

Abstract
Csk (C-terminal Src kinase) takes part in a highly specific high-affinity interaction via its Src homology 3 (SH3) domain with the proline-enriched tyrosine phosphatase PEP in hematopoietic cells. The solution structure of the Csk-SH3 domain complexed with a 25-residue peptide from the proline/glutamate/serine/threonine rich (PEST) domain of PEP reveals the basis for this highly specific peptide-recognition motif involving an SH3 domain. Two hydrophobic residues Ile 625 and Val 626 on a proline-rich sequence, in the non-catalytic PEST domain of PEP are recognized by a highly specific interaction involving three residues Ala 40, Thr 42 and Lys 43 on the SH3 domain of Csk. Ile 625 and Val 626 lie C-terminal to the conventional proline-rich SH3 domain recognition sequence on PEP. This interaction is required, in addition to the classic polyproline helix (PPII) recognition by the Csk-SH3 domain, for the association between Csk and PEP in vivo. NMR relaxation analysis suggests that Csk-SH3 has different dynamic properties in the various subsites important for peptide recognition.
Csk (C-terminal Src kinase) phosphorylates Src and Src family kinases (SFKs) in the C-terminal tyrosine residue (Y527 in Src) in vivo, thus playing a major role in the down regulation of SFKs1,2. Csk is ubiquitously expressed in mammalian cells3 and has been found to be crucial in the regulation of neural development4, T-cell development and regulation5, and cytoskeletal organization6. Csk is a modular protein that shares a significant sequence homology with Src (37 %). Like SFKs, Csk possesses SH3 (Src homology domain 3), SH2 (Src homology domain 2) and catalytic kinase domains. However, it lacks both the N-terminal myristoylation signal, and the tyrosine residues in the activation loop and C-terminal tail. These elements are essential for the regulation of the activity of SFKs7. This indicates a different regulatory mechanism for Csk compared to SFKs.
Csk associates with the protein tyrosine phosphatase, PEP (Proline enriched phosphatase), through its SH3 domain in hematopoietic cells8 . This highly specific interaction acts as a negative regulator of antigen receptor-mediated signal transduction in T-cells9-11. PEP interacts with the SH3 domain of Csk through a proline-rich sequence of its non-catalytic PEST (Proline/Glutamate/Serine/Threonine rich) domain referred to as PEP-3BP1 (where 3BP denotes SH3 binding peptide)8,12 (Fig. 1b). However, the proline-rich segment, though necessary, is insufficient for PEP to bind to Csk12. The hydrophobic sequence of residues (colored blue in Fig. 1b) C-terminal to the proline-rich region in PEP-3BP1 is necessary for the interaction. We provide here a structural basis for this novel, highly specific interaction, extending beyond the polyproline core binding motif, involving an SH3 domain and its natural ligand.
Fig. 1.

Sequence alignment for several SH3 domains involved with proline-rich peptide binding along with proline-rich peptides from tyrosine phosphatases, PEP and PTP-PEST. Amino acids are represented by single letter codes.
a, Sequence alignment (accession codes in brackets) for Csk from Homo sapiens (P41240), Csk from Xenopus laevis (AAC05835), Chk from Homo sapiens (NP002369). Csk from Mus musculus (AAA18766) is identical to that from Homo sapiens for the depicted range. Src from Homo sapiens (P12391) has been included for comparison. The residues that are important for the recognition of the PPII helix are shown in red and those involved in recognition of the hydrophobic sequence C-terminal to the PPII helix are shown in blue. These residues are conserved in all mammalian Csk’s and are not conserved Csk homologues. The sequence in the linker region between the SH3 and SH2 domains important for the activation of the kinase domain of Csk is shown in green (A. Shekhtman, R. Ghose, D. Wang, P. A. Cole & D. Cowburn, submitted). This sequence is absent in Src.
b, Sequence alignment (accession codes in brackets) of proline-rich sequences from the non-catalytic PEST domains of the phosphatases PEP (accession code P29352) and PTP-PEST (NP_035333). The conserved proline-rich sequence is shown in red and those that are important for the high-affinity interaction with Csk-SH3 are shown in blue12.
Solution structure of the Csk-SH3/PEP-3BP1 complex
In order to elucidate the nature of the interaction of the SH3 domain of Csk with PEP, we studied the solution structure and dynamics of the SH3 domain (residues 1-83 of Csk’s 450 residues) complexed with a 25-residue peptide (PEP-3BP1) from the PEST domain of PEP. A crystal structure exists for the free Csk SH3 domain13, showing that the free Csk-SH3 domain exists as a dimer of dimers. The quality of the NMR spectra in solution indicates slow exchange between oligomers (not shown). Addition of PEP-3BP1 produced well-resolved spectra characteristic of a monomer (vide infra) in solution. Evaluation of the 15N {1H'} steady-state nOe indicated that nine residues (1-9) in the N-terminus, thirteen residues (71-83) in the C-terminus of the SH3 domain, and the first seven residues in the N-terminus of PEP-3BP1 were unstructured and hence excluded from the final structure calculation of the complex.
The structural characteristics of the 25 calculated structures of the complex, which had the lowest value of the target function, are summarized in Table 1 and the salient structural features of the complex are shown in Fig.2. The solution structure of the Csk-SH3 domain in the complex is similar to other SH3 domains14 and essentially identical to that of the monomers in the crystal structure13. The rmsd between the solution structure and the monomers in the crystal structure (1CSK) is 0.85 Å for backbone and 1.40 Å for all heavy atoms in the ordered regions. Csk-SH3 has 5 β-strands (12-16, 35-41, 47-51, 57-61, 65-67) organized in an anti-parallel β-barrel structure with a 310 helix (62-64) between the final two β-strands.
Table 1.
