

Abstract
PURPOSE
Non–small-cell lung cancer (NSCLC) with STK11mut has inferior outcomes to immune checkpoint inhibitors (ICIs). Using multiomics, we evaluated whether a subtype of STK11mut NSCLC with a uniquely inflamed tumor immune microenvironment (TIME) harboring TP53 comutations could have favorable outcomes to ICIs.
PATIENTS AND METHODS
NSCLC tumors (N = 16,896) were analyzed by next-generation sequencing (DNA-Seq/592 genes). A subset (n = 5,034) underwent gene expression profiling (RNA-Seq/whole transcriptome). Exome-level neoantigen load for STK11mut NSCLC was obtained from published pan-immune analysis. Tumor immune cell content was obtained from transcriptome profiles using the microenvironment cell population (MCP) counter. ICI data from POPLAR/OAK (n = 34) and the study by Rizvi et al (n = 49) were used to model progression-free survival (PFS), and a separate ICI-treated cohort (n = 53) from Dana-Farber Cancer Institute (DFCI) was used to assess time to treatment failure (TTF) and tumor RECIST response for STK11mutTP53mut versus STK11mutTP53wt NSCLC.
RESULTS
Overall, 12.6% of NSCLC tumors had a STK11mut with the proportions of tumor mutational burden (TMB)-high (≥10 mut/Mb), PD-L1 ≥50%, and microsatellite instability-high being 38.3%, 11.8%, and 0.72%, respectively. Unsupervised hierarchical clustering of STK11mut (n = 463) for stimulator of interferon-gamma (STING) pathway genes identified a STING-high cluster, which was significantly enriched in TP53mut NSCLC (P < .01). Compared with STK11mutTP53wt, tumors with STK11mutTP53mut had higher CD8+T cells and natural killer cells (P < .01), higher TMB (P < .001) and neoantigen load (P < .001), and increased expression of MYC and HIF-1A (P < .01), along with higher expression (P < .01) of glycolysis/glutamine metabolism genes. Meta-analysis of data from OAK/POPLAR and the study by Rizvi et al showed a trend toward improved PFS in patients with STK11mutTP53mut. In the DFCI cohort, compared with the STK11mut TP53wt cohort, the STK11mutTP53mut tumors had higher objective response rates (42.9% v 16.7%; P = .04) and also had longer TTF (14.5 v 4.5 months, P adj = .054) with ICI.
CONCLUSION
STK11mut NSCLC with TP53 comutation is a distinct subgroup with an immunologically active TIME and metabolic reprogramming. These properties should be exploited to guide patient selection for novel ICI-based combination approaches.
BACKGROUND
Lung cancer is the leading cause of cancer death in the United States, accounting for a projected 127,070 deaths in 2023.1 The development of immune checkpoint inhibitors (ICIs) has transformed the treatment landscape for many tumor types, including lung cancer. ICIs alone or combined with chemotherapy have significantly improved survival in patients with advanced and early-stage non–small-cell lung cancer (NSCLC).2-4 Understanding which factors within the tumor microenvironment contribute to the unique aspect of some individuals with NSCLC deriving longer-term durable benefit with ICI is currently the focus of extensive research.
CONTEXT
Key Objective
STK11mut is believed to negatively influence response to immune checkpoint inhibitors (ICIs) in non–small-cell lung cancer (NSCLC). Whether certain comutations such as TP53mut that can lead to genomic instability can have a more activated stimulator of interferon-gamma (STING) pathway signature in STK11mut NSCLC and enhance outcomes to ICIs in NSCLC is an area of active interest.
Knowledge Generated
Using a multiomics approach, we identified a differential impact of TP53 comutation on the tumor immune microenvironment (TIME) in STK11mut NSCLC. Compared with STK11mutTP53wt, STK11mutTP53mut tumors had an immunologically active TIME with a pronounced STING signature and metabolic reprogramming with higher expression of glycolysis/glutamine metabolism genes. Using clinical data sets, we observed improved clinical outcomes with respect to survival and response rates in NSCLC tumors with STK11mutTP53mut compared with STK11mutTP53wt.
Relevance
Implementing a multiomics approach can help in identifying and exploiting therapeutic vulnerabilities associated with distinct comutation subsets within STK11mut NSCLC.
Mutations of the serine-threonine kinase 11 (STK11) gene are believed to influence response to ICIs in NSCLC negatively. STK11, also known as liver kinase B1 (LKB1), is a tumor suppressor gene that encodes a serine-threonine kinase located on chromosome 19p13.3.5 LKB1 activates the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway and, in turn, regulates cellular metabolism, growth, and polarity.6 Under stress conditions, LKB1 promotes survival by facilitating metabolic reprogramming from an anabolic to a catabolic state.7 Loss of STK11 results in reduced phosphorylation of AMPK and TSC1, activating mTORC1, which drives growth and survival pathways. STK11 point mutations have been observed in 10%-15% of lung adenocarcinomas,8 and several studies have indicated they confer poorer outcomes to ICI therapy in NSCLC.9-12 Previous immune gene expression analysis of STK11mut NSCLC from The Cancer Genome Atlas (TCGA) has demonstrated enrichment in wound healing pathways, with the upregulation of angiogenic gene signatures that mediate resistance to immunotherapy.13
TP53 is a commonly mutated tumor suppressor gene that arrests cells in the G1 phase in response to double-strand DNA breaks.14 Mutations in TP53 lead to genomic instability and are likely to increase tumor mutational burden (TMB) and neoantigen load,15 both of which have been associated with improved response to ICIs. Several studies have also shown that TP53-mutated NSCLC tumors express higher levels of PD-L1 protein than wild-type tumors.16,17 In addition, TP53 may have a differential impact and potential interactions with STK11, given that TP53 is a known transcriptional regulator of STK11.18 Some recent studies have demonstrated a better prognosis of STK11mut NSCLC with TP53 comutations.19 Conversely, in KRASmut lung adenocarcinomas, concurrent TP53mut is associated with an inferior prognosis. How TP53 mutations transcriptionally modulate the immune microenvironment in STK11mut NSCLC has not been well understood.
Additionally, the cyclic GMP-AMP synthase (cGAS) stimulator of interferon gamma (STING) pathway genes has emerged as a critical pathway in the induction of robust immune defense programs.20 By regulating macrophage polarization and T-cell differentiation, the STING pathway acts as a bridge between innate and adaptive immunity and augments antitumor immunity. Thus, the cGAS-STING pathway represents a promising biomarker and target for immunotherapeutic approaches by converting tumor immune microenvironment (TIME) from cold to hot.21,22 Interestingly, STK11mut NSCLC has increased the availability of S-adenosylmethinione, which leads to epigenetic inactivation of the STING pathway.23 Whether certain comutations can have a more activated STING pathway signature in STK11mut NSCLC and enhance outcomes to ICIs in NSCLC is an area of active interest.
We used real-world data from Caris Life Sciences and TCGA and transcriptomic analysis to identify a distinct immune inflamed (STING-high) phenotype of STK11mut NSCLC enriched in TP53mut. We subsequently evaluated the impact of these TP53 co-occurring mutations on the TIME, metabolic phenotype, and therapeutic vulnerability of STK11mut NSCLC to ICI. We then used publicly available data from clinical trials and real-world data sets to explore the clinical significance of our findings.
PATIENTS AND METHODS
A total of 16,896 NSCLC tumors were molecularly profiled at Caris Life Sciences (Phoenix, AZ) between February 2015 and January 2020. Next-generation sequencing was performed on genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples using the NextSeq platform (Illumina, Inc, San Diego, CA). For detailed methodology on next generation sequencing (NGS), refer to Appendix 1. For TMB calculations, we excluded all variants previously described as germline alterations according to dbSNP and 1KG databases, and TMB was then measured (592 genes and 1.4 megabases [MB] sequenced per tumor) by counting all nonsynonymous exonic missense and truncating mutations found per tumor. TMB was adjusted by dividing by a factor of 1.2 to ensure that the fraction of TMB-H matched the observed published clinical data.24 A cutoff point of ≥10 mutations/MB was used on the basis of the KEYNOTE-158 trial.25 Microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) status reporting is described in Appendix 1.
