

Abstract

Evidence supports boosting nicotinamide adenine dinucleotide (NAD+) to counteract oxidative stress in aging and neurodegenerative disease. One approach is to enhance the activity of nicotinamide phosphoribosyltransferase (NAMPT). Novel NAMPT positive allosteric modulators (N-PAMs) were identified. A cocrystal structure confirmed N-PAM binding to the NAMPT rear channel. Early hit-to-lead efforts led to a 1.88-fold maximum increase in the level of NAD+ in human THP-1 cells. Select N-PAMs were assessed for mitigation of reactive oxygen species (ROS) in HT-22 neuronal cells subject to inflammatory stress using tumor necrosis factor alpha (TNFα). N-PAMs that increased NAD+ more effectively in THP-1 cells attenuated TNFα-induced ROS more effectively in HT-22 cells. The most efficacious N-PAM completely attenuated ROS elevation in glutamate-stressed HT-22 cells, a model of neuronal excitotoxicity. This work demonstrates for the first time that N-PAMs are capable of mitigating elevated ROS in neurons stressed with TNFα and glutamate and provides support for further N-PAM optimization for treatment of neurodegenerative diseases.
Keywords: NAMPT, NAD+, Oxidative stress, Neuroinflammation, Aging, Allosteric activation
Neurodegenerative disease (ND) and aging pose a massive burden on human health and lack effective treatments. One of the most prominent risk factors for developing ND is age.1 Brain aging affects mitochondrial function, oxidative stress response, neuroinflammation, DNA repair, autophagy, aberrant protein accumulation, Ca2+ homeostasis, neuroplasticity, neurogenesis, and energy metabolism.2
Reactive oxygen species (ROS) are produced as a normal part of human physiology, primarily in mitochondria, where they serve as signaling molecules.3 Unchecked, under oxidative stress (OS), ROS cause widespread damage.4 Most aspects of ND are aggravated by OS. For example, OS is known to be an early event in Alzheimer’s disease (AD) pathogenesis.5 There is an urgent need for therapies that address OS in the brain, and antioxidants have been studied in this context with mixed results.6 Boosting cellular nicotinamide adenine dinucleotide (NAD+) is an alternative approach.
NAD+ is present in every cell in the human body and fulfills numerous physiological roles in energy production as a cofactor for hundreds of enzymes and as a substrate for enzymes with NADase activity.7 In humans, NAD+ is biosynthesized via three distinct pathways (Figure 1): the Preiss Handler pathway from nicotinic acid (NA); the de novo pathway from tryptophan (Trp); and the salvage pathway from nicotinamide (NAM).8 NAM is produced by NAD+ catabolism as a result of the NADase activity. The NAM recycled by the salvage pathway is converted into nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), and NMN is then transformed into NAD+ by nicotinamide mononucleotide adenylyltransferase (NMNAT). The salvage pathway dominates cellular NAD+ synthesis in mammals, and NAMPT catalyzes the rate-limiting step, therefore controlling the cellular NAD+ supply.9
Figure 1.

NAD+ salvage pathway and associated systems.
With its vital place in cell physiology, it is no surprise that NAD+ modulation may be an appropriate therapeutic intervention. Fully depleting NAD+ by inhibiting its biosynthesis can be cytotoxic and thus is being studied for cancer treatment.10
Augmenting NAD+ improves cell function in multiple disease models and has, therefore, recently garnered interest for the treatment of ND, diabetes, cardiovascular disease, metabolic disease, and aging.7,11 Supporting evidence comes from in vitro and in vivo studies.12,13 NAD+ supplementation, usually via its biosynthetic precursors (NAM/NR/NMN), has been shown to improve mitochondrial function, Ca2+ homeostasis, and neuronal plasticity while attenuating oxidative stress, neuroinflammation, and aberrant protein accumulation.14−19 Evidence from dietary supplements in clinical trials is supportive.20
An alternative strategy for augmenting cellular NAD+ is to enhance its biosynthesis. Evidence indicates that NAMPT expression and function decline with age.21 This presents a potential limitation for the efficacy of supplementation with NAM. A solution to the limits imposed by rate-limiting NAMPT activity is to increase enzyme catalytic turnover of NAM. The use of a NAMPT activator can be envisaged, either alone or in combination with supplementation.
Small molecule NAMPT activation is a nascent field of research. The first reported NAMPT activators, the P7C3 series, showed neuroprotective and proneurogenic activity in animal models.22,23 However, their binding to NAMPT is problematic because no cocrystal structure has been published, and activation of recombinant NAMPT was not observed.24 Gardell et al. described SBI-797812, a “NAMPT booster”; although again, no structure with NAMPT has been published. This line of development led to structurally similar activators.24−27 The phenolic NAT compounds were reported to have neuroprotective activity and, like the NAMPT boosters, activate recombinant NAMPT.28,29 Most recently, our group reported a mechanism of allosteric modulation of NAMPT, along with the discovery of the NP-A1 series of NAMPT-positive allosteric modulators (N-PAMs).30,47
The research described herein sought to expand the chemical space of N-PAMs and to characterize their efficacy in enhancing cellular NAD+ levels with a particular emphasis on their potential as CNS therapeutics capable of addressing OS in the context of aging and ND.
We performed a high-throughput screen (HTS) of 22 000 compounds to identify those that augment NAMPT enzymatic activity.31 Screening used a coupled three-enzyme assay to detect conversion of NAM to NAD+, as previously described by our group.30 Our activators bind to the rear allosteric channel of NAMPT and are, therefore, termed N-PAMs. We envisioned the use of N-PAMs in ND and, therefore, chose an HTS hit series to pursue with desirable predicted brain penetration, as calculated by the central nervous system multiparameter optimization (CNS MPO) score. The hit NP-A3-B2 was selected, and initial hit-to-lead optimization was explored. We also established assays to assess the mitigation of neuronal OS.
