

Abstract
Background:
Genetic aberrations in the NFκB pathway lead to primary immunodeficiencies with various degrees of severity. We previously demonstrated that complete ablation of the RelB transcription factor, a key component of the alternative pathway, results in an early manifested combined immunodeficiency requiring stem cell transplantation.
Objective:
To study the molecular basis of a progressive severe autoimmunity and immunodeficiency in three patients.
Methods:
Whole exome sequencing was performed to identify the genetic defect. Molecular and cellular techniques were utilized to assess the variant impact on NFκB signaling, canonical and alternative pathway crosstalk as well as the resultant effects on immune function.
Results:
Patients presented with multiple autoimmune progressive severe manifestations encompassing the liver, gut, lung, and skin, becoming debilitating in the second decade of life. This was accompanied by a deterioration of the immune system, demonstrating an age-related decline in naïve T cells and responses to mitogens, accompanied by a gradual loss of all circulating CD19+ cells. Whole exome sequencing identified a novel homozygous c.C1091T (P364L) transition in RELB. The P364L RelB protein was unstable, with extremely low expression, but retained some function and could be transiently and partially upregulated following Toll receptor stimulation. Stimulation of P364L patient fibroblasts resulted in a marked rise in a cluster of pro-inflammatory hyper-expressed transcripts consistent with the removal of RelB inhibitory effect on RelA function. This is likely the main driver of autoimmune manifestations in these patients.
Conclusion:
Incomplete loss of RelB provided a unique opportunity to gain insights into NFκB’s pathway interactions as well as the pathogenesis of autoimmunity. The P364L RelB leads to gradual decline in immune function with progression of severe debilitating autoimmunity.
Keywords: RelB, autoimmunity, combined immunodeficiency, naïve T cells, memory B cells, NFκB pathway
1.1. INTRODUCTION
A growing number of primary immune deficiencies have been shown to arise from mutations in NFκB signaling pathway components [1–9]. The nuclear factor kappa-B (NFκB) family of transcription factors consist of five members: RelA, RelB, Rel (c-Rel) and two precursor proteins NFκB1 (p105) and NFκB2 (p100) that are processed into p50 and p52, respectively [10, 11]. Two NFκB signaling pathways are recognized leading to gene transcription: the classical, mediated by RelA (and c-Rel) and the alternative, mediated by RelB. Whereas the classical pathway is activated by many stimuli (e.g. TNFR, TLR, TCR), direct activation of the non-canonical alternative pathway is observed downstream of only a few receptors (e.g. TWEAKR, LTβR, CD40 and BAFF-R), all of which simultaneously activate the RelA-mediated pathway.
In the classical pathway, IκB proteins bind and sequester RelA and c-Rel in the cytoplasm, preventing activation. Stimulation-induced phosphorylation of IκB by Inhibitor of NFκB kinase subunit 1 (IKK1, IKKα) and IKK2 (IKKβ) IκB kinases initiates IκB degradation, freeing NFκB complexes to enter the nucleus. In contrast, the alternative pathway is regulated by binding of NFκB2 p100 to RelB, with activation of this complex requiring inducible processing of p100 to p52. Although the two independently activated NFκB pathways are recognized, cross-regulation has been proposed [12]. While directly activated by few receptors, RelB is commonly co-expressed with RelA and is hypothesized to play an important role in regulating RelA activity.
Human NFκB defects generally affect both adaptive and innate immunity. Hypomorphic mutations in IKKγ (NEMO) cause a variety of phenotypes with or without typical features of ectodermal dysplasia. Immune aberrations vary greatly from patient to patient even when carrying the same mutation; some demonstrating a predominantly T cell or combined immunodeficiency, with others lacking only the ability to produce specific antibodies. Anhidrotic ectodermal dysplasia with immunodeficiency can also arise from autosomal dominant IκBα gain-of-function mutations. Ablative mutations of IKK2 significantly impair both innate and adaptive immunity and patients present with features of severe combined immunodeficiency. Haplo-insufficiencies of RelA are also associated with variable phenotypes, ranging from lymphoproliferative disease and autoimmune cytopenia to TNF-dependent chronic mucocutaneous ulceration [13, 14]. In the alternative pathway, monoallelic mutation of NFκB2 p100 inhibiting the production of p52 from p100 has been shown to be associated with hypogammaglobulinemia, alopecia, and endocrinopathies [4, 15, 16].
Previously, we identified complete RelB deficiency in three children who presented very early in infancy (7 days-4 months) with severe infections and failure to thrive [17, 18]. Complete RelB defcient patients showed both B and T cell defects. These patients had a dysplastic thymus and demonstrated an abnormal accumulation of peripheral CD45RO+ T cells, a greatly skewed TCR repertoire and a depressed in vitro response to mitogens. Although B cell numbers were normal to elevated, the CD27+ B cell memory population was absent, with low specific antibody production necessitating replacement with intravenous immunoglobulin. As a result of this combined immunodeficiency, all three patients underwent hematopoietic stem cell therapy at a young age [19].
We describe here the first known incomplete RelB deficiency in three patients. In contrast to RelB null deficiency, these patients experience gradually developing severe auto-inflammatory phenomena and progressively declining immunity in the second decade of life. The predominance of auto-inflammatory manifestations appear to arise from compromised RelB-mediated inhibition of RelA-induced pro-inflammatory transcriptional activity, supporting the hypothesis of cross-regulation of the NFκB canonical and alternative pathways in a human model, and providing insights into the pathogenesis of autoimmunity.
1.2. METHODS
1.2.1. Whole Exome Analysis
Genomic DNA was extracted from peripheral blood by the Puregene kit (Gentra, Minneapolis, USA), according to the manufacturer’s instructions. Whole exome sequencing was performed on the patients’ DNA. Samples were enriched with Sureselect Human All Exome v.2 kit which was targeted 50Mb (Agilent, Santa Clara, CA, USA). Sequencing was carried out on HiSeq2000 (Illumina, San diego, CA, USA) as 100-bp paired –end runs. Image analysis and base calling were performed with the Genome Analyzer Pipeline version 1.5 with default parameters. Reads were mapped to the human reference genome sequence (assembly GRCh37/hg19) using the Burrows-Wheeler Alignment Tool (BWA) version 0.5.8c, and allelic variants were detected using the Genome Analysis Toolkit (GATK). Dataset files including the annotated information were analyzed using ANNOVAR according to the dbSNP database (build 135) and the NHLBI exome variant database with the following filtering steps: variant type including missense, nonsense, and splice-site; not within segmental duplications; minor allele frequency (MAF) less than 0.01; SIFT score <0.05 when available; PolyPhen2 score >0.85 when available.
1.2.2. Transcriptome Analysis
Fibroblasts derived from patients and age-matched controls were grown in cell culture and stimulated with poly(I:C) (10µg/ml, Invivogen). Cells were harvested at indicated times and RNA was prepared with TRIZOL and DNAse1 treatment by standard methods. Strand-specific libraries were generated from 200 ng total RNA using the KAPA Stranded mRNA sequencing and Library Preparation Kit (Illumina). cDNA libraries were single-end sequenced (50bp) on an Illumina HiSeq 2000. Reads were aligned to the human genome (NCBI51/hg38) with TopHat v1.3.3 and allowed one alignment with up to two mismatches per read. mRNA RPKM values were calculated by dividing mapped exonic reads by the length of the spliced product. Downstream analysis used genes that had an RPKM≥3 in at least one sample, and showed stimulus-induction of ≥ 4-fold with a false discovery rate p-value of ≤0.01 at at least one timepoint, but were not differentially expressed in unstimulated conditions >2-fold. The log2 of the Counts per million (CPM) for all samples associated with each gene were z-scored and used for k-means clustering with k=2. Differential expression between patient and control samples was determined with a cut-off of 4-fold and false discover rate op-value <0.01. Motif analysis was done by HOMER using −1000 to +300 relative to the transcription start site of each gene.
1.2.3. Flow Cytometry
Peripheral blood cell phenotypes were determined by flow cytometry analysis using a four laser BD analyzer LSR II CFI BGRV (Becton Dickinson, Franklin Lakes, NJ). Flow cytometry was performed at The SickKids-UHN Flow and Mass Cytometry Facility, Toronto, with funding from the Ontario Institute for Cancer Research, McEwen Centre for Regenerative Medicine and the Canada Foundation for Innovation. For a list of antibodies used, see Fig E1 in the Online Repository.
