. 19. For example, a clade was identified as absent from industrialized (or non-industrialized) populations if and only if there were no MAGs within any SGB represented in the clade from the population category in the complete set of MAGs reported in ref. 19.
Categorization of human population lifestyles
For analyses of gut microbiota extinctions from humans, human populations were categorized into ‘industrialized’ and ‘non-industrialized’. For all human populations besides the archaic human population, these categories were based on those provided by previous studies19,37. All populations previously categorized as ‘westernized’ were categorized here as ‘industrialized’. The archaic human population20 included here was categorized as ‘non-industrialized’.
Calculation of per-gene dN/dS ratios from co-diversifying bacterial clades
Code used to identify signatures of natural selection in co-diversifying gut bacterial genomes is available at https://github.com/CUMoeller-Lab/Sanders-etal-2022-analysis. Open reading frames were extracted from bacterial genomes using getorf in EMBOSS v6.5.766 with the option ‘-Table 11’. CORFs for each co-diversifying bacterial clade (Mantel r > 0.75) were identified using CoreCruncher v167 with ‘-score 80’ and ‘-freq 100’. CORFs were then translated with transeq in EMBOSS. Translated CORFs were aligned with MAFFT using default settings. Aligned translated CORFs and unaligned CORFs were used to generate codon-based DNA alignments with pal2nal.v14 using the setting ‘-codontable 11’. Codon-based alignments for each CORF were concatenated and used to build a phylogenetic tree per co-diversifying clade with RAxML. The tree for each individual clade was rooted on the basis of the outgroup relationships for that clade in the combined marker-gene-based phylogeny used for testing co-diversification above. Co-diversified clades whose individually calculated species tree could not be reconciled with the outgroup pattern from the all-bacteria tree were not considered further.
Changes in dN/dS within individual clades were tested using the Branch Model mode of CodeML68 as implemented in GWCodeML v169. Foreground branches for testing were defined as those branches leading to a monophyletic grouping of 100% of either Homo- or Pan-derived bacteria for a given clade. When clades contained separate monophyletic groups of Homo- and Pan-derived bacteria, those branches were tested as separate operations (that is, Pan vs other primates+Homo, or Homo vs other primates+Pan.).
Statistics and reproducibility
No statistical method was used to predetermine sample size. Sampling was based on availability of existing faecal collections previously reported41–46. No data were excluded from the analyses. No experiments were conducted, so no randomization was performed, and investigators were not blinded to allocation during outcome assessment. For parametric statistical tests shown in Extended Data Figs. 4–6, data distributions were log transformed to conform to assumptions regarding homoscedasticity. Underlying data for these figures are presented in Supplementary Tables 3 and 6. For all non-parametric tests, data met assumptions of the statistical tests used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Map of sampling locations.
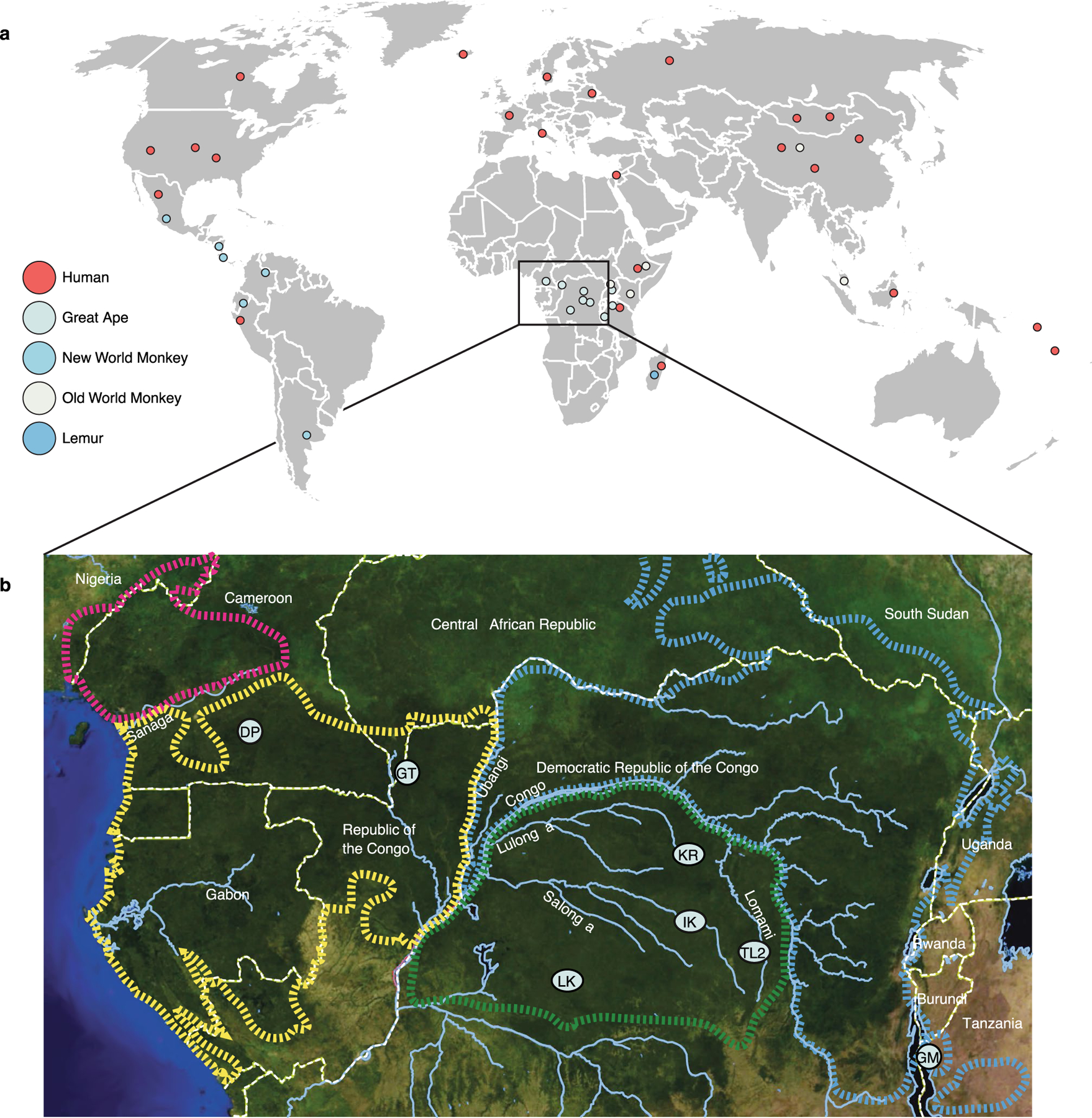
a, Map shows sampling locations for human, great ape, new world monkey, old world monkey, and lemur fecal samples. Circles correspond to individual populations sampled as indicated by the key. b, Map of equatorial Africa shows sampling locations for Pan fecal samples sequenced for this study. Two-letter codes correspond to those associated with host IDs in Supplementary Table 1. DP = Doumo Pierre; IK = Ikela; GT = Goualougo Triangle; TL2 = Tshuapa-Lomami-Lualaba; GM = Gombe; LK = Lui-kotal; KR = Kokolopori..