A high-resolution (1.79 Å) cocrystal structure was obtained, which demonstrated allosteric binding of the hit to the NAMPT rear channel (Figure 2A,B). The cocrystal structure of NAMPT with NP-A3-B2 and NAM provided valuable information for our initial optimization efforts that sought to enhance protein–ligand interactions.
Figure 2.

NP-A3-B2:NAMPT cocrystal structure demonstrating NP-A3-B2 bound to the rear channel. Depicted in three views. (A) The entire structure showing NAMPT monomers in cyan and orange and NP-A3- B2 in purple, (B) the NAM-bound active site and NP-A3-B2-bound rear channel, and (C) NP-A3-B2 chemical structure and specific interactions with NAMPT amino acid residues: R1 methoxyphenyl hydrogen bonding network with waters and NAM and potential π–π interaction with His 191, R2 furfuryl interactions with a water and polar Arg 349 and Lys 189, and R3 tetrahydropyran in the solvent-exposed rear channel opening in both 3D and 2D representations, for clarity.
NP-A3-B2 was divided into three regions: R1, methoxyphenyl; R2, furfuryl; and R3, tetrahydropyran.
Region R1 can be subdivided into the methoxy moiety and phenyl moiety because of the unique environments immediately surrounding them (Figure 2C). The methoxy group sits adjacent to the active site and, on the basis of 1.8–1.9 Å distances, is participating in a hydrogen bonding network between two water molecules, valine-242 carbonyl, aspartate-219, serine-275, and NAM. The phenyl moiety is surrounded by the lipophilic side chains of valine-242, isoleucine-309, isoleucine-351, and alanine-370. The only polar side chain adjacent to the phenyl moiety is histidine-191 that sits 3.7 Å away and is orthogonal to the plane of the benzene ring in a possible edge-to-face π–π interaction.
Furfuryl R2 is oriented into a polar pocket made up of lysine-189, arginine-349, and the carbonyls of other residues. The furfuryl carbonyl hydrogen bonds to a water molecule that, in turn, hydrogen bonds to arginine-349. Distances from the furan ring to the lysine and arginine are 3.6 and 3.8 Å, respectively. Notably, there is no evidence for an interaction with lysine-189, although this has been reported to be a key interaction for NAMPT activation.29
Finally, tetrahydropyran R3 sits at the interface between the allosteric channel and the external solvent and is surrounded by proline-273, proline-307, valine-242, and tyrosine-188. The crystal structure indicates that NP-A3-B2 does not have any significant polar interactions with NAMPT in this region. Our SAR hypothesis is that the bulky tetrahydropyran acts as an anchor, which stabilizes the molecule at an effective depth in the channel.
Cocrystal structure analysis revealed that the methoxyphenyl and furfuryl groups take part in a number of interactions that could be optimized. We hypothesized that adding ligand–protein interactions in the R1 and R2 regions would increase binding affinity and result in increased potency and efficacy. Thus, we prioritized this for the initial SAR investigation and designed our synthetic methods with this in mind.
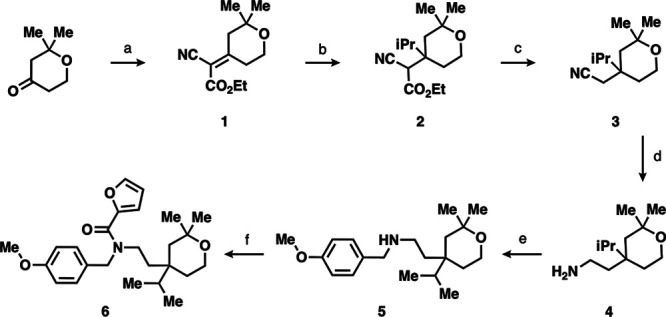
We designed a synthetic route to verify the hit by resynthesis and make changes in R1 and R2 near the end of the route to efficiently produce analogues. The six-step synthetic route began with the Knoevenagel condensation between ethyl cyanoacetate and 2,2-dimethyltetrahydropyran-4-one to produce the double α,β-unsaturated carbonyl and nitrile containing 1 (Scheme 1).32 Next, using Grignard, the isopropyl group was installed on 1 to yield 1,4-conjugate addition product 2. Decarboxylation gave the nitrile 3 that was reduced to the key primary amine intermediate 4 with cobalt chloride and sodium borohydride. Secondary amine 5 was achieved by reductive amination with 4-methoxybenzaldehyde. Finally, NP-A3-B2 synthesis was completed via coupling with HATU to afford amide 6. This route was successfully scaled up from milligram to decagram quantities.
Scheme 1. (a) EtOAcCN, NH4OAc, PhMe, Dean Stark Trap, 130 °C, 24 h, 80%, 1:1 E/Z Isomers; (b) iPrMgBr, THF, rt to 70 °C, 2 h, 61%; (c) KOH, DMF, 150 °C, 2 h, 78%; (d) CoCl2, NaBH4, MeOH, 0 °C to rt, 24 h, 67%; (e) (1) 1,4-Methoxybenzaldehyde, 10% AcOH, THF, rt, 24 h and (2) NaBH4, rt, 5 h, 30%; (f) 2-Furoic Acid, HATU, DIPEA, DMF, rt, 24 h, 45%.