For RNA-sequencing, FFPE specimens (n = 5,034) with a minimum of 10% of tumor content in the area for micro-dissection was required to enable enrichment and extraction of tumor-specific RNA. For detailed methodology on RNA extraction, see Appendix 1. Raw data were demultiplexed by Illumina Dragen BioIT accelerator, trimmed, counted, polymerase chain reaction duplicates removed, and aligned to human reference genome hg19 by STAR aligner.26 Transcripts per million molecules (TPMs) were generated using Salmon.27 The microenvironment cell population-counter (MCP-counter) was used to quantify the abundance of immune and stromal cell populations as previously described.28 Hierarchical clustering (ward D) for lung adenocarcinomas was performed on the basis of a previously used method of using expression of genes primarily associated with the STING pathway (CCL5, CXCL10, and MB21D1 otherwise known as cGAS).29
Immunohistochemistry (IHC) was performed on whole FFPE sections. The primary PD-L1 antibody clone was 22c3 (Dako). For detailed methodology on IHC, see Appendix 1.
The prevalence of molecular alterations in different groups was compared using chi-square or Fisher exact tests. In addition, TMB and gene expression (TPM) were compared among different cohorts using the Kruskal-Wallis nonparametric test. An unadjusted P value of <.05 was considered a trending difference; P values were further corrected for multiple comparisons using the Benjamini-Hochberg method to avoid type I error. An adjusted P value (ie, q value) of <.05 was considered a significant difference.
For TCGA data, mutational profiles generated by the MuTect2 somatic variation calling workflow were obtained for 1,059 NSCLC samples (n = 567, adenocarcinoma; n = 492, squamous carcinoma) from UCSC Xena, along with transcriptomic profiles (gene-level transcription estimates, normalized counts) data.30 In addition, data on TMB predicted neoantigen burden, tumor immune infiltrate, and interferon-gamma (IFN-γ) response signature for TCGA data were retrieved from a previous publication31 and compared between STK11mut versus STK11wt NSCLC and between STK11mut tumors stratified by TP53 mutation status (STK11mut TP53 mut v STK11mut TP53wt) NSCLC using the Wilcoxon rank sum test.
The immune gene panel profile included CXCL10, CXCL7, CCL5, CCL3, IL10, IL6, CGAS, CD8, CD3, CD274, LAG3, TIM3, CTLA4, IDO1, ICOS, GZMB, human leukocyte antigen genes, beta-2 microglobulin CD40, LAG3, IFN-γ, TNF receptor superfamily member four gene (TNFRSF4 [also known by the aliases OX40 and CD134]), TNF receptor superfamily member nine gene (4-1BB and CD137), HAVCR2, MB21D1, PPBP, and BRD4.
Because subsequent treatments can often confound overall survival,32 we used progression-free survival (PFS) and time to treatment failure (TTF) as relevant end points for outcomes associated with ICI therapy. PFS data from the OAK and POPLAR trials were obtained from previously published data sets.33 The POPLAR and OAK trials evaluated atezolizumab versus docetaxel in patients with previously treated NSCLC.34,35 Additionally, clinical data from the cohort in the study by Rizvi et al36, which included patients with advanced NSCLC treated with anti–PD-1 or anti–PD-L1 therapy and profiled by targeted NGS, were extracted from cBioPortal (accessed June 2020). Patients who received a combination of anti–cytotoxic T-lymphocyte–associated-antigen 4 (CTLA-4) therapy were excluded from this analysis to make this cohort comparable with the OAK/POPLAR cohort where single-agent ICI was used. Kaplan-Meier curves were used to model PFS in both the POPLAR/OAK and Rizvi cohorts, and the log-rank test was used for comparison. Objective response rate (ORR) comparisons were performed as reported in the respective data set. A Cox proportional hazards regression model was used to calculate hazard ratios (HRs) and 95% CI for PFS. HR and 95% CI from the Cox proportional hazards model were used to pool outcomes for PFS and generate a Forest plot.
We also used a real-world data set from Dana-Farber Cancer Institute (DFCI) comprising metastatic NSCLC treated with first-line ICI or chemoimmunotherapy harboring STK11mut TP53wt (n = 32) or STK11mut TP53mut (n = 21). We defined TTF as the time from initiation of first-line ICI-based therapy to initiation of second-line treatment due to disease progression or death, whichever comes first. The HR and P value were derived using a Cox multivariable model accounting for the ± use of chemotherapy with ICI. For the DFCI cohort, RECIST-based responses were obtained from computed tomography images of tumors.
We used SQLite37 to manage data and R38 with ggpubr39 on RStudio40 to analyze data and generate plots. The survival analysis for individual studies was conducted in R, version 3.4.1. The forest plot was generated in R version 4.0.0.
RESULTS
Molecular Landscape of STK11mut NSCLC and Relevant Immunotherapy Biomarkers
To study STK11mut NSCLC, we used a real-world data set of NSCLC samples previously submitted to Caris Life Sciences for molecular profiling. In total, 2,137 (12.6%) of 16,896 tumor samples had STK11 mutations, a majority (81%; n = 1,731) of which were adenocarcinomas. Frameshift alterations were the most common alterations (34%, Fig 1A). A higher proportion of men had STK11mut NSCLC versus women (13.5% v 11.7%; P < .001). Approximately 3.6% (n = 78/2,137) were classified as squamous histology. The median age for STK11mut NSCLC was slightly lower than STK11wt (67 v 69 years; P < .01). After KRAS mutation (50%), the most commonly comutated gene with STK11 was TP53 in 46% of STK11mut NSCLC (Fig 1B). The most common copy number amplifications in STK11mut NSCLC were identified in the MYC gene (3.5%). Using the Caris data, we characterized STK11mut NSCLC for biomarkers generally associated with better clinical outcomes to ICIs in NSCLC (Fig 1C): TMB, MSI, and PD-L1 expression. We identified that approximately 38% of STK11mut tumors had TMB-H, 34% of STK11mut tumors were PD-L1 ≥1%, and 12% were PD-L1 ≥50%. MSI was only detected in 0.7% of STK11mut tumors. Notably, STK11mut NSCLC had significantly lower PD-L1 positivity (≥1%) and PD-L1 ≥50% compared with tumors with STK11wt tumors (Fig 1C). However, no differences in TMB-H were observed for STK11mut versus STK11wt (38 v 36%; P > .1). Among STK11mut NSCLC, the most common KRAS subtype was G12C (39%).
FIG 1.

(A) Distribution of mutations in STK11. Frameshift alterations were the most common (33%). (B) The 10 most common comutations with STK11 mutations in NSCLC as assessed by NGS. (C) Frequency of biomarkers associated with responsiveness to immune checkpoint inhibitors in STK11mut NSCLC, including TMB-H, MSI/dMMR, and elevated PD-L1 expression. PD-L1 expression was significantly lower for STK11mut versus STK11wt NSCLC in the Caris cohort (% are rounded to the closest whole number as appropriate). dMMR, mismatch repair deficient; MSI-H, microsatellite instability-high; NGS, next generation sequencing; NSCLC, non–small-cell lung cancer; STK11, serine-threonine kinase 11; TMB, tumor mutational burden.