Structural characteristics for 25 lowest energy structures of the Csk-SH3/PEP-3BP1 complex 1
| NMR Constraints2 | Csk-SH3 | PEP-3BP1 |
|---|---|---|
| Total nOes | 805 | 313 |
| Intraresidue | 360 | 209 |
| Short range | 294 | 104 |
| Long range | 151 | – |
| Dihedral constraints (ϕ,ψ) | 101 | 25 |
| Coupling constants 3J(H’Hα) | 73 | 19 |
| Hydrogen-bonding restraints | 124 | |
| Intermodular nOes | 42 | |
| Coordinate precision3 (Å) | ||
| Backbone heavy atoms | 0.68 ± 0.09 (0.32 ± 0.07) | |
| All heavy atoms | 1.14 ± 0.11 (0.73 ± 0.10) | |
| Backbone dihedral angle statistics4 (%) | ||
| Most favored | 83.6 | |
| Additional favored | 15.9 | |
| Generously favored | 0.4 | |
| Disallowed | 0.1 | |
There was one distance violation >0.5 Å (0.51 Å) and no dihedral angle violations >6° in the ensemble of 25 structures.
NMR constraints for Csk-SH3 (1–83) and PEP-3BP1 (1–25). Short range constraints (1 ≤ i <5), long range constraints (i > 5).
Coordinate precision for Csk-SH3 (10–69) and PEP-3BP1 (8–25) with respect to the mean structure. The coordinate precision for regions with secondary structure (Csk-SH3: 12–16, 35–41, 47–51, 57–61, 62–64 and 65–67, and PEP-3BP1: 17–19) is shown in brackets.
Dihedral angle characteristics from PROCHECK50.
Fig. 2.
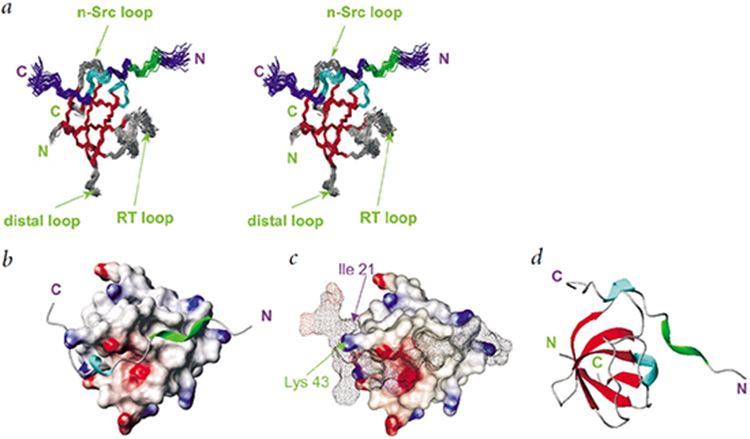
Solution structure of the Csk-SH3-PEP-3BP1 complex. All structures were drawn using MOLMOL48.
a, Stereo view of a cluster of 25 lowest target function structures of the Csk-SH3-PEP-3BP1 complex. (β-strands are shown in red, 310-helices in cyan and the PPII helix in green. The SH3 domain is labeled in green; PEP-3BP1, in purple. Important loops on Csk-SH3 are labeled. Residues 11–68 Csk-SH3 and 8–25 in PEP-3BP1 are shown,
b, Surface representation of Csk-SH3 showing the grooves on which PEP-3BP1 (shown as a ribbon) sits. Regions of positive and negative charges are shown in blue and red, respectively.
c, Surface-on-surface representation of the Csk-SH3-PEP-3BP1 complex. The clamping of lie 21 from PEP-3BP1 on the finger formed by Lys 43 on Csk-SH3 is indicated.
d, Ribbon diagram for the Csk-SH3PEP-3BP1 complex with Csk-SH3 (green) and PEP-3BP1 (blue).
The peptide (PEP-3BP1) forms the characteristic left-handed type II polyproline helix (PPII) involving the residues 10-13. An unusual characteristic of the peptide structure is the presence of a 310 helix, C-terminal to the PPII region, involving residues 17-19. This is the first time that a region of secondary structure has been located beyond the PPII region in an SH3 binding peptide. This structural feature is necessary to position the hydrophobic sequence of residues on PEP-3BP1 onto a hydrophobic patch on the surface of the SH3 domain. This hydrophobic interaction is crucial in providing the high-affinity (Kd=0.8 μM compared to Kd >10 μM for most SH3/peptides interactions15) and high specificity that characterizes the association between PEP-3BP1 and the SH3 domain of Csk.
Characteristics of the peptide-recognition surface
There are two distinct peptide recognition surfaces on Csk-SH3 – the first one is the well-known polyproline helix binding surface16 shown in Fig. 3a. The second, shown in Fig. 3b, is the hydrophobic patch which recognizes the C-terminal sequence of hydrophobic residues on PEP-3BP1 and which is responsible for the specificity of the recognition of PEP by Csk.
Fig. 3.
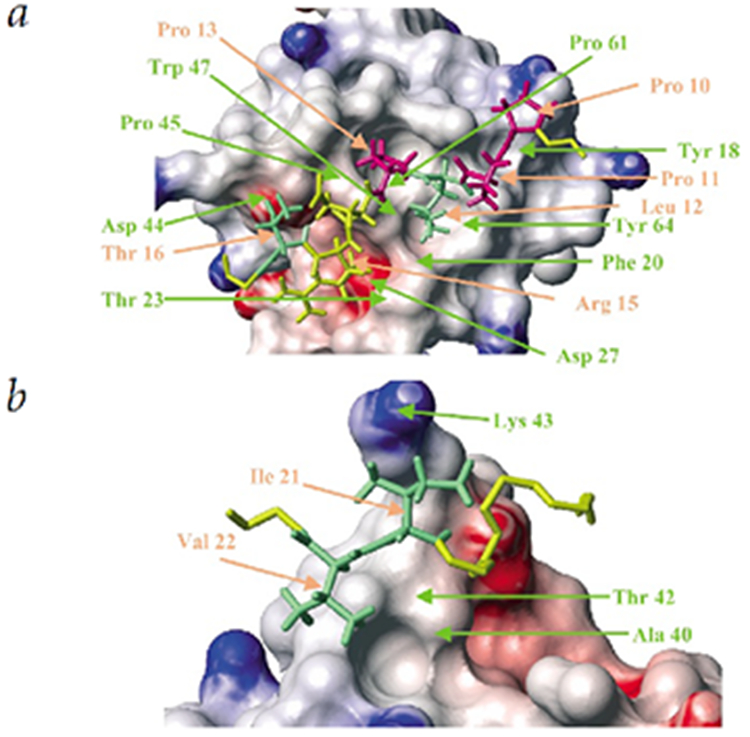
Detailed view of the two distinct surfaces on Csk-SH3 involved in peptide recognition. SH3 residues are labeled in green and PEP-3BP1 residues in orange.