Derivatization was initiated by using alternate benzaldehyde reagents with 1° amine 4 and carboxylic acid reagents with 2° amine 5. Analogues in the methoxyphenyl R1 region were designed to explore increased hydrophobic surface area, the addition of a hydrogen bond donor, or fluorination of potential metabolic oxidation sites (Table 2). Furthermore, the methoxy was replaced with a dimethylamino bioisostere. Analogues in the furfuryl R2 region were designed to assess varying heteroatoms and positions around the ring, addition of a halogen substituent, or addition of heteroatoms to the ring (Table 1). Addition of Br- and Cl- substituents in JGB-1-122 and JGB-1-135 were tolerated, although with loss of potency, and were compatible with docking studies (Figure S1). Expansion to six-membered heterocycles was explored, as well.
Table 2. Compound Structure with NAMPT Enzyme and THP-1 Cell Activity: R1 and R3 regions.

Table 1. Compound Structure with NAMPT Enzyme and THP-1 Cell Activity: R2 region.

Changes to the aliphatic bulk in region R3 were made to test the hypothesis that this solvent-exposed moiety acts as an anchor, which holds the rest of the molecule at a particular depth in the channel. The size was decreased by removing the isopropyl group, all substituents on the tetrahydropyran, and the entire region (Table 2).
Initial screening in THP-1 human monocytes (3 μM, 24 h) indicated that JGB-1-134, -127, -137, and -155 had significantly greater ability to raise NAD+ levels compared with the parent compound NP-A3-B2 (data not shown). Follow-up full concentration–response studies in this system highlighted JGB-1-155 as the top performer with an 88% increase in NAD+ (1.88-fold) over control. This was a significant improvement from NP-A3-B2 that induced an 18% NAD+ increase (1.18-fold) at 30 μM (Figure 3A). The data indicate that JGB-1-155, -137, and -127 have improved potency in THP-1 cell cultures.
Figure 3.

NAD+ measured in THP-1 cells (A) in response to compounds and (B) in response to NAMPT inhibitor FK866 in the presence of compounds. (C) NAMPT enzyme activity in response to compounds.
FK866 is a highly selective and potent NAMPT inhibitor. In the presence of JGB-1-137 and JGB-1-155, the concentration response for FK866 was right-shifted, and higher FK866 concentrations ablated the effects of activators (Figure 3B). These data are compatible with the cellular effects of our activators being mediated by direct binding and activation of NAMPT.
Analogues were also assessed in the coupled enzyme assay, used for HTS, to measure Amax and EC50 values (Figure 3C). JGB-1-147 exhibited inhibitory activity (Table 1), whereas only JGB-1-155 exhibited slight potency gains in the enzyme assay over the initial hit by having an EC50 of 3.29 μM relative to 4.06 μM for NP-A3-B2. The 1.85-fold Amax of the hit was surpassed only by JGB-1-137 with an Amax of 2.08-fold.
The results from the cell-based NAD+ and cell-free NAMPT enzyme assays led to the selection of compounds for study in the cell-based assays modeling aspects of cellular ND and aging: JGB-1-127, -134, -137, -155, and NP-A3-B2 were selected.
We set out to design assays representing the connection between OS and the pathophysiology of aging and ND. Assay development highlighted the neuronal HT-22 mouse hippocampal cell line as the most robust for these purposes and inflammatory cytokine tumor necrosis factor alpha (TNFα) as being a reliable and disease-relevant method for inducing ROS generation, which is relevant to neuroinflammation. The mitochondrion is the primary ROS-producing organelle and is especially ROS-sensitive, thereby reflecting cell-wide OS. For this reason, the fluorogenic MitoSOX Red Superoxide Indicator dye, which localizes to mitochondria, was used to quantify ROS levels.
TNFα is a cytokine released by immune cells to initiate inflammation in response to perceived threats. Microglia serve as the primary immune cells of the brain and carefully watch the CNS for specific patterns indicating danger.33 Chronic neuroinflammation is a prominent feature of aging and ND in which activated microglia assume an inflammatory (M1)-dominant phenotype in response to oxidative stress and other aging-associated stressors.34,35 M1 microglia release proinflammatory cytokines, like TNFα, that can be neurotoxic, exacerbate further microglial activation, and cause increased amyloid-β production in astrocytes.36 Specifically, TNFα can induce ROS production in neurons via TNF receptor 1 (TNFR1) and subsequent NFκB-promoted gene expression of ROS-generating enzymes, such as iNOS and NOX2.37 This has been demonstrated in HT-22 cell cultures, which further supports our use of the cell line to model the interaction between TNFα and OS.38
Nicotinamide riboside (NR) treatment in AD mouse models has been shown to prevent oxidative DNA damage, decrease measures of activated astrocytes and microglia, and inhibit the upregulation of proinflammatory pathways, including the release of TNFα.34,39 In a model of vascular dementia, 8 weeks of daily intraperitoneal NAD+ administration attenuated microglial activation and TNFα mRNA expression and blunted increased ROS in the hippocampus and cortex.40 The influence of NAD+ on these systems is established; however, this has not been shown for NAMPT activators.