For activation analyses, purified PBL were stimulated with soluble anti-CD3 (UCHT1) 2µg/ml plus anti-CD28 (CD28.2) 10µg/ml, or PMA (Sigma Aldrich) (50ng/ml) for 22–24 hours and harvested for staining.
1.2.4. Cytokine Determinations
Post-Ficoll peripheral blood mononuclear cells were cultured with PHA or anti-CD3 and anti-CD28 or PMA plus Ionomycin for 48 hours or as indicated, and culture supernatants collected for analysis by ELISA (R&D Systems – Bio Techne Canada). Serum levels of BAFF were determined using the BAFF/BLyS/TNFSF13B Quantikine ELISA kit (R&D Systems).
1.2.5. Western Blotting
Whole cell lysates were prepared in RIPA buffer and analyzed by Western blotting. Cytoplasmic and nuclear fractions were prepared using the Pierce NE-PER subcellular fractionation kit (ThermoFisher Scientific, Waltham, MA), as per the manufacturer’s instructions. Anti- RelA, c-Rel, IκBα, NFkB1 p100, actin, Rcc1, Giα, C-terminal RelB (sc-226) and N-terminal RelB antibody (sc-38007) were purchased from Santa Cruz Biotechnology Inc (TX). Anti N-terminal RelB (04-1077) was also purchased from EMD Millipore. Where necessary, expression levels were quantified by densitometry scanning and normalized by comparison with matching control protein levels. All blots were repeated at least twice.
1.2.6. NFκB DNA binding Assay
Nuclear lysates of cells were prepared with the Pierce NE-PER subcellular fractionation kit (ThermoFisher Scientific). Matching amounts of lysate were diluted in 1% NP-40 lysis buffer and NFκB pull-down performed by addition of agarose-conjugated double stranded DNA NFκB gel shift consensus binding site for NFκB complexes at 4°C for 4 hours (sc-2505- Santa Cruz Biotechnology Inc, Dallas, TX). Specificity controls were performed with agarose-conjugated mutant NFκB binding double stranded oligonucleotides (sc-2511). TNF-alpha and LIGHT/TNFSF14 were purchased from R&D Systems (MN).
1.2.7. Transfection
HEK293 cells were transfected using Lipofectamine 2000 (ThermoFisher Scientific) as per manufacturer’s instructions. RELB cDNA in pCMV6 vector was from Origene Technologies (Rockville, MD). The P364L mutation was introduced using the QuikChange XL site directed mutagenesis kit (Agilent - Santa Clara, CA) and confirmed by Sanger sequencing. Luciferase assays were performed by transfection of pNFKB Tluc16-DD and control pEF1α Tluc16-DD (Thermo Fisher scientific) and induction of luciferase activity assayed using the TurboLuc Luciferase One-Step Glow Assay Kit (Thermo Scientific).
1.2.8. RNA preparation and Analysis
RNA was purified from cells using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany) and analyzed with the Genechip PrimeView Human Gene Expression Array (ThermoFisher Scientific) at the Centre for Applied Genomics at SickKids Hospital, Toronto, with funding through Genome Canada and the Ontario Genomics Institute. RelB RNA levels were compared using quantitative PCR with Superscript IV VILO (ThermoFischer Scientific) transcribed first strand cDNA by reference to a standard curve using SYBR Select Master Mix (Applied Biosystems) with the RelB primers RELB-F AGGCAGGTACGTGAAAGGCAAT, RELB-R GCTCTGCGACAAGGTGCAGA, and normalized with endogenous control primers GAPDH-F GGCAAATTCCATGGCACCGT, GAPDH-R CACGACGTACTCAGCGCCAGCAT.
1.2.9. REB Approval
All patient studies were approved by the SickKids Research Ethics Board (Protocol no. 1000005598).
1.3. RESULTS
1.3.1. Clinical Presentation
Patient (Pt) 1, currently 18 years old, is a female born at term after a normal pregnancy and delivery to consanguineous parents (first cousins) of Iranian Jewish descent. During the first 3 years of life she experienced upper respiratory and middle ear infections. Chronic diarrhea at age 3 prompted an endoscopy, which revealed increased numbers of apoptotic cell bodies in the basal cells of the duodenal glandular crypts (Fig 1I–J). Villus atrophy and the presence of intra-epithelial lymphocytic infiltrates supported the notion of autoimmune enteritis. Apoptotic cells in the stem cell region at the neck of the gastric glands were also noted (not shown), consistent with autoimmune injury to the gastric mucosa. Subsequently, due to persistent rise in liver enzymes, a liver biopsy was performed. H&E and Masson staining showed stage 2–3 portal fibrosis with extensive portal infiltration of inflammatory cells (Fig 1A–D). The infiltrates consisted predominately of CD4+ T cells as well as plasma cells. Together, these findings demonstrate advanced chronic autoimmune liver disease. The changes were progressive over time as evidenced by a second biopsy performed at an older age.
Figure 1. Histopathology of liver, duodenum, and lung.
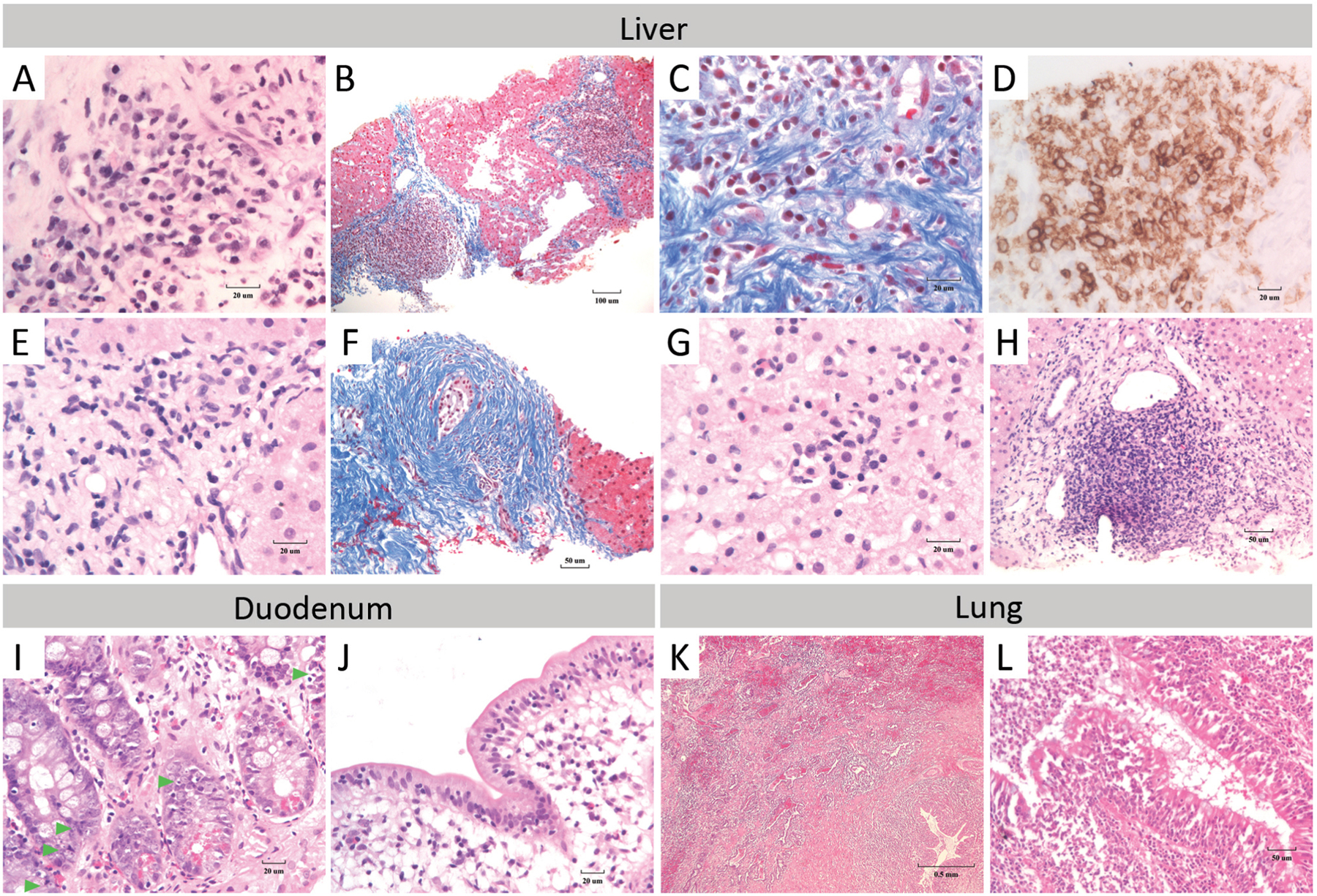
In Pt 1, (A) stage 2–3 liver portal fibrosis with localization of plasma cells in autoimmune hepatitis-like regions are shown (H&E). (B) Masson connective tissue stain demonstrates collagen deposition within the portal tract and a bridging portal-portal tract pattern in the liver. (C) Portal tissue contains heavy infiltrates of plasma cells (Masson stain). (D) Plasma cells stained with CD138 are present in autoimmune hepatitis-like regions in the liver. In Pt 2, (E) H&E staining reveals plasma cells within the lymphocyte rich hepatitis infiltrates, consistent with the finding of autoimmune hepatitis. (F) Masson staining shows stage 3 to early stage 4 portal fibrosis. Liver fibrotic areas have concentric pattern of collagen deposition around a bile duct, a pattern that is often associated with sclerosing cholangitis. Inflammatory lymphoid infiltrate with plasma cells are shown within the (G) hepatic lobule and (H) portal tract (H&E stain). (I) Abnormal presence of apoptotic activity in the basal cells of the duodenal glandular crypts of Pt 1 are shown (H&E). Green arrowheads indicate apoptotic bodies. (J) Intra-epithelial lymphocyte counts of 45 lymphocytes per 100 enterocytes with some villous atrophy coupled with the presence of marginal lengthening of the duodenal crypt (favor Marsh scale: 1). (K) Lung biopsy of Pt 1 demonstrates florid acute and chronic bronchiolitis with organizing interstitial fibrosis in the pre-bronchiolar pulmonary parenchyma (H&E) and (L) bronchiolar epithelium and lumen.