a, Polyproline helix recognition surface. The proline residues on PEP-3BP1 involved in PPII helix formation, namely Pro 10, Pro 11 and Pro 13 are shown in pink. The important hydrophobic residues Leu 12 and Thr 16 are shown in green. The charged residue Arg 15, shown in yellow, is involved in charge-charge interaction with Asp 27 on Csk-SH3. The PPII helix sits on distinct grooves on the Csk-SH3 surface formed mainly by the aromatic residues Tyr 18, Phe 20, Trp 47 and Tyr 64. The PEP-3BP1 residues Pro 10, Leu 12, Pro 13, Arg 15 and Thr 16 are crucial for peptide binding.
b, Hydrophobic specificity pocket. The γ- and δ-methyls of Ile 21 on PEP-3BP1 wrap around the finger formed by Lys 43 on Csk-SH3 and the γ2-methyl of Val 22 inserts into a cavity formed by the methyl groups of Ala 40 and Thr 42 on Csk-SH3.
The polyproline helix-binding surface is seen in detail in Fig. 3a. The surface of the protein shows distinct grooves formed by the aromatic residues (shown in red in Fig. 1a) Tyr 18, Phe 20, Trp 47 and Tyr 64 in which the hydrophobic residues that constitute the PPII helix of PEP-3BP1 sit. It was shown by alanine substitution that the residues Pro 10, Leu 12, Pro 13, Arg 15 and Thr 16 in the sequence PPPLPERT (9-16, shown in red in Fig. 1b) were crucial for the binding of PEP-3BP1 to Csk-SH3 whereas the mutations P9A, P11A and E14A had no effect on the peptide binding12. The hydrophobic residues Pro 10, Leu 12 and Pro 13 sit on grooves formed on the surface of Csk-SH3 by the sidechains of the aromatic residues Tyr 18, Phe 20, Trp 47 and Tyr 64 along with those of the hydrophobic residues Thr 23, Pro 61 and Ala 62. The C' oxygen of Pro 13 is within hydrogen-bond distance of the indole Hε of Trp 47 on Csk-SH3 in the calculated structures. The interaction is further stabilized by the interaction of the positively charged sidechain of Arg 15, immediately adjacent to the polyproline helix, with the negatively charged groove formed by the sidechain of the conserved Asp 27 on Csk-SH3. Thr 16 on PEP-3BP1 makes further hydrophobic contacts though its γ-methyl group with Pro 45 on Csk-SH3. The mutation T16A destabilizes this interaction, as the length of the sidechain is not sufficient to extend beyond the charged surface provided by Asp 44 to contact Pro 45. The residues Pro 9, Pro 11 and Glu 14 on PEP-3BP1 all point away from the surface of the protein making no stabilizing contacts and hence mutation of these residues to Ala has no effect on peptide binding. The orientation of PEP-3BP1 on the Csk-SH3 surface is that of a class II ligand in which positively charged Arg residue lies C-terminal to the PPPLP region positioning the ligand as shown in Fig. 2b.17-20.
The novel feature of peptide recognition by Csk-SH3 is the recognition of the hydrophobic sequence on PEP-3BP1 (IV) shown in blue in Fig. 1b. This sequence is required for the high affinity and high specificity recognition by Csk-SH3. PEP also possesses another proline-rich sequence PEP-3BP2, however this sequence, though identical to PEP-3BP1 in its proline-rich part (PPPLPERT) binds Csk-SH3 with an affinity that is more than a hundred-fold less12. The two residues C-terminal to the proline-rich sequence, Ile 21 and Val 22, equivalent to Ile 625 and Val 626 on full-length PEP, are crucial for this high-affinity interaction12. The mutations I21A and V22A reduced the binding affinity 10-fold and 5-fold respectively, whereas the simultaneous mutations of both residues completely abolished binding12. These results are consistent with the association of Csk with full-length PEP in vivo12. It was later found that Csk also associates with a ubiquitously expressed protein tyrosine phosphatase, PTP21 through a proline-rich sequence in its non-catalytic PEST domain. The peptide, PTP-3BP2, shown in Fig. 1b bears a very high sequence homology to PEP-3BP1 with Ile 21 replaced by Val and Val 22 replaced by Leu. These seemingly minor mutations reduced the affinity of PTP-3BP2 to Csk-SH3 by factor greater than 10 compared to PEP-3BP121.