Extended Data Fig. 2 |. Histogram of number of significant nodes detected after permuting host labels.
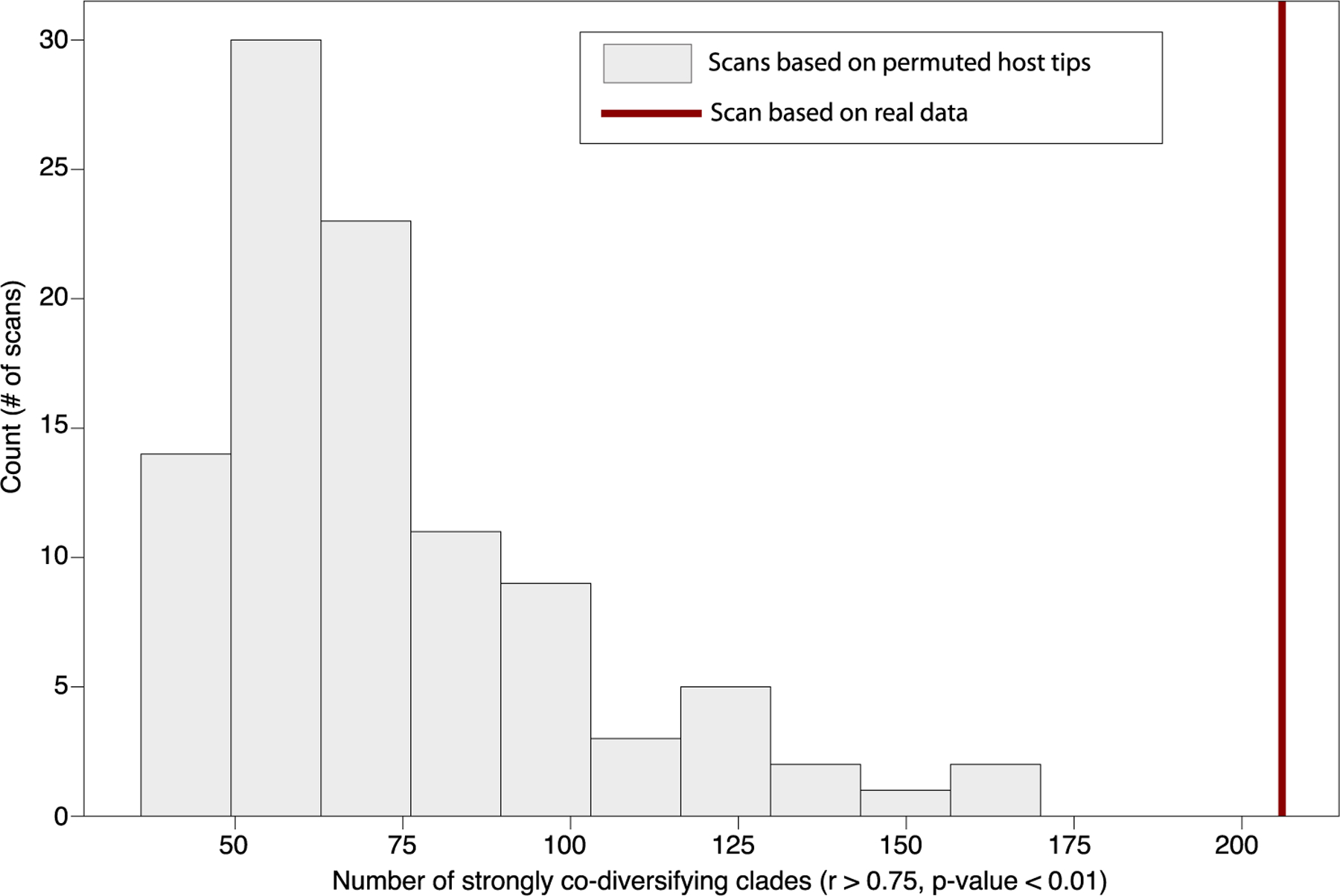
X axis indicates number of significant nodes (Mantel test p < 0.01, r > 0.75) recovered in the co-diversification scan after permuting labels of host tree but retaining symbiont tree labels and all other structure in the dataset. Results of 100 random permutations are shown. Value for unpermuted dataset is shown as a vertical red line..
Extended Data Fig. 3 |. Number of significant nodes detected after removing MAGs from individual host species.