At the age of 8 years she underwent a resection of the right lower lobe of her lungs because of chronic unresolved changes. H&E staining revealed acute and chronic bronchiolitis with organizing interstitial fibrosis in the peri-bronchial parenchyma (Fig 1K–L). While Mycobacterium tuberculosis was identified in the resected lobe, the florid inflammatory changes suggested an aberrantly exaggerated inflammatory response.
Over the years, Pt 1 suffered multiple chronic cutaneous lesions which rarely grew various fungal species. Lesions became secondarily infected by bacteria resulting in cutaneous abscess or cellulitis. Skin biopsy of a lesion revealed diffuse inflammatory infiltrates consisting of T cells and plasma cells. Despite supportive treatment, the patient currently appears frail, underweight, and short.
Pt 2, the younger sister of Pt 1, is currently 12 years old. She too was born at term after a normal pregnancy and delivery. Her history is remarkable for an episode of severe and prolonged chicken pox complicated by pneumonitis and secondary skin infections occurring at 2 years of age. She suffered chronic cutaneous lesions starting at age 3 that appeared autoinflammatory in nature as response to antimicrobial treatment was inadequate. At 10 years of age, a liver biopsy was performed to evaluate a prolonged and progressive increase in liver enzymes. Remarkably, similar to her older sister, the biopsy demonstrated stage 3–4 portal fibrosis with massive infiltrates of lymphocytes and plasma cells (Fig 1E–H). Concentric peri-bile ductal fibrosis was noted, consistent with sclerosing cholangitis. She too is delayed in her linear growth and puberty.
Pt 3 is the younger male sibling of P1 and P2 and is currently 4 years old. He was born after an uncomplicated pregnancy and delivery. He appears healthier than his older sisters, and so far, his growth and development have been appropriate for age. Of special note is an episode of severe pneumonitis occurring at 1 year of age due to adenovirus infections which required prolonged hospitalization.
In addition to the noted progression of symptoms in individual patients, the age of the patients appeared correlated with the severity of their symptoms, the youngest being healthiest and the oldest manifesting the most severe disease, in line with a slowly progressive illness.
1.3.2. P364L mutation of RelB
Whole exome sequencing revealed a homozygous change within the coding region of the RELB gene of all three patients (Fig 2A), which was not present in public databases. The c.C1091T transition resulted in a predicted amino acid substitution of leucine for proline at position 364 (P364L) within the translated protein. Sanger sequencing of genomic DNA confirmed the presence of the homozygous mutation in all three patients, whereas both parents and an unaffected sibling were all heterozygous. Proline 364 falls within the IPT (Immunoglobulin-like fold, Plexins, Transcription factors) domain of RelB that mediates dimerization of RelB with other members of the NFκB family [20, 21]. Although P364 is not within the dimerization interface itself, it is a highly conserved residue across NFκB family members (Fig 2B), raising the possibility that this leucine - proline substitution could affect RelB integrity and function.
Figure 2. Structural analysis and expression of the P326L RelB mutant.
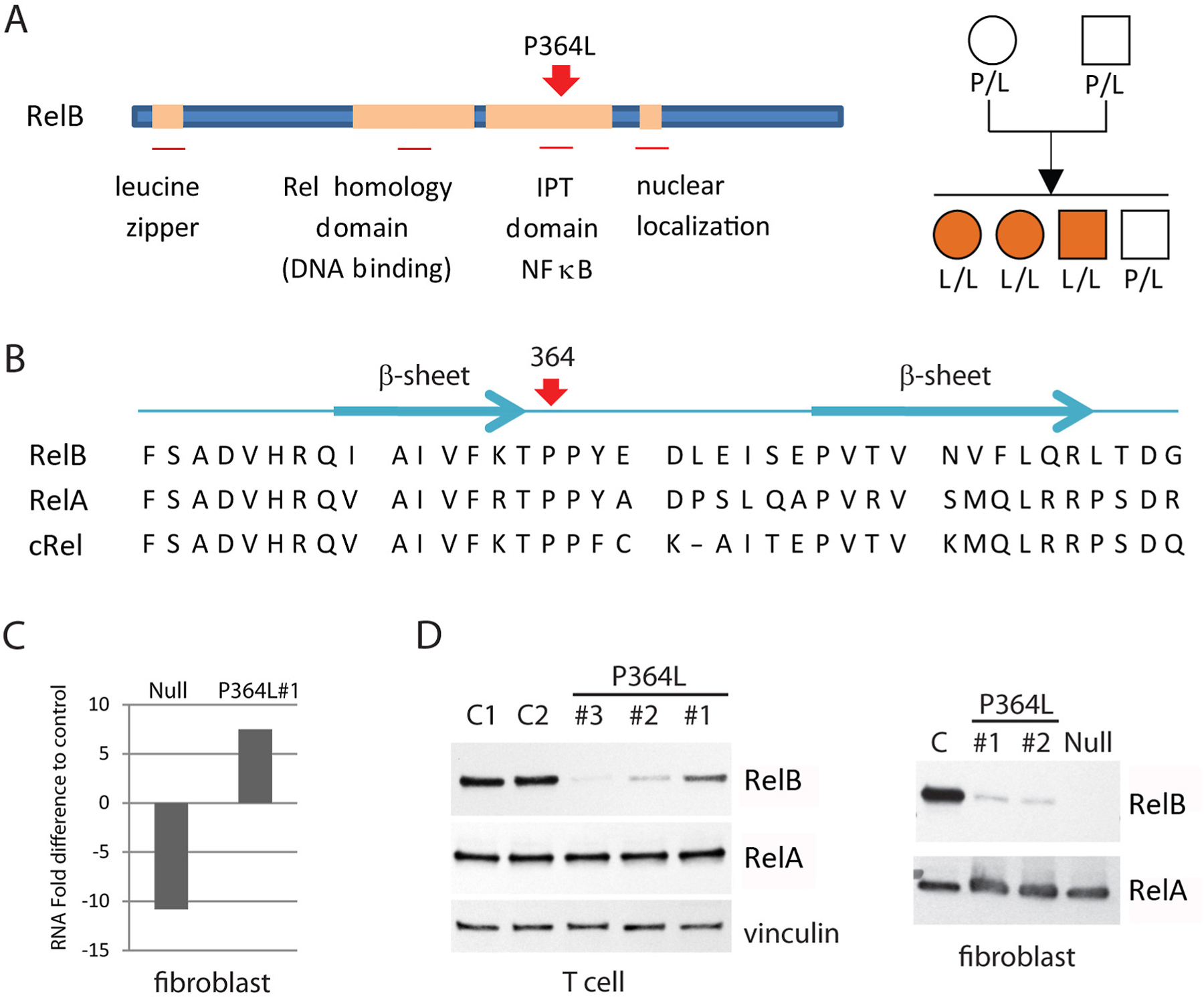
(A) Schematic diagram of RelB structure indicating functional domains, including the position of the P364L mutation (left), and pedigree of the affected family (right). Circles represent female subjects and squares denote male subjects. Orange symbols represent affected patients. IPT=Immunoglobulin-like fold, Plexins, Transcription factors. (B) Position P364 is highly conserved in IPT NFκB transcription factors. Human RelB, RelA and c-Rel sequences with secondary structural elements adjacent to P364 indicated. (C) Levels of RelB RNA in RelB null and P364L patient fibroblasts relative to normal control cells. (D) Western blot of RelB and control RelA protein expression in patient and normal primary cells as well as fibroblasts. C = control; Null = complete RelB deficient patient fibroblast.