The solution structure of the PEP-3BP1/Csk-SH3 complex provides clear structural insight into the highly specific interaction involving the residues Ile 21 and Val 22 on PEP-3BP1. Fig.2b shows that the 310-helix involving residues Pro 17, Glu 18 and Ser 19 creates an almost a 90º turn in the peptide chain and serves to position the PEP-3BP1 hydrophobic patch onto Csk-SH3. The 310-helix of PEP-3BP1 apparently has only a structural role and provides no direct stabilizing interactions with the Csk-SH3. The mutations P17A, E18A and S19A have no effect on peptide binding since the ability to form a 310-helix is not hindered12. Fig. 3b reveals in detail the interaction involving Ile 21, which clamps onto the sidechain of Lys 43 of Csk-SH3. The γ and δ-methyl groups of Ile 21 form a fork around the finger provided by the sidechain of Lys 43, and the γ2-methyl of Val 22 is inserted into a rather shallow cavity formed by the methyl groups of Ala 40 and Thr 42 on Csk-SH3. The methyl groups of Val 22 are well resolved in the NMR spectra indicating highly restricted rotation about the χ1 dihedral angle, consistent with the ensemble of NMR structures. Rotations about the χ1 and χ21 dihedral angles in Ile 21 are also restricted. Mutation12 of Ile 21 to Ala abolishes the possibility of the clamping onto the finger produced by Lys 43 on Csk-SH3 and upsets the optimal positioning of Val 22 onto the hydrophobic pocket. The mutation I21V (occurring naturally in PTP-3BP2, see Fig. 1b) would result in non-optimal clamping as the distance between the γ-methyls of Val is insufficient to clamp onto the Lys 43 finger. Furthermore, the hydrophobic cavity formed by Ala 40 and Thr 42 on Csk-SH3, where the γ2-methyl of Val 22 of PEP-3BP1 is positioned, is not deep enough to accommodate a Leu sidechain, thus the mutation V22L, occurring naturally in PTP-3BP2, results in decreased affinity21. The residues Ala 40, Thr 42 and Lys 43 in Csk-SH3, deemed important for the high-affinity interaction between Csk-SH3 and PEP-3BP1, while widely conserved in all mammalian Csk's, are substituted with Glu, Cys and Glu, respectively, in Chk (Csk homologous kinase)22,23 which does not interact with PEP21.
Understanding the nature of the two interaction surfaces also reveals the reason why the addition of PEP-3BP1 disrupts the oligomerization of the Csk-SH3 in solution. The two dimer interfaces in the published crystal structure of free Csk-SH3 involve a large majority of the key residues that constitute the two surfaces (the PPII-recognition surface and the hydrophobic specificity patch) that interact with PEP-3BP113. The solution structure of Csk-SH3 domain in complex with PEP-3BP1 does not show significant differences at these key residues when compared with the monomers in the corresponding crystal structure.
Dynamics of the Csk-SH3/PEP-3BP1 complex on multiple timescales
NMR provides a unique way to study not only the structure but also the dynamics of biomolecules in solution with behavior on a wide range of timescales24. In solution, both the overall rotational hydrodynamics of the entire molecule as well as the local motion of groups contribute to the relaxation of NMR active nuclei. Fast motion on the ps-ns timescale, typical of a covalent bond, has a direct influence on the relaxation of the various NMR active species in solution and their effects can be studied using an analysis of the spin-lattice relaxation rates (R1), spin-spin relaxation rates (R2) and heteronuclear nuclear Overhauser enhancements (nOe). This data is usually analyzed using the Lipari-Szabo formalism25 which yields an "bond order parameter" S2, which is 0 for a bond completely disordered on the ps-ns timescale, and 1 when a bond is fully rigid on this timescale. Slow motion on the ms-μs timescale can be detected from interpretation of the spin-spin relaxation rates (R2). Detailed knowledge of dynamics on several timescales is crucial to full functional characterization of a system.
Relaxation analysis indicated an axially symmetric overall diffusion tensor 26 with D∥/D⊥ =1.54±0.13 and a rotational correlation time (τC) of 10.5±0.7 ns. This implies that the complex tumbles in solution as a cylinder. The τC obtained from hydrodynamic calculations27,28 for the twenty-five NOE-determined structures including the disordered termini and a hydration shell of 3.1 Å was 10.3 ns. The calculated rotational diffusion tensor was axially symmetric with D∥/D⊥= 1.62. A similar calculation excluding the disordered tails also produced an approximately axially symmetric rotational diffusion tensor (1.0:0.9:0.8) with D∥/D⊥= 1.14 and τC = 5.1 ns. In the case of the unsolvated protein, the corresponding values were τC = 9.0 ns, D∥/D⊥= 1.70 for the full complex including the tails, and τC = 4.4 ns, D∥/D⊥= 1.18 for the core. The difference in τC values is too large to be caused solely by the difference in molecular weight (28%). This analysis confirms that both the anisotropy and the long correlation time, in comparison to a compact sphere of similar molecular weight, arise from the presence of the disordered termini. The excellent agreement between the hydrodynamic calculations and experimentally determined hydrodynamic behavior indicate that the complex is a monomeric species and that there is no significant contribution of oligomers to the solution properties, as measured by NMR relaxation. The monomer state was further confirmed by observing no indication of aggregation; the NMR spectra were essentially unchanged by dilution to 100μM concentration and no peaks corresponding to oligomers was seen in the NOESY-HSQC spectra. The orientation of the principal axis of the diffusion tensor is shown in Fig. 4a. The similarity of the R2av (the average between the in phase and antiphase decay rates, vide infra) values measured in the ordered regions of both Csk-SH3 and PEP-3BP1 indicates a common rotational correlation time.
Fig. 4.

Backbone dynamics of the Csk-SH3/PEP-3BP1 complex determined by 15N relaxation measurements.
a, Orientation of the principal axis of the rotation diffusion tensor of the Csk-SH3/PEP-3BP1 complex as determined from 15N relaxation data. The principal axes of the inertia tensor of the Csk-SH3 core (11-68) are also shown.
b, Order parameters for the Csk-SH3 domain. The regions of secondary structure are indicated – β-sheets in red and the 310-helix in cyan. The residues (11-68) in the protein core indicated by solid circles were analyzed using an axially-symmetric diffusion tensor26. The residues in the N- and C-terminal tails (1-10, 69-83) were analyzed using an isotropic diffusion tensor and are indicated by open circles. Orange circles indicate the Lipari-Szabo25 order parameter S2 whereas blue circles indicate the order parameter for fast motion S2f 47 for residues exhibiting fast motion on multiple timescales. The residues Ala 40 and Thr 42, required for forming hydrophobic contacts with the peptide have unusually low order parameters and are circled. Residues showing ΔR2av values in range 4.0 s−1 to 6.5 s−1 are indicated by blue squares while those with ΔR2av > 6.5 s−1 are indicated by red squares.