STING Pathway Expression in STK11mut NSCLC Identifies a Proinflammatory Subtype Enriched in TP53mut
Using the Caris cohort, we next evaluated the whole transcriptome-derived expression of immune signatures that could define a distinct subset of STK11mut NSCLC with a proinflammatory tumor microenvironment. We used the activity of the STING pathway as a surrogate for immune activation and sensitivity to ICI-based therapy.29,41 Activation of the STING pathway induces a type I IFN response that drives the expression of multiple chemokines, such as CXCL10 and CCL5.42 Unsupervised hierarchical clustering was performed in STK11mut NSCLC using RNA-seq expression for CCL5, CXCL10, and MB21D1(cGAS). Specifically, in adenocarcinomas with STK11mut (n = 463), we found that high STING activity was enriched in tumors with TP53 comutation (Fig 2). Consistent with this observation, there was an increase in the expression of genes related to T-cell chemotaxis and immune cell cytotoxicity in STK11mutTP53mut NSCLC samples compared with STK11mutTP53wt (Figs 3A-3G). These individual genes included the proinflammatory chemokines CCL5 (P < .01) and CXCL10 (P < .01), the stimulatory cytokine IFN-γ (P < .01), GZB (P < .01)—essential for the cytotoxicity of CD8+ T cells and natural killer (NK) cells—and the inflammatory cytokine IL-6 (P < .01).
FIG 2.

Hierarchical clustering of STK11mut adenocarcinoma (n = 463) for STING pathway genes (CCL5, CXCL10, cGAS) in the Caris cohort identified a STING-high and a STING-low cluster. The STING-high cluster was significantly enriched in TP53mut (48 v 32%; P < .01; the STING genes [top] are log2 transformed, not scaled, color key –3 to 3. The immune genes from Chen et al [bottom] are log10 transformed, not scaled, color key –4 to 4. All expression values are in TPM). cGAS, cyclic GMP-AMP synthase; mut, mutation; STING, stimulator of interferon-gamma; TPM, transcripts per million; wt, wild type.
FIG 3.

(A-C) MCP analysis in the Caris cohort showed higher CD8 T cell and NK cell and lower myeloid dendritic cell infiltration in STK11mut/TP53mut versus STK11mut/TP53wt (P < .01). (D-J) Immune-gene expression in the Caris cohort revealed significantly higher expression of genes related to T-cell chemotaxis and immune cell cytotoxicity (CCL5, CXCL10, GZB, IFNγ, IL-10, IL-6) in the STK11mut/TP53mut versus STK11mut/TP53wt cohort. MCP, microenvironment cell population; TPM, transcripts per million.
STK11mut NSCLC with TP53 Comutation Displays an Immunologically Active Tumor Microenvironment with Metabolic Reprogramming
On the basis of our results that TP53 co-occurring mutations in STK11mut NSCLC are associated with a STING-high phenotype, we re-examined the aforementioned biomarkers commonly associated with response to ICI therapy. STK11mut versus STK11wt NSCLC did not differ in median TMB (8 v 8 mut/Mb; P > .1) or neoantigen load (154.5 v 165; P > .1). By contrast, when considering TP53 comutation, the median TMB (11 v 8 mut/Mb; P < .001) and neoantigen load (263 v 134; P < .001) were statistically higher in the STK11mutTP53mut compared with the STK11mutTP53wt NSCLC tumors (Fig 4).
FIG 4.

The median neoantigen load (A; TCGA: 154 v 165) and TMB (B; Caris: 10 v 10 mut/Mb, P > .1) was not significantly different for STK11mut versus STK11wt. The median neoantigen load (C; TCGA: 263 v 134; P < .001) and TMB (D; Caris: 11 v 8 mut/Mb) was higher in the STK11mut/TP53mut tumors versus STK11mut/TP53wt NSCLC. NSCLC, non–small-cell lung cancer; TCGA, The Cancer Genome Atlas; TMB, tumor mutational burden.
Given the increased TMB and neoantigen load in the comutated tumors, we next explored whether this increase in immune response-associated biomarkers was associated with an increase in immune cell infiltration in these samples. On the basis of the MCP-counter method, we first tested for infiltration of CD8+ cytotoxic T cells (Figs 3H-3J). There was a significantly greater infiltration of CD8+ cytotoxic cells in STK11mutTP53mut NSCLC samples than in STK11mutTP53wt tumors (P < .01, Fig 3A). Similarly, there was greater infiltration of NK cells—known to play a role in tumor suppression43—in the STK11mutTP53mut samples than in the STK11mutTP53wt ones (P < .01, Fig 3B). Interestingly, the recruitment of myeloid dendritic cells was notably decreased in STK11mutTP53mut tumors (P < .01, Fig 3C). These myeloid dendritic cells that are CD11C+ CD123−, also known as conventional dendritic cells (cDCs), have a specialized role in antigen uptake and priming various subsets of adaptive immunity.44
We next asked whether STK11mutTP53mut tumors demonstrate differences in metabolic profile. We found differential enrichment of glycolysis and glutamine metabolism genes in STK11mut TP53mut NSCLC samples as opposed to STK11mutTP53wt controls, including MYC (P < .01), HIF-1A (P < .01), HK2 (P < .01), LDHA (P < .01), ALDOA (P < .01), and others (Fig 5).
FIG 5.

Median expression (TPM) of MYC and HIF-α were increased in the STK11mut/TP53mut versus STK11mut/TP53wt (P < .01) along with differentially higher expression (P < .01) of genes associated with both glycolysis (HK2, LDHA, ALDOA) and glutamine metabolism (GOT2, PPAT2). HIF-α, hypoxia-inducible factor α; TPM, transcripts per million.
Taken together, these data demonstrate that comutation with TP53 is associated with an immunogenic TIME and metabolic reprogramming in STK11mut NSCLC tumors.
STK11mutTP53mut NSCLC and Clinical Outcomes With ICIs
Given the evidence suggesting that STK11mutTP53mut tumors have increased immune cell infiltration and activity, we looked at publicly available and institutional clinical data sets to assess whether patients with STK11mutTP53mut NSCLC would have better clinical outcomes to ICIs than patients with STK11mut TP53wt NSCLC. To test this, we analyzed data from the OAK and POPLAR clinical trials.45 Using this data set, we identified a limited number of patients with STK11mutTP53mut (n = 14) and STK11mutTP53wt (n = 20) NSCLC treated with atezolizumab (Fig 6A). Although there was no statistically significant difference in median PFS between these groups (Fig 5; HR, 1.88 [95% CI, 0.89 to 3.97]; P = .098), there was a trend toward improved PFS in the STK11mutTP53mut group, with long-term PFS benefit only observed in the STK11mutTP53mut group. The disease control rate (complete response [CR] + partial response [PR] + stable disease [SD]) was 46.1% in the STK11mutTP53mut versus 16.6% in the STK11 mut cohort (χ2, 3.18; P = .07). We also performed an analysis of PFS using data previously published by Rizvi et al,36 examining patients with advanced NSCLC treated with anti–PD-1/PD-L1 therapy alone and found a numerical trend favoring long-term benefit (tail of the curve) in patients with STK11mutTP53mut NSCLC (Fig 6B; HR, 1.27 [95% CI, 0.66 to 2.44]). We then generated a forest plot and performed a meta-analysis of these studies using a random-effects model, again suggesting a trend toward inferior benefit for patients with STK11mutTP53wt NSCLC (Fig 6C; HR, 1.50 [95% CI, 0.92 to 2.46]). The durable benefit, defined in this study as a PR or SD that lasted >6 months, was 25% in the STK11mut/TP53 mut cohort versus 18.7% in the STK11mut/TP53wt cohort.
FIG 6.