Quantitative PCR analysis of mRNA revealed that RelB message levels were higher in P364L cells than normal control and RelB null patient cells (Fig 2C). However, Western blot analysis showed a marked reduction in RelB protein expression in all patients relative to controls, although the amount remaining appeared somewhat variable (Fig 2D). This suggested that the P364L mutation may destabilize the RelB protein, resulting in its degradation. Unlike previously identified RelB deficient patients, in whom a complete ablation of RelB expression occurred, the presence of residual P364L RelB protein suggested that despite significant RelB deficiency, some function, normal or otherwise, was likely to remain, and that this contributes to the very different clinical presentation of these patients.
1.3.3. Age-related decline in B and T cell compartments
In vitro T cell responses to anti-CD3 or PHA declined over time in all three patients (Table 1). Flow cytometry analyses showed that CD4+ and CD8+ peripheral T cells were present within normal range both for absolute numbers and CD4:CD8 ratio (Table 1). However, detailed analysis revealed a highly abnormal percentage of CD45RO+ cells within the CD4+ peripheral T cell pool to the extent of complete absence of naïve peripheral CD4+ T cells (CD45RO-CCR7+) in Pt 1, severely reduced numbers in Pt 2, and already a significant reduction in the younger Pt 3, indicating the strong correlation with age (Fig 3A). A similar pattern was observed in the CD8+ peripheral T cell pool (Fig 3B). Analysis of CCR7 co-expression demonstrated that the CD45RO+ cells were of both central memory and effector memory phenotypes within the CD4+ population, and predominantly effector memory and terminally differentiated memory cells within the CD8+ pool. Flow cytometry also revealed that the Treg compartment (CD3+CD4+CD127-CD25+) was expanded in all three patients (Fig 3C).
Table 1:
Immune function of the patients
| Patient 1 | Patient 2 | Patient 3 | Normal range | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 2.5 | 7 | 16 | 1 | 2 | 12 | 1 | 3 | |
| Markers (cells/µL) | |||||||||
| CD3 | 3277 | 2537 | 2318 | N/D | 2696 | 1637 | 2428 | 5518 | 700-4200 |
| CD4 | 3072 | 1577 | 1532 | N/D | 2081 | 1049 | 1862 | 4647 | 300-2000 |
| CD8 | 409 | 926 | 764 | N/D | 614 | 525 | 567 | 653 | 300-1800 |
| CD19/20 | 2185 | 411 | 23 | N/D | 2648 | 107 | 1255 | 1579 | 200-1600 |
| CD56 | 203 | 308 | 139 | N/D | 473 | 98 | 604 | 726 | 90-900 |
| Mitogen responses (% of control) | |||||||||
| PHA | N/D | 28 | 5 | N/D | 100 | 39 | 120 | 18 | >50% |
| aCD3 | N/D | 64 | <1 | N/D | 90 | 37 | 10 | 3 | >50% |
| Immunoglobulin (g/L) | |||||||||
| IgG | 18.3 | 9.1 | 5.61 | 19.2 | 16.8 | 12.9 | 15.3 | 10.6 | 5.0–14.6 |
| IgA | 0.2 | 0.75 | 0.24 | 0.375 | 3.38 | 0.45 | 0.17 | 1.53 | 0.3–2.0 |
| IgM | 4.9 | 0.8 | 0.82 | 1.06 | 1.22 | 0.72 | 4.09 | 0.23 | 0.2–2.5 |
Figure 3. Premature accumulation of mature activated T cells.
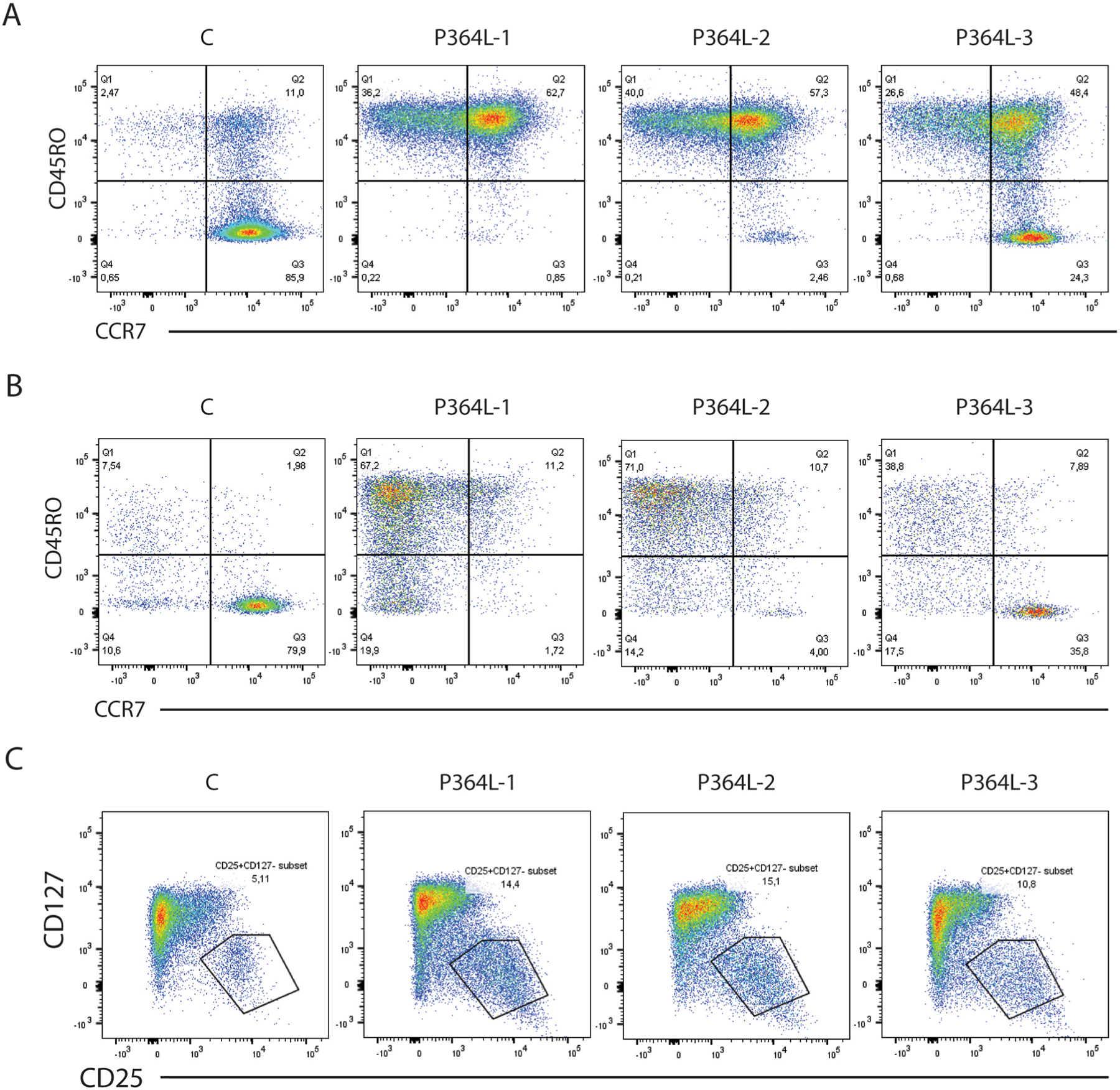
(A) Staining of CD3+CD4+ normal and patient T cells for expression of CD45RO and CCR7 reveals accelerated age-dependent loss of naïve CD45RO- CCR7+ T cells, with accumulation of CD45RO+CCR7+ central memory and CD45RO+CCR7- effector memory cells. C = normal control subject, 20 years old; P364L-1 (P1), 18 years old; P364L-2 (P2), 12 years old; P364L-3 (P3), 4 years old. (B) Staining of CD3+CD8+ normal and patient T cells for expression of CD45RO and CCR7 reveals a loss of naïve CD8+ T cells. (C) Expansion of the CD3+CD4+CD127- CD25+ Treg compartment in P364L RelB patients.
