

Abstract
The binding of iodine-labelled plasminogen to Helicobacter pylori CCUG 17874 was characterized. Inhibition of the binding was observed after preincubation of H. pylori cells with nonradiolabelled plasminogen, lysine, or the lysine analogue ɛ-aminocaproic acid. Fragments of plasminogen, kringles 1 to 3, kringle 4, and mini-plasminogen, were also studied as potential inhibitors. Mini-plasminogen caused total inhibition of the plasminogen binding, while the other fragments caused only partial inhibition. These findings suggest that H. pylori binds specifically the fifth kringle structure of the plasminogen molecule. Plasminogen binding to H. pylori seems to be independent of culture media and independent of the presence of the cytotoxin-associated CagA antigen. Immunoblot analysis identified two plasminogen binding proteins of 57 and 42 kDa. Scatchard plot analysis revealed one binding mechanism with a Kd value of 7 × 10−7 M. Conversion of H. pylori cell-bound plasminogen to plasmin in the presence of a tissue-type plasminogen activator was demonstrated by digestion of the chromogenic substrate S-2251. No activation was noted when plasminogen or tissue-type plasminogen activator was incubated with H. pylori cells alone. Formation of H. pylori cell surface-bound plasmin may be important to provide a powerful proteolytic mechanism for gastric tissue penetration in type B gastritis and peptic ulcer disease, since plasmin degrades not only fibrin but also extracellular matrix proteins such as various collagens and fibronectin.
Human gastric disorders such as type B gastritis and peptic ulcer disease are associated with the pathogen Helicobacter pylori (8, 20). H. pylori is known to interact with gastric mucins and binds to gastric epithelial cells via specific surface proteins (4, 9, 10, 39). H. pylori also interacts with extracellular matrix (ECM) proteins, such as laminin, collagen type IV, and vitronectin, associated with subepithelial basement membranes (31, 38, 44), which can be exposed after disruption of the gastric epithelial cells. These interactions may be important for the development of subepithelial tissue damage in chronic type B gastritis and gastric and duodenal ulcers. We previously reported that H. pylori interacts with plasminogen (15, 32) and have now further defined the characteristics of binding and activation of plasminogen to plasmin on the cell surface of H. pylori CCUG 17874.
Plasminogen is a plasma and extracellular matrix glycoprotein and is composed of a 92-kDa single chain in its native form. Activators such as urokinase (uPA) and tissue type plasminogen activator (tPA) convert plasminogen to plasmin, which is an active form of the molecule composed of one A chain and one B chain connected by two disulfide bridges (7, 43). The A chain consists of five kringle (or loop) structures with pronounced internal homology. These kringles have lysine binding sites, which are responsible for the binding to fibrin. The main function of plasminogen is to mediate fibrinolysis in normal hemostasis, a process in which fibrin is degraded to fibrin fragments. However, plasmin may also degrade ECM proteins such as collagens to matrix fragments. All of these plasmin activities are controlled by specific inactivators, such as type I plasminogen activator inhibitor (PAI-1), which regulates pericellular plasmin generation by inhibiting uPA and tPA (43).
Plasminogen receptors are present on leukocytes, platelets, and the cell surfaces of several bacterial pathogens such as group A, C, and G streptococci, Staphylococcus aureus, Neisseria meningitidis, and Borrelia burgdorferi (13, 16, 18, 19, 26, 30, 40–42). Cell surface-bound plasminogen is easily activated to plasmin, which might enable bacterial pathogens binding plasminogen or plasmin to utilize the ECM digestive properties of plasmin to penetrate infected tissues (18, 24). In the case of H. pylori, a similar mechanism may thus contribute to the maintenance of chronic type B gastritis.
MATERIALS AND METHODS
Bacterial strains.