The hit, selected analogues and previously reported NAMPT booster SBI-797812 were screened against 1 μg/mL of TNFα (Figure 4A). All NP-A3 series N-PAMs showed positive trends, and NP-A3-B2, JGB-1-137, and -155 significantly suppressed ROS at 40 μM. SBI-797812 had no significant effect on ROS at any concentration tested. Consistent with its relative NAD+ enhancement in THP-1 cells, JGB-1-155 outperformed all other analogues in ROS attenuation. Relative to TNFα-treated cells, this comprised a 0.63-fold ROS reduction (vs 0.82-fold for NP-A3-B2). Following the identification of JGB-1-155 as the most efficacious ROS inhibitor, we made more detailed measurements. This revealed the significant reduction of TNFα-induced ROS by 20 μM and 40 μM JGB-1-155 (Figure 4B). The MitoSOX signal induced by TNFα and its reduction by JGB-1-155 treatment are seen in cell imaging (Figure 4C).
Figure 4.
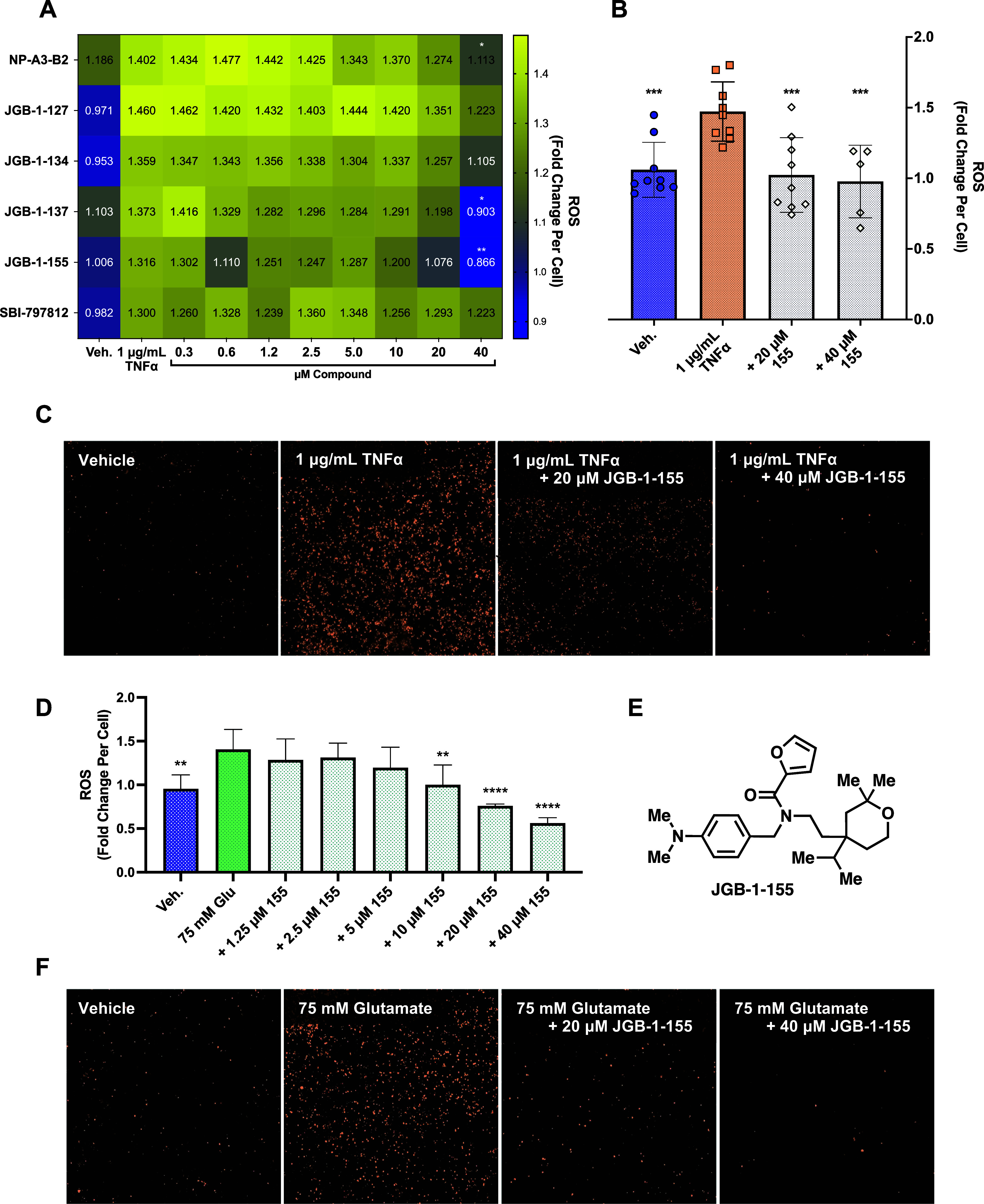
(A) Heatmap of dose–responses of select compounds screened against TNFα-induced ROS elevation in HT-22. (B) Isolated confirmation assay of JGB-1-155 under the same conditions as (A). (C) Images from wells in (B). (D) JGB-1-155 dose–response in HT-22 stressed with glutamate (E) structure of JGB-1-155. (F) Images taken from wells in (D). Statistical significance is relative to the insult (TNFα or glutamate) in (A), (B), and (D).
We became interested in assessing the ROS-mitigating effects of JGB-1-155 against additional insults in HT-22 cells. Glutamate-mediated excitotoxicity is a significant feature of the synaptic and neuronal dysfunction present in aging and many neuropathological conditions, including AD.41 Excitotoxicity involves the abnormal influx of Ca2+ into postsynaptic neurons and can contribute to OS by stimulating ROS-generating enzymes and suppressing antioxidant enzymes, like superoxide dismutase (SOD).42 NAM and NAD+ have provided neuroprotection in HT-22 cells and cortical neurons challenged with glutamate.18,43,44 However, the specific mitigation of ROS in cells stressed with glutamate via NAD+ enhancement has not been demonstrated, and furthermore, such neuroprotection has not been shown with N-PAMs and NAMPT activators. Evaluating activity against OS in neurons induced by two different stressor pathways, TNFα and glutamate, is a valuable measure of the generalizable ability of N-PAMs to protect against OS.
