

Abstract
Friedreich’s ataxia (FRDA) is an autosomal-recessive disorder primarily attributed to biallelic GAA repeat expansions that reduce expression of the mitochondrial protein frataxin (FXN). FRDA is characterized by progressive neurodegeneration, with many patients developing cardiomyopathy that progresses to heart failure and death. The potential to reverse or prevent progression of the cardiac phenotype of FRDA was investigated in a mouse model of FRDA, using an adeno-associated viral vector (AAV8) containing the coding sequence of the FXN gene. The Fxnflox/null::MCK-Cre conditional knockout mouse (FXN-MCK) has an FXN gene ablation that prevents FXN expression in cardiac and skeletal muscle, leading to cardiac insufficiency, weight loss, and morbidity. FXN-MCK mice received a single intravenous injection of an AAV8 vector containing human (hFXN) or mouse (mFXN) FXN genes under the control of a phosphoglycerate kinase promoter. Compared to vehicle-treated FXN-MCK control mice, AAV-treated FXN-MCK mice displayed increases in body weight, reversal of cardiac deficits, and increases in survival without apparent toxicity in the heart or liver for up to 12 weeks postdose. FXN protein expression in heart tissue was detected in a dose-dependent manner, exhibiting wide distribution throughout the heart similar to wild type, but more speckled. These results support an AAV8-based approach to treat FRDA-associated cardiomyopathy.
Keywords: Friedreich’s ataxia, frataxin, cardiomyopathy, adeno-associated viral vector, AAV, gene therapy, phosphoglycerate kinase, PGK, promoter
Graphical abstract
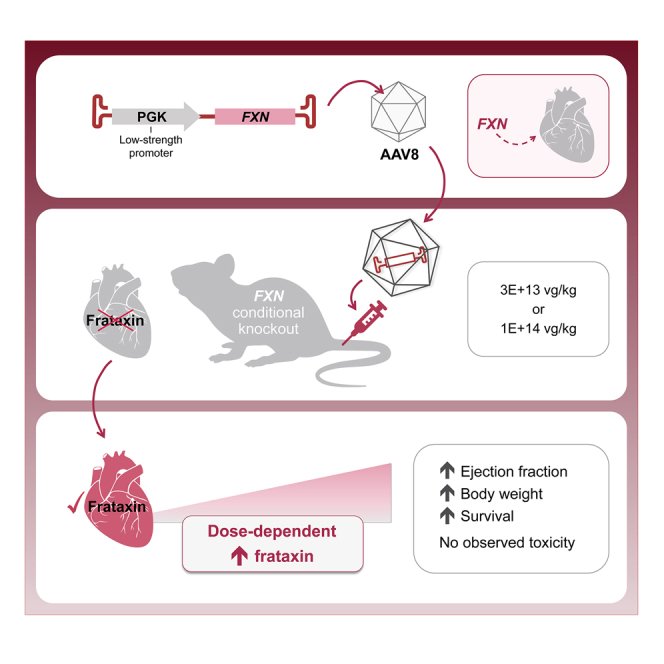
Fonck and colleagues report that AAV8-mediated systemic administration of FXN transgene significantly improved survival, body weight, heart morphology, cardiac function, cardiac injury biomarkers, and expression of mitochondrial electron transport chain complexes of Friedreich’s ataxia without toxicity observed in the heart and liver of a FXN conditional knockout mouse model.