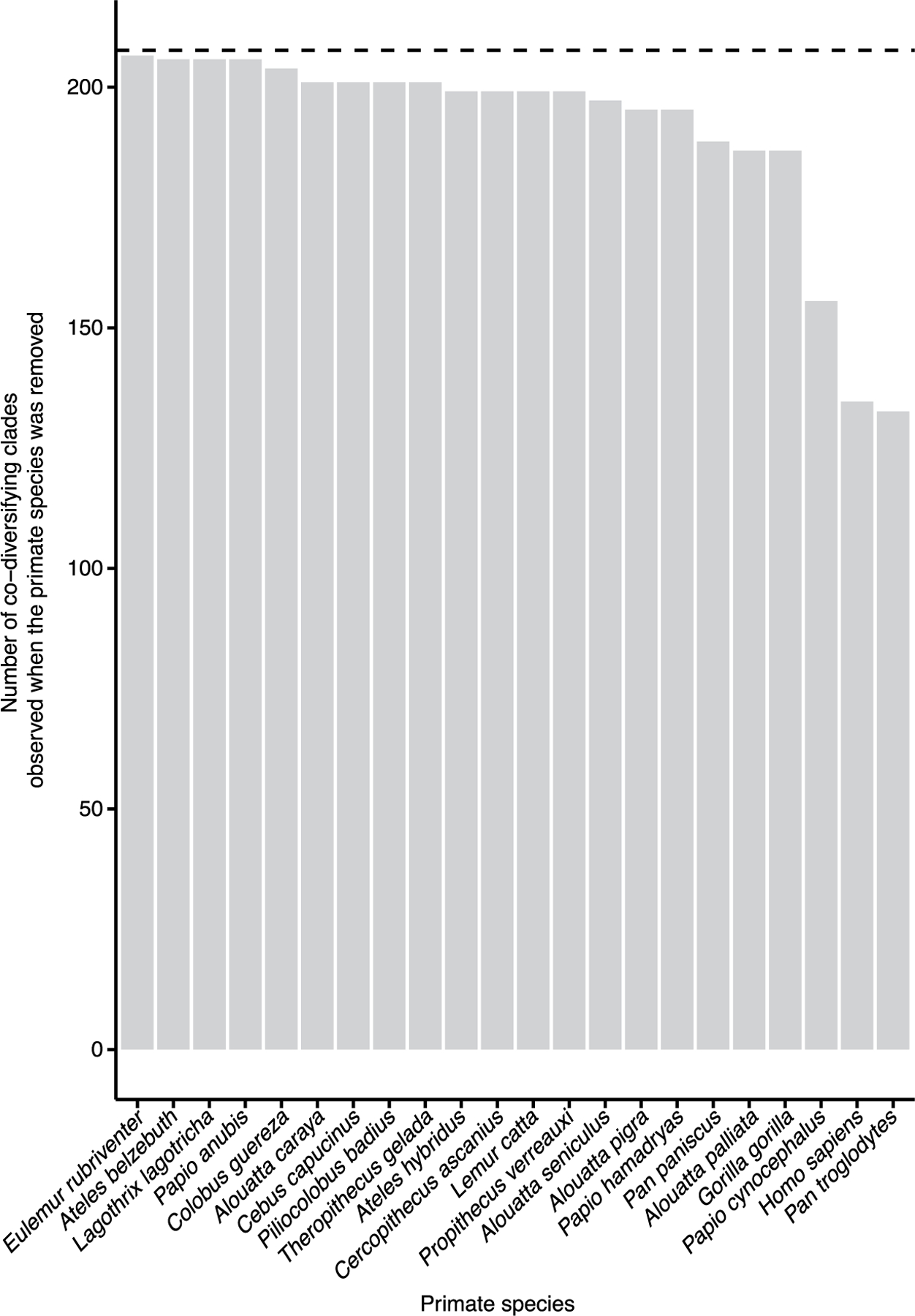
X axis indicates the host species whose MAGs were removed from that dataset before performing sensitivity analyses in which scans for co-diversification were performed after removing individual host species. The number of co-diversifying clades (Mantel test p < 0.01, r > 0.75) detected in each scan are shown. Value for the full dataset is shown as a horizontal dashed line..
Extended Data Fig. 4 |. Depths of co-diversifying bacterial clades corroborate known ages of host clades.