The dimethylamino analogue JGB-1-155 was tested against a 75 mM glutamate insult and exhibited the same dose-dependent ROS-reducing activity seen against TNFα. JGB-1-155 treatments of 20 μM and 40 μM decreased ROS levels below that of the vehicle-treated control, which can be explained by the background levels of oxidative stress in vehicle (DMSO)-treated neurons (Figure 4D). Images taken from the assay demonstrate this effect further (Figure 4F).
We posit that N-PAMs produce an antioxidant effect through the support of NAD+-dependent SIRTs. SIRT1 is known to mediate numerous survival and adaptive pathways in the brain, such as OS response, mitochondrial biogenesis, and anti-inflammatory effects.45 NAD+-induced ROS abrogation has been demonstrated to be dependent on SIRT1 in a model of chronic cerebral hypoperfusion.40 Similarly, NMN was shown to prevent ROS generation via SIRT1 in the hippocampi of septic mice.46 Direct chemical antioxidant activity is another possible mechanism for the observed effects of our N-PAMs on HT-22 cells. However, JGB-1-127 and JGB-1-134 contain the same potential antioxidant groups and did not inhibit ROS generation.
Insight into SAR was provided by NAMPT enzyme activation and activity in THP-1 and HT-22 cells, which will help guide future synthesis (Figure 5A). Compounds with a single 4-position substituent on the R1 benzene ring had superior activity in both NAMPT activation and THP-1 cells. Additionally, the 4-dimethylamino in JGB-1-155 improved activity in both THP-1 and HT-22 cell cultures over the 4-methoxy compounds. Thiophene replacement of the R2 furan moiety, such as in JGB-1-137, significantly improved NAD+ production in THP-1 and increased efficacy in NAMPT. All R2 changes adding atoms beyond an unsubstituted five-membered ring directly bound to the carbonyl were detrimental. Increasing size by extending the furan with homologation, expanding to a six-membered heterocycle, and adding a halogen substituent all undermined activity. The R2 ring is somewhat amenable to heteroatom position alteration or addition, as many of these analogues retained activity, but changing polarity in this way did not increase enzyme activation, and the imidazole in JGB-1-147 inhibits NAMPT. While the potency does not compare with the numerous nanomolar NAMPT inhibitors, such as the archetypal FK866, JGB-1-147 represents a novel chemotype and potentially a novel mechanism of action. The majority of NAMPT inhibitors contain N-heterocycles that occupy the NAM binding pocket (Figure 2B), which is an unlikely binding pose for JGB-1-147. This also provides an additional example of inhibitor–activator conversion, such as that seen in the development of SBI-797812.24 Notably, the largest gains in activity were not dependent on optimizing the interaction of the R2 region with lysine-189. Structural changes attempting to enhance the interaction between the ligand and lysine-189 only improved activity for JGB-1-127.
Figure 5.

(A) SAR summary for NP-A3-B2 optimization efforts. (B) Tetrahydropyran bulk-dependent activity.
We hypothesized that the bulky tetrahydropyran R3 group acted as an anchor to stabilize the compound in the NAMPT allosteric channel. This contribution to activity was confirmed by complete voiding of activity upon its removal. Furthermore, the importance of bulk in this region is well demonstrated by the size-dependent increase in activity from no R3 group at all to mono-, to tri-, to tetra-substituted tetrahydropyran (Figure 5B). Additional bulky tetrahydropyran derivatives can add further support to the observed trend, such as those with isobutyl, isopentyl, neopentyl, cyclopentyl, and adamantyl groups in place of the isopropyl moiety.
We set out to improve potency and efficacy of the HTS hit, NP-A3-B2, and to assess neuroprotective activity in aging- and ND-relevant assays. The early lead compound, JGB-1-155, achieved a promising improvement in increased cellular NAD+ levels and neuronal ROS mitigation, which supports further optimization of the NP-A3-B2 hit series.
Acknowledgments
Terry Moore is thanked for guidance and manuscript comments.
Glossary
Abbreviations
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NMN
nicotinamide mononucleotide
- NR
nicotinamide riboside
- NAMPT
nicotinamide phosphoribosyltransferase
- N-PAM
NAMPT-positive allosteric modulator
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor alpha
- ND
neurodegenerative disease
- OS
oxidative stress
- PARP
poly ADP ribose polymerase
- CD38
cluster of differentiation 38
- SARM1
sterile α and TIR motif containing 1
- SIRT
sirtuin
- NADase
NAD+ catabolic enzyme
- CNS
central nervous system
- HTS
high-throughput screen
- AD
Alzheimer’s disease
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00391.
Synthetic procedures, characterization, purity, experimental methods for crystallography and bioassay, and docking images (PDF)
Accession Codes
NAMPT:NP-A3-B2:NAM, PDB: 8TM7.
Author Contributions
The manuscript was written by J.G.-B. and edited by G.R.J.T. Crystallography was conducted by K.R. Enzyme assays and THP-1 assays were conducted by K.R., M.A.-B., S.R.M., and C.P. HT-22 assays were conducted by J.G.-B. and mentored by L.T. Synthesis was conducted by J.G.-B., mentored by T.D., and assisted by V.W. and G.R.V. E.T.M.A. conducted CAMD. The overall project was directed by G.R.J.T.