c, Heteronuclear steady-state 15N-{1H'} nOe for PEP-3BP1. The PPII helix is indicated in green and the 310-helix in cyan. Val 22 shows a low nOe value and is circled. Residues showing ΔR2av values in range 4.0 s−1 to 6.5 s−1 are indicated by blue squares while those with ΔR2av > 6.5 s−1 are indicated by red squares.
d, Close-up of the interacting methyl groups on Ala 40 and Thr 42 on Csk-SH3 and Val 22 on PEP-3BP1, the NH' groups belonging to these residues exhibit enhanced motion on the ps-ns timescale, and are indicated in red. Val 41 does not take part in this interaction and its NH' group (shown in yellow) has a high order parameter.
e, Residues which show motion on the ms timescale all lie on the PPII helix recognition surface on Csk-SH3. Residues showing ΔR2av values in range 4.0 s−1 to 6.5 s−1 are colored blue while those with ΔR2av > 6.5 s−1 are shown in red. Prolines residues are indicated in yellow. The residues on Csk-SH3 (green) and PEP-3BP1 (purple) that showed ΔR2av > 6.5 s−1, are labeled. Additionally, the Gly 58 and Ile 60 on Csk-SH3 also had ΔR2av > 6.5 s−1. The figure was prepared using the program GRASP50.
Dynamic behavior on several timescales is seen in the Csk-SH3/PEP-3BP1 complex. The N- and C-terminal tails of Csk-SH3 are disordered on the ps-ns timescale while the regions with secondary structure are generally ordered (average S2 = 0.86±0.15). Some disordered residues are seen in the top of the RT, n-Src and distal-loops (indicated in Fig. 1a) (average S2 for loops = 0.80±0.16). These segments are not critical to the recognition face in the complex. The residues Ala 40 and Thr 42, lying at the end of the third β-strand and crucial in the recognition of Val 22 on the peptide, have low 15NH' order parameters (circled in blue in Fig. 4b). These residues also have steady-state 15N{1H'} nOe's that are far lower (0.32 and 0.18 respectively) than average (0.72±0.13 for regions with secondary structure, 0.67±0.03 for loops) confirming that these low order parameters are not artifacts of analysis. It is interesting to note that Val 22 on PEP-3BP1 also has a lower 15N{1H'} nOe (0.37 at 500 MHz, Fig. 4c) than the ordered regions (0.67±0.03 at 500 MHz). Fig. 4d shows a closer view of the interacting methyl groups of Ala 40 and Thr 42 on Csk-SH3 and Val 22 on PEP-3BP1. The apparently well-packed sidechain methyl groups for these two residues which are crucial in providing specificity to the recognition of PEP-3BP1 by Csk-SH3, stand in apparent contrast to the experimentally determined dynamic character of the backbone. The methyl groups of Val 41 on Csk-SH3 do not take part in this interaction and Val 41 has a high 15NH' order parameter. One can speculate that the loss of sidechain entropy on complex formation is compensated by increased flexibility of the backbone for these residues. A detailed study of sidechain dynamics would provide a better understanding of these effects29. However, it is extremely difficult to evaluate these results in a quantitative fashion in terms of thermodynamic quantities, as performed by Zidek et. al.30 and Lee et. al.29, since no data for the unligated state is available. As previously stated, free Csk-SH3 is oligomeric and the dimerization interfaces occlude the two peptide-recognition surfaces. Thus any point mutations on these interfaces to disrupt oligomerization would necessarily affect peptide binding.
Several residues on both Csk-SH3 and PEP-3BP1 exhibit apparent motion in the ms timescale. The residues are associated exclusively with the PPII helix-binding surface of Csk-SH3 (Fig. 4e). These motions are characterized by large differences in relaxation rates, ΔR2av (see Materials and Methods)31. The largest ΔR2av value on PEP-3BP1 is seen on Leu 12, which lies in the center of the polyproline helix. The large ΔR2av values in this region may result from conformational mobility of the peptide on the surface of Csk-SH3 and of the residues of Csk-SH3 themselves in the ms timescale. This region is very rich in aromatic residues contributing to significant differences in the chemical shift environments of the various conformers and resulting in very large ΔR2av values. However, the indole imino Hε of Trp 47, that is hydrogen-bonded with the C' atom of Pro 13 on PEP-3BP1, shows no measurable ΔR2av and has a 15N{1H'} steady-state nOe value that is similar to that seen for the core residues of PEP-3BP1. The residues that form the 310-helix on PEP-3BP1 also show some large ΔR2av values. Ile 21 on PEP-3BP1 shows a very small but measurable ΔR2av value while Val 22, disordered on the ps-ns timescale, shows no measurable ΔR2av value indicating that the two peptide-recognition surfaces have significantly different dynamic properties. In fact, none of the residues with measurable ΔR2av show S2 values that deviate by more than one standard deviation from the mean value for the Csk-SH3 core.
This clear separation of timescales in dynamics between the two peptide- recognition surfaces may provide insight into the specificity of peptide recognition. Slow ms-scale motions have been described as "locally-collective"24 usually involving several atoms as opposed to ns scale motions that are local and involve fast bond librations and dihedral angle transitions. Thus, the ms scale motions are expected to involve larger changes in local structure (and hence of chemical shift) and regions which exhibit this kind of motion would be more tolerant of the nature of the ligand associated with it as long as the gross features required for interaction are present. This is indeed true for the interaction of SH3 domains with PPII core sequences. These interactions are rather non-specific and characterized by relatively low binding affinities (Kd > 10μM)15. A similar argument has been made to correlate the presence of slow motion in the DNA binding domain of the Transition-state regulator AbrB in Bacillus subtilis32 and its non-specificity in DNA sequence recognition. Highly specific interactions that involve rather small, localized features, on the other hand, would be less tolerant of large conformational variations.