Patients also suffered progressive loss of the entire B cell compartment. Two of the P364L RelB patients demonstrated a marked reduction in CD19+ B cell numbers (Fig 4A), which appeared to be strongly age-dependent, with the youngest (4 yrs) showing normal levels, whilst the eldest (18 yrs) had few cells remaining, in line with studies suggesting a role for RelB in promoting B cell survival, through its action downstream of the survival promoting BAFF receptor. A further reduction in CD27+ memory populations was observed, even in the 4yr old (Fig 4B). These age-correlated anomalies coincided with the gradual decline of the patients’ ability to produce specific antibodies in response to vaccination and necessitated immunoglobulin replacement therapy.
Figure 4. Age-dependent loss of B cell compartment with reduced B cell memory.
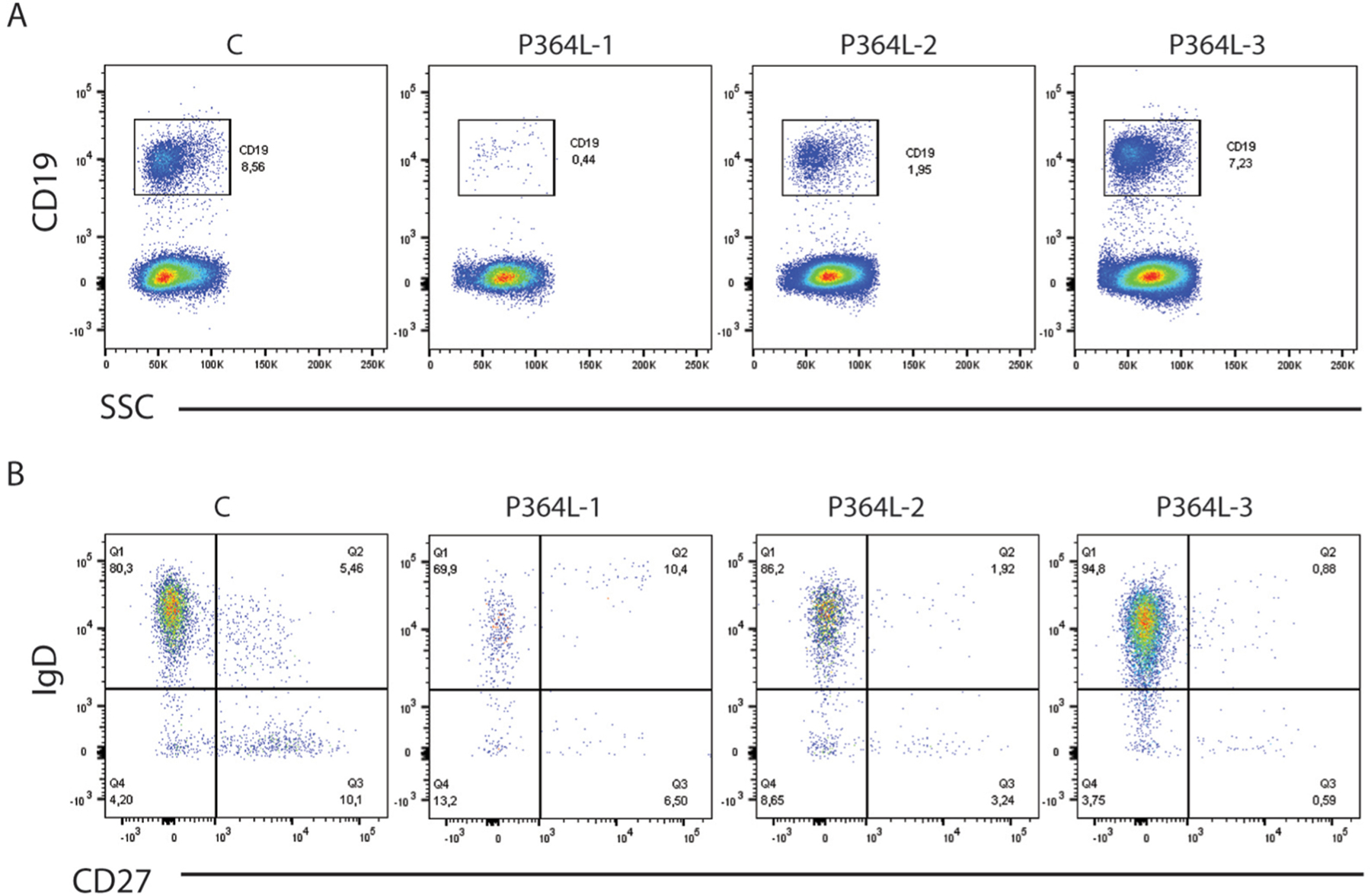
(A) Percentage of CD19+ cells in total lymphocytes. SSC: side scatter. (B) Staining for CD19+ B cells for IgD and CD27 expression reveals decreased IgD+CD27+ memory and IgD-CD27+ class switched memory B cells. C = normal control subject, 20 years old; P364L-1 (P1), 18 years old; P364L-2 (P2), 12 years old; P364L-3 (P3), 4 years old.
1.3.4. P634L RelB Stability and Function
The level of P346L RelB was extremely low in patient’s lymphocytes and fibroblasts. This is likely due to rapid degradation as treatment of patient fibroblasts with inhibitors to block proteasome-mediated protein degradation (MG132 or Lactocystin) rapidly increased levels of the P364L RelB protein (Fig 5A). Indeed, a variety of stimuli could increase dramatically mutant RelB expression that rapidly declined upon removal of the TLR4 ligand LPS (Fig 5C). Similarly, Pam3CSK4 (TLR2 ligand) or poly(I:C) (TLR3 ligand) induced a transient P364L RelB spike in expression (Fig 5D).
Figure 5. P364L RelB instability and TLR-mediated induction.
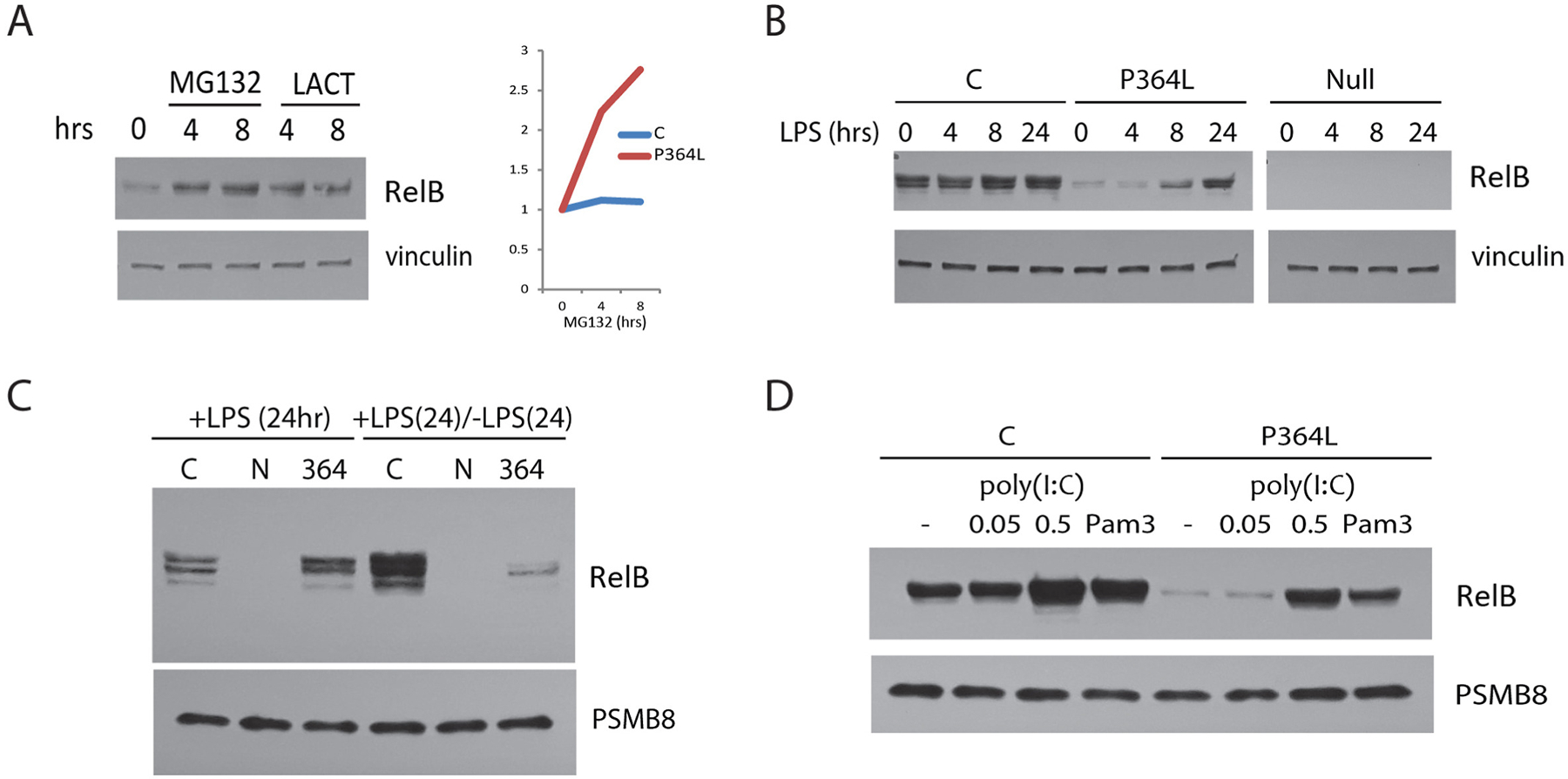
(A) Brief treatment with MG132 or Lactocystin proteasomal inhibitors results in a rapid increase in P364L RelB protein expression in patient cells (left panel). In comparison, normal cells (C) demonstrate little change in RelB protein level (right panel). (B) LPS treatment up-regulates RelB protein expression in P364L RelB patient fibroblasts. (C) Removal of LPS after 24 hours, results in rapid loss of the induced P364L RelB protein to pre-stimulation levels. (D) Activation of TLR2 and TLR3 by Pam3CSK4 (Pam3) and poly(I:C), respectively, can also increase P364L RelB expression.