Introduction
Friedreich’s ataxia (FRDA) is an autosomal-recessive disorder caused by biallelic GAA trinucleotide repeat expansions of the frataxin (FXN) gene.1 This intronic mutation diminishes FXN protein expression, which in most patients ranges from 2% to 30% of normal levels.1,2 FXN is a highly conserved mitochondrial protein that is involved in the assembly of the iron-sulfur complex, an important component in electron transport chain complexes I and II.3 Reduced FXN levels can result in iron accumulation in mitochondria with consequent oxidative damage and decreased ATP production, all of which can ultimately lead to cell death in vulnerable tissues,4,5 such as the central and peripheral nervous systems, heart, and pancreas.5 With an estimated prevalence of 1 in 40,000, FRDA is the most commonly inherited ataxia among people of European ancestry.6,7
Although neurological deficits are the hallmark of FRDA, the disease causes multisystemic deficits, including cardiomyopathy and glucose intolerance.5 FRDA onset occurs typically during late childhood to adolescence, with disease progression sometimes leading to premature death between 35 and 45 years of age.5,8,9 Heart disease in FRDA initially presents as concentric and hypertrophic cardiomyopathy, eventually progressing to dilated cardiomyopathy over time; some patients suffer embolic strokes and mural thrombi in the left ventricle.5,9 Histological changes in the left ventricle typically consist of cellular hypertrophy, diffuse fibrosis, and focal myocardial necrosis.10,11 The most frequent cause of death in FRDA results from congestive heart failure or arrhythmia (59%).9
Given that FRDA is a monogenic loss-of-function disease, one promising disease-modifying therapeutic approach to treat FRDA is adeno-associated viral vector (AAV)-mediated gene transfer.12 Previously published studies using AAV-mediated gene therapy administered to cardiac or neurological mouse models of FRDA have shown therapeutic benefit.13,14,15 In terms of identifying minimally therapeutic levels of AAV-mediated FXN expression, it is worth noting that heterozygous FXN-knockout mice are indistinguishable from their wild-type (WT) littermates. Likewise, human heterozygous carriers manifest no clinical symptoms.16 These observations suggest that expressing half of the endogenous FXN level may be sufficient to prevent or halt disease progression. In addition, recent reports have shown that FXN overexpression can cause toxicity both in vitro and in vivo.17,18,19
In the present study, and to avoid potential toxicity due to high FXN expression, a relatively low-strength, nonviral, phosphoglycerate kinase (PGK) promoter was selected.20 AAV8 vectors were designed to contain single-stranded DNA with codon-optimized (co) human (hFXNco) or mouse FXN (mFXNco) genes, which were evaluated in a cardiac mouse model of FRDA, the Fxnflox/null::MCK-Cre conditional knockout (FXN-MCK).21 Due to FXN protein deficiency in both heart and skeletal muscle, these FXN-MCK mice exhibit progressive weight loss associated with cardiac deficits leading to morbidity at approximately 10 weeks of age (WOA). With the AAV treatment, all FXN-MCK mice survived through 10 WOA with improved cardiac function and morphology, with FXN protein expressed in a widely distributed pattern throughout the heart, similar to that of WT but more speckled.
Results
Vector and viral constructs
To investigate the potential of AAV-based gene therapy to treat FRDA-associated cardiomyopathy, an AAV8 vector containing co cDNAs of either hFXNco or mouse FXN (mFXNco) genes were designed under the control of a human 3-PGK promoter: AAV8-hPGK-hFXNco (AAV-hFXN) and AAV8-hPGK-mFXNco (AAV-mFXN) vectors (Table S1). The DNA construct was originally subcloned into a proprietary plasmid including the viral inverted terminal repeat (ITR). RNA processing was supported by the presence of an intron and a polyadenylation signal, both from the simian virus 40 (SV40) (Figure 1A).22
Figure 1.

AAV FXN expressing construct and in vitro testing
(A) Structure of the AAV FXN-expressing construct. (B) Representative IF image of C2C12 cells treated with 1E+7 vg/cell AAV-hFXN or untransduced negative control of FXN (green) and nucleus (blue). Scale bars represent 100 μm. (C) Dose-response curve of FXN IF intensity to AAV-hFXN MOI (vg/cell) fit with 4PL equation to determine EC50 value. (D) Log-log plot of AAV-hFXN-driven FXN mRNA expression as a function of AAV-hFXN MOI (vg/cell). (E) Correlation plot of mRNA and protein expression following AAV-hFXN transduction. (F) Western blot probing for FXN (green) and GAPDH loading control (red), imaged on the LI-COR Odyssey system. Graphs were plotted using GraphPad Prism. IF images were taken on an Odyssey IX73 fluorescence microscope. IF, immunofluorescence; MOI, multiplicity of infection; polyA, polyadenylation signal.