H. pylori CCUG 17874 was obtained from the Culture Collection, University of Gothenburg, Gothenburg, Sweden. CagA-negative H. pylori strains, G12, G 50, G104, G198, were originally isolated at the hospital in Grosseto, Italy (45), and were obtained from Thomas Borén, Department of Oral Biology, Umeå University, Umeå, Sweden. The H. pylori strains were grown on agar supplemented with horse blood (GAB-Camp medium) and incubated for 2 to 3 days at 37°C under microaerophilic conditions (37). To compare the influence on plasminogen binding of different culture media, H. pylori CCUG 17874 was also grown for 24 h at 37°C under microaerophilic conditions in GB broth supplemented with 5% horse serum (36). After being harvested, the bacteria were washed twice in 0.07 M phosphate-buffered saline (PBS) (pH 7.2), centrifuged at 1,000 × g for 20 min, and resuspended to a final concentration of 109 cells ml−1 in PBS.
Binding assay.
Plasminogen (Sigma, St. Louis, Mo.) was labelled with 125I (Amersham, Little Chalfont, United Kingdom) by a modified chloramine-T method with Iodobeads (Pierce, Rockford, Ill.) (25). Aprotinin, an inhibitor of plasmin (Bayer, Leverkusen, Germany), was added at 100 KIU ml−1 to all buffers containing plasminogen. The binding assay was performed as described previously (29). Briefly, radiolabelled plasminogen (50 μl, containing approximately 3 × 104 cpm) in PBS (pH 7.2) containing 1% bovine serum albumin (BSA) (Boehringer GmbH, Mannheim, Germany) was incubated with 100 μl of a bacterial cell suspension (108 cells) at 20°C for 1 h. After the addition of 2 ml of ice-cold PBS containing 0.1% Tween 20 (Kebo Lab, Spånga, Sweden), the mixture was centrifuged at 1,000 × g for 20 min. The supernatant was aspirated, and the radioactivity in the pellet was counted in a 1260-Multigamma counter (LKB-Wallac, Turku, Finland). The amount of protein bound was expressed as a percentage of the total amount of 125I-protein added to 108 bacteria. The binding assays were usually performed at pH 7.2, but they were also carried out at pH 2, 4, 6, 9, and 11 to investigate the pH dependence of plasminogen binding to H. pylori cells. All the tests were performed three times in duplicate.
Inhibition assay.
H. pylori CCUG 17874 cell suspensions (100 μl of a suspension of 109 cells ml of PBS−1) were preincubated for 1 h with 1 × 107 to 7.6 × 107 nM concentrations of the following inhibitors: unlabelled plasminogen (Sigma), ɛ-aminocaproic acid (EACA) (Sigma), lysine (Sigma), or the plasminogen fragments kringles 1 to 3 (K1–3), kringle 4 (K4), and mini-plasminogen (mini-plg). These fragments were a kind gift from Björn Wiman, Department of Clinical Chemistry, Karolinska Hospital, Stockholm, Sweden. BSA (10 to 50 mg ml−1) and fibrinogen (Sigma) (1 to 3 mg ml−1) were also used as inhibitors, as described above. Thereafter, radiolabelled plasminogen was added and the binding assay was performed as described above.
Scatchard plot analysis.
H. pylori cells (108 bacteria) were incubated with increasing amounts of plasminogen (1 to 50 μg of a mixture of 125I-labelled and unlabelled plasminogen in PBS–1% BSA) for 1 h at room temperature. The experiments were performed in duplicate. The amount of cell-bound plasminogen was determined and plotted against the amount of plasminogen added, to achieve a saturation curve. Finally, the proportion of bound to free plasminogen was plotted against the amount of bound plasminogen, and the Kd value was calculated (17, 34).
SDS-PAGE and immunoblot assay.
Surface proteins of H. pylori CCUG 17874 that had been grown on GAB-Camp agar were extracted by distilled water. Before extraction, the bacteria were harvested and washed twice in PBS (1,000 × g for 10 min). The bacteria were then resuspended in sterile distilled water (10 ml g of bacteria−1), incubated for 30 min at 20°C, and centrifuged (12,100 × g for 20 min). As a control, the same extraction procedure was performed with GAB-Camp agar without bacteria.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed under reducing conditions (21) in a Mini-protean II slab gel vertical electrophoresis apparatus (Bio-Rad, Richmond, Calif.) with 12% separating gels. The proteins were transferred to Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore Intertech, Bedford, Mass.) by Western blotting (27). The membranes were incubated in blocking buffer I for 15 min (27). Strips of the membranes were incubated for 1.5 h at room temperature with plasminogen (1 μg ml−1) diluted in a washing buffer (PBS–1% skim milk powder) (Oxoid, Basingstoke, England). The strips were then rinsed three times for 5 min each in washing buffer and incubated for 2 h at 20°C with rabbit anti-human plasminogen antibodies (diluted 1:2,000 in washing buffer) (DAKO A/S, Glostrup, Denmark). The strips were rinsed as above and incubated with peroxidase-conjugated swine anti-rabbit immunoglobulins (1:1,000) (DAKO) for 1 h at 20°C. After being washed as above, the strips were incubated with 50 mM sodium acetate buffer (pH 5.0) containing 0.04% 3-amino-9-ethylcarbazole (Sigma) and 0.015% H2O2.