Discussion
The structural studies presented here show that interactions involving residues distant from the standard PPII recognition surface play a major role in specific recognition by Csk-SH3 of its natural ligand. The highly specific, high-affinity interaction between PEP-3BP1 and the SH3 domain of Csk involves two hydrophobic residues which lie C-terminal to the PPII helix region, namely Ile 21 (Ile 625 in full-length PEP) and Val 22 (Val 626 in full-length PEP) which interact with three residues on Csk-SH3, namely Ala 40, Thr 42 and Lys 43. These residues lie at the end of the second β-strand and the start of the n-Src loop on Csk-SH3. The high-affinity and high-specificity accorded to the interaction of an SH3 domain with its peptide ligand by residues distant from the PPII helix region on the peptide is also present in the SFK/Nef interactions. HIV-1 Nef binds the SH3 domains of Hck (Hematopoetic cell kinase) with sub-micromolar affinity (Kd ~ 0.3μM in the context of the entire protein) but not the highly homologous Fyn (Fgr and yes-related novel) kinase, with the reverse being true for HIV-2 Nef and SIV Nef33. This specificity has been attributed to a sequence of three residues C-terminal to the proline-rich sequence, namely, Trp 113, Thr 117 and Gln 118 on HIV-1 Nef that are mutated to Tyr, Glu and Glu, respectively in HIV-2 Nef and SIV Nef34. These residues lie on the A and B α-helices forming a pocket that accommodates the sidechains of a single residue, 9635, located on the RT-loop of the SH3 domain. This residue is Arg 96 in Fyn and packs in the more negatively charged pocket provided by HIV-2 Nef and SIV Nef, in contrast to Ile 96 in Hck, which packs more efficiently in the hydrophobic pocket of HIV-1 Nef. It is clear that the details of the interactions determining the specificity of HIV/SIV Nef towards SH3 domains in SFKs are quite different from that in the case of the association of PEP with Csk-SH3. The peptide recognition surfaces in the two cases, namely, HIV-1 Nef/Fyn-SH3 (R96I mutant)35 and Csk-SH3/PEP-3BP1, are depicted in Fig. 5. The total contact surfaces, 1,245 Å2 in the former and 1,130 Å2 in the latter, are quite similar. These, however, are much larger than the canonical PPII recognition surface (780 Å2 in Src-SH336). It is clear that the PPII recognition surface (shown in cyan in Fig. 5) is quite similar in the two interactions while the specificity pockets (shown in red in Fig. 5) are quite different, both in general location, and in characteristics.
Fig. 5.

Comparison of the peptide recognition surfaces in the HIV-1 Nef/Fyn-SH3 (R96I)33 mutant and Csk-SH3/PEP interactions. Two orientations of the surfaces are shown. The canonical PPII recognition surface is shown in cyan and the specificity pocket is in red. The PPII recognition pocket is very similar in the two cases while the specificity pockets are quite different. The residues that form the canonical PPII recognition site are labeled in blue. The figures were prepared using the program GRASP50. The residues that form the specificity pocket in Fyn-SH3 are labeled in brown and those that form the specificity pocket in Csk-SH3 are labeled in green.
a, HIV-1 Nef/Fyn-SH3 (R96I). The residues Ile 96 and Glu 94 form the specificity pocket. Ile 96 provides most of the binding energy to the interaction of the mutant Fyn-SH3 and HIV-1 Nef. Glu 116 corresponds to Lys 43 on Csk-SH3.
b, Csk-SH3/PEP-3BP1. The residues Ala 40, Thr 42 and Lys 43 are deemed crucial for specific recognition of PEP-3BP1. His 21 and Thr 23 correspond to Glu 94 and Ile 96 on Fyn-SH3.
Materials and Methods
Sample Preparation
The gene for the SH3 domain of Csk (residues 1-83) was cloned into the expression vector PGEX-3Xb (Pharmacia), over expressed in E. coli BL21 and purified using the methods described in detail elsewhere (A. Shekhtman, R. Ghose, D. Wang, P. A. Cole & D. Cowburn, submitted). A synthetic cDNA encoding PEP-3BP1 was prepared with optimal codon usage for E. coli, subcloned into the HindIII-BamHI sites of pTMHa expression vector37 and purified as previously described38. Four NMR samples of Csk-SH3/PEP-3BP1, [U 15N]-Csk-SH3/PEP-3BP1, [U 15N, 13C]-Csk-SH3/PEP-3BP1, Csk-SH3/[U 15N, 13C]-PEP-3BP1, were prepared in aqueous buffer (20 mM Tris-HCl pH 7.2, 200 mM NaCl, 1 mM DTT, 5mM EDTA, 2 mM ADP, 0.02% NaN3). The samples ranged in concentration from 800μM to 1.0 mM. A final sample in D2O was prepared by multiple concentration steps of [U 15N]-Csk-SH3/PEP-3BP1 by adding deuterated buffer yielding a 500 μM sample. Direct fluorescence titration studies showed a Kd of 0.8 μM for PEP-3BP1 binding to the Csk-SH3 domain, consistent with previous measurements using a competitive assay, of 0.7μM12.
NMR Spectroscopy
NMR spectra were recorded on Bruker DMX spectrometers operating at 1H frequencies of 500 MHz or 600 MHz and equipped with triple-resonance probes with z-axis magnetic field gradient capability. All spectra were collected at 20° C and processed using the NmrDraw39 suite of programs, and analyzed using the program Pipp40 and in-house software.