Although RelB levels were typically much lower than controls, P364L RelB could be seen to undergo nuclear localization in patient fibroblasts stimulated with the Lymphotoxin-β-receptor (LTβR, TNFRSF14) ligand LIGHT (TNFSF14) (directly activating both classical and alternative NFκB signaling pathways [22]) (Fig 6A). Furthermore, both RelA and P364L RelB in nuclear extracts of patient EBV B cell lines demonstrated the ability to bind an NFκB consensus DNA sequence (Fig 6A, 6B).
Figure 6. Retention of functional properties by P364L RelB.
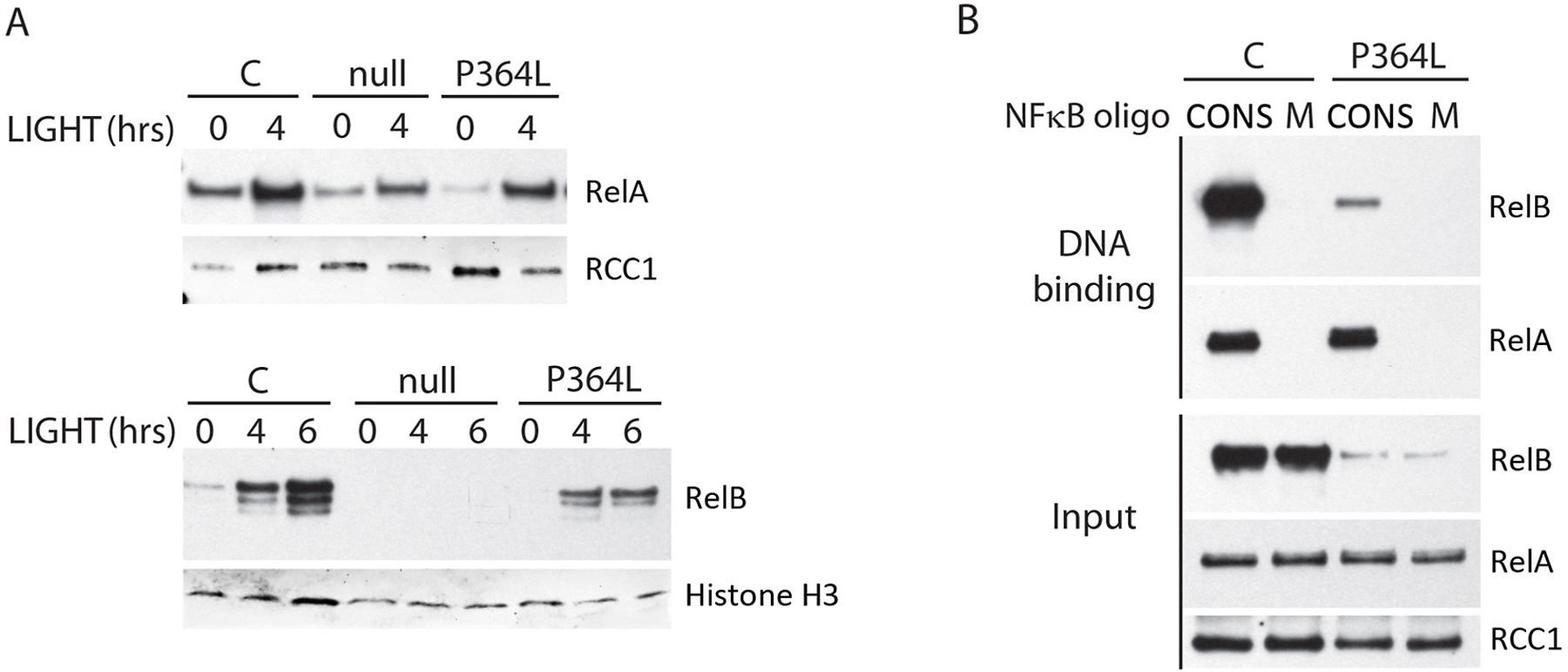
(A) RelA and P364L RelB both undergo nuclear transit in response to LIGHT in patient fibroblasts. Nuclear lysates of LIGHT-stimulated patient (P364L) and control (C) cells (100ng/ml) were immunoblotted for RelB. Null = complete RelB deficient patient fibroblasts. (B) P364L RelB in nuclear lysates of patient EBV-B cell lines binds a double-stranded consensus NFκB DNA binding sequence, but not a mutated control sequence. CONS = double stranded NFκB consensus sequence oligonucleotide. M = mutated non-functional double stranded NFκB consensus sequence oligonucleotide. Blotting of input nuclear lysate for RCC1 was used as a loading control.
While retaining some functionality and transiently responding to a group of stimulated receptors, the very low endogenous level at which P364L RelB was expressed in patient cells suggested that it was unlikely to be effective in transmitting alternative pathway signaling, or importantly, in negatively regulating RelA.
1.3.5. Hyper activation downstream of the classical pathway.
Most NFκB activity in cells is due to activation of the RelA protein as a part of the canonical NFκB pathway. RelB is directly activated by only a limited number of cell surface receptors; however, it is co-expressed with RelA in many cell types. Although not activated in the canonical pathway, the presence of RelB is thought to act as a negative regulator of RelA activity through direct hetero-dimerization and we have used this human model to test the hypothesis that classical pathway activation may be dysregulated.
The pathogen-associated molecular pattern (PAMP) poly(I:C) used for modeling innate immune responses, activates both the interferon-response factor 3 (IRF3) pathway and the RelA-mediated NFκB pathway through Toll like receptor 3 (TLR3), but not the alternative RelB-mediated pathway. Stimulation of P364L patient fibroblasts with poly(I:C) followed by RNA-seq analysis revealed a set of hyper-expressed genes in cluster A of our analysis, while the majority of genes comprised in the larger cluster B were equivalently induced. Analysis of genes within these clusters revealed an essentially normal type 1 interferon gene expression response, while induction of a subset of NFκB target inflammatory genes was elevated in both patient samples examined (Fig 7A). This suggested that the innate immune response may skew towards inflammation, consistent with the significant auto-inflammatory phenomena seen in the patients. Regulatory motif analysis revealed IRF and ISRE as the top result of normally expressed genes within cluster B, while NFκB was the top hit for genes in hyper-expressed cluster A (Fig 7B). Among these hyper-expressed inflammatory genes in cluster A were NFκB activators Cgas, Irak2, and Traf1, which is also known to activate MAPK/JNK signaling [23, 24]. Additionally, several NFκB target genes were also among the hyper-expressed genes in cluster A such as negative regulators of NFκB: Nfkbia, Nfkbie, Nfkbiz, and Tnfaip3, and the pro-inflammatory cytokine, Ccl2. Interestingly, Icam1, another activator and target gene of MAPK/JNK signaling [25], was also hyper-expressed in both patients and downstream MAPK signal transducer, Junb, and downstream MAPK transcription factor, Egr1, was hyper-expressed along with Egr1-target gene, Tnfaip3 [26, 27].
