

1. Introduction
Cells have developed adaptive mechanisms to respond to stresses such as reduced oxygen levels and changes in nutrient availability. A well-characterized mechanism for adaptation involves upregulation of hypoxic inducible factor (HIF), whose protein levels are regulated by oxygen-dependent prolyl hydroxylase domain enzymes (PHDs). PHDs are members of a super-family of enzymes called 2-oxoglutarate-, iron-, and oxygen-dependent oxygenases (2-OGDOs). For the vast majority of 2-OGDOs, 2-oxoglutarate (2-OG) binding to the active site is followed by O2 binding, and the target and co-factors binding the 2-OGDO. This leads to the oxidative decarboxylation of 2-OG, hydroxylation of the target, and release of succinate and CO2.[1] In addition to regulating the HIF transcription factor, enzymes within this super-family also target histones and ribosomal proteins. A new and lesser-known member of this family is 2-oxoglutarate- and iron-dependent oxygenase domain-containing protein 1 (OGFOD1). OGFOD1 hydroxylates a specific proline on ribosomal protein s23 (RPS23), thereby modulating translation.[2–5] OGFOD1 homologues in yeast, fruit flies, and humans have been demonstrated to regulate translation, especially translation termination. In human cardiomyocytes, OGFOD1 deletion, and subsequent loss of RPS23 prolyl hydroxylation, suppresses expression of cytoskeletal proteins, nucleic acid binding proteins, mRNA splicing proteins, and chaperones; while up-regulating proteins functioning in glycolytic processes, TCA cycle, cardiac muscle contraction, and ATP metabolism and biosynthesis.[3, 6] Together, these data highlight OGFOD1’s regulation of key processes, including the cytoskeleton, protein synthesis and folding, and energy metabolism. Regulating these essential processes in cardiomyocytes makes OGFOD1 an intriguing potential regulator of cardiac hypertrophy.
In the present study, we investigated OGFOD1 expression in human heart failure, where we found OGFOD1 to be upregulated. Cardiac hypertrophy, which can lead to heart failure, relies on an increase in protein synthesis to support increased cardiac load.[7–9] Therefore, the finding that OGFOD1 was higher in failing human hearts combined with the role of OGFOD1 in regulating translation prompted an examination of OGFOD1’s role in cardiac hypertrophy. Here, we induce hypertrophy in OGFOD1-knockout (KO) mice and wildtype (WT) mice using isoproterenol (ISO), and find that OGFOD1 deletion protects mice against ISO-induced hypertrophy. To understand pathways that may be mis-regulated at the translational level upon OGFOD1 deletion, we did RNA-sequencing (RNAseq), and correlated the results with our murine proteomics[6] to identify targets differentially regulated at the RNA and protein levels. We found that OGFOD1 regulated the expression of proteins functioning in DNA damage repair, unfolded protein response, and response to oxygen levels, supporting a key role for OGFOD1 in the cardiac stress response.
2. Methods
2.1. Human heart samples
Human left ventricular myocardium was obtained from explanted cardiomyopathic hearts from patients undergoing heart transplantation. The discarded tissue samples received an exemption following institution review board guidelines. Non-failing hearts were from organ donors who were excluded from transplant due to age or technical problems.
2.2. Mice
Ogfod1-KO mice were generated as previously described.[2] All animal studies were completed in a manner consistent with the recommendations established by the Guide for the Care and Use of Laboratory Animals (National Institutes of Health), and all animal protocols were approved by the National Heart, Lung and Blood Institute’s Animal Care and Use Committee. All mice are on a C57BL/6N background from Taconic farms. Age-matched Ogfod1-WT and Ogfod1-KO male mice aged 3–5 months were used for this study. Echocardiography and isoproterenol treatment details are provided in supplemental methods.
2.4. RNA extraction
RNA was extracted from N = 5 mouse hearts from each of the following 4 groups: WT + vehicle, WT + ISO, KO + vehicle, and KO + ISO using RNeasy RNA extraction kit (Qiagen). RNA was extracted from human hearts using Qiagen’s RNeasy mini kit, and DNase treated on-column following the manufacturer’s instructions. RNA concentration was determined by Nanodrop.
2.5. RNA sequencing
Libraries were prepared using TruSeq stranded total RNA sample preparation kit (illumina). Libraries were sequenced using paired-end 150-bp reads on an Illumina Novaseq 6000. Raw sequencing reads were trimmed for adapters using TrimGalore (v0.6.2). Trimmed reads were mapped to the mouse reference genome GRC m38 with HISAT2 (v2.1.0). Gene based quantification was performed with featureCounts (v1.6.3) with GENCODE v23. Differential expression analysis was implemented using limma (v3.40.6). Gene set enrichment analysis was carried out using gage (v2.34).[10] The RNA seq data is publicly available in GEO via accession #GSE217755. Enrichment terms were determined using Metascape. See supplemental methods for references.