Plasminogen activation.
Bacterial cells (109) were incubated for 2 h at +4°C with 50 μg of plasminogen or 250 μl of fresh human plasma in 1.5 ml of PBS containing 1% BSA in the presence or absence of 100 ng of tPA (Calbiochem, La Jolla, Calif.). The influence of α2-antiplasmin was tested by including 70 μg of α2-antiplasmin (Calbiochem) per ml in the combination of bacteria, plasminogen, and tPA. The bacteria were then washed twice in PBS–1% BSA and finally resuspended in 500 μl of the same buffer. Substrate buffer containing PBS, 80 μM chromogenic substrate S-2251 (H-d-Valyl-l-leucyl-l-lysine-p-nitroaniline dihydrochloride) (Chromogenix, Mölndal, Sweden), and 0.4 M NaCl was added to each sample (2.5 ml). After incubation for 2.5 h at 37°C, the samples were read in a spectrophotometer at 405 nm. Substrate in the presence of plasmin, plasminogen, or tPA was used as the control.
RESULTS
H. pylori CCUG 17874 was used for the characterization of plasminogen binding. Binding of radiolabelled plasminogen to H. pylori CCUG 17874 was totally inhibited by lysine and the lysine analogue EACA (50% inhibition at approximately 0.5 mM), as well as by plasminogen (50% inhibition at 1 μM) (Fig. 1). The plasminogen fragments K1–3, which consists of the first three kringles in the plasminogen molecule, K4, the fourth kringle, and mini-plg, the fifth kringle and the C-terminal part of the plasminogen molecule, were also used to characterize the binding of plasminogen to H. pylori CCUG 17874. Mini-plg was the most potent inhibitor, causing total inhibition and reaching 50% inhibition at an inhibitor concentration of 1 μM, while K1–3 and K4 caused only partial inhibition of the interaction (Fig. 1). BSA and fibrinogen did not influence the binding (data not shown). The binding was pH dependent with an optimum at pH 7 (Fig. 2). No difference in the plasminogen binding capacity of H. pylori CCUG 17874 was observed after growth on agar or in broth (data not shown). The CagA-negative H. pylori G12, G104, G50, and G198 bound plasminogen to the same extent (13 to 23%) as did the CagA-positive strain CCUG 17874 (data not shown), indicating that plasminogen binding is CagA independent.
FIG. 1.

Inhibition of 125I-plasminogen binding, expressed as a percentage of the binding capacity in control experiments, to H. pylori CCUG 17874 after a 1-h preincubation of the bacteria (108 cells) with different inhibitors (100 μl, 1 to 108 nM): unlabelled plasminogen, EACA, lysine, or plasminogen fragments (K1–3, K4, or mini-plg). See Materials and Methods for details.
FIG. 2.

Binding of 125I-plasminogen to H. pylori CCUG 17874 at different pH values as described in Materials and Methods.
Scatchard plot analysis of the plasminogen binding to H. pylori CCUG 17874 revealed a straight line, indicating a single interaction. The Kd value obtained was 7 × 10−7 M (Fig. 3).
FIG. 3.

Scatchard plot analysis of plasminogen binding to H. pylori CCUG 17874. H. pylori cells were incubated with increasing amounts of plasminogen. A Kd value of 7 × 10−7 M was calculated. See Materials and Methods for details.