Figure 7. Elevated subset of NFκB transcripts in response to classical pathway activation.
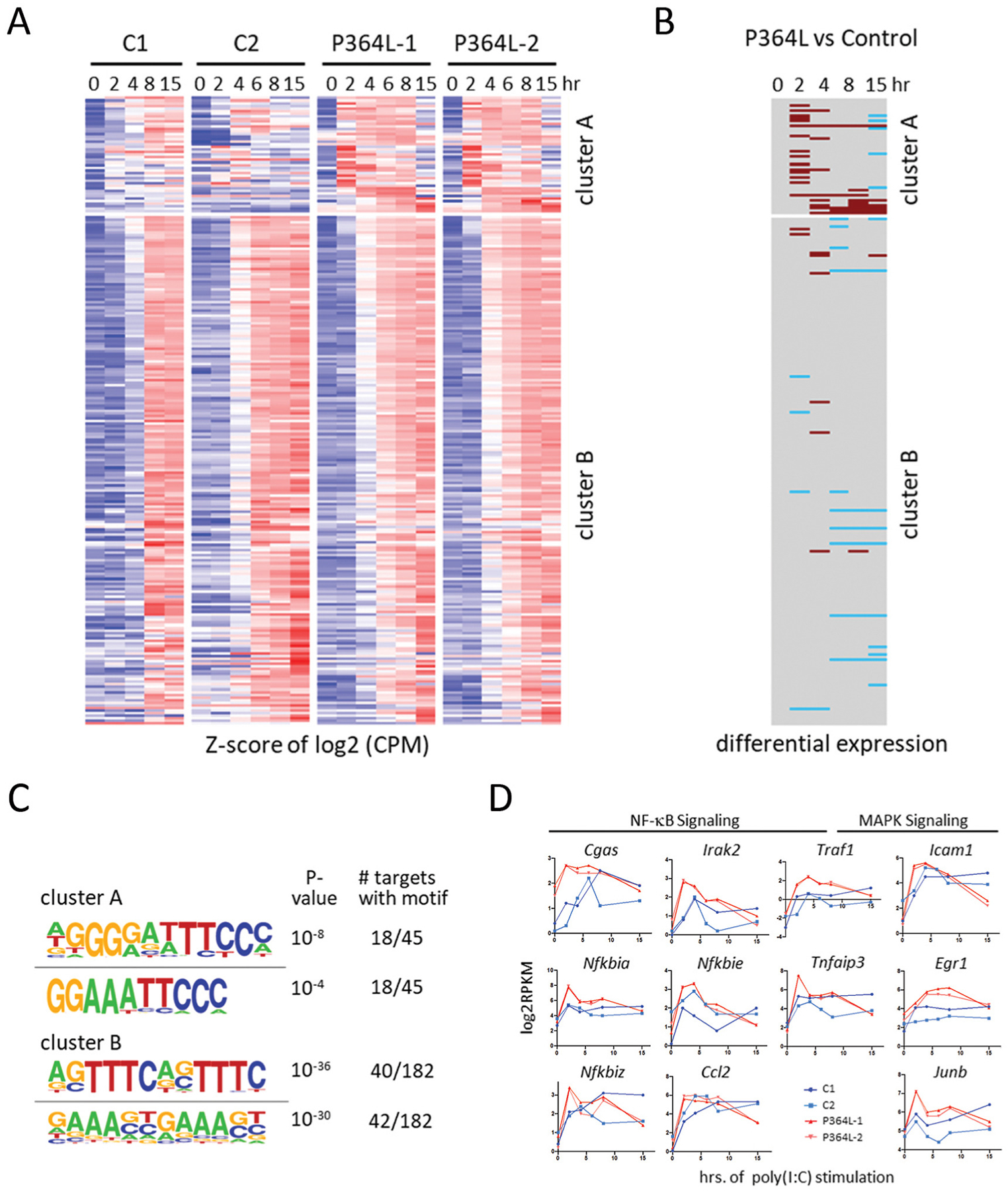
(A) Heatmap of z-scored log2 transcript levels based on RNA-seq timecourse analysis of poly(I:C)-stimulated fibroblasts derived from indicated patients or healthy controls. Data was clustered by k-means into 2 clusters. (B) Genes and timepoints that show differential expression of >4 fold with a false discovery rate p-value of <0.01 are indicated. Hyper- and hypo-expression in patient fibroblasts is indicated in brown and blue, respectively. (C) Motif-analysis of regulatory regions (−1000 to +300 from TSS) yields the indicated top two motifs for clusters A and B. (D) Some of the genes in cluster A.
Similar results were obtained by studying LIGHT ligand induced RelB and RelA dependent gene expression (Fig E2–E4, Online repository). Dramatic abnormal upregulation of a subset of RelA-dependent pro-inflammatory cytokines and chemokines including CXCL8, CXCL5, CCL5 and CXCL6 was revealed uniquely in P364L RelB fibroblasts (Fig E2, Online Repository). This again strongly suggested a loss of RelB negative regulation of RelA
Together, these data suggested that there is hyper-activation of classical NFκB and MAPK signaling, potentially skewing the innate immune response towards hyper-inflammation, and consistent with models predicting reduced negative cross-regulation of RelA by RelB.
1.4. DISCUSSION
We have described here three siblings suffering predominately from severe autoimmune manifestations involving the liver, gut, lung, and skin, as well as repeated infection, and the range of patient ages (4 to 18 years) has helped define a pattern of age-dependent deterioration of clinical features and progressive failure of the immune system. The oldest patient (Pt 1, 18yrs) at her most recent evaluation was found to be very short, underweight, and suffering from chronic diarrhea as well as chronic severe lung, liver, and skin manifestations. Her sister (Pt 2), at the age of 12 years, has already begun developing liver disease identical to her older sister, but is neither as short nor frail. Thus far, the youngest sibling (Pt 3), age 4 years, has been growing and developing and has had only one severe infectious episode, although flow cytometry analyses suggest he is likely to progress down the same path as his siblings. Together with the longitudinal long-term follow-up, this has highlighted the progressive nature of inflammation and autoimmunity in these patients. Paralleling their time-dependent clinical deterioration, age-dependent declines in circulating naïve T cells, in vitro T cell proliferation, circulating CD19+ B cells and CD27+ memory B cells were all documented in P354L RelB patients. Consistent with this, a decline in the ability to respond to vaccination by the production of specific antibodies was observed, necessitating immunoglobulin replacement.
WES revealed a homozygous mutation in RelB (P364L) in all three patients while parents carried one abnormal allele each. Evaluation of P364L RelB protein by Western blotting revealed markedly reduced levels compared to control, but not a complete absence of RelB as observed in RelB null cases [17, 18].
RelB is considered an intrinsically unstable protein in resting cells, requiring interaction with NFκB p100 and other binding partners for protection from proteolytic degradation [28]. The P364L mutation falls within the IPT (Immunoglobulin-like fold, Plexins, Transcription factors) domain of RelB, which mediates dimerization of RelB with other members of the NFκB family. It is possible that this mutation, while not residing directly within the dimerization interface of the RelB IPT domain, may still interfere with protein-protein interactions - wherein stabilization of RelB within complexes is limited compared to wild-type species. Treatment with inhibitors of proteasome-mediated degradation rapidly increased P364L RelB protein levels in patient cells, consistent with this idea.
We have shown that under conditions of TLR stimulation, expression of P364L RelB protein was significantly induced, possibly due to improved interaction with its partners resulting in increased stability. This effect was, nevertheless, both delayed and transient, suggesting that RELB dependent responses would be at best, only partially restored. Even under optimal conditions, restoring P364L RelB to near wild type levels in vitro prior to stimulation, fibroblasts responses to LIGHT were not normalised. It is plausible to hypothesize that this minimal restoration of RelB function over a null background may be sufficient to delay the rapid decline in immune function.