Resonance Assignment and Constraint Generation
Nearly complete backbone and sidechain assignments for Csk-SH3 and PEP-3BP1 were obtained from HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, HBHA(CO)NH41, CC(CO)NH and HCC(CO)NH experiments42. The peak intensities measured from 15N-edited NOESY-HSQC (100ms and 120ms in H2O)41, 13C-edited NOESY-HSQC41 (100ms, 120ms in H2O and 120 ms in D2O) spectra were converted into distance constraints using in-house calibration program that mimics the CALIBA function of the program DYANA v1.543. The program TALOS44 was used to generate ϕ and ψ constraints from the H', Cα, Cβ and C’ chemical shifts in the ordered regions of the complex. ϕ angles were also obtained from 3J(H'Hα) coupling from the HNHA experiment41.
Relaxation Experiments
Conventional R1, R2 (CPMG) and 15N{1H'} steady-state nOe experiments were collected on [U 15N] –Csk/PEP-3BP1 complex at 500 MHz and 600 MHz and a 15N-{1H'} steady-state nOe experiment on the Csk-SH3/[U 15N,13C]-PEP-3BP1 complex (the 13C-15N dipolar coupling does not have a measurable effect) at 500 MHz, using previously described protocols28. The relaxation delays were 4 ms (×2), 500 ms (×3) for R1 experiments and 8 ms (×2) and 200 ms (×3) for the R2 experiments for an optimal sampling of the relaxation curve45. A CPMG pulse repetition time of 1 ms was used for the R2 measurements. Recycle times used were 1.5 s (R1, R2), 7.5 s (nOe reference) and 5 s (with 2.5 s saturation time, nOe). The relaxation rates were extracted from mono-exponential fits of the relaxation data using in-house programs utilizing the ODRPACK library46 with the errors of the fitted parameters representing the 95 % confidence bounds obtained from the covariance matrix of the fits. The dynamics on the fast ps-ns timescale were obtained by standard analyses of the R1, R2 and nOe data using the Lipari-Szabo25 and extended Lipari-Szabo47 models as previously described using the DYNAMICS28 package.
The influence of dynamics in the ms timescale on the relaxation properties was determined using the relaxation-compensated CPMG experiment31. Delays between CPMG pulses (τcp) of 1 ms (8, 24, 40, 56, 96, 112), 2 ms (16, 16, 32, 48, 96, 128), 10 ms (0, 40, 80, 160, 280, 360) and 20 ms (0, 80, 160, 240, 320, 400) on the [U 15N] –Csk/PEP-3BP1 sample at 500 MHz and 1 ms (0, 4, 8, 40, 60, 100), 6 ms (0, 40, 120, 160, 200, 280, 360) and 12 ms (0, 24, 48, 96, 120, 168, 216) at 600 MHz on the Csk-SH3/[U 15N,13C]-PEP-3BP1 sample along with recycle delays of 1.5 s, were used. The relaxation delays, in ms, used are indicated in brackets. The relaxation rates R2av (average between the in phase and antiphase relaxation rates) measured with τcp=1 ms correlated extremely well with the rates obtained from the R2 (CPMG) experiment. No attempt was made to extract the exact rates (Rex) from the limited data. Instead those residues which showed a statistically significant difference (ΔR2av) greater than 4.0 s−1 between the relaxation rates measured using τcp=1 ms and τcp=10 ms were identified as displaying dynamics on the ms timescale.
Structure Calculations
Structure calculations were performed using the DYANA v1.543 package using experimentally determined distance and angular constraints (ϕ, ψ) constraints. In the final calculation, 124 hydrogen-bonding constraints (2 upper limits and 2 lower limits corresponding to each hydrogen bond) were also included. Residues 1-9 and 70-83 in Csk-SH3 and residues 1-7 in PEP-3BP1 were excluded (as flexible) from the final structure calculations of the complex. Structure calculations were performed by using random structures generated by initially placing the N-terminus of the PEP-3BP1 peptide at distance of 90 Å from the C-terminal end of the Csk-SH3 domain. In all, 1,000 structures were generated and subjected to 4,000 steps of torsional angle dynamics at an elevated temperature followed by 16,000 steps of simulated annealing. Finally the resulting structures were subjected to 8,000 steps of conjugate gradient minimization. The 25 structures that had the lowest value for the target function were considered to represent the structure of the Csk-SH3/PEP-3BP1 complex.
Co-ordinates
The coordinates have been deposited in the Protein Data Bank (accession code 1JEG).
Acknowledgements
R.G. would like to thank David Fushman for useful discussions and for providing the DYNAMICS package and Patrick Loria for providing the relaxation compensated CPMG pulse sequence. The authors thank P. A. Cole for useful discussions. This work has been supported by a grant from the National Institutes of Health, and a fellowship from the National Cancer Institute to A.S.