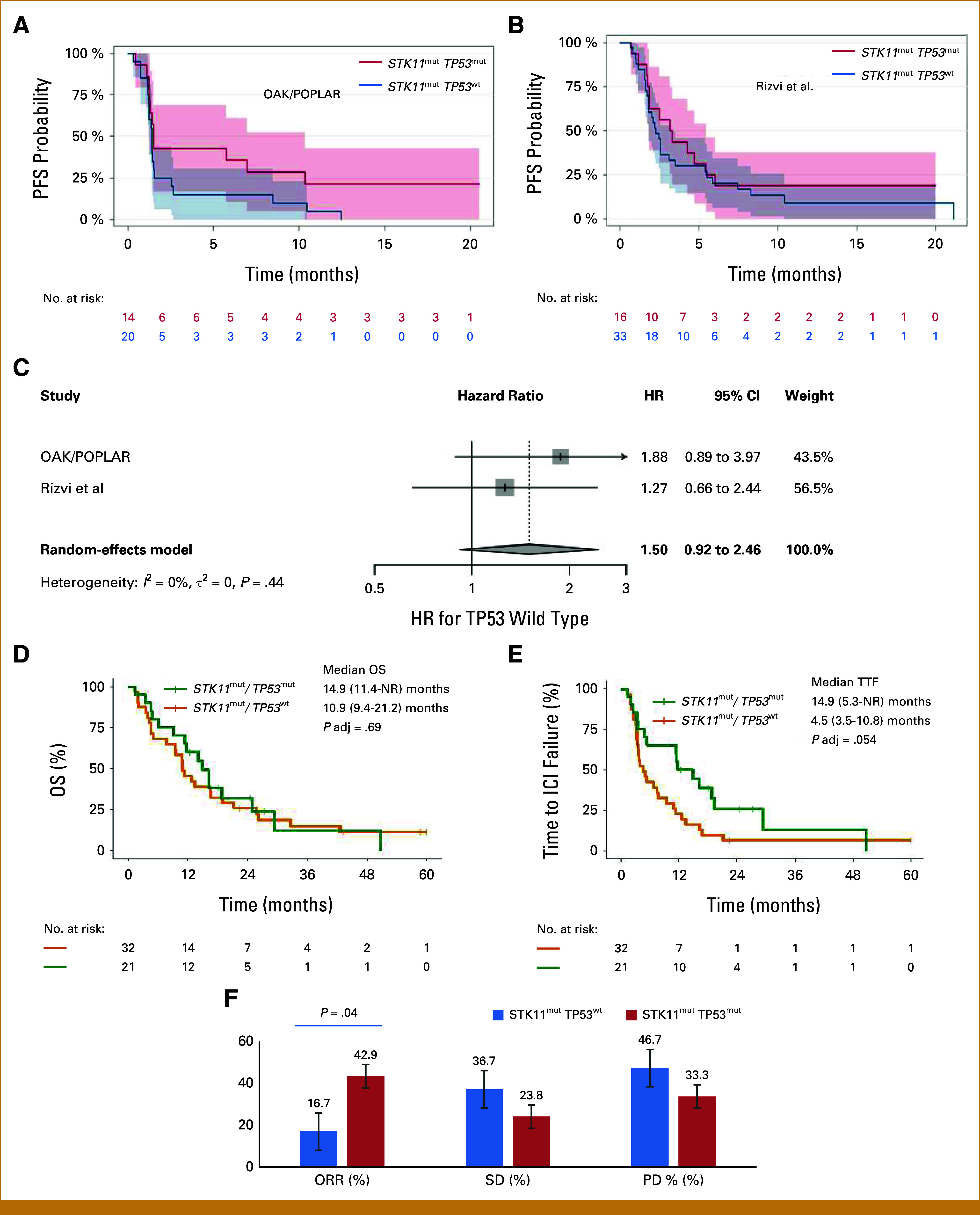
(A) In the combined OAK/POPLAR cohort, median PFS (HR, 1.88 [95% CI, 0.89 to 3.97]; P = .098) was not statistically different between STK11mut/TP53mut versus STK11mut/TP53wt. However, the 15-month PFS was 21% in the STK11mut/TP53mut versus 0% in the STK11mut/TP53wt. (B) In the Rizvi et al cohort, there was a numerical trend for long-term ICI benefit in the STK11mut TP53mut NSCLC. (C) Combined analysis of PFS in both the OAK/POPLAR cohort and Rizvi et al cohort. HRs and 95% CI from the Cox proportional hazards model were used to pool outcomes for PFS and generate a forest plot. (D and E) DFCI cohort with similar OS but significantly improved TTF for the comutated cohort on frontline ICI-based therapies (P adj = .054). (F) Bar graph showing the proportion of patients in the DFCI cohort whose tumors had an ORR, PD, or had SD in the STK11mutTP53wt (blue) and STK11mutTP53mut (red) cohorts. ORR was higher for STK11mut TP53mut versus STK11mut TP53wt (P = .04). A numerically higher % of PD was seen in the STK11mut TP53wt cohort. Note that response was evaluated by RECIST, and data were available to 30 of 32 patients in the STK11mutTP53wt cohort. DFCI, Dana-Farber Cancer Institute; ICI, immune checkpoint inhibitor; NSCLC, non–small-cell lung cancer; NR, not reached; HR, hazard ratio; ORR, objective response rate; OS, overall survival; PD, progressive disease; PFS, progression-free survival; SD, stable disease; TTF, time to treatment failure.
Appendix Table A1 summarizes relevant demographic and clinical characteristics for the DFCI cohort. No significant differences were observed for the STK11mutTP53wt group compared with the STK11mutTP53mut group. In the DFCI cohort (Figs 6D and 6E), although there was no meaningful difference in the overall survival (OS) for STK11mutTP53wt versus STK11mutTP53mut, we observed a numerically higher TTF (14.5 v 4.5 months, P-adj = .054) on first-line ICIs or chemotherapy ± ICIs for the comutated cohort compared with STK11mut alone. In the STK11mutTP53mut group (n = 21 patients), all events for treatment failure except for one (18 of 19 failures) were due to death. Since most patients died before receiving subsequent treatment, median OS and median TTF were largely overlapping for this group. We also observed higher ORR (Appendix Table A2) in the STK11mutTP53mut group (n = 21) compared with the STK11mutTP53wt group (n = 30; ORR: 42.9% v 16.7%, respectively; P = .04 [Fig 6F]). Although our clinical data sets were limited by the fact that we included disparately treated cohorts and had a small sample size, these data support improved response rates and suggested some hints of enhanced benefit in both first-line and second-line ICI-based therapies in STK11mutTP53mut NSCLC.
DISCUSSION
We used molecular data from nearly 17,000 NSCLC tumor samples to characterize the molecular and immune phenotype in STK11mut NSCLC. We identified a differential impact of TP53 comutation on the TIME of STK11mut NSCLC, which could improve the therapeutic selection of ICI-based therapies. To our knowledge, these are the most extensive data sets to date evaluating the TIME and expression profiles in STK11mut NSCLC.