To better understand the putative mechanism leading to a prominent clinical course of severe autoimmunity we have used several models. We have used the PAMP poly(I:C), which activates innate immune responses through both the IRF3 and the RelA mediated pathways, to stimulate patient fibroblasts and analysed the resultant gene expression by RNA-seq. The analysis revealed clusters of proinflammatory hyper-expressed transcripts within the NFκB activator category as well as NFκB target genes. This strongly suggested that the innate immune response may skew towards inflammation.
In a second model, we have stimulated patient fibroblasts with LIGHT which allows the investigation of both classical and alternative NFκB activation. LIGHT activation has been implicated before in playing a role in the pathogenesis of autoimmune disorders like rheumatoid arthritis where it has been shown to induce pro-inflammatory cytokines and chemokines in synoviocytes 30,31. Stimulation of P364L fibroblasts with LIGHT resulted in hyper-induction of a variety of pro-inflammatory genes.
While most NFκB activity in the cell in response to multiple stimuli is mediated by RelA in the canonical pathway, only five receptors stimulate RelB directly in the alternative pathway. However, it was previously thought that RelB presence may act as a negative regulator of RelA activity. Together, the data from both our models demonstrating hyper-activation of the RelA dependent classical pathway strongly supports the hypothesis predicting reduced negative cross-regulation of RelA by RelB.
In sum, we have shown here that reduced cellular levels of RelB caused by the P364 mutation leads to dysregulation of the NFκB main pathway with marked skewing towards pro-inflammation. This provides evidence, albeit indirect, for cross-talk between the alternative and classical NFκB pathways, and provides insights into the development of severe autoimmune disorder and declining immune system in these patients.
Supplementary Material
HIGHLIGHTS.
Homozygous P364L RelB mutation is associated with severe autoimmunity and combined immunodeficiency, involving an age-dependent decline in T cell function and inability to produce specific antibodies
The P364L mutation results in the retention of some residual RelB protein that can be upregulated and retains some functional activities
Over time, P364L RelB naïve T cells and all B cells are lost
ACKNOWLEDGEMENTS
This work was supported by the Immunodeficiency Canada Distinguished Professorship in Immunology (CMR), the Program for Immunogenomics, the Canadian Centre for Primary Immunodeficiency, the Jeffrey Modell Foundation, and Immunodeficiency Canada.
ABBREVIATIONS
- AU
arbitrary unit
- BAFF
B-cell activating factor
- BAFF-R
BAFF receptor
- CCR7
C-C chemokine receptor type 7
- EBV
Epstein-Barr virus
- HEK293
human embryonic kidney 293
- IKK1
IκB kinase alpha
- IKK2
IκB kinase beta
- IFN-γ
interferon gamma
- IL-2
interleukin-2
- IPT
immunoglobulin-like fold, Plexins, Transcription factors
- NEMO
NFκB Essential Modulator
- NFκB
nuclear factor kappa-B
- NFκB1
nuclear factor kappa-B subunit 1
- NFκB2
nuclear factor kappa-B subunit 2
- LIGHT
tumor necrosis factor superfamily member 14
- LPS
lipopolysaccharide
- LTβR
lymphotoxin beta receptor
- NEMO
NFkB Essential Modulator
- PHA
phytohemagglutinin
- Poly(I:C)
Polyinosinic:polycytidylic acid
- SCID
severe combined immunodeficiency
- TCR
T cell receptor
- TCRγδ
Gamma-delta T cell receptor
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor alpha
- TNFR
tumor necrosis factor receptor
- TNFRSF12A
TNF Receptor Superfamily Member 12A
- TNFRSF14
TNF Receptor Superfamily Member 14
- TRAF
TNF receptor associated factor
- TRAF1
TNF receptor associated factor 1
- TRAF2
TNF receptor associated factor 2
- TRAF3
TNF receptor associated factor 3
REFERENCES
- [1].Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest, 2003;112:1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet, 2001;27:277–85. [DOI] [PubMed] [Google Scholar]
- [3].Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J et al. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J Exp Med, 2004;200:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood, 2014;124:2964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lopez-Granados E, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat, 2008;29:861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol, 2004;113:725–33. [DOI] [PubMed] [Google Scholar]
- [7].Pannicke U, Baumann B, Fuchs S, Henneke P, Rensing-Ehl A, Rizzi M et al. Deficiency of innate and acquired immunity caused by an IKBKB mutation. The New England journal of medicine, 2013;369:2504–14. [DOI] [PubMed] [Google Scholar]
- [8].Picard C, Casanova JL, Puel A Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev, 2011;24:490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schimke LF, Rieber N, Rylaarsdam S, Cabral-Marques O, Hubbard N, Puel A et al. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J Clin Immunol, 2013;33:1088–99. [DOI] [PubMed] [Google Scholar]
- [10].Millet P, McCall C, Yoza B RelB: an outlier in leukocyte biology. Journal of leukocyte biology, 2013;94:941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sun SC Non-canonical NF-kappaB signaling pathway. Cell research, 2011;21:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Oeckinghaus A, Hayden MS, Ghosh S Crosstalk in NF-kappaB signaling pathways. Nature immunology, 2011;12:695–708. [DOI] [PubMed] [Google Scholar]
- [13].Comrie WA, Faruqi AJ, Price S, Zhang Y, Rao VK, Su HC et al. RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. J Allergy Clin Immunol, 2018;141:1507–10 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Badran YR, Dedeoglu F, Leyva Castillo JM, Bainter W, Ohsumi TK, Bousvaros A et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J Exp Med, 2017;214:1937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. American journal of human genetics, 2013;93:812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA Novel NFKB2 mutation in early-onset CVID. J Clin Immunol, 2014;34:686–90. [DOI] [PubMed] [Google Scholar]
- [17].Sharfe N, Merico D, Karanxha A, Macdonald C, Dadi H, Ngan B et al. The effects of RelB deficiency on lymphocyte development and function. Journal of autoimmunity, 2015;65:90–100. [DOI] [PubMed] [Google Scholar]
- [18].Merico D, Sharfe N, Hu P, Herbrick J-A, Roifman CM RelB deficiency causes combined immunodeficiency. LymphoSign Journal, 2015;2:147–55. [Google Scholar]
- [19].Ovadia A, Dinur Schejter Y, Grunebaum E, Kim VH, Reid B, Schechter T et al. Hematopoietic stem cell transplantation for RelB deficiency. J Allergy Clin Immunol, 2017;140:1199–201 e3. [DOI] [PubMed] [Google Scholar]
- [20].Ryseck RP, Novotny J, Bravo R Characterization of elements determining the dimerization properties of RelB and p50. Mol Cell Biol, 1995;15:3100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vu D, Huang DB, Vemu A, Ghosh G A structural basis for selective dimerization by NF-kappaB RelB. J Mol Biol, 2013;425:1934–45. [DOI] [PubMed] [Google Scholar]
- [22].Norris PS, Ware CF The LTβR Signaling Pathway. Madame Curie Bioscience Database [Internet], Austin (TX): Landes Bioscience; 2000–2013. [Google Scholar]
- [23].Meylan E, Tschopp J IRAK2 takes its place in TLR signaling. Nature Immunology, 2008;9:581–2. [DOI] [PubMed] [Google Scholar]
- [24].Mendoza-Rodríguez M, Arévalo Romero H, Fuentes-Pananá EM, Ayala-Sumuano J-T, Meza I IL-1β induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Letters, 2017;390:39–44. [DOI] [PubMed] [Google Scholar]
- [25].Lebedeva T, Dustin ML, Sykulev Y ICAM-1 co-stimulates target cells to facilitate antigen presentation. Current Opinion in Immunology, 2005;17:251–8. [DOI] [PubMed] [Google Scholar]
- [26].Shan J, Dudenhausen E, Kilberg MS Induction of early growth response gene 1 (EGR1) by endoplasmic reticulum stress is mediated by the extracellular regulated kinase (ERK) arm of the MAPK pathways. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 2019;1866:371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Colombo GI, Lai T-Y, Wu S-D, Tsai M-H, Chuang EY, Chuang L-L et al. Transcription of Tnfaip3 Is Regulated by NF-κB and p38 via C/EBPβ in Activated Macrophages. PLoS ONE, 2013;8:e73153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fusco AJ, Savinova OV, Talwar R, Kearns JD, Hoffmann A, Ghosh G Stabilization of RelB requires multidomain interactions with p100/p52. J Biol Chem, 2008;283:12324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.