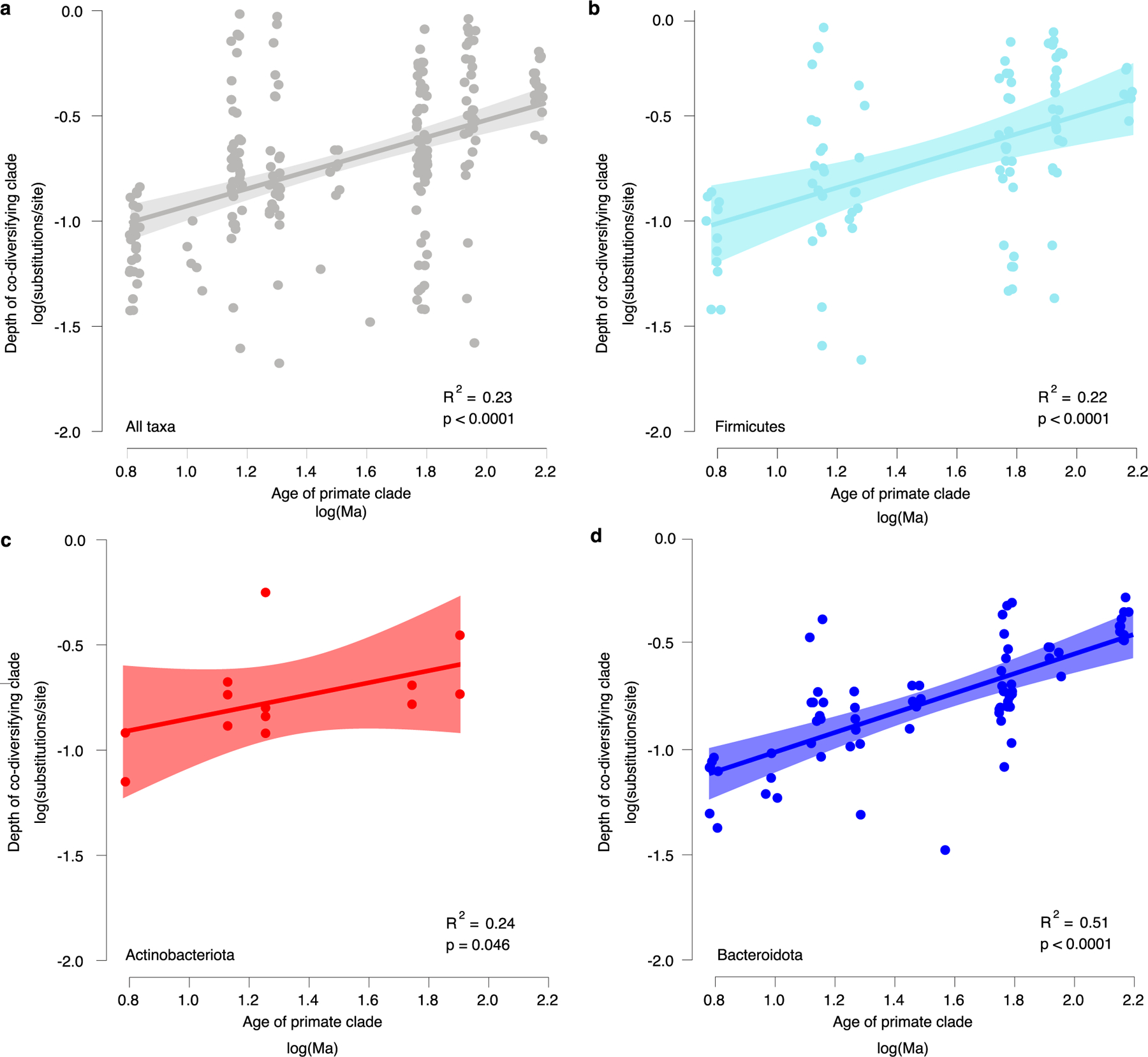
a, Scatter plot and regression line show the positive association between the depths of co-diversifying bacterial clades based on protein divergence of bac120 single-copy core genes and the known ages of their corresponding host clades based on timetree.org (df = 204; t = 6.03; unadjusted p-value = 7.36e-09). Each point corresponds to a co-diversifying bacterial clade. b–d, Scatter plots show relationships for Firmicutes (df = 72; t = 8.58; unadjusted p-value = 9.16e-11) (b), Actinobacteriota (df = 11; t = 2.25; unadjusted p-value = 0.046) (c), and Bacteroidota (df = 91; t = 4.38, unadjusted p-value = 3.17e-05) (d). Colours denote bacterial phyla as in Fig. 1b. In a–d, bands represent 99% confidence intervals, centre lines indicate best-fit regression, and p-values represent results of two-sided Student’s t-tests..
Extended Data Fig. 5 |. Depths of non-co-diversifying bacterial clades and ages of host clades.
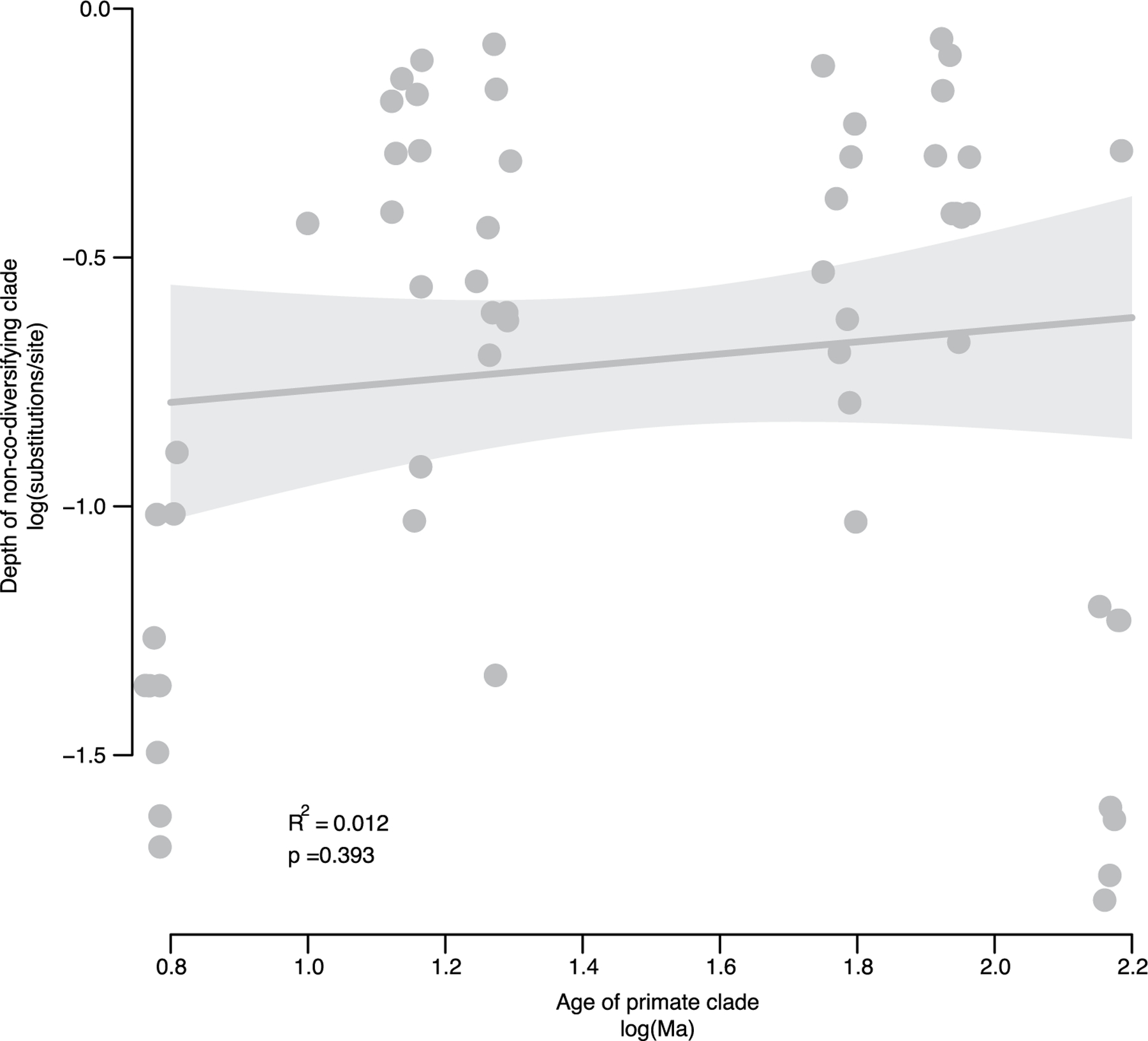
Scatter plot and regression line show the association between the depths of strongly non-co-diversifying bacterial clades (r < 0) based on protein divergence of bac120 single-copy core genes and the known ages of their corresponding host