Previous work has identified STK11mut as one of the most prevalent genomic drivers of inherent resistance to ICIs.46 Skoulidis et al12 demonstrated that STK11mut predicts poor outcomes with ICIs in KRAS-mutant lung adenocarcinoma. More recent real-world data have indicated that STK11mut may primarily have a negative prognostic impact, independent of the KRAS mutational status.47-49 Although exploratory analysis of the KEYNOTE-042 trial demonstrated that STK11mut NSCLC did not particularly have worse outcomes than its STK11wt counterparts, this analysis was not stratified for the TP53mut status of the tumor.50 Furthermore, a recent multicenter real-world data demonstrated a lack of benefit of adding ICIs to chemotherapy versus chemotherapy alone in STK11mut NSCLC,51 bringing into question current conventional approaches of anti–PD-1 with chemotherapy in managing this hard-to-treat subset of NSCLC. Interestingly, the addition of anti–CTLA-4 to anti-PD-1 seems to have added value on the basis of a recent post hoc analysis of the POSEIDON trial that showed that anti–PD-L1 with anti–CTLA-4 and chemotherapy as first-line therapy had somewhat improved benefit with a 10% higher PFS at 12 months that anti–PD-L1 with chemotherapy in STK11mut NSCLC.52 Similarly, the Checkmate 9LA demonstrated a trend toward improved OS in STK11mut NSCLC treated with anti–PD-1 + anti–CTLA-4 and chemotherapy compared with chemotherapy alone.53
Previous work has shown that STK11mut NSCLC is associated with lower PD-L1 expression, decreased chemokine signaling, lower NK cell activation, and suppressed T-cell transcriptional signatures, ultimately promoting a suppressed TIME.54 To identify comutations capable of rescuing this phenotype in STK11mut NSCLC, we analyzed the STING pathway as a surrogate for immune activation and sensitivity to ICI-based therapy, especially since the downstream type I IFN signaling from this pathway acts as a bridge between innate immunity and adaptive immunity driving antitumor immune responses.29,41,55 Our analysis showed that TP53mut is associated with increased STING pathway activity among STK11mut NSCLC, particularly in adenocarcinomas, a finding observed by another group, albeit in a smaller data set.29 In addition, using TCGA analysis of NSCLC, others have demonstrated the presence of an IFN-γ subtype in STK11mutTP53mut tumors. This subtype is known to have an M1 predominant macrophage polarization and higher T-cell repertoire diversity, which are thought to increase susceptibility to ICIs.13
Additionally, in STK11mut NSCLC, we demonstrated that mutations in TP53 are associated with an immune-activated phenotype on the basis of a higher neoantigen load and an increased infiltration of cytotoxic lymphocytes. These data align with recently published data suggesting the same in TP53mut NSCLC versus TP53wt regardless of STK11 status.56 Although we observed higher recruitment of both CD8+ T cells and NK cells in the TIME of STK11mutTP53mut NSCLC, cDCs infiltration was somewhat attenuated compared with STK11mutTP53wt tumors. cDCs are involved in antigen uptake and CD8+ T-cell priming, thus crucial to orchestrating cancer immunity.57 Since cDCs are generally recruited to the TIME by chemokines produced by NK cells, it is unclear from our data why STK11mutTP53mut NSCLC are deficient in cDCs despite having elevated NK cell infiltration. These intriguing findings and their potential therapeutic implications merit further exploration.
Our findings in the TIME were corroborated to some extent by our exploratory PFS analysis from the data of ICI-treated NSCLC in OAK/POPLAR and the study by Rizvi et al, where STK11mut NSCLC with TP53mut had a nonsignificant suggestion of an improved durable benefit (PR or SD ≥6 months) and clinical benefit rate (CR + PR + SD) than STK11mut TP53wt tumors. Interestingly, the pooled estimate of the HR from the two studies (OAK/POPLAR and Rizvi et al) for patients with TP53wt tumors was 1.5, suggesting some level of benefit in the TP53mut subset. However, the CI crossed 1 (Fig 6B), likely due to the small sample size. Furthermore, in the DFCI cohort, we observed an approximately three times longer time-to-treatment failure in the STK11mut TP53mut cohort in patients with NSCLC treated with frontline ICI or ICI-based therapies (Fig 6C), albeit with no differences in OS. In this same cohort, however, patients with STK11mut TP53mut tumors had higher ORRs by RECIST compared with STK11mut TP53wt tumors (42.9% v 16.7%; P = .04; Fig 6F) These data suggest that further studies are warranted to determine if the more favorable TIME in STK11mut TP53mut NSCLC is genuinely associated with clinical benefit to ICI-based therapies, as indicated by our limited data set.
Loss of LKB1 because of inactivating mutations in STK11 is known to lead to a higher level of HIF-1A expression, thereby shifting glucose metabolism to the glycolytic pathway.58 Furthermore, LKB1 loss can also lead to increased dependence on glutamine metabolism, rendering these tumors susceptible to glutaminase inhibition.59 Our analysis of the glycolytic and glutamine pathways (Fig 5) in STK11mut NSCLC suggests that TP53 comutation may potentiate these known effects of STK11 mutations on metabolic reprogramming, thereby shifting cellular metabolism away from glucose dependence. Furthermore, we also observed a higher expression of HIF-1A in the STK11mut TP53mut tumors. These findings suggest there may be value in targeting glycolysis and glutamine dependence in STK11mut tumors with TP53 comutations. Early efforts in this direction are currently underway that involve novel combination approaches targeting immune-metabolic pathways.60,61
Previous studies have reported the highest frequency of TP53 mutations in patients with NSCLC who smoke, along with higher rates of tumor frameshift mutations.62 Some other studies have elucidated that smoking is associated with higher rates of TP53 polymorphism and tumor chromosomal abnormalities.63,64 These changes could inevitably translate into a higher TMB and neoantigen load. In our limited DFCI data set, we did not observe a significant difference in smoking status for TP53mut versus TP53wt tumors with STK11mut. Unfortunately, within the Caris cohort, we could not tease out whether the smoking status of patients in the STK11mut TP53mut NSCLC subset is a confounder that could contribute to an increased immune inflamed TIME.
Notably, as a limitation, our analysis does not account for mutations in KRAS or PD-L1 status. Furthermore, although glycolysis and glutamine uptake in the tumor are putative mechanisms implicated in immune evasion,65 our data suggest that comutation of TP53 in STK11mut NSCLC increases tumor immunogenicity despite an increase in glycolysis and glutamine metabolism. Thus, further work is needed to better understand the functional interplay of the tumor immune profile with intratumoral metabolism in STK11mut NSCLC. Owing to the limitations associated with publicly available data, we included two disparate data sets for our clinical outcomes. The OAK/POPLAR data include patients treated with ICIs in the second line, whereas the DFCI data had patients treated in the first line. Subsequent studies examining the clinical impact of TP53 comutation in STK11mut NSCLC with high or low TMB in the context of chemoimmunotherapy versus ICIs alone are merited.
Taken together, our study suggests that comutation with TP53 defines a distinct phenotypic subtype of STK11mut NSCLC harboring an immunologically active TIME with features of unique metabolic reprogramming. Practically, once validated in larger settings, a comutation-based treatment selection strategy whereby doublet ICIs with HIF1A inhibitors or anti–PD-(L)-1 with STING agonists in patients with STK11mutTP53mut NSCLC should be explored. We demonstrate that implementing a multiomics approach to identify and exploit underlying therapeutic vulnerabilities associated with distinct comutation subsets within STK11mut NSCLC needs to be extensively studied. Given that we have not been able to test our findings formally in a first-line prospective trial, currently, our findings are hypothesis generating and should not necessarily affect treatment decisions in the current paradigm. However, our findings provide a framework for evaluating future personalized precision medicine-based immuno-oncology combination approaches in STK11 TP53 comutated NSCLC.
APPENDIX 1. Supplemental Methods
Methods
NGS
The minimum purity allowed was 20%. A custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with >99% confidence on the basis of allele frequency and amplicon coverage, with an average sequencing depth of coverage of approximately 800 and an analytic sensitivity to detect variants with a variant allele frequency of ≥5%. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as pathogenic, presumed pathogenic, variant of unknown significance, presumed benign, or benign, according to the American College of Medical Genetics and Genomics standards. When assessing mutation frequencies of individual genes, pathogenic and presumed pathogenic were counted as mutations.
RNA
Qiagen RNA formalin-fixed paraffin-embedded, tissue extraction kit was used for extraction, and the RNA quality and quantity were determined using the Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized/purified cDNA targets, and the bait-target complexes were amplified in a postcapture polymerase chain reaction. The Illumina NovaSeq 6500 was used to sequence the whole transcriptome from patients to an average of 60M reads. Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies, Santa Clara, CA) was used for fusion detection.