This study is supported by NIH grant RF1AG067771. J.G.-B. was supported, in part, by NIH T32AG57468. This research used resources of the Advanced Photon Source, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
The authors declare the following competing financial interest(s): G.R.J.T. is an inventor on patents owned by the University of Illinois.
Special Issue
Published as part of ACS Medicinal Chemistry Lettersvirtual special issue “Celebrating the 60th Anniversary of the MIKIW Meeting-in-Miniature”.
Supplementary Material
References
- Hou Y.; Dan X.; Babbar M.; Wei Y.; Hasselbalch S. G.; Croteau D. L.; Bohr V. A. Ageing as a risk factor for neurodegenerative disease. Nature Reviews Neurology. 2019, 15 (10), 565–581. 10.1038/s41582-019-0244-7. [DOI] [PubMed] [Google Scholar]
- Mattson M. P.; Arumugam T. V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metabolism. 2018, 27 (6), 1176–1199. 10.1016/j.cmet.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H.; Jones D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nature Reviews Molecular Cell Biology. 2020, 21 (7), 363–383. 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- Rizzo A. M.; Berselli P.; Zava S.; Montorfano G.; Negroni M.; Corsetto P.; Berra B.. Endogenous Antioxidants and Radical Scavengers. In Advances in Experimental Medicine and Biology; Springer US, 2010; pp 52–67. [DOI] [PubMed] [Google Scholar]
- Uddin M. S.; Kabir M. T.. Oxidative Stress in Alzheimer’s Disease: Molecular Hallmarks of Underlying Vulnerability. In Biological, Diagnostic and Therapeutic Advances in Alzheimer’s Disease: Non-Pharmacological Therapies for Alzheimer’s Disease; Ashraf G. M., Alexiou A., Eds.; Springer Singapore, 2019; pp 91–115. [Google Scholar]
- Walia V.; Kaushik D.; Mittal V.; Kumar K.; Verma R.; Parashar J.; Akter R.; Rahman M. H.; Bhatia S.; Al-Harrasi A.; Karthika C.; Bhattacharya T.; Chopra H.; Ashraf G. M. Delineation of Neuroprotective Effects and Possible Benefits of AntioxidantsTherapy for the Treatment of Alzheimer’s Diseases by Targeting Mitochondrial-Derived Reactive Oxygen Species: Bench to Bedside. Molecular Neurobiology. 2022, 59 (1), 657–680. 10.1007/s12035-021-02617-1. [DOI] [PubMed] [Google Scholar]
- Rajman L.; Chwalek K.; Sinclair D. A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metabolism. 2018, 27 (3), 529–547. 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E. S.; Schramm V. L. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry. 2008, 47 (42), 11086–11096. 10.1021/bi801198m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo J. R.; Grimm A. A.; Imai S.-i. The NAD Biosynthesis Pathway Mediated by Nicotinamide Phosphoribosyltransferase Regulates Sir2 Activity in Mammalian Cells. J. Biol. Chem. 2004, 279 (49), 50754–50763. 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Sampath D.; Zabka T. S.; Misner D. L.; O’Brien T.; Dragovich P. S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacology & Therapeutics. 2015, 151, 16–31. 10.1016/j.pharmthera.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Kang B. E.; Choi J.-Y.; Stein S.; Ryu D. Implications of NAD+ boosters in translational medicine. European Journal of Clinical Investigation. 2020, 50 (10), e13334. 10.1111/eci.13334. [DOI] [PubMed] [Google Scholar]
- Wang X.; He H.-J.; Xiong X.; Zhou S.; Wang W.-W.; Feng L.; Han R.; Xie C.-L. NAD+ in Alzheimer’s Disease: Molecular Mechanisms and Systematic Therapeutic Evidence Obtained in vivo. Frontiers in Cell and Developmental Biology. 2021, 9, 668491. 10.3389/fcell.2021.668491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J.; Baur J. A.; Imai S. I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metabolism. 2018, 27 (3), 513–528. 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Pitta M.; Jiang H.; Lee J.-H.; Zhang G.; Chen X.; Kawamoto E. M.; Mattson M. P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiology of Aging. 2013, 34 (6), 1564–1580. 10.1016/j.neurobiolaging.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu A.; Diwan V.; Kaur H.; Bhateja D.; Singh C. K.; Sharma S.; Padi S. S. V. Nicotinamide reverses behavioral impairments and provides neuroprotection in 3-nitropropionic acid induced animal model of Huntington’s disease: implication of oxidative stress-poly(ADP-ribose) polymerase pathway. Metabolic Brain Disease. 2018, 33 (6), 1911–1921. 10.1007/s11011-018-0297-0. [DOI] [PubMed] [Google Scholar]