clades based on timetree.org (df = 53; t = 0.86; unadjusted p-value = 0.393). Each point corresponds to a bacterial clade. The non-codiversifying clades were derived from host species spanning the same epochs as in Extended Data Fig. 3. Bands represent 99% confidence intervals, centre line represents best-fit regression, and p-value represents result of two-sided Student’s t-tests. In contrast to results displayed in Extended Data Fig. 3 based on co-diversifying clade depths, non-co-diversifying clade depths were not significantly positively associated with known ages of the corresponding host clades..
Extended Data Fig. 6 |. Rates of genomic evolution vary among bacterial phyla.
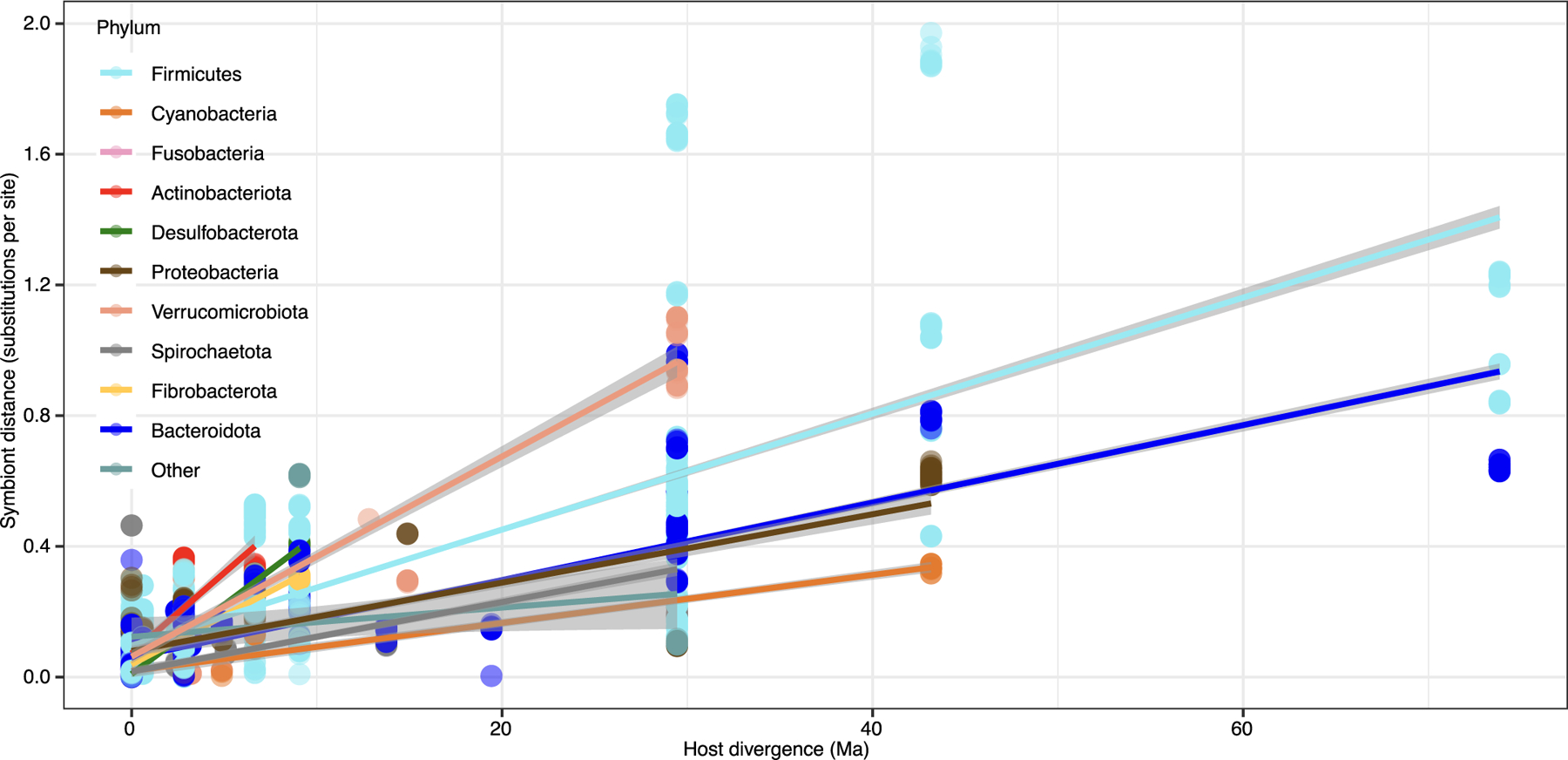
Scatter plot and regression lines show the positive relationships within co-diversifying bacterial clades between DNA substitutions per site of bacterial lineages and divergence time of host species from which the lineages were recovered. Each point represents a comparison between two co-diversifying bacterial lineages. Points and lines are coloured based on bacterial phyla as indicated by the key and corresponding to Fig. 1. Bands represent 95% confidence intervals and centre lines represent best-fit regression..