AAV treatment efficiently transduces mouse and human cell lines and leads to robust expression of mature FXN
C2C12 (immortalized mouse myoblast) cells were transduced by the AAV-hFXN vector, and FXN expression was detected in the cytoplasm by immunofluorescence (IF) (Figure 1B). Quantification of IF analysis revealed dose-dependent FXN protein expression with a half-maximal effective concentration (EC50) at a MOI of 1.16E+5 viral genomes (vg)/cell and a plateau at 1E+6 vg/cell (Figure 1C). AAV-hFXN-driven FXN mRNA also showed a dose-dependent response, although without a plateau (Figure 1D). FXN mRNA production and protein expression were strongly correlated with a coefficient of determination (R2) value of 0.94 (Figure 1E). To confirm that FXN expressed from the AAV-hFXN cDNA was correctly processed, western blot analysis was performed. The full-length 210-amino acid form of hFXN is expressed in the cytoplasm of cells, rapidly translocating to the mitochondria, where it undergoes conversion to the intermediate form (42–210) and then the mature form (81–210) through the action of a mitochondrial processing peptidase.23,24 The size of mature hFXN is ∼14 kDa.23 Western blot results not only confirmed dose-dependent FXN expression at MOIs of 1E+6 or 1E+7 vg/cell, but also showed the 14-kDa fully mature form of FXN (Figure 1F).25 The antibody used detects all three FXN forms, including full, intermediate, and mature species, but cannot reliably quantify relative amounts. To confirm that AAV-hFXN could also transduce human cell lines, AC16 (immortalized human cardiomyocyte) cells were transduced with AAV-hFXN at MOIs of 1E+5, 1E+6, or 1E+7 vg/cell. Imaging of transduced AC16 cells showed that FXN was localized to the cytoplasm and quantitative IF demonstrated MOI concentration-dependent FXN expression (Figures S1A and S1B).
AAV treatment prolongs survival and maintains body weight
FXN-MCK mice, a tissue-specific FXN-knockout in cardiac and skeletal muscle, was selected as an animal model to evaluate the potential for phenotypic rescue of FRDA cardiomyopathy by AAV-hFXN and AAV-mFXN (Figure 2A; Table S1). The FXN-MCK mouse phenotype includes functional cardiac deficits, such as decrease of ejection fraction, which are likely associated with progressive weight loss and morbidity (Figure 2B). All of the vehicle-treated FXN-MCK mice became moribund and had to be sacrificed before 10 WOA, consistent with the published literature and The Jackson Laboratory’s historical data.21,26 Strikingly, all of the FXN-MCK mice treated at 6 WOA with AAV-hFXN or AAV-mFXN at doses of 3E+13 vg/kg or 1E+14 vg/kg survived until the planned necropsy at 10 WOA (cohort 1; 4 weeks postdose), as did all of the vehicle-treated WT control mice (Figure 2B). Animals in cohort 2 were designed to be continually monitored beyond 10 WOA until one animal reached humane endpoint, at which time the entire cohort would be euthanized (Figure 2A; Table S2). One animal in the AAV-mFXN lower-dose group (3E+13 vg/kg) reached humane endpoint at 18 WOA; therefore, the entire cohort 2 was euthanized for the second time point at 12 weeks postdose despite their body condition scores (BCSs) being within the normal range (Figure 2A).27 AAV-treated FXN-MCK mice showed a statistically significant improvement in average body weight compared to vehicle-treated FXN-MCK mice (Figures 2C and 2D). No significant body weight differences were noted between AAV-treated FXN-MCK mice and sex-matched WT controls. No body weight data were collected from vehicle-treated FXN-MCK mice after 10 WOA due to mortality.
Figure 2.

Survival, body weight, cardiac imaging, and cardiac biomarkers
(A) In vivo mouse proof of concept study design: FXN-MCK mice were intravenously dosed at 6 WOA. Cohort 1 mice were euthanized at 10 WOA. Cohort 2 mice were monitored beyond 10 WOA until one animal in the cohort reached humane endpoint and the entire cohort was euthanized, which happened at 18 WOA. (B) The probability of survival was significantly lower in vehicle-treated FXN-MCK mice compared to AAV-treated FXN-MCK mice and WT control mice. Data are represented as the age at which mice were euthanized if the animals displayed poor conditions, as described in Materials and methods. (C and D) Average body weights per sex and treatment group ± SD. Significant differences noted by Student’s t test when comparing vehicle-treated FXN-MCK mice to sex-matched WT controls starting at 6 WOA in males (p < 0.001) and 7 WOA in females (p < 0.05). No consistent significant differences found when comparing AAV-hFXN- or AAV-mFXN-treated FXN-MCK mice to sex-matched WT controls. (E) Significant reduction in average percentage ejection fraction was noted in vehicle-treated FXN-MCK mice at 9–10 WOA compared to the vehicle-treated WT control (p < 0.0001), AAV-hFXN-treated (p < 0.0001), and