SDS-PAGE gels of a water extract of H. pylori CCUG 17874, transferred to PVDF membranes and stained with amido black, showed a complex mixture of proteins (Fig. 4, lane 1). An immunoblot of the same water extract, produced with plasminogen and antibodies to plasminogen, revealed two proteins binding to plasminogen. The molecular masses of these proteins were 42 and 57 kDa (lane 2). Extracts of GAB-Camp agar without bacterial growth, analyzed in the same way as the bacterial extract, did not show any plasminogen binding protein (data not shown).
FIG. 4.
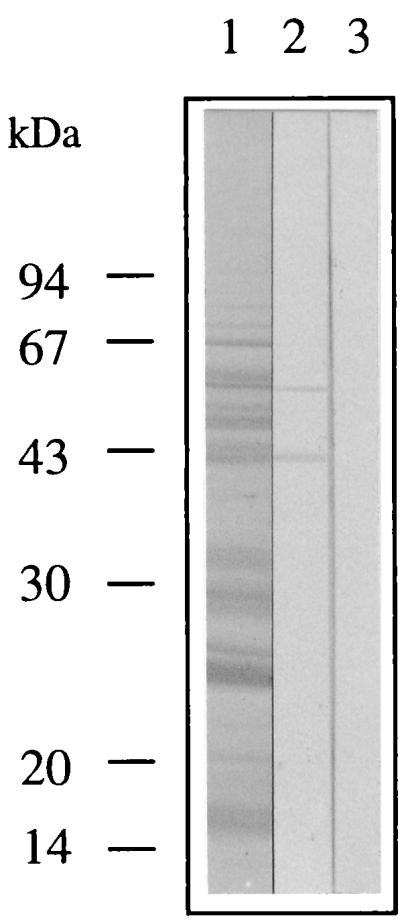
Surface proteins of H. pylori CCUG 17874 extracted with water, separated by SDS-PAGE (12% polyacrylamide), and transferred to a PVDF membrane. Lanes: 1, the water extract profile was stained with amido black; 2, the separated proteins were incubated with plasminogen and subsequently detected with anti-plasminogen antibodies and peroxidase-conjugated antibodies; 3, control experiment in which the separated proteins were incubated with primary and secondary antibodies.
The conversion of plasminogen to plasmin was studied by measuring the breakdown of the chromogenic substrate S-2251. When H. pylori CCUG 17874 cells were incubated with plasminogen or plasma in the presence of tPA, digestion of the substrate could be observed (Fig. 5). No reaction was seen when the bacteria were incubated with plasminogen or plasma in the absence of tPA (Fig. 5). The plasmin activity was not influenced by the presence of α2-antiplasmin (data not shown). H. pylori incubated with plasminogen or tPA alone did not cause any digestion of the substrate, nor did incubation of plasminogen or tPA alone with the substrate do so. The activity of H. pylori cell-bound plasminogen increased with increasing numbers of bacterial cells (data not shown).
FIG. 5.

Activation of plasminogen to plasmin on the surface of H. pylori CCUG 17874. H. pylori cells (109) were incubated with 50 μg of plasminogen (plg) or 250 μl of plasma in the presence or absence of 100 μg of tPA. Plasmin activity was indicated by digestion of the chromogenic substrate S-2251. Plasmin in the presence of substrate was used as a positive control. See Materials and Methods for details.
DISCUSSION
We have previously shown that H. pylori surface lectins interact with carbohydrate ligands on cells and in ECM and that H. pylori binds laminin and vitronectin in a sialic acid-specific manner (31, 38, 44). In an earlier study of plasminogen binding to H. pylori, it was shown that this binding was not inhibited by simple carbohydrates or substances such as transferrin and heparin and that the interaction was not sialic acid dependent (32), although plasminogen was reported to interact with a sialic acid-specific lectin of tissue-invading S-fimbriated Escherichia coli (28). We previously reported that plasminogen binding seems to be a common phenomenon among H. pylori strains, and in a comparison of spiral and coccoid forms, we observed that plasminogen binding to H. pylori CCUG 17874 increased when coccoid forms of the bacteria were used (15). We now propose that plasminogen binding to H. pylori is independent of CagA antigen expression. H. pylori CCUG 17874 was selected for the present investigation, since it is a well-characterized strain and has been used in previous studies of hemagglutinating properties and of plasminogen, vitronectin, and laminin binding (22).