2.6. RT-qPCR
CDNA was synthesized from the RNA template using Bio-Rad’s iScript™ cDNA synthesis kit (Biorad #1708890). Synthesized cDNA was used as a template for primer-specific quantitative PCR amplification using Roche SYBR-Green (Sigma # 4913850001) on the Stratagene Mx3005P Realtime PCR System (Agilent Technologies). Additional details and primer sequences can be found in supplemental methods.
2.7. Western blot
Mouse and human hearts were homogenized in Radioimmunoprecipitation assay buffer (ThermoFisher #89901) amended with protease inhibitors (Roche #04-693-159-001). Hearts were homogenized by mechanical disruption on the Precellys® 24 with Cryolys (Bertin Technologies) tissue homogenizer chilled to 4°C. Protein lysates were separated by SDS-PAGE, transferred to blotting membranes, and probed with primary and secondary antibodies to determine specific protein abundances. The specificity of the OGFOD1 antibodies were validated using western blot on either WT and KO mouse cardiac lysates (Figure S3A) or in lysates from human induced cardiomyocytes (Figure S3D). All unedited blots can be found in Figure S1. Additional western blot details are provided in the supplemental methods.
2.8. Statistics
A Student’s T-test was used to compare 2 groups with Gaussian distribution. The Mann-Whitney test was used to compare 2 groups without Gaussian distribution. Analysis of Variance (ANOVA) was used to compare more than 2 groups. A P-value less than 0.05 was considered significant. All tests were applied assuming unequal sample variation.
3. Results and Discussion
We assessed OGFOD1 mRNA expression in non-failing and failing human hearts and found OGFOD1 mRNA (Figure 1A) and protein (Figure 1B, S1, S3) were significantly upregulated in human failing hearts, with changes at the mRNA and protein level each showing an approximate 2-fold increase. To investigate the role of OGFOD1 in hypertrophy, we treated wildtype (WT) mice and OGFOD1-knockout (KO) mice with 21 mg/kg/day isoproterenol (ISO) released steadily over 2 weeks by osmotic minipump. ISO-treated KO mice showed significantly less hypertrophy than ISO-treated WT mice (Figure 1C). This protection was also evident in the heart weight to body weight (HW/BW) ratios (Figure 1D), in the posterior wall thickness (Figure 1E), and in the heart weights as determined by echocardiography (Figure 1F). Interestingly, neither the ejection fraction (Figure 1G) nor the fractional shortening (Figure 1H) changed significantly with this ISO treatment, suggesting that the hearts had mild hypertrophy which had not yet led to contractile dysfunction. The finding that OGFOD1 loss is protective in ISO-induced hypertrophy prompted us to assess OGFOD1 expression levels in response to ISO treatment where we found that OGFOD1 levels were unchanged (Figure S3B, S3C). Two weeks of β-adrenergic stimulation induced by ISO treatment is less chronic and less severe than human heart failure, so OGFOD1 accumulating in human heart failure, but not in ISO-induced hypertrophy in mice, may be due to differences in the severity of the model or disease state. More work is needed to investigate how changes in the levels of OGFOD1’s substrates, such as 2-OG, or inhibitory end-products, such as succinate, can impact OGFOD1 activity.
Figure 1.