Extended Data Fig. 7 |. COG pathways enriched in Pan MAGs from co-diversifying clades missing from humans.
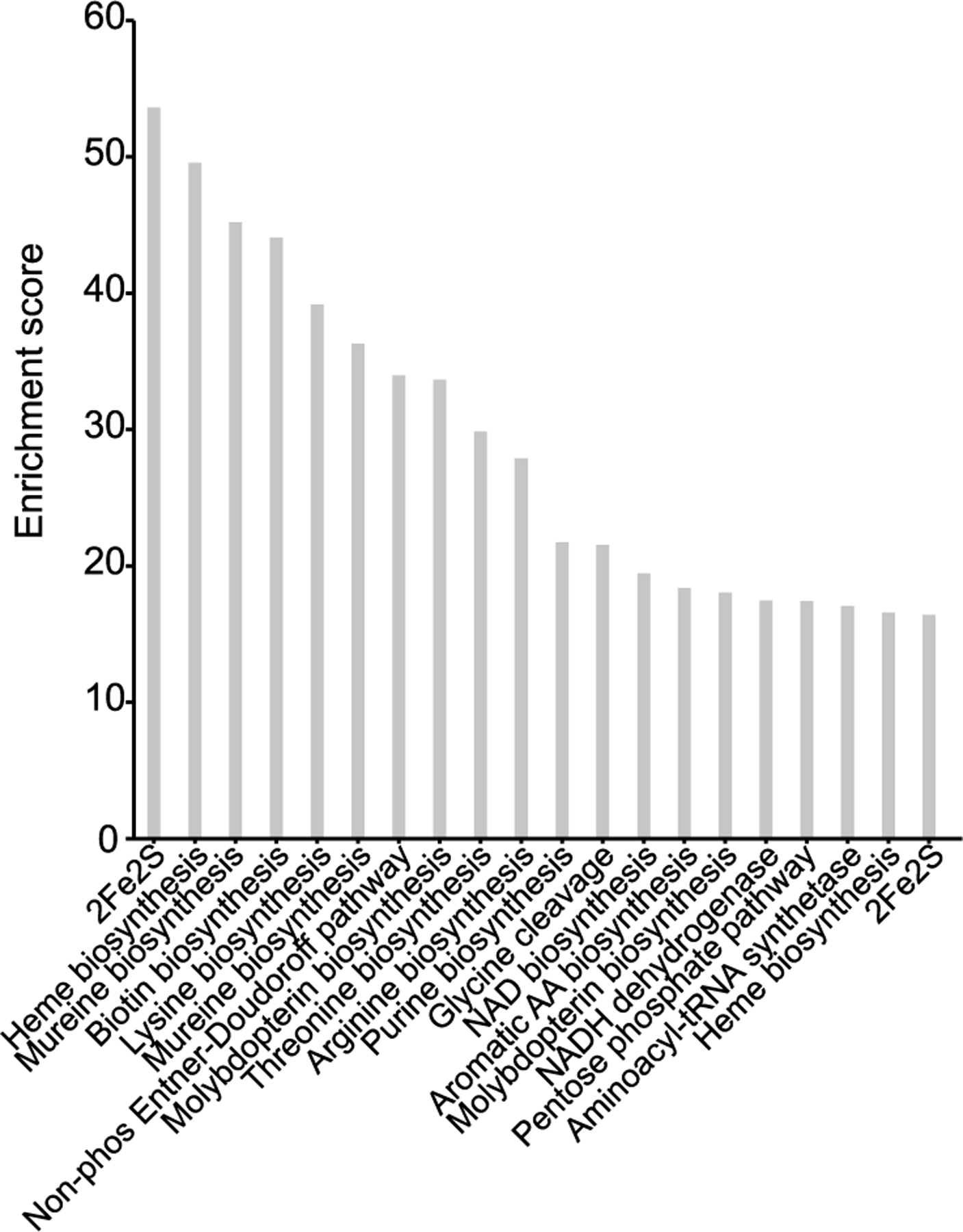
Bar plots show the enrichment scores of COG pathways identified as significantly overrepresented in Pan MAGs from co-diversifying clades missing from humans relative to Pan MAGs from co-diversifying clades present in humans. Enrichment scores were calculated as the Rao test statistic for equality of proportions as implemented in Anvi’o anvi-compute-functional-enrichment. Only the top 20 COG pathways are shown in the figure. For a full list see Supplementary Table 5..
Extended Data Fig. 8 |. Genomic signatures of selection in human and chimpanzee gut bacteria.