In the present study, plasminogen binding to H. pylori CCUG 17874 was inhibited by lysine and the lysine analogue EACA, which suggests that lysine is important for binding of plasminogen to H. pylori, a phenomenon that has been shown for several other bacterial species (2, 18, 19, 42). Inhibition experiments performed with plasminogen fragments show that interaction between H. pylori CCUG 17874 and plasminogen occurs at the fifth kringle of the plasminogen molecule. From these results, we can conclude that the binding of plasminogen to H. pylori is highly specific. Although studies on binding kinetics and Scatchard plot analysis demonstrated one single specific binding mechanism with a moderate binding affinity, Kd = 7 × 10−7 M (Fig. 3), two proteins were shown to recognize plasminogen in immunoblots. Further studies must be performed to elucidate whether there is a correlation between these plasminogen binding proteins of H. pylori and any of the other plasmin/plasminogen binding proteins that have so far been cloned (2, 14, 23, 35).
Although H. pylori survives passage through the low pH in gastric juice, this pathogen prefers the environment close to the epithelial surface, which is embedded in mucus and where the pH is higher. It is therefore not surprising to find that plasminogen binding to H. pylori CCUG 17874 was pH dependent with an optimum at pH 7 (Fig. 2).
Three main functions of plasminogen receptors have been postulated. First, plasminogen receptors may assemble plasminogen and plasmin in a defined microenvironment; second, activation of plasminogen increases when the molecule is bound to a cell surface receptor; and, finally, receptor-bound plasmin is protected from inactivation by α2-antiplasmin (26). It is possible that by binding plasminogen, bacterial pathogens will benefit from the fact that cell-bound plasminogen can be activated and plasmin is protected from inactivators. In this way, the ECM-digestive properties of plasmin may be used by the pathogen for tissue penetration. Moreover, it was shown that Borrelia burgdorferi requires plasminogen to disseminate in ticks and to increase spirochetemia in mice (5). We can now add H. pylori to the list of gram-positive and gram-negative bacterial pathogens that have the ability to invade cutaneous tissues and mucosal surfaces and have been shown to bind human plasminogen and plasmin and to activate plasminogen to plasmin (1, 6, 13, 18, 42). Conversion of plasminogen to plasmin on the surface of H. pylori was observed in the presence of plasminogen or plasmin only when tPA had been added. It seems that the concentration of tPA in plasma was insufficient to initiate the conversion of plasminogen without the addition of more tPA. A similar result was reported by Kuusela and Saksela in a study of Staphylococcus aureus (18). It was explained by the removal of enzymatically active tPA as a result of the formation of irreversible complexes between tPA and its inhibitor PAI-1 during plasma storage, and it was suggested that the conditions in vivo might be more favorable for plasminogen conversion (18). It should also be considered, for H. pylori, that the conditions at the gastric epithelium in vivo probably are different from those in an in vitro plasma environment.
Although the main physiological role of plasminogen is fibrinolysis, as shown by experiments with mice deficient in plasminogen and/or fibrinogen (3), plasminogen has also been proposed to be involved in several physiological processes, such as wound healing and tissue remodelling, as well as in tissue invasion of tumor cells via degradation of extracellular matrix (33). Concerning H. pylori, it has been reported that H. pylori infection influences the activity of plasminogen activators, increasing uPA activity and decreasing tPA activity (12). This correlated with previously detected alterations in gastric carcinoma (34a). The changes in activity of the plasminogen activators were reversed when the H. pylori infection was eradicated (11). These findings support the possibility that H. pylori is important in the development of gastric cancer. Whether the formation of H. pylori cell surface-bound plasmin is of any importance in the development of gastric cancer remains to be investigated, but it may be important to provide a powerful proteolytic mechanism for gastric tissue penetration in type B gastritis and peptic ulcer disease.
ACKNOWLEDGMENTS
This study was supported by a grant from the Swedish Medical Research Council (16 × 04723) to T. Wadström and a grant from Kungliga fysiografiska sällskapet, Lund, Sweden, to M. Pantzar. The study was also supported by grants from the Magnus Bergvalls foundation and the Crafoord foundation and the Medical Faculty, University of Lund, Lund, Sweden.