AAV-mFXN-treated groups (p < 0.001). No significant differences were observed between WT controls and AAV-hFXN- or AAV-mFXN-treated FXN-MCK mice at 18–19 WOA. (F) Data represented as average left ventricular (LV) mass normalized to body weight per treatment group at each time point. Data were analyzed using 1-way ANOVA. Significant difference between vehicle-treated FXN-MCK mice compared to WT control at 6 WOA (p < 0.05) and at 9–10 WOA (p < 0.0001). Significant differences also noted between vehicle-treated FXN-MCK mice and AAV-mFXN- and AAV-hFXN-treated FXN-MCK mice at 9–10 WOA (p < 0.0001). No significant differences noted between AAV-hFXN- or AAV-mFXN-treated FXN-MCK mice and age-matched WT controls at 18–19 WOA. (G) LV mass normalized to body weight of FXN-MCK mice and WT mice before treatment. Significant difference between vehicle-treated FXN-MCK mice compared to WT control at 6 WOA (Student’s t test; p < 0.01). (H) Myosin light chain serum level as average values per treatment group. Data analyzed by 1-way ANOVA compared to vehicle control at each time point. Significant differences were noted in myosin light chain levels when comparing vehicle-treated FXN-MCK mice to WT controls (p < 0.05) or to AAV-hFXN- and AAV-mFXN-treated FXN-MCK mice (p < 0.05) at 9–10 WOA. No significant differences noted between AAV-hFXN- or AAV-mFXN-treated FXN-MCK mice and age-matched WT controls at 18–19 WOA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
AAV treatment corrects cardiac phenotype
Cardiac function was monitored by echocardiography (high-frequency ultrasound). Significant reductions in ejection fraction were noted in vehicle-treated FXN-MCK mice at 9–10 WOA compared with WT controls (p < 0.0001). FXN-MCK mice that received either AAV-hFXN or AAV-mFXN at both lower and higher doses showed a significant increase in ejection fraction at 9–10 WOA compared to the vehicle-treated FXN-MCK mice (p < 0.001), which was maintained until the final measurement at 18–19 WOA (Figure 2E). There were no significant differences between WT controls and AAV-treated FXN-MCK mice at the final time point.
Average heart weight normalized to body weight was used as a measure of AAV-mediated efficacy given that FXN-MCK mice develop cardiac hypertrophy and have relatively low body weight.21,26 The average heart weight normalized to body weight was significantly (p < 0.0001) higher in vehicle-treated FXN-MCK mice compared with AAV-treated FXN-MCK mice and sex-matched WT control mice at 9–10 WOA (Figure S2A). There were no significant differences in normalized heart weights between AAV-treated FXN-MCK mice and age-matched WT controls at 18–19 WOA (Figure S2A). Significant improvements in left ventricular mass normalized to body weight (p < 0.0001) and fractional shortening (p < 0.001) from echocardiography were also observed in AAV-treated FXN-MCK mice compared to vehicle-treated FXN-MCK mice at 9–10 WOA. No significant differences were observed between AAV-treated FXN-MCK mice and WT controls at 18–19 WOA (Figures 2F and S2B). More important, there was a significant difference in the left ventricular mass normalized to body weight between vehicle-treated FXN-MCK mice (4.6 mg/g) and WT control mice (3.1 mg/g) before 6 WOA (p < 0.01) (Figure 2G), indicating that the cardiac abnormality in FXN-MCK mice developed before AAV treatment. Ejection fraction was also reduced from 55% to 47% in WT control mice compared to FXN-MCK mice at 6 WOA, although the difference was not statistically significant (Figure S2C). Together, these results show that AAV treatment resulting in de novo FXN expression in FXN-MCK mice enabled the rescue of important aspects of the cardiac phenotype.
To further investigate potential improvements of cardiac condition and to assess the safety of the AAV treatment, terminal serum cardiac biomarkers were measured. Myosin light chain, a biomarker that reflects myocardial damage, increased significantly to 1.5 ng/mL in vehicle-treated FXN-MCK mice, compared to the WT level of 0.04 ng/mL (p = 0.038). Animals treated with AAV-hFXN or AAV-mFXN at a dose level of 1E+14 vg/kg showed reductions to 0.096 or 0.01 ng/mL, respectively (p = 0.019 and p = 0.014, respectively) (Figure 2H). Vehicle-treated FXN-MCK mice exhibited a cardiac troponin (cTnI) level of 0.46 ng/mL, whereas it was undetectable in the WT mice. Following treatment with AAV-hFXN or AAV-mFXN at a dose of 1E+14 vg/kg, there was a trend toward cTnI reduction of 0.05 and 0.2 ng/mL, respectively. However, it is noteworthy that no statistically significant differences were detected (p = 0.67 and p = 0.92, respectively), potentially due to the presence of outliers in the dataset (Figure S2D). No significant differences were observed in terminal serum creatine kinase or fatty acid binding protein 3 levels across all groups at 9–10 WOA (Figure S2E and S2F).