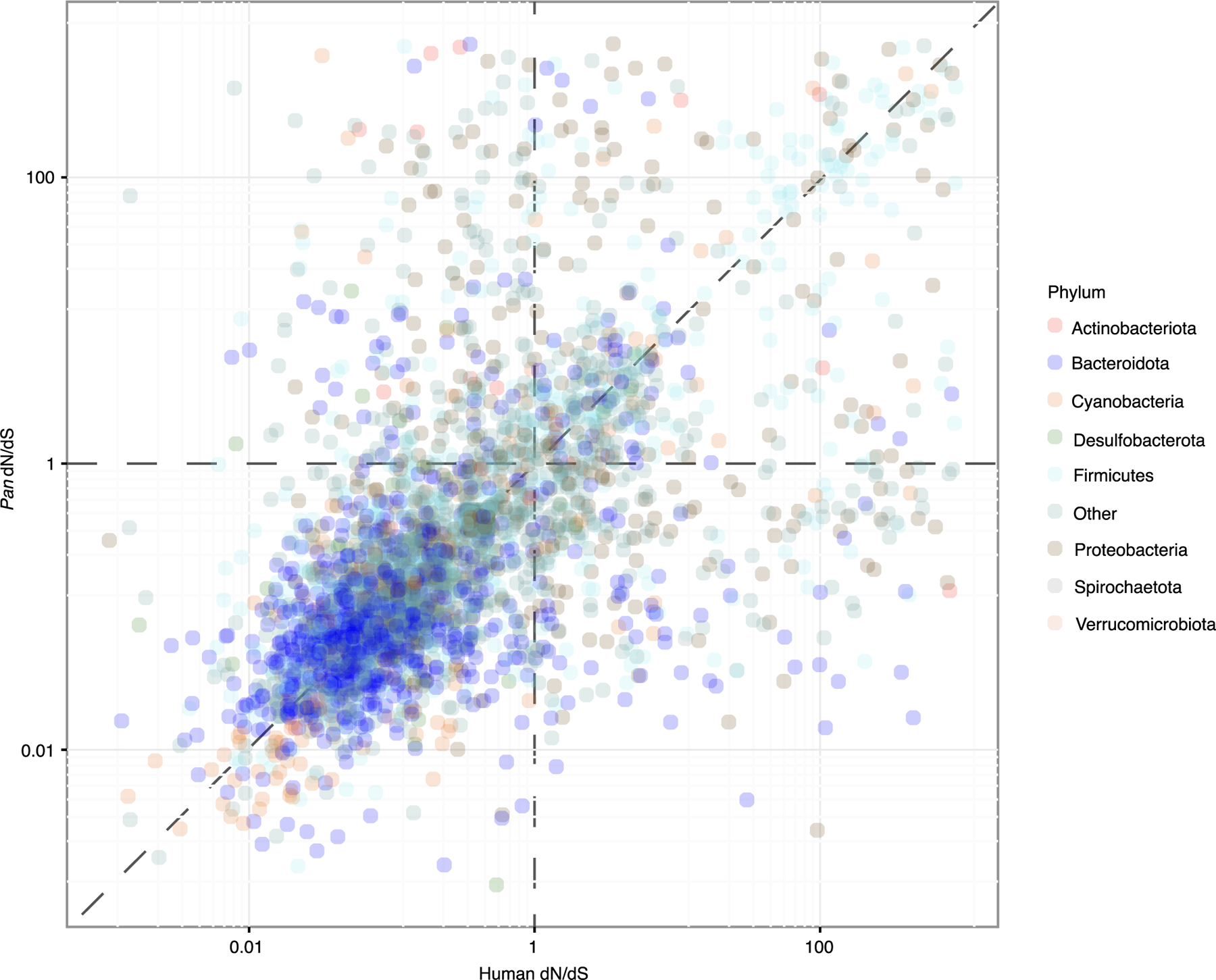
Scatter plot shows the relationship between per-CORF dN/dS values in humans and Pan. Points correspond to individual CORFs from co-diversifying bacterial lineages detected in human and Pan. Dashed vertical and horizontal lines correspond to the dN/dS expectation under neutral evolution, and dashed diagonal line corresponds to a 1-to-1 relationship between dN/dS values in humans and Pan. Points are coloured based on bacterial phyla as in Fig. 1 and as indicated in the key..
Supplementary Material
Acknowledgements
We thank W. Yan for assistance with DNA extractions from chimpanzee faecal samples and H. Ochman for comments on the manuscript. Primate cartoons were created with BioRender.com. Funding was provided by National Institutes of Health grant R35 GM138284 (A.H.M.) and grant R01 AI050529 (B.H.H.).
Footnotes
Code availability
Code used for co-diversification and selection analyses is available at https://github.com/CUMoellerLab.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41564-023-01388-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-023-01388-w.
Peer review information Nature Microbiology thanks Ruth Ley, Jonathan Clayton and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
All sequence data generated in this study have been deposited to the National Center for Biotechnology Information Sequence Read Archive under accessions PRJNA842693 (Nanopore data) and PRJNA842587 (Illumina data). All bacterial genome assemblies generated in this study are available at Dryad under accession at https://doi.org/10.5061/dryad.00000006x. Previously published data from humans and non-human primates analysed in this study are available from http://opendata.lifebit.ai/table/?project=SGB and the European Nucleotide Archive (accession PRJEB35610).