- Harlan B. A.; Pehar M.; Sharma D. R.; Beeson G.; Beeson C. C.; Vargas M. R. Enhancing NAD+ Salvage Pathway Reverts the Toxicity of Primary Astrocytes Expressing Amyotrophic Lateral Sclerosis-linked Mutant Superoxide Dismutase 1 (SOD1). J. Biol. Chem. 2016, 291 (20), 10836–10846. 10.1074/jbc.M115.698779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M.; Mitchell; Sarah J.; Fang; Evandro F.; Iyama T.; Ward T.; Wang J.; Dunn; Christopher A.; Singh N.; Veith S.; Hasan-Olive Md M.; Mangerich A.; Wilson Mark A.; Mattson Mark P.; Bergersen Linda H.; Cogger Victoria C.; Warren A.; Le Couteur David G.; Moaddel R.; Wilson David M.; Croteau Deborah L.; de Cabo R.; Bohr Vilhelm A. A High-Fat Diet and NAD+ Activate Sirt1 to Rescue Premature Aging in Cockayne Syndrome. Cell Metabolism. 2014, 20 (5), 840–855. 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh P.-O. Nicotinamide attenuates the injury-induced decrease of hippocalcin in ischemic brain injury. Neurosci. Lett. 2013, 545, 6–10. 10.1016/j.neulet.2013.04.010. [DOI] [PubMed] [Google Scholar]
- Hosseini L.; Farokhi-Sisakht F.; Badalzadeh R.; Khabbaz A.; Mahmoudi J.; Sadigh-Eteghad S. Nicotinamide Mononucleotide and Melatonin Alleviate Aging-induced Cognitive Impairment via Modulation of Mitochondrial Function and Apoptosis in the Prefrontal Cortex and Hippocampus. Neuroscience. 2019, 423, 29–37. 10.1016/j.neuroscience.2019.09.037. [DOI] [PubMed] [Google Scholar]
- Brakedal B.; Dölle C.; Riemer F.; Ma Y.; Nido G. S.; Skeie G. O.; Craven A. R.; Schwarzlmüller T.; Brekke N.; Diab J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metabolism. 2022, 34 (3), 396–407. 10.1016/j.cmet.2022.02.001. [DOI] [PubMed] [Google Scholar]
- Imai S.-i. The NAD World 2.0: the importance of the inter-tissue communication mediated by NAMPT/NAD+/SIRT1 in mammalian aging and longevity control. npj Systems Biology and Applications. 2016, 2 (1), 1–9. 10.1038/npjsba.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper A. A.; Xie S.; Capota E.; Estill S. J.; Zhong J.; Long J. M.; Becker G. L.; Huntington P.; Goldman S. E.; Shen C.-H.; Capota M.; Britt J. K.; Kotti T.; Ure K.; Brat D. J.; Williams N. S.; MacMillan K. S.; Naidoo J.; Melito L.; Hsieh J.; De Brabander J.; Ready J. M.; McKnight S. L. Discovery of a Proneurogenic, Neuroprotective Chemical. Cell. 2010, 142 (1), 39–51. 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Han T.; Nijhawan D.; Theodoropoulos P.; Naidoo J.; Yadavalli S.; Mirzaei H.; Pieper A. A.; Ready J. M.; McKnight S. L. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014, 158 (6), 1324–1334. 10.1016/j.cell.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell S. J.; Hopf M.; Khan A.; Dispagna M.; Hampton Sessions E.; Falter R.; Kapoor N.; Brooks J.; Culver J.; Petucci C.; Ma C. T.; Cohen S. E.; Tanaka J.; Burgos E. S.; Hirschi J. S.; Smith S. R.; Sergienko E.; Pinkerton A. B. Boosting NAD(+) with a small molecule that activates NAMPT. Nature Communications. 2019, 10 (1), 3241. 10.1038/s41467-019-11078-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkerton A. B.; Sessions E. H.; Hershberger P.; Maloney P. R.; Peddibhotla S.; Hopf M.; Sergienko E.; Ma C.-T.; Smith L. H.; Jackson M. R.; Tanaka J.; Tsuji T.; Akiu M.; Cohen S. E.; Nakamura T.; Gardell S. J. Optimization of a urea-containing series of nicotinamide phosphoribosyltransferase (NAMPT) activators. Bioorg. Med. Chem. Lett. 2021, 41, 128007. 10.1016/j.bmcl.2021.128007. [DOI] [PubMed] [Google Scholar]
- Akiu M.; Tsuji T.; Iida K.; Sogawa Y.; Terayama K.; Yokoyama M.; Tanaka J.; Asano D.; Honda T.; Sakurai K.; et al. Discovery of DS68702229 as a Potent, Orally Available NAMPT (Nicotinamide Phosphoribosyltransferase) Activator. Chem. Pharm. Bull. 2021, 69 (11), 1110–1122. 10.1248/cpb.c21-00700. [DOI] [PubMed] [Google Scholar]
- Tang S.; Garzon Sanz M.; Smith O.; Krämer A.; Egbase D.; Caton P. W.; Knapp S.; Butterworth S. Chemistry-led investigations into the mode of action of NAMPT activators, resulting in the discovery of non-pyridyl class NAMPT activators. Acta Pharmaceutica Sinica B 2023, 13 (2), 709–721. 10.1016/j.apsb.2022.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Liu M.; Zu Y.; Yao H.; Wu C.; Zhang R.; Ma W.; Lu H.; Xi S.; Liu Y.; Hua L.; Wang G.; Tang Y. Optimization of NAMPT activators to achieve in vivo neuroprotective efficacy. Eur. J. Med. Chem. 2022, 236, 114260. 10.1016/j.ejmech.2022.114260. [DOI] [PubMed] [Google Scholar]
- Yao H.; Liu M.; Wang L.; Zu Y.; Wu C.; Li C.; Zhang R.; Lu H.; Li F.; Xi S.; Chen S.; Gu X.; Liu T.; Cai J.; Wang S.; Yang M.; Xing G.-G.; Xiong W.; Hua L.; Tang Y.; Wang G. Discovery of small-molecule activators of nicotinamide phosphoribosyltransferase (NAMPT) and their preclinical neuroprotective activity. Cell Research. 2022, 32 (6), 570–584. 10.1038/s41422-022-00651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K. M.; Shen Z.; Gordon-Blake J.; Lee H.; Laham M. S.; Krider I. S.; Christie N.; Ackerman-Berrier M.; Penton C.; Knowles N. G.; Musku S. R.; Fu J.; Velma G. R.; Xiong R.; Thatcher G. R. J. Mechanism of Allosteric Modulation of Nicotinamide Phosphoribosyltransferase to Elevate Cellular NAD+. Biochemistry. 2023, 62 (4), 923–933. 