References
- 1.Erpel T & Courtneidge SA Src Family Protein Tyrosine Kinases and Cellular Signal Transduction Pathways. Current Opinion Cell Biol. 7, 176–182 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Superti-Furga G & Courtneidge SA Structure-function relationships in Src family and related protein tyrosine kinases. Bioessays 17, 321–330 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Sabe H et al. Molecular cloning and expression of chicken C-terminal Src kinase: lack of stable association with c-Src protein. Proc. Natl. Acad. Sci. U S A 89, 2190–4 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imamoto A & Soriano P Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell 73, 1117–24 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Chow LM, Fournel M, Davidson D & Veillette A Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature 365, 156–60 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Thomas SM, Soriano P & Imamoto A Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature 376, 267–71 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Harrison SC & Eck MJ Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Cloutier JF & Veillette A Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. 15, 4909–18 (1996). [PMC free article] [PubMed] [Google Scholar]
- 9.Vang T et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent Protein Kinase Inhibits Signaling through the T Cell Receptor. J. Exp. Med 193, 497–508. (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gjorloff-Wingren A, Saxena M, Williams S, Hammi D & Mustelin T Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur. J. Immunol 29, 3845–54 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Cloutier JF & Veillette A Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J. Exp. Med 189, 111–21 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregorieff A, Cloutier JF & Veillette A Sequence requirements for association of protein-tyrosine phosphatase PEP with the Src homology 3 domain of inhibitory tyrosine protein kinase p50(csk). J. Biol. Chem 273, 13217–22 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Borchert TV, Mathieu M, Zeelen JP, Courtneidge SA & Wierenga RK The crystal structure of human CskSH3: structural diversity near the RT-Src and n-Src loop. FEBS Lett. 341, 79–85 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Cowburn D & Kuriyan J SH2, SH3 and PH domains. in Signal Transduction (eds. Heldin C-H & Purton M) 127–142 (Chapman & Hall, London, 1996). [Google Scholar]
- 15.Mayer BJ SH3 domains : complexity in moderation. J. Cell Sci 114, 1253–1263 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Yu H et al. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76, 933–45 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Yu H et al. Solution structure of the SH3 domain of Src and identification of its ligand-binding site. Science 258, 1665–8 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Lim WA, Richards FM & Fox RO Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372, 375–9 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Lim WA & Richards FM Critical residues in an SH3 domain from Sem-5 suggest a mechanism for proline-rich peptide recognition. Nature Struct. Biol 1, 221–225 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Weng Z et al. Structure-function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol. Cell. Biol 15, 5627–5634 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson D, Cloutier JF, Gregorieff A & Veillette A Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J. Biol. Chem 272, 23455–62 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Davidson D, Chow LM & Veillette A Chk, a Csk family tyrosine protein kinase, exhibits Csk-like activity in fibroblasts, but not in an antigen-specific T-cell line. J. Biol. Chem 272, 1355–62 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Grgurevich S et al. The Csk-like proteins Lsk, Hyl, and Matk represent the same Csk homologous kinase (Chk) and are regulated by stem cell factor in the megakaryoblastic cell line MO7e. Growth Factors 14, 103–15 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Kay LE Protein dynamics from NMR. Nature Struct. Biol. 5, 513–517 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Lipari G & Szabo A Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. J. Am. Chem. Soc 104, 4559–4570 (1982). [Google Scholar]
- 26.Ghose R, Fushman D & Cowburn D Determination of the rotational diffusion tensor of macromolecules in solution from NMR relaxation data with a combination of exact and approximate methods - application to the determination of interdomain orientation in multidomain proteins. J. Magn. Reson 149, 204–217 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Garcia de la Torre J, Huertas ML & Carrasco B HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson 147, 138–46 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Fushman D, Cahill S & Cowburn D The main chain dynamics of the dynamin pleckstrin homology (PH) domain in solution: Analysis of 15N relaxation with monomer/dimer equilibration. J. Mol.. Biol 266, 173–194 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Lee AL, Kinnear SA & Wand J Redistribution and loss of sidechain entropy upon complex formation of a calmodulin-peptide complex. Nature Struct. Biol 7, 72–77 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Zidek L, Novotny MV & Stone M Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nature Struct. Biol 6, 1118–1121 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Loria JP, Rance M & Palmer AGPI A Relaxation-Compensated Carr-Purcell-Meiboom-Gill Sequence for Characterizing Chemical Exchange by NMR Spectroscopy. J. Am. Chem. Soc 121, 2331–2332 (1999). [Google Scholar]
- 32.Vaughn JL, Feher VA, Bracken C & Cavanagh J The DNA-binding domain in the Bacillus subtilis transition-state regulator AbrB employs significant motion for promiscuous DNA recognition. J. Mol. Biol 305, 429–439 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Lee CH et al. A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 14, 5006–15 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collette Y et al. HIV-2 and SIV Nef proteins target different Src family SH3 domains than does HIV-1 Nef because of a triple amino acid substitution. J. Biol. Chem 275, 4171–6. (2000). [DOI] [PubMed] [Google Scholar]
- 35.Lee C-H, Saksela K, Mirza UA, Chait BT & Kuriyan J Crystal Structure of the Conserved Core of HIV-1 Nef Complexed with a Src Family SH3 Domain. Cell 85, 931–942 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Feng S, Chen JK, Yu H, Simon JA & Schreiber SL Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science 266, 1241–1247 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Staley JP & Kim PS Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic tripsin inhibitor. Protein Sci. 3, 1822–1832 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu W, Ji H & Lu M Trimerization specificity in HIV-1 gp41 : Analysis with a GCN4 Leucine Zipper Model. Biochemistry 38, 5378–5385 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–93 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Garrett DS, Powers R, Gronenborn AM & Clore GM A common sense approach to peak picking in two-, three- and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson 95, 214–220 (1991). [DOI] [PubMed] [Google Scholar]
- 41.Cavanagh J, Fairbrother WJ III, A.J.P. & Skelton NJ Protein NMR Spectroscopy, (Academic Press, San Diego, 1996). [Google Scholar]
- 42.Logan TM, Olejniczak ET, Xu RX & Fesik SW A general method for assigning NMR spectra of denatured proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J. Biomol. NMR 3, 225–31 (1993). [DOI] [PubMed] [Google Scholar]
- 43.Guentert P, Mumenthaler C & Wuthrich K Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol 273, 283–298 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Cornilescu G, Delaglio F & Bax A Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Jones JA Optimal sampling strategies for the measurement of relaxation times in proteins. J. Magn. Reson 126, 283–6 (1997). [Google Scholar]
- 46.Boggs PT, Byrd RH, Rogers TE & Schnabel RB User's Reference Guide for ODRPACK 2.01-Software for Weighted Orthogonal Distance Regression; NIST IR4834, (U.S. Government Printing Office: Washington, DC, 1992). [Google Scholar]
- 47.Clore GM et al. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc 112, 4989–4936 (1990). [Google Scholar]
- 48.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R & Thornton JM AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–86 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Koradi R, Billeter M & Wuthrich K A program for display and analysis of macromolecular structures. J. Mol. Graph 14, 51–55 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Nicholls A, Sharp KA & Honig B Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11, 281–96 (1991). [DOI] [PubMed] [Google Scholar]