We thank Janna Holmgren, Pär Aleljung, and Meeme Utt for skillful technical support and Björn Wiman, Department of Clinical Chemistry, Karolinska Hospital, Stockholm, Sweden, for kindly providing us with plasminogen fragments.
REFERENCES
- 1.Ben Nasr A, Wistedt A, Ringdahl U, Sjöbring U. Streptokinase activates plasminogen bound to human group C and G streptococci through M-like proteins. Eur J Biochem. 1994;222:267–276. doi: 10.1111/j.1432-1033.1994.tb18865.x. [DOI] [PubMed] [Google Scholar]
- 2.Berge A, Sjöbring U. PAM, a novel plasminogen-binding protein from Streptococcus pyrogenes. J Biol Chem. 1993;268:25417–25424. [PubMed] [Google Scholar]
- 3.Bugge T H, Kombrinck K W, Flick M J, Daugherty C C, Danton M J, Degen J L. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–719. doi: 10.1016/s0092-8674(00)81390-2. [DOI] [PubMed] [Google Scholar]
- 4.Clyne M, Drumm B. Adherence of Helicobacter pylori to primary human gastrointestinal cells. Infect Immun. 1993;61:4051–4057. doi: 10.1128/iai.61.10.4051-4057.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coleman J L, Gebbia J A, Piesman J, Degen J L, Bugge T H, Benach J L. Plasminogen is required for efficient dissemination of Borrelia burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell. 1997;89:1111–1119. doi: 10.1016/s0092-8674(00)80298-6. [DOI] [PubMed] [Google Scholar]
- 6.Coleman J L, Sellati T J, Testa J E, Kew R R, Furie M B, Benach J L. Borrelia burgdorferi binds plasminogen, resulting in enhanced penetration of endothelial monolayers. Infect Immun. 1995;63:2478–2484. doi: 10.1128/iai.63.7.2478-2484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danø K, Andreasen P A, Gröndahl-Handen J, Kristensen P, Nielsen L S, Skriver L. Plasminogen activators, tissue degradation and cancer. Adv Cancer Res. 1985;44:139–266. doi: 10.1016/s0065-230x(08)60028-7. [DOI] [PubMed] [Google Scholar]
- 8.Dunn B E, Cohen H, Blaser M. Helicobacter pylori. Clin Microbiol Rev. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans D G, Evans D J, Jr, Graham D Y. Adherence and internalization of Helicobacter pylori by HEP-2 cells. Gastroenterology. 1992;102:1557–1567. doi: 10.1016/0016-5085(92)91714-f. [DOI] [PubMed] [Google Scholar]
- 10.Falk P, Roth K A, Borén T, Westblom U, Gordon J I, Normark S. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc Natl Acad Sci USA. 1993;90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Götz, J. M., J. W. Ravensbergen, H. W. Verspaget, I. Biemond, C. F. M. Sier, G. J. A. Offerhaus, C. B. H. M. Lamers, and R. A. Veenendaal. 1996. The effect of treatment of Helicobacter pylori infection on gastric mucosal plasminogen activators. Fibrinolysis 10(Suppl. 2):85–89.