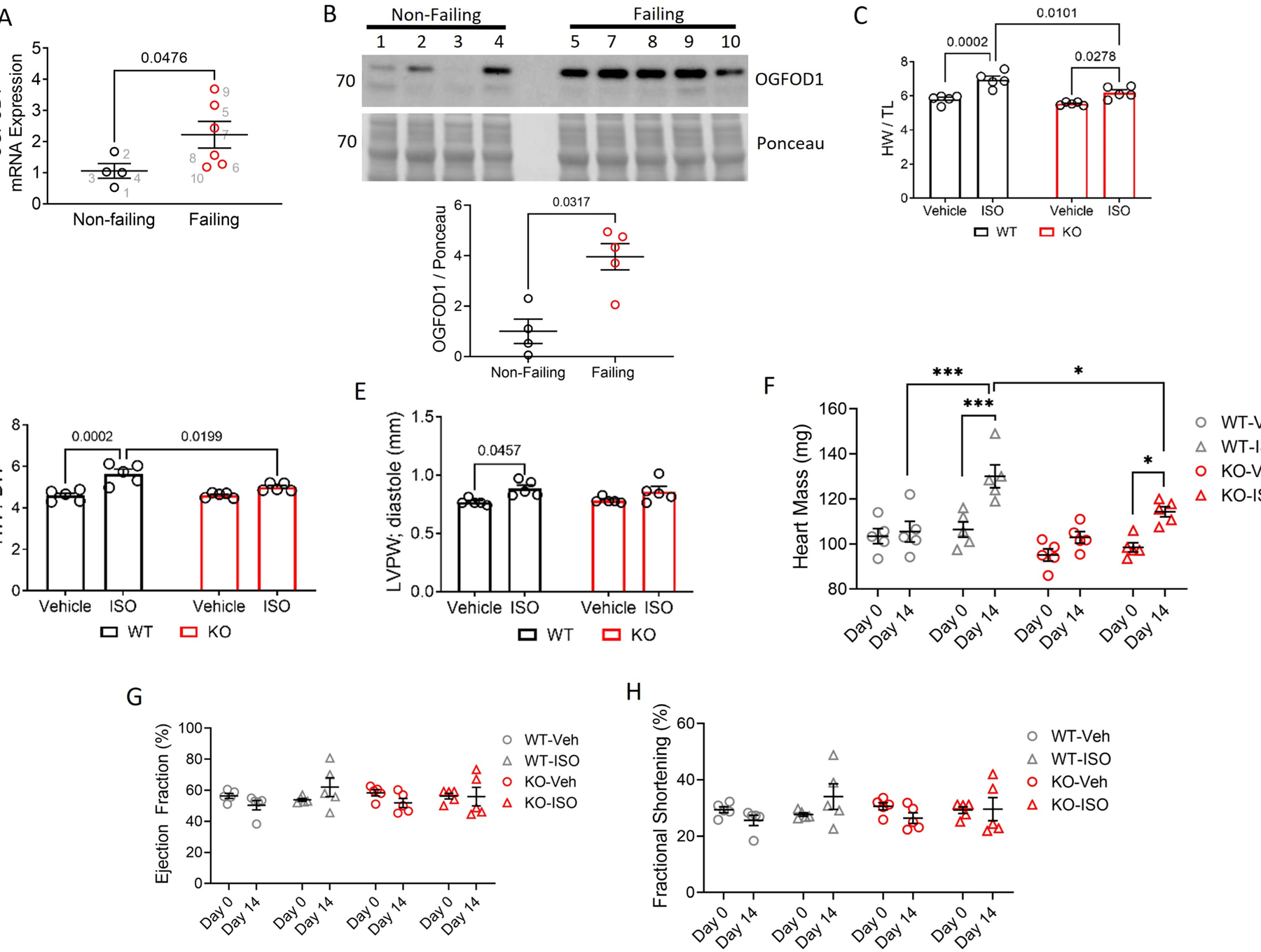
OGFOD1 in human heart failure, cardiac hypertrophy, and translation regulation. A-B. OGFOD1 mRNA (A) and protein (B) abundance in male non-failing and failing human hearts. N = 4–6 biological replicates per group. Data in panel A are representative of 3 independent experiments. Each number in panel A denotes a biological replicate, and matches the sample with the same number in panel B. C-D. Heart weight to tibia length (C) and heart weight to body weight (D) ratios in vehicle- and ISO-treated mice. E-H. Left ventricular posterior wall thickness in diastole (E), heart mass (F), ejection fraction (G), fractional shortening (H) as determined by echocardiography. Data shown as mean ± standard error. N = 5 biological replicates per group. Mann-Whitney test (A-B), Two-way ANOVA (C-E), or Three-way ANOVA with Tukey’s multiple comparisons test (F-H) was used to determined significance. A P-value less than 0.05 was considered significant. *P < 0.05 **P < 0.005 ***P < 0.0005.