AAV treatment results in functional FXN expression
To demonstrate AAV8 transduction, qPCR analysis was performed to measure vector copy number (VCN) in the following tissues. As expected, heart tissue from vehicle-treated FXN-MCK mice and WT mice contained no vector DNA. For cohort 2 vehicle-treated FXN-MCK mice, qPCR analysis was performed at 10 WOA and not at the scheduled necropsy at 18 WOA, due to mortality. There was no significant difference in heart VCN levels between animals dosed with AAV-mFXN or AAV-hFXN at 1E+14 vg/kg at any postdose time point (Figures 3A and 3B). Animals treated with AAV-hFXN vector at 1E+14 vg/kg showed similar transduction as assessed by VCN from the two time points, 1.5 vg/ diploid genome (dg) at 4 weeks postdose and 1.29 vg/dg at 12 weeks postdose. Similar results were obtained with AAV-mFXN treatment at 1E+14 vg/kg from the two time points, 1.12 vg/dg at 4 weeks postdose and 1.58 vg/dg at 12 weeks postdose. As expected, mice treated with a lower dose of 3E+13 vg/kg AAV-mFXN vector had a lower VCN of 0.77 vg/dg at 4 weeks postdose and 0.3 vg/dg at 12 weeks postdose. Differences in heart VCN between the lower and higher AAV-mFXN doses were only statistically significant in cohort 2 at 12 weeks postdose (p = 0.003). Together, these data suggest a trend by which tissue transduction with AAV-FXN is dose dependent but not time dependent (within 12 weeks postdose) in the hearts of FXN-MCK mice (Figures 3A and 3B; Tables S3 and S4).
Figure 3.

AAV transduction and FXN expression in the FXN-MCK mouse model
(A and B) VCN per group in mouse heart samples at 4 weeks postdosing (A) and 12 weeks postdosing (B). (C and D) FXN protein expression per group in mouse heart samples at 4 weeks postdosing (C) and 12 weeks postdosing (D). (B and D) All of the animals in cohort 2 survived until 18 WOA, except vehicle-treated FXN-MCK mice, which were terminated at 10 WOA due to mortality phenotype. Data are mean ± SD, where columns represent means, error bars represent SDs, and each point represents an individual animal; data were analyzed by mixed-effects analysis 1-way ANOVA of Tukey multiple comparisons test. (E) Representative western blot demonstrated a protein band at the same size as the rhFXN detected in heart tissue protein samples from FXN-MCK mice receiving AAV-hFXN at 1E+14 vg/kg at 4 weeks postdosing and 12 weeks postdosing. (F) Associated with FXN deficiency, FXN-MCK mice have a downregulation of immunoreactive levels of complexes I and II compared with WT. Expression of AAV FXN restores complex I and II levels to near normal. ∗p < 0.05; ∗∗p < 0.01. ND, nondetectable; ns, not significant.
In terms of FXN protein expression, an average of 169.7 and 165.5 ng/mg of endogenous FXN was measured in the hearts of WT mice at 9–10 WOA and 18–19 WOA (Figures 3C and 3D; Tables S3, S5, and S6). In contrast, AAV-driven FXN protein expression in FXN-MCK mice, at any of the doses tested, was higher or similar compared to WT levels, in a dose-dependent manner. In the hearts of FXN-MCK mice, a higher dose of 1E+14 vg/kg AAV-hFXN resulted in FXN protein expression 7.3-fold higher compared to WT levels at 4 weeks postdose (p = 0.0047), which increased to 17.9-fold at 12 weeks postdose (p < 0.0001). Similar levels of FXN in heart tissue were measured following AAV-mFXN treatment at a higher dose of 1E+14 vg/kg, reaching 12- or 21.2-fold increases compared to WT levels at 4 or 12 weeks postdose (both p < 0.0001), respectively. There was no significant difference in expression between AAV-hFXN and AAV-mFXN at the same dose of 1E+14 vg/kg at either 4 weeks (p = 0.1442) or 12 weeks postdose (p = 0.4509). At the lower dose level of 3E+13 vg/kg, FXN expression in heart tissue was found to be 3-fold higher than WT levels at 4 weeks postdose, although the difference was not statistically significant. At 12 weeks postdose, the expression was increased to 5.1-fold above WT levels (Figures 3C and 3D; Table S5). FXN protein expression levels were also increased in AAV-treated FXN-MCK mice in the cerebellum, dorsal root ganglion (DRG), liver, and spinal cord at 12 weeks postdose (Figure S3A; Table S5).