- 12.Götz J M, Vergouwe Y, Verspaget H W, Biemond I, Sier C F M, Lamers C B H W, Veenendaal R A. Gastric mucosal plasminogen activators in Helicobacter pylori infection. Dig Dis Sci. 1996;41:1577–1582. doi: 10.1007/BF02087903. [DOI] [PubMed] [Google Scholar]
- 13.Hu L T, Perides G, Noring R, Klempner M S. Binding of human plasminogen to Borrelia burgdorferi. Infect Immun. 1995;63:3491–3496. doi: 10.1128/iai.63.9.3491-3496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L T, Pratt S D, Perides G, Katz L, Rogers R A, Klempner M S. Isolation, cloning, and expression of a 70-kilodalton plasminogen binding protein of Borrelia burgdorferi. Infect Immun. 1997;65:4989–4995. doi: 10.1128/iai.65.12.4989-4995.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khin M M, Ringnér M, Aleljung P, Wadström T. Binding of human plasminogen and lactoferrin by Helicobacter pylori coccoid forms. J Med Microbiol. 1996;45:433–439. doi: 10.1099/00222615-45-6-433. [DOI] [PubMed] [Google Scholar]
- 16.Klempner M S, Noring R, Epstein M P, McCloud B, Hu R, Limentani S A, Rogers R A. Binding of human plasminogen and urokinase-type plasminogen activator to the Lyme disease spirochete, Borrelia burgdorferi. J Infect Dis. 1995;171:1258–1265. doi: 10.1093/infdis/171.5.1258. [DOI] [PubMed] [Google Scholar]
- 17.Klotz I M. Number of receptor sites from Scatchard graphs: facts and fantasies. Science. 1982;217:1247–1249. doi: 10.1126/science.6287580. [DOI] [PubMed] [Google Scholar]
- 18.Kuusela P, Saksela O. Binding and activation of plasminogen at the surface of Staphylococcus aureus. Eur J Biochem. 1990;193:759–765. doi: 10.1111/j.1432-1033.1990.tb19397.x. [DOI] [PubMed] [Google Scholar]
- 19.Kuusela P, Ullberg M, Saksela O, Kronvall G. Tissue-type plasminogen activator-mediated activation of plasminogen on the surface of group A, C, and G streptococci. Infect Immun. 1992;60:196–201. doi: 10.1128/iai.60.1.196-201.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labigne A, de Reuse H. Determinants of Helicobacter pylori pathogenicity. Infect Agents Dis. 1996;5:191–202. [PubMed] [Google Scholar]
- 21.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Lelwala-Guruge J, Ljungh Å, Wadström T. Haemagglutinating patterns of Helicobacter pylori. APMIS. 1992;100:908–913. [PubMed] [Google Scholar]
- 23.Lottenberg R, Broder C C, Boyle D P, Kain S E, Schroeder B L, Curtiss R., III Cloning, sequence analysis, and expression in Escherichia coli of a streptococcal plasmin receptor. J Bacteriol. 1992;174:5204–5210. doi: 10.1128/jb.174.16.5204-5210.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lottenberg R, Minning-Wenz D, Boyle M D P. Capturing host plasmin(ogen): a common mechanism for invasive pathogens. Trends Microbiol. 1994;2:20–24. doi: 10.1016/0966-842x(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 25.Markwell M A K. A new solid state reagent to iodinate proteins. Anal Biochem. 1982;125:427–432. doi: 10.1016/0003-2697(82)90025-2. [DOI] [PubMed] [Google Scholar]
- 26.Miles L A, Plow E F. Plasminogen receptors: ubiquitous sites for cellular regulation of fibrinolysis. Fibrinolysis. 1988;2:61–71. [Google Scholar]
- 27.Nilsson I, Ljungh Å, Aleljung P, Wadström T. Immunoblot assay for serodiagnosis of Helicobacter pylori infections. J Clin Microbiol. 1997;35:427–432. doi: 10.1128/jcm.35.2.427-432.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parkkinen J, Hacker J, Korhonen T. Enhancement of tissue plasminogen activator-catalyzed plasminogen activation by Escherichia coli S fimbriae associated with neonatal septicaemia and meningitis. Thromb Haemostasis. 1991;65:483–486. [PubMed] [Google Scholar]
- 29.Paulsson M, Wadström T. Vitronectin and type I collagen binding of Staphylococcus aureus and coagulase negative staphylococci. FEMS Microbiol Immunol. 1990;65:55–62. doi: 10.1111/j.1574-6968.1990.tb03479.x. [DOI] [PubMed] [Google Scholar]