Immunohistochemistry
A board-certified pathologist evaluated all immunohistochemistry (IHC) results independently. The primary PD-L1 antibody clone was 22c3 (Dako). Tumor Proportion Score (TPS) was measured as the percentage of viable tumor cells showing partial or complete membrane staining at any intensity. The tumor was considered PD-L1–positive if TPS was ≥1% and PD-L1 high if TPS was ≥50%.
Microsatellite instability testing
Multiple test platforms were used to determine the microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) status of the tumors, including fragment analysis (FA, Promega, Madison, WI), IHC (MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 antibodies; and PMS2, EPR3947 antibody [Ventana Medical Systems, Inc, Tucson, AZ]), and NGS (>2,800 target microsatellite loci were examined and compared with the reference genome from the University of California, Santa Cruz Genome Browser database). The three platforms generated highly concordant results, as previously reported. In the rare cases of discordant results, the MSI-H or MMR status of the tumor was determined in the order of IHC, FA, and NGS.
Compliance Statement
This study was conducted per the guidelines of the Declaration of Helsinki, the Belmont Report, and the US Common Rule. In keeping with 45 CFR 46.101(b)(4), this study's analysis of the Caris cohort used retrospective, deidentified data. Therefore, this study is considered institutional review board (IRB) exempt, and no patient consent was necessary. Appropriate IRB approval was obtained for the Dana-Farber Cancer Institute (DFCI) cohort. The DFCI institutional review board granted a waiver of informed consent for this cohort of patients.
FIG A1.

RNA expression comparing cGAS (MB21D1) for TP53mut versus TP53wt in tumors with STK11mut NSCLC. Higher expression (transcripts per million) was observed in the TP53mut cohort versus the TP53wt cohort (P < .001). cGAS, cyclic GMP-AMP synthase; mut, mutation; NSCLC, non–small-cell lung cancer; wt, wild type.
TABLE A1.
Baseline Demographic and Clinical Characteristics of Patients in the DFCI Cohort
| Characteristic | STK11mutTP53wt (n = 32) | STK11mutTP53mut (n = 21) | P |
|---|---|---|---|
| Age, years, median (95% CI) | 62.69 (59.65 to 65.74) | 65.96 (61.46 to 70.45) | — |
| Sex, No. (%) | |||
| Male | 19 (59.4) | 13 (61.9) | |
| Female | 13 (40.6) | 8 (38.1) | .85 |
| Race, No. (%) | |||
| White | 29 (90.6) | 14 (66.7) | |
| Black or African American | 2 (6.2) | 2 (9.5) | |
| Other/not reported | 1 (3.1) | 5 (23.8) | .12 |
| Histology, No. (%) | |||
| Adenocarcinoma | 29 (90.6) | 14 (66.7) | |
| Squamous | 1 (3.1) | 3 (14.3) | |
| Mixed | 1 (3.1) | 0 (0.0) | |
| NOS/poorly differentiated | 1 (3.1) | 4 (19.1) | .07 |
| Smoking status, No. (%) | |||
| Current/former | 30 (93.7) | 20 (95.2) | |
| Never | 2 (6.2) | 1 (4.8) | 1 |
| ICI therapy, No. (%) | |||
| Anti–PD-1/anti–PDL1 based | 29 (90.6) | 20 (95.2) | |
| Anti–PD-1 + anti–CTLA-4 | 3 (9.375) | 1 (4.76) | .84 |
| TMB, No. (%) | |||
| TMB-H (≥10) | 13 (40.63) | 16 (76.19) | .09 |
NOTE. There were no significant differences in demographic characteristics (sex, race, or smoking status) or clinical features (histology, ICI therapy, TMB-H, OS, or TTF) between STK11mutTP53wt and STK11mutTP53mut patients.
Abbreviations: CTLA-4, cytotoxic T-lymphocyte–associated-antigen 4; DFCI, Dana-Farber Cancer Institute; ICI, immune checkpoint inhibitor; NOS, not otherwise specified; OS, overall survival; TMB, tumor mutational burden; TTF, time to treatment failure.
TABLE A2.
RECIST-Based ORRs in the DFCI Cohort
| Response | STK11mutTP53wt (n = 32) | STK11mutTP53mut (n = 21) | P |
|---|---|---|---|
| ORR, % | 16.67 | 42.86 | .04 |
| CR, No. (%) | 0 (0.00) | 0 (0.00) | |
| PR, No. (%) | 5 (16.67) | 9 (42.86) | |
| SD, No. (%) | 11 (36.67) | 5 (23.81)/ | .33 |
| PD, No. (%) | 14 (46.67) | 7 (33.3) | .12 |
| Not available | 2 | 0 | — |
| Total available | 30 | 21 | — |
Abbreviations: CR, complete response; DFCI, Dana-Farber Cancer Institute; ORR, objective response rate; PD, progressive disease; PR, partial response; SD, stable disease.
AUTHOR CONTRIBUTIONS
Conception and design: Abdul Rafeh Naqash, Charalampos S. Floudas, Hirva Mamdani
Administrative support: Abdul Rafeh Naqash, Mark M. Awad
Provision of study materials or patients: Abdul Rafeh Naqash, Mark M. Awad, Luis E. Raez, Jorge J. Nieva, Hossein Borghaei, Hirva Mamdani
Collection and assembly of data: Abdul Rafeh Naqash, Charalampos S. Floudas, Amin H. Nassar, Elio Adib, Khalil Choucair, Joanne Xiu, Biagio Ricciuti, Joao V. Alessi, Mark M. Awad, Gilberto Lopes, Jorge J. Nieva, Stephen V. Liu
Data analysis and interpretation: Abdul Rafeh Naqash, Charalampos S. Floudas, Etan Aber, Asaf Maoz, Elio Adib, Khalil Choucair, Yasmine Baca, Mark M. Awad, Chul Kim, Julia Judd, Luis E. Raez, Gilberto Lopes, Jorge J. Nieva, Hossein Borghaei, Naoko Takebe, Patrick C. Ma, Balazs Halmos, David J. Kwiatkowski, Stephen V. Liu
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Abdul Rafeh Naqash
This author is a Social Media Editor for JCO Precision Oncology. Journal policy recused the author from having any role in the peer review of this manuscript.