To understand whether both human and mouse vectors could express the functional transgene containing the mature form of FXN in vivo, we performed western blot analysis on heart tissues. Protein bands of similar size (∼14 kDa) to the positive control,25 a recombinant human mature form of FXN (rhFXN), were detected in every heart tissue sample from AAV-treated FXN-MCK mice, but not in vehicle-treated FXN-MCK mice (Figures 3E and S3B–S3E). These results suggest that both AAV-driven mFXN and full-length hFXN could be cleaved by the mitochondrial processing peptidase to convert into the mature mitochondrial form.23 To test the functional contribution of AAV-mFXN and AAV-hFXN at a cellular level, the immunoreactive levels of the mitochondrial electron transport chain complexes were examined. Associated with FXN deficiency, vehicle-treated FXN-MCK mice had a downregulation of complexes I and II compared with the vehicle-treated WT mice. In the same heart tissues in which the AAV-driven mature mitochondrial form of FXN was detected, the expression of complexes I and II were restored with the AAV treatment demonstrating functionality of the restored FXN (Figures 3F and S3F).
AAV treatment results in broad distribution of expressed FXN in the heart
Immunohistochemistry (IHC) for FXN confirmed FXN expression in the hearts of AAV-treated FXN-MCK mice with a broad distribution pattern. Cardiac tissue from vehicle-treated WT mice displayed a diffuse light level of positivity that was fairly uniform throughout the heart, whereas AAV-treated FXN-MCK hearts showed regional variability, with a greater staining intensity in many areas than was observed in vehicle-treated WT hearts. FXN expression was not detected in vehicle-treated FXN-MCK mice (Figure 4A).
Figure 4.

FXN protein distribution in the heart
(A) Heart tissue samples showing FXN expression (IHC) of vehicle-treated WT and FXN-MCK mice and AAV-treated FXN-MCK mice. The images in the bottom row depict the magnification of an area from the sample directly above it (blue box). Scale bars represent 1 mm (top row) and 100 μm (bottom row). (B) HALO analysis to semiquantify the mean percentage of areas exhibiting FXN stains in strong, moderate, and weak intensity, and total (adding all percentages of strong, moderate, and weak) of all mice. The data below the graph show the mean total percentages of areas exhibiting FXN stains at any intensity in each group, corresponding to the bars above; SDs are provided in Table S7.
For semiquantitative analysis, expression levels of FXN+-stained areas were defined as weak, moderate, and strong by the HALO algorithm. The percentage of area exhibiting weak FXN expression was 35.1% of the total heart section area analyzed in FXN-MCK mice that received 3E+13 vg/kg AAV-mFXN. Following a higher dose of 1E+14 vg/kg, areas corresponding to weak FXN expression were greater, 41.0% and 40.7% for AAV-mFXN and AAV-hFXN treatment, respectively, percentages that were similar to the WT endogenous FXN level, at 43.3% (Figure 4B; Table S7). The percentage of area exhibiting moderate FXN expression was similar or slightly greater in FXN-MCK mice that received 3E+13 vg/kg AAV-mFXN at 12.5% compared to vehicle-treated WT mice at 10.0%. Following a higher dose of 1E+14 vg/kg, the areas corresponding to moderate FXN expression increased to 20.3% and 23.0% for AAV-mFXN and AAV-hFXN treatment, respectively (Figure 4B). The percentage of areas exhibiting strong FXN expression were greater for all of the treatment groups, at 11.0% for 3E+13 vg/kg AAV-mFXN treatment and 17.5% and 20.7% for 1E+14 vg/kg AAV-mFXN and AAV-hFXN treatment, respectively, compared to vehicle-treated WT mice at 1.1% (Figure 4B). Overall, the total percentage of areas exhibiting FXN expression were similar between AAV-treated FXN-MCK mice that received the lower dose of 3E+13 vg/kg at 58.6% and the vehicle-treated WT mice at 54.4%, and increased to 78.8% and 84.5% when treated at a higher dose of 1E+14 vg/kg AAV-mFXN and AAV-hFXN, respectively (Figure 4B). Consistent with the ELISA data, the IHC results showed dose-dependent FXN protein expression, and semiquantitative image analysis revealed a broad distribution of FXN transgene expression similar to WT endogenous expression, although it was more speckled.