- 30.Perides G, Noring R, Klempner M S. Inhibition of Borrelia burgdorferi-bound fibrinolytic enzymes by α2-antiplasmin, PAI-1 and PAI-2. Biochem Biophys Res Commun. 1996;219:690–695. doi: 10.1006/bbrc.1996.0296. [DOI] [PubMed] [Google Scholar]
- 31.Ringnér M, Paulsson M, Wadström T. Vitronectin binding by Helicobacter pylori. FEMS Microbiol Immunol. 1992;105:219–224. doi: 10.1111/j.1574-6968.1992.tb05904.x. [DOI] [PubMed] [Google Scholar]
- 32.Ringnér M, Valkonen K H, Wadström T. Binding of vitronectin and plasminogen to Helicobacter pylori. FEMS Immunol Med Microbiol. 1994;9:29–34. doi: 10.1111/j.1574-695X.1994.tb00470.x. [DOI] [PubMed] [Google Scholar]
- 33.Rømer J, Bugge T H, Pyke C, Lund L R, Flick M J, Degen J L, Danø K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–292. doi: 10.1038/nm0396-287. [DOI] [PubMed] [Google Scholar]
- 34.Scatchard G. The attraction of proteins for small molecules and ions. Ann N Y Acad Sci. 1949;51:660–672. [Google Scholar]
- 34a.Sier C F, Verspaget H W, Griffiôen G, Ganesh S, Vloedgraven H J, Lamers C B. Plasminogen activators in normal tissue and carcinomas of the human oesophagus and stomach. Gut. 1993;34:80–85. doi: 10.1136/gut.34.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sjöström I, Gröndahl H, Falk G, Kronvall G, Ullberg M. Purification and characterisation of a plasminogen-binding protein from Haemophilus influenzae. Sequence determination reveals identity with aspartase. Biochim Biophys Acta. 1997;1324:182–190. doi: 10.1016/s0005-2736(96)00218-0. [DOI] [PubMed] [Google Scholar]
- 36.Soltész L V, Mårdh P-A. Serum-free liquid medium for Neisseria gonorrhoea. Curr Microbiol. 1980;4:45–49. [Google Scholar]
- 37.Soltész V, Schalén C, Mårdh P A. New selective medium for Campylobacter pylori. In: Kaijser B, Falsen E, editors. Proceedings of the Fourth International Workshop on Campylobacter infections. 1988. pp. 433–436. [Google Scholar]
- 38.Trust T J, Doig P, Emödy L, Kienle Z, Wadström T, O’Toole P. High-affinity binding of the basement membrane proteins collagen type IV and laminin to the gastric pathogen Helicobacter pylori. Infect Immun. 1991;59:4398–4404. doi: 10.1128/iai.59.12.4398-4404.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzouvelekis L S, Mentis A F, Makris A M, Spiliadis C, Blackwell C, Weir D M. In vitro binding of Helicobacter pylori to human gastric mucin. Infect Immun. 1991;59:4252–4254. doi: 10.1128/iai.59.11.4252-4254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ullberg M, Karlsson I, Wiman B, Kronvall G. Two types of receptors for human plasminogen on group G streptococci. APMIS. 1992;100:21–28. doi: 10.1111/j.1699-0463.1992.tb00835.x. [DOI] [PubMed] [Google Scholar]
- 41.Ullberg M, Kronvall G, Karlsson I, Wiman B. Receptors for human plasminogen on gram-negative bacteria. Infect Immun. 1990;58:21–25. doi: 10.1128/iai.58.1.21-25.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ullberg M, Kuusela P, Kristiansen B-E, Kronvall G. Binding of plasminogen to Neisseria meningitidis and Neissera gonorrhoea and formation of surface associated plasmin. J Infect Dis. 1992;166:1329–1334. doi: 10.1093/infdis/166.6.1329. [DOI] [PubMed] [Google Scholar]
- 43.Vaheri A, Stephens R W, Salonen E-M, Pöllänen J, Tapiovaara H. Plasminogen activation at the cell surface matrix interface. Cell Differ Dev. 1990;32:255–262. doi: 10.1016/0922-3371(90)90038-x. [DOI] [PubMed] [Google Scholar]
- 44.Valkonen K H, Ringnér M, Wadström T. High affinity binding of laminin by Helicobacter pylori. FEMS Immunol Med Microbiol. 1993;7:29–38. doi: 10.1111/j.1574-695X.1993.tb00378.x. [DOI] [PubMed] [Google Scholar]
- 45.Xiang Z, Censini S, Bayeli P F, Telford J L, Figura N, Rappuoli R, Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]