10.1021/acs.biochem.2c00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z.; Ratia K.; Krider I.; Ackerman-Berrier M.; Penton C.; Musku S. R.; Gordon-Blake J. M.; Laham M. S.; Christie N.; Ma N.; Fu J.; Xiong R.; Courey J. M.; Velma G. R.; Thatcher G. R. J. Synthesis, Optimization, and Structure−Activity Relationships of Nicotinamide Phosphoribosyltransferase (NAMPT) Positive Allosteric Modulators (N-PAMs). J. Med. Chem. 2023, 66 (24), 16704–16727. 10.1021/acs.jmedchem.3c01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Blake J.; Karumudi B.; Ratia K.; Knopp R. C.; Dye K.; Ben Aissa M.; Thatcher G. R. P1-083: Novel NAMPT activators attenuate neurotoxicity and neuroinflammation associated with neurodegeneration. Alzheimer’s & Dementia. 2019, 15, P266–P267. 10.1016/j.jalz.2019.06.108. [DOI] [Google Scholar]
- Stevenson G. I.; Garavelas A.; Cosgrove K. L.; Reynolds K. A.; Franken N. C.; Whittell L. R.; Wijesekera H. P.. Tetrahydropyran-4-ylethylamino- or tetrahydropyranyl-4-ethyloxy-pyrimidines or -pyridazines as isoprenylcysteincarboxymethyl transferase inhibitors. US WO2014041349 A1, 2014.
- Uddin M. S.; Kabir M. T.; Mamun A. A.; Barreto G. E.; Rashid M.; Perveen A.; Ashraf G. M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. International Immunopharmacology. 2020, 84, 106479. 10.1016/j.intimp.2020.106479. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Lautrup S.; Cordonnier S.; Wang Y.; Croteau D. L.; Zavala E.; Zhang Y.; Moritoh K.; O’Connell J. F.; Baptiste B. A.; Stevnsner T. V.; Mattson M. P.; Bohr V. A. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E1876–E1885. 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Wang H.; Yin Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Frontiers in Aging Neuroscience. 2022, 14, 815347. 10.3389/fnagi.2022.815347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Reyes R. E.; Nava-Mesa M. O.; Vargas-Sánchez K.; Ariza-Salamanca D.; Mora-Muñoz L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Frontiers in Molecular Neuroscience. 2017, 10, 00427. 10.3389/fnmol.2017.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R.; Maier O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxidative Medicine and Cellular Longevity. 2015, 2015, 610813. 10.1155/2015/610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Lu Y.; Wang R.; Xu T.; Lei X.; Jin H.; Gao X.; Xie Y.; Liu X.; Zeng J. MiR-29c Inhibits TNF-α-Induced ROS Production and Apoptosis in Mouse Hippocampal HT22 Cell Line. Neurochem. Res. 2023, 48 (2), 519–536. 10.1007/s11064-022-03776-w. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Wei Y.; Lautrup S.; Yang B.; Wang Y.; Cordonnier S.; Mattson M. P.; Croteau D. L.; Bohr V. A. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proceedings of the National Academy of Sciences. 2021, 118 (37), e2011226118 10.1073/pnas.2011226118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Zhang J.; Zheng Y.; Zhang Y.; Zhang X. J.; Wang H.; Du Y.; Guan J.; Wang X.; Fu J. NAD+ improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. Journal of Neuroinflammation. 2021, 18 (1), 207. 10.1186/s12974-021-02250-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira R. G.; Cerpa W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cellular and Molecular Neurobiology. 2021, 41 (7), 1413–1430. 10.1007/s10571-020-00924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armada-Moreira A.; Gomes J. I.; Pina C. C.; Savchak O. K.; Gonçalves-Ribeiro J.; Rei N.; Pinto S.; Morais T. P.; Martins R. S.; Ribeiro F. F.; Sebastião A. M.; Crunelli V.; Vaz S. H. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Frontiers in Cellular Neuroscience. 2020, 14, 00090. 10.3389/fncel.2020.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Gharavi R.; Pitta M.; Gleichmann M.; Mattson M. P. Nicotinamide Prevents NAD+ Depletion and Protects Neurons Against Excitotoxicity and Cerebral Ischemia: NAD+ Consumption by SIRT1 may Endanger Energetically Compromised Neurons. NeuroMolecular Medicine. 2009, 11 (1), 28–42. 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Li H.; Ding S. The Effects of NAD+ on Apoptotic Neuronal Death and Mitochondrial Biogenesis and Function after Glutamate Excitotoxicity. International Journal of Molecular Sciences. 2014, 15 (11), 20449–20468. 10.3390/ijms151120449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F.; Wijaya L.; Tang B. L. SIRT1 in the brain-connections with aging-associated disorders and lifespan. Frontiers in Cellular Neuroscience. 2015, 9, 64. 10.3389/fncel.2015.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.-r.; Liu Q.; Zhu C.-l.; Sun X.-y.; Sun C.-y.; Yu C.-m.; Li P.; Deng X.-m.; Wang J.-f. β-Nicotinamide mononucleotide activates NAD+/SIRT1 pathway and attenuates inflammatory and oxidative responses in the hippocampus regions of septic mice. Redox Biology. 2023, 63, 102745. 10.1016/j.redox.2023.102745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.