FXN expression was not associated with cardiotoxicity
To evaluate histological changes that constitute efficacy and distinguish those from potential changes that may represent test article-induced toxicity, H&E staining on tissue sections with microscopic evaluation was done. Microscopic changes in the hearts of vehicle-treated FXN-MCK mice were consistent with the cardiomyopathy that is expected in an animal model of FRDA, including degenerating cardiomyocytes that were present in all of the animals evaluated (5/5 animals scored), mononuclear cell infiltration (4/5), myocardial fibrosis (3/5), and atrial thrombi (2/5). Minimal mononuclear cell infiltration was noted in vehicle-treated WT mice (2/4). In contrast, FXN-MCK mice that received AAV-hFXN at 1E+14 vg/kg or AAV-mFXN at 3E+13 vg/kg or 1E+14 vg/kg showed reduced cardiomyocyte degeneration (0/17), mononuclear cell infiltration (5/17), myocardial fibrosis (0/17), or atrial thrombi (1/17) at 4 weeks postdose, indicating efficacy (Table 1).
Table 1.
Histology evaluation for efficacy and safety of AAV treatment in the FXN-MCK mouse model
| Group |
1 |
4 |
7 |
10 |
13 |
|---|---|---|---|---|---|
| Genotype |
Wild type |
FXN-MCK |
FXN-MCK |
FXN-MCK |
FXN-MCK |
| Treatment | Vehicle | Vehicle | AAV-mFXN 3E+13 vg/kg | AAV-mFXN 1E+14 vg/kg | AAV-hFXN 1E+14 vg/kg |
| Males | |||||
| No. (animals examined) | 2 | 3 | 3 | 3 | 2 |
| Heart (no. examined) | N = 2 | N = 3 | N = 3 | N = 3 | N = 2 |
| Degeneration, cardiomyocyte | (0)a | (3) | (0) | (0) | (0) |
| Minimal | 0 | 1 | 0 | 0 | 0 |
| Moderate | 0 | 2 | 0 | 0 | 0 |
| Thrombus, atrial | (0) | (1) | (0) | (0) | (0) |
| Mild | 0 | 1 | 0 | 0 | 0 |
| Fibrosis, myocardial | (0) | (2) | (0) | (0) | (0) |
| Mild | 0 | 2 | 0 | 0 | 0 |
| Infiltration, mononuclear cell | (2) | (3) | (1) | (1) | (2) |
| Minimal | 2 | 3 | 1 | 1 | 2 |
| Liver (no. examined) | N = 2 | N = 3 | N = 3 | N = 3 | N = 2 |
| Vacuolation, centrilobular | (0) | (1) | (0) | (0) | (0) |
| Minimal | 0 | 1 | 0 | 0 | 0 |
| Females | |||||
| No. (animals examined) | 2 | 3 | 3 | 3 | 3 |
| Heart (no. examined) | N = 2 | N = 2b | N = 3 | N = 3 | N = 3 |
| Degeneration, cardiomyocyte | (0)a | (2) | (0) | (0) | (0) |
| Minimal | 0 | 1 | 0 | 0 | 0 |
| Mild | 0 | 1 | 0 | 0 | 0 |
| Thrombus, atrial | (0) | (1) | (0) | (1) | (0) |
| Mild | 0 | 1 | 0 | 1 | 0 |
| Fibrosis, myocardial | (0) | (1) | (0) | (0) | (0) |
| Minimal | 0 | 1 | 0 | 0 | 0 |
| Infiltration, mononuclear cell | (0) | (1) | (0) | (1) | (0) |
| Minimal | 0 | 1 | 0 | 1 | 0 |
| Liver (no. examined) | N = 2 | N = 3 | N = 3 | N = 3 | N = 3 |
| Vacuolation, centrilobular | (0) | (2) | (0) | (0) | (0) |
| Minimal | 0 | 2 | 0 | 0 | 0 |
Select microscopic findings in the hearts and livers of cohort 1 males and females euthanized 4 weeks postdose.
The numbers in parentheses represent the total counts of animals exhibiting findings.
For animal numbers, see Materials and methods.