Because OGFOD1 is known to regulate translation,[2, 3, 5] we investigated OGFOD1’s role in regulating translation in the heart to identify targets that may be important in susceptibility to hypertrophy. We did RNA-sequencing (RNAseq) on transcripts isolated from mouse hearts (Figure S2), and correlated the results with our murine proteomics[6] to identify targets differentially regulated at the RNA and protein levels. There were 3861 identified proteins and 15605 identified transcripts, and 3519 were identified in both the RNAseq and the proteomics datasets. Of these 3519 identified gene products, 3000 were unchanged at the RNA or protein levels upon OGFOD1 deletion. Of the remaining 519 that were changed in OGFOD1-KO hearts compared to WT hearts, 5 were changed in opposing directions at the RNA level versus the protein level, and only 21 were changed in the same direction at the RNA and protein levels (Figure 2A). The remaining factors were changed at only the RNA level or only the protein level. 335 gene products were changed significantly at the RNA level, but not the protein level (Figure 2B, 2C). These changes indicate OGFOD1 may play a role in coupling mRNA and protein level abundances. When we analyzed these gene products based upon their up- or downregulation, the downregulated factors were significantly enriched in branched chain amino acid degradation and fatty acid oxidation, supporting a role for OGFOD1 in regulating metabolic processes. Upregulated factors were significantly enriched in smooth muscle contraction. 158 factors were changed at the protein level, but not the RNA level (Figure 2D) in KO hearts. According to Metascape enrichment analysis, these proteins were most significantly enriched in the term Response to Oxygen Levels consistent with a role for OGFOD1 in stress (Figure 2E).[11] We again separately analyzed the factors that were upregulated versus those that were downregulated. Downregulated proteins were enriched in metabolic process, and upregulated proteins were enriched in response to oxygen. Interestingly, this group of upregulated proteins (Figure 2D, 2E) included an increase in heat shock proteins, several DNA damage repair enzymes and vacuolar ATPase subunits. Altogether, these results support a role for OGFOD1 in regulating translation in the heart, as a potential mechanism for altering susceptibility to cardiac hypertrophy. However, because the OGFOD1-KO mouse has global OGFOD1 deletion, rather than cell-type specific OGFOD1 deletion, we are unable to attribute the observed protection directly to cardiomyocytes. Future investigations are needed to identify specific cell types responsible for the observed protection in cardiac hypertrophy.
Figure 2.
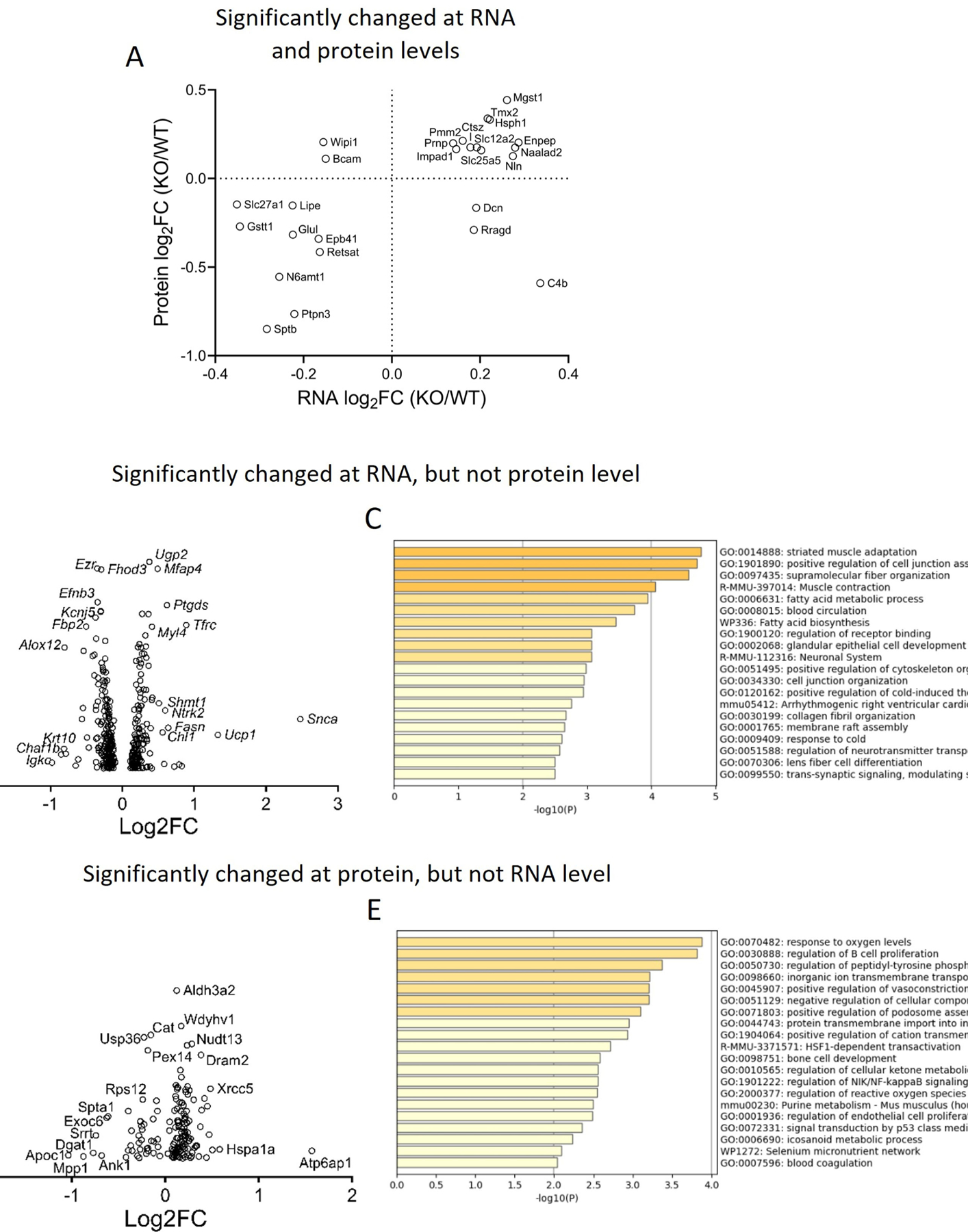
Expression of factors identified in both the RNAseq and proteomics datasets. A. RNA versus protein expression changes for factors that were significantly changed at both the RNA and protein levels. B, D. Expression changes plotted against P-values for factors changed at the RNA level, but not the protein level (B); or factors changed at the protein level, but not the RNA level (D). C, E. Enrichment terms for factors identified in panel B and panel D, respectively.