Consulting or Advisory Role: foundation me
Travel, Accommodations, Expenses: Foundation Medicine, Jazz Pharmaceuticals (Inst), Binacea
Asaf Maoz
Research Funding: Novartis (Inst)
Patents, Royalties, Other Intellectual Property: Patent applications related to macrophage-based cancer immunotherapy
Elio Adib
Employment: Amgen
Joanne Xiu
Employment: Caris Life Sciences
Yasmine Baca
Employment: Caris Life Sciences
Biagio Ricciuti
Honoraria: Targeted Oncology
Consulting or Advisory Role: Regeneron, AstraZeneca, Amgen, Guidepoint Inc
Mark M. Awad
Consulting or Advisory Role: Merck, Pfizer, Bristol Myers Squibb, Foundation Medicine, Novartis, Gritstone Bio, Mirati Therapeutics, EMD Serono, AstraZeneca, Instil Bio, Regeneron, Janssen, Affini-T Therapeutics
Research Funding: Genentech/Roche (Inst), Lilly (Inst), AstraZeneca (Inst), Bristol Myers Squibb (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb Foundation
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1127368
Chul Kim
Consulting or Advisory Role: Janssen, Novartis, PierianDx, AstraZeneca, Sanofi, Diffusion Pharmaceuticals, Mirati Therapeutics, Jazz Pharmaceuticals, Arcus Biosciences, Daiichi Sankyo, Eisai
Research Funding: AstraZeneca, Novartis, Karyopharm Therapeutics, Bristol Myers Squibb, Regeneron, Debiopharm Group, Janssen, Genentech/Roche, Daiichi Sankyo
Luis E. Raez
Consulting or Advisory Role: AstraZeneca, Novocure, BMS, Lilly, Bayer
Research Funding: Genentech/Roche (Inst), Merck Serono (Inst), Novartis (Inst), Pfizer (Inst), Syndax (Inst), Loxo (Inst), Merck (Inst), Bristol Myers Squibb (Inst), Guardant Health (Inst), Heat Biologics (Inst), Amgen (Inst), Calithera Biosciences (Inst), Daiichi Sankyo/UCB Japan (Inst), NantHealth (Inst), Anheart Therapeutics (Inst), Natera (Inst)
Gilberto Lopes
Stock and Other Ownership Interests: Lucence Diagnostics, Xilis, Biomab, Morphometrix, CDR-Life
Honoraria: Boehringer Ingelheim, Blueprint Medicines, AstraZeneca, Merck, Janssen
Consulting or Advisory Role: Pfizer, AstraZeneca
Research Funding: Merck Sharp & Dohme (Inst), EMD Serono (Inst), AstraZeneca (Inst), AstraZeneca, Blueprint Medicines (Inst), Tesaro (Inst), Bavarian Nordic (Inst), NOVARTIS (Inst), G1 Therapeutics (Inst), adaptimmune (Inst), BMS (Inst), GlaxoSmithKline (Inst), AbbVie (Inst), Rgenix (Inst), Pfizer (Inst), Roche (Inst), Genentech (Inst), Lilly (Inst), Janssen (Inst), Lucence, Xilis, E.R. Squibb Sons, LLC
Travel, Accommodations, Expenses: Boehringer Ingelheim, Pfizer, E.R. Squibb Sons, LLC, Janssen, Seagen, Celgene, Ipsen, Pharmacyclics, Merck, AstraZeneca
Other Relationship: Mirati Therapeutics
Jorge J. Nieva
Stock and Other Ownership Interests: Epic Sciences, Cansera, Quantgene, Indee P/L
Consulting or Advisory Role: AstraZeneca, Naveris, AADi, Bioatla, Mindmed, ANP Technologies
Research Funding: Merck (Inst), Genentech (Inst)
Patents, Royalties, Other Intellectual Property: Patent Pending—movement and unexpected healthcare encounters
Hossein Borghaei
Stock and Other Ownership Interests: Sonnet, Rgenix, Nucleai
Honoraria: Bristol Myers Squibb, Celgene, Axiom Biotechnologies, Pfizer, Amgen, Regeneron, Daiichi Sankyo/UCB Japan
Consulting or Advisory Role: Bristol Myers Squibb, Lilly, Genentech, Pfizer, Boehringer Ingelheim, EMD Serono, Novartis, Merck, AstraZeneca, Genmab, Regeneron, BioNTech, AbbVie, PharmaMar, Takeda, Amgen, Sonnet, Rgenix, Beigene, Jazz Pharmaceuticals, Mirati Therapeutics, Guardant Health, Janssen Oncology, ITeos Therapeutics, Natera, Oncocyte, Puma Biotechnology, BerGenBio, Bayer, IO Biotech
Research Funding: Bristol Myers Squibb (Inst), Lilly (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Lilly, Clovis Oncology, Celgene, Genentech, Novartis, Merck, Amgen, EMD Serono, Regeneron
Other Relationship: University of Pennsylvania, Takeda, Incyte, Novartis, SpringWorks Therapeutics
Patrick C. Ma
Honoraria: AstraZeneca, BeiGene
Research Funding: AstraZeneca (Inst), Bristol Myers Squibb (Inst), AbbVie (Inst), Merck (Inst), Apollomics (Inst), OncoC4 (Inst), Genmab (Inst), BeiGene (Inst), Mirati Therapeutics (Inst), Roche/Genentech (Inst), Elevation Oncology (Inst), Calithera Biosciences (Inst)
Balazs Halmos
Consulting or Advisory Role: AstraZeneca, Genentech/Roche, Pfizer, Takeda, Novartis, Merck, Bristol Myers Squibb, Turning Point Therapeutics, Apollomics, Janssen Oncology, Veracyte, BeiGene, Arcus Biosciences, Merus, Lilly, Bayer
Research Funding: Merck (Inst), AstraZeneca (Inst), Mirati Therapeutics (Inst), Boehringer Ingelheim (Inst), Roche/Genentech (Inst), Pfizer (Inst), Takeda (Inst), AbbVie (Inst), Bristol Myers Squibb (Inst), GlaxoSmithKline (Inst), Blueprint Medicines (Inst), Novartis (Inst), Advaxis (Inst), Janssen Oncology (Inst), Elevation Oncology (Inst), Daiichi Sankyo/AstraZeneca (Inst), Amgen
David J. Kwiatkowski
Consulting or Advisory Role: Genentech/Roche, AADI, Slingshot Insights, Guidepoint Global, Bridgebio, William Blair
Research Funding: AADi, Revolution Medicines, Genentech/Roche
Stephen V. Liu
Consulting or Advisory Role: Genentech, Bristol Myers Squibb, AstraZeneca, Takeda, Regeneron, Guardant Health, Janssen Oncology, MSD Oncology, Jazz Pharmaceuticals, Daiichi Sankyo/UCB Japan, Turning Point Therapeutics, Elevation Oncology, Novartis, Eisai, Gilead Sciences, Sanofi, Catalyst Pharmaceuticals, Candel Therapeutics, Merus, AbbVie, Amgen, Boehringer Ingelheim, Mirati Therapeutics, Pfizer
Research Funding: Genentech/Roche (Inst), Merck (Inst), Alkermes (Inst), Turning Point Therapeutics (Inst), RAPT Therapeutics (Inst), Merus (Inst), Elevation Oncology (Inst), Nuvalent, Inc (Inst), Gilead Sciences (Inst), AbbVie (Inst), Ellipses Pharma (Inst)
Travel, Accommodations, Expenses: Caris Life Sciences
Hirva Mamdani
Consulting or Advisory Role: Zentalis, MorphoSys, Seagen, AstraZeneca, Genentech, AstraZeneca, Daiichi Sankyo/Astra Zeneca
Research Funding: AstraZeneca (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1. Siegel RL, Miller KD, Wagle NS, et al. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 3. Tsukagoshi M, Yokobori T, Yajima T, et al. Skeletal muscle mass predicts the outcome of nivolumab treatment for non-small cell lung cancer. Medicine (Baltimore) 2020;99:e19059. doi: 10.1097/MD.0000000000019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Netterberg I, Bruno R, Chen YC, et al. Tumor time-course predicts overall survival in non-small cell lung cancer patients treated with atezolizumab: Dependency on follow-up time. CPT Pharmacometrics Syst Pharmacol. 2020;9:115–123. doi: 10.1002/psp4.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao N, Wilkerson MD, Shah U, et al. Alterations of LKB1 and KRAS and risk of brain metastasis: Comprehensive characterization by mutation analysis, copy number, and gene expression in non-small-cell lung carcinoma. Lung Cancer. 2014;86:255–261. doi: 10.1016/j.lungcan.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pons-Tostivint E, Lugat A, Fontenau JF, et al. STK11/LKB1 modulation of the immune response in lung cancer: From biology to therapeutic impact. Cells. 2021;10:3129. doi: 10.3390/cells10113129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laderian B, Mundi P, Fojo T, et al. Emerging therapeutic implications of STK11 mutation: Case series. Oncologist. 2020;25:733–737. doi: 10.1634/theoncologist.2019-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hasegawa T, Yanagitani N, Ninomiya H, et al. Association between the efficacy of pembrolizumab and low STK11/LKB1 expression in high-PD-L1-expressing non-small-cell lung cancer. In Vivo. 2020;34:2997–3003. doi: 10.21873/invivo.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gainor JF, Rizvi H, Jimenez Aguilar E, et al. Clinical activity of programmed cell death 1 (PD-1) blockade in never, light, and heavy smokers with non-small-cell lung cancer and PD-L1 expression ≥50. Ann Oncol. 2020;31:404–411. doi: 10.1016/j.annonc.2019.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 12. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malhotra J, Ryan B, Patel M, et al. Clinical outcomes and immune phenotypes associated with STK11 co-occurring mutations in non-small cell lung cancer. J Thorac Dis. 2022;14:1772–1783. doi: 10.21037/jtd-21-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soussi T, Wiman KG. TP53: An oncogene in disguise. Cell Death Differ. 2015;22:1239–1249. doi: 10.1038/cdd.2015.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donehower LA, Soussi T, Korkut A, et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep. 2019;28:1370–1384.e5. doi: 10.1016/j.celrep.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cortez MA, Ivan C, Valdecanas D, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst. 2016;108:djv303. doi: 10.1093/jnci/djv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ji M, Liu Y, Li Q, et al. PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/KRAS mutations. Cancer Biol Ther. 2016;17:407–413. doi: 10.1080/15384047.2016.1156256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Co NN, Iglesias D, Celestino J, et al. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer. 2014;120:3457–3468. doi: 10.1002/cncr.28854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bange E, Marmarelis ME, Hwang WT, et al. Impact of KRAS and TP53 co-mutations on outcomes after first-line systemic therapy among patients with STK11-mutated advanced non-small-cell lung cancer. JCO Precis Oncol. doi: 10.1200/PO.18.00326. 10.1200/PO.18.00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Decout A, Katz JD, Venkatraman S, et al. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21:548–569. doi: 10.1038/s41577-021-00524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang M, Chen P, Wang L, et al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. 2020;13:81. doi: 10.1186/s13045-020-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ou L, Zhang A, Cheng Y, et al. The cGAS-STING pathway: A promising immunotherapy target. Front Immunol. 2021;12:795048. doi: 10.3389/fimmu.2021.795048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dan Q, Yang Y, Ge H. cGAS-STING pathway as the target of immunotherapy for lung cancer. Curr Cancer Drug Targets. 2023;23:354–362. doi: 10.2174/1568009623666221115095114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann Oncol. 2019;30:44–56. doi: 10.1093/annonc/mdy495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–1365. doi: 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 26. Dobin A, Davis CA, Schlesinger F, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patro R, Duggal G, Love MI, et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Della Corte CM, Sen T, Gay CM, et al. STING pathway expression identifies NSCLC with an immune-responsive phenotype. J Thorac Oncol. 2020;15:777–791. doi: 10.1016/j.jtho.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48:812.e4–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zietemann VD, Schuster T, Duell TH. Post-study therapy as a source of confounding in survival analysis of first-line studies in patients with advanced non-small-cell lung cancer. J Thorac Dis. 2011;3:88–98. doi: 10.3978/j.issn.2072-1439.2010.12.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441–1448. doi: 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 34. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837–1846. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 36. Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hipp R. SQLite. 2020. [Google Scholar]
- 38.R-Core-Team . A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; [Google Scholar]
- 39.Kassambara A.Ggpubr: ‘ggplot2’ Based Publication Ready Plots.
- 40.Rstudio-Team . Rstudio: Integrated Development for R. Rstudio. PBC; [Google Scholar]
- 41. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barber GN. STING: Infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 44. Rhodes JW, Tong O, Harman AN, et al. Human dendritic cell subsets, ontogeny, and impact on HIV infection. Front Immunol. 2019;10:1088. doi: 10.3389/fimmu.2019.01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mazieres J, Rittmeyer A, Gadgeel S, et al. Atezolizumab versus docetaxel in pretreated patients with NSCLC: Final results from the randomized phase 2 POPLAR and phase 3 OAK clinical trials. J Thorac Oncol. 2021;16:140–150. doi: 10.1016/j.jtho.2020.09.022. [DOI] [PubMed] [Google Scholar]
- 46. Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shire NJ, Klein AB, Golozar A, et al. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS One. 2020;15:e0238358. doi: 10.1371/journal.pone.0238358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papillon-Cavanagh S, Doshi P, Dobrin R, et al. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open. 2020;5:e000706. doi: 10.1136/esmoopen-2020-000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosellini P, Amintas S, Caumont C, et al. Clinical impact of STK11 mutation in advanced-stage non-small cell lung cancer. Eur J Cancer. 2022;172:85–95. doi: 10.1016/j.ejca.2022.05.026. [DOI] [PubMed] [Google Scholar]
- 50. Cho BC, Lopes G, Kowalski DM, et al. Abstract CT084: Relationship between STK11 and KEAP1 mutational status and efficacy in KEYNOTE-042: Pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. Cancer Res. 2020;80 suppl 16; abstr CT084. [Google Scholar]
- 51. Skoulidis F, Arbour KC, Hellmann MD, et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J Clin Oncol. 2019;37 suppl 15; abstr 102. [Google Scholar]
- 52. Peters S, Cho B, Luft A, et al. OA15.04 association between KRAS/STK11/KEAP1 mutations and outcomes in POSEIDON: Durvalumab ± Tremelimumab + Chemotherapy in mNSCLC. J Thorac Oncol. 2022;17(suppl 9):S39–S41. [Google Scholar]
- 53. Paz-Ares LG, Ciuleanu TE, Cobo M, et al. First-line nivolumab plus ipilimumab with chemotherapy versus chemotherapy alone for metastatic NSCLC in CheckMate 9LA: 3-year clinical update and outcomes in patients with brain metastases or select somatic mutations. J Thorac Oncol. 2023;18:204–222. [Google Scholar]
- 54. Yau E, Glenn S, Papinicalou-Sengos A, et al. P1. 04-10 Identification of immunotherapy targets in STK11 mutant non-squamous non-small cell lung cancer. J Thorac Oncol. 2019;14:S442. [Google Scholar]
- 55. Li A, Yi M, Qin S, et al. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol. 2019;12:35. doi: 10.1186/s13045-019-0721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Biton J, Mansuet-Lupo A, Pécuchet N, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin Cancer Res. 2018;24:5710–5723. doi: 10.1158/1078-0432.CCR-18-0163. [DOI] [PubMed] [Google Scholar]
- 57. Böttcher JP, Reis e Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. 2018;4:784–792. doi: 10.1016/j.trecan.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Faubert B, Vincent EE, Griss T, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci USA. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Galan-Cobo A, Sitthideatphaiboon P, Qu X, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019;79:3251–3267. doi: 10.1158/0008-5472.CAN-18-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Momcilovic M, Shackelford DB. Targeting LKB1 in cancer—Exposing and exploiting vulnerabilities. Br J Cancer. 2015;113:574–584. doi: 10.1038/bjc.2015.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sumbly V, Landry I. Unraveling the role of STK11/LKB1 in non-small cell lung cancer. Cureus. 2022;14:e21078. doi: 10.7759/cureus.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Halvorsen AR, Silwal-Pandit L, Meza-Zepeda LA, et al. TP53 mutation spectrum in smokers and never smoking lung cancer patients. Front Genet. 2016;7:85. doi: 10.3389/fgene.2016.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aldakheel FM, Abuderman AA, Alali BH, et al. Smoking and P53 polymorphism association with chromosomal aberration in lung cancer. J King Saud Univ Sci. 2021;33:101533. [Google Scholar]
- 65. Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: Emerging concepts and therapeutic opportunities. Biochim Biophys Acta Rev Cancer. 2017;1868:212–220. doi: 10.1016/j.bbcan.2017.04.002. [DOI] [PubMed] [Google Scholar]