Off-target tissues, including liver, sciatic nerve, quadriceps muscle, spinal cord, diaphragm, DRG, and brain, were evaluated for potential test article-induced toxicity at 4 and 12 weeks postdose. In the livers of vehicle-treated FXN-MCK mice, centrilobular hepatocytes were pale and vacuolated (3/6). Centrilobular vacuolation was not observed in AAV-hFXN- or AAV-mFXN-treated mice, which is also consistent with the rescue of cardiac deficits or lesions driven by the absence of FXN in these FXN-MCK mice. Microscopic changes in the sciatic nerve, quadriceps muscle, spinal cord, diaphragm, DRG, ovary, and testis were not identified in the vehicle-treated FXN-MCK mice. Microscopic changes were not observed in these selected tissues from FXN-MCK mice following the administration of AAV-hFXN or AAV-mFXN at either time point, except that vacuolation in the DRG was noted in 4/5 of the AAV-hFXN-treated FXN-MCK mice and 5/6 of the AAV-mFXN-treated FXN-MCK mice (both at 1E+14 vg/kg), compared with 0/4 of the lower-dose (3E+13 vg/kg) AAV-mFXN treated FXN-MCK mice and 1/4 of vehicle-treated WT mice (Table S8). This vacuolation was characterized by the presence of multiple clear vacuoles along axons coursing through the ganglia, often surrounding or associated with presumptive Schwann cell nuclei. In general, vacuolation observations in the absence of DRG cell body degeneration should be considered inconclusive. The administration of AAV vector at a dose as low as 3E+13 vg/kg effectively alleviated cardiac phenotypes in the FXN-MCK mice without inducing additional toxicity in the off-target selected tissues.
To evaluate additional pathologies associated with FXN insufficiency, such as iron accumulation and fibrosis, we conducted iron staining and Masson’s trichrome staining.28 Abnormal iron accumulation was observed in the hearts of vehicle-treated FXN-MCK mice but not in the hearts of WT or AAV-treated mice (Figure 5A). Consistent with the microscopic evaluation, Masson’s trichrome staining revealed cardiac fibrosis in vehicle-treated FXN-MCK mice (6/6), which was nearly absent in FXN-MCK mice that received AAV treatment at 4 weeks postdose (3/17) (Figure 5B). In summary, these results support the role of AAV-mediated FXN expression in the prevention or resolution of cardiac pathology without toxic side effects in this mouse model of FRDA.
Figure 5.

Histological improvement in the heart with the AAV treatment
(A) Heart tissue samples stained with Prussian blue that identifies areas of iron accumulation; scale bars represent 100 μm. Images in the upper right of the second column depict a magnification of an area from the smaller red box (arrow) in the vehicle-treated FXN-MCK mice. (B) Representative Masson trichrome stains for fibrosis of heart tissue samples, with the images in the bottom row depicting the magnification of an area from the sample directly above it. Scale bars represent 1 mm (top row) and 100 μm (bottom row).
Discussion
This study assessed the efficacy and safety of AAV vectors containing the FXN gene at doses of 3E+13 or 1E+14 vg/kg administered to conditional knockout mice (FXN-MCK) that model the cardiopathic manifestations of FRDA. The AAV8 serotype was selected based on internal studies, which demonstrated similar cardiac transduction between AAV8 and AAV9 (data not shown). This choice is also supported by relevant publications regarding AAV8 tropism in the heart,29,30 as well as a relatively lower level of preexisting immunity against AAV8 in the general population compared with other AAV serotypes.31 Two expression vectors were tested, one expressing hFXN (AAV-hFXN) and the other expressing mFXN (AAV-mFXN), to evaluate possible pharmacodynamic differences in species-specific FXN in mice. The amino acid sequence of the co human FXN transgene was confirmed to be identical to the human FXN consensus sequence. Significant improvements in body weight, heart morphology, cardiac function, and cardiac injury biomarkers, in addition to an extension in the mean of the probability of survival, were observed in FXN-MCK mice that received AAV-based gene therapy compared to vehicle-treated FXN-MCK mice. No differences were observed between the AAV-hFXN and AAV-mFXN vectors in expression levels, correction of cardiac phenotype, prevention of early mortality, or histopathological evaluation. These results support the hypothesis that de novo FXN expression in vulnerable organs, such as the heart, can prevent the progression of FRDA-like cardiac deficits and their associated morbidity, even when administering the AAV vector after early aspects of FRDA pathophysiology, such as deficits in ejection fraction, have already manifested.
In addition to the neurological symptoms that develop during FRDA, the majority of patients develop severe cardiomyopathies that eventually progress to heart failure.9,32 Given the severity and monogenic nature of the disease and the lack of disease-modifying treatments available, a gene transfer approach using an AAV vector containing the FXN gene was considered a promising therapeutic strategy to treat FRDA.12 Published studies using AAV constructs containing the FXN gene administered in various FRDA animal models with diverse routes of administration resulted in improved outcomes regarding cardiomyopathy, neurodegeneration, and lifespan extension.13,14,15 However, some of these studies that used constructs with a strong constitutive synthetic promoter, such as CAG, a cytomegalovirus early enhancer element fused to the chicken β-actin gene promoter, resulted in what appear to be FXN overexpression