Supplementary Material
4. Acknowledgements
This work was funded by the NHLBI-NIH Intramural Research Program (ZO1-HL002066), and done in collaboration with the Animal Surgery and Resources Core, Murine Phenotyping Core, DNA Sequencing and Genomics Core, iPSC Core, and Bioinformatics and Computational Biology Core.
Footnotes
Disclosures
None.
References
- [1].Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ, 2-Oxoglutarate-Dependent Oxygenases, Annu Rev Biochem 87 (2018) 585–620. [DOI] [PubMed] [Google Scholar]
- [2].Singleton RS, Liu-Yi P, Formenti F, Ge W, Sekirnik R, Fischer R, Adam J, Pollard PJ, Wolf A, Thalhammer A, Loenarz C, Flashman E, Yamamoto A, Coleman ML, Kessler BM, Wappner P, Schofield CJ, Ratcliffe PJ, Cockman ME, OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation, Proc Natl Acad Sci U S A 111(11) (2014) 4031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stoehr A, Kennedy L, Yang Y, Patel S, Lin Y, Linask KL, Fergusson MM, Zhu J, Gucek M, Zou J, Murphy E, The ribosomal prolyl-hydroxylase OGFOD1 decreases during cardiac differentiation and modulates translation and splicing, JCI Insight 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wehner KA, Schutz S, Sarnow P, OGFOD1, a novel modulator of eukaryotic translation initiation factor 2alpha phosphorylation and the cellular response to stress, Mol Cell Biol 30(8) (2010) 2006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Katz MJ, Acevedo JM, Loenarz C, Galagovsky D, Liu-Yi P, Perez-Pepe M, Thalhammer A, Sekirnik R, Ge W, Melani M, Thomas MG, Simonetta S, Boccaccio GL, Schofield CJ, Cockman ME, Ratcliffe PJ, Wappner P, Sudestada1, a Drosophila ribosomal prolyl-hydroxylase required for mRNA translation, cell homeostasis, and organ growth, Proc Natl Acad Sci U S A 111(11) (2014) 4025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Harris M, Sun J, Keeran K, Aponte A, Singh K, Springer D, Gucek M, Pirooznia M, Cockman ME, Murphy E, Kennedy LM, Ogfod1 deletion increases cardiac beta-alanine levels and protects mice against ischemia-reperfusion injury, Cardiovasc Res (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, Vanwagner LB, Tsao CW, Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association, Circulation 141(9) (2020). [DOI] [PubMed] [Google Scholar]
- [8].Zuhlke V, Du Mesnil de R, Gudbjarnason S, Bing RJ, Inhibition of protein synthesis in cardiac hypertrophy and its relation to myocardial failure, Circ Res 18(5) (1966) 558–72. [DOI] [PubMed] [Google Scholar]
- [9].Heineke J, Molkentin JD, Regulation of cardiac hypertrophy by intracellular signalling pathways, Nat Rev Mol Cell Biol 7(8) (2006) 589–600. [DOI] [PubMed] [Google Scholar]
- [10].Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ, GAGE: generally applicable gene set enrichment for pathway analysis, BMC Bioinformatics 10 (2009) 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK, Metascape provides a biologist-oriented resource for the analysis of systems-level datasets, Nat Commun 10(1) (2019) 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.