References
- 1.Bello MG, Knight R, Gilbert JA & Blaser MJ Preserving microbial diversity. Science 362, 33–34 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Sonnenburg ED & Sonnenburg JL The ancestral and industrialised gut microbiota and implications for human health. Nat. Rev. Microbiol 17, 383–390 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Groussin M, Mazel F & Alm EJ Co-evolution and co-speciation of host-gut bacteria systems. Cell Host Microbe 28, 12–22 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Davenport ER et al. The human microbiome in evolution. BMC Biol 15, 127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moeller AH et al. Cospeciation of gut microbiota with hominids. Science 353, 380–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishida AH & Ochman H Captivity and the co-diversification of great ape microbiomes. Nat. Commun 12, 5632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garud NR, Good BH, Hallatschek O & Pollard KS Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol 17, e3000102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grieneisen L et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science 373, 181–186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodrich JK et al. Human genetics shape the gut microbiome. Cell 4, 789–799 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooper LV, Littman DR & Macpherson AJ Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonnenburg JL & Bäckhed F Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz Heijtz R et al. Normal gut microbiota modulates brain development and behavior. Proc. Natl Acad. Sci. USA 108, 3047–3052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youngblut ND et al. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun 10, 2200 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang DD et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y-W, Simmons BA & Singer SW MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Alneberg J et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Sieber CMK et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol 3, 836–843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manara S et al. Microbial genomes from non-human primate gut metagenomes expand the primate-associated bacterial tree of life with over 1000 novel species. Genome Biol 20, 299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasolli E et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wibowo MC et al. Reconstruction of ancient microbial genomes from the human gut. Nature 594, 234–239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaumeil P-A, Mussig AJ, Hugenholtz P & Parks DH GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minh BQ et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hommola K, Smith JE, Qiu Y & Gilks WR A permutation test of host-parasite cospeciation. Mol. Biol. Evol 26, 1457–1468 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Stecher G, Suleski M & Hedges SB TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol 34, 1812–1819 (2017). [DOI] [PubMed] [Google Scholar]
- 25.de Vienne DM et al. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol 198, 347–385 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Duchêne S et al. Genome-scale rates of evolutionary change in bacteria. Microb. Genom 2, e000094 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menardo F, Duchêne S, Brites D & Gagneux S The molecular clock of Mycobacterium tuberculosis. PLoS Pathog 15, e1008067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rascovan N et al. Emergence and spread of basal lineages of Yersinia pestis during the neolithic decline. Cell 176, 295–305.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Ochman H, Elwyn S & Moran NA Calibrating bacterial evolution. Proc. Natl Acad. Sci. USA 96, 12638–12643 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki TA et al. Codiversification of gut microbiota with humans. Science 377, 1328–1332 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moeller AH et al. Rapid changes in the gut microbiome during human evolution. Proc. Natl Acad. Sci. USA 111, 16431–16435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moeller AH The shrinking human gut microbiome. Curr. Opin. Microbiol 38, 30–35 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Yatsunenko T et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blaser MJ The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol 17, 461–463 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Sonnenburg JL & Sonnenburg ED Vulnerability of the industrialised microbiota. Science 366, eaaw9255 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Pamer EG Resurrecting the intestinal microbiota to combat antibiotic-resistant pathogens. Science 352, 535–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Almeida A et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol 39, 105–114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson ND et al. Metagenomic assembly through the lens of validation: recent advances in assessing and improving the quality of genomes assembled from metagenomes. Brief. Bioinform 20, 1140–1150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Navarre WW et al. PoxA, yjeK, and elongation factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol. Cell 39, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wrangham RW et al. The raw and the stolen: cooking and the ecology of human origins. Curr. Anthropol 40, 567–594 (1999). [PubMed] [Google Scholar]
- 41.Keele BF et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313, 523–526 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keele BF et al. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460, 515–519 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rudicell RS et al. Impact of simian immunodeficiency virus infection on chimpanzee population dynamics. PLoS Pathog 6, e1001116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y et al. Eastern chimpanzees, but not bonobos, represent a simian immunodeficiency virus reservoir. J. Virol 86, 10776–10791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu W et al. Wild bonobos host geographically restricted malaria parasites including a putative new Laverania species. Nat. Commun 8, 1635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bibollet-Ruche F et al. CD4 receptor diversity in chimpanzees protects against SIV infection. Proc. Natl Acad. Sci. USA 116, 3229–3238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quick J The ‘Three Peaks’ faecal DNA extraction method for long-read sequencing v2 (protocols.io.7rshm6e). protocols.io, 10.17504/protocols.io.7rshm6e (2019). [DOI]
- 49.Mölder F et al. Sustainable data analysis with Snakemake. F1000Research 10, 33 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10 (2011). [Google Scholar]
- 51.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome Res 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gurevich A, Saveliev V, Vyahhi N & Tesler G QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown CT & Irber L sourmash: a library for MinHash sketching of DNA. J. Open Source Softw 1, 27 (2016). [Google Scholar]
- 55.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolmogorov M et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaser R, Sović I, Nagarajan N & Šikić M Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clayton JB et al. Captivity humanizes the primate microbiome. Proc. Natl Acad. Sci. USA 113, 10376–10381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Houtz JL, Sanders JG, Denice A & Moeller AH Predictable and host-species specific humanization of the gut microbiota in captive primates. Mol. Ecol 30, 3677–3687 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eren AM et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Revell LJ phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 3, 217–223 (2012). [Google Scholar]
- 62.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ranwez V, Douzery EJP, Cambon C, Chantret N & Delsuc F MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol 35, 2582–2584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rice P, Longden I & Bleasby A EMBOSS: the European molecular biology open software suite. Trends Genet 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Harris CD, Torrance EL, Raymann K & Bobay L-M CoreCruncher: fast and robust construction of core genomes in large prokaryotic data sets. Mol. Biol. Evol 38, 727–734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Z PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Macías LG, Barrio E & Toft C GWideCodeML: a Python package for testing evolutionary hypotheses at the genome-wide level. G3 10, 4369–4372 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data generated in this study have been deposited to the National Center for Biotechnology Information Sequence Read Archive under accessions PRJNA842693 (Nanopore data) and PRJNA842587 (Illumina data). All bacterial genome assemblies generated in this study are available at Dryad under accession at https://doi.org/10.5061/dryad.00000006x. Previously published data from humans and non-human primates analysed in this study are available from http://opendata.lifebit.ai/table/?project=SGB and the European Nucleotide Archive (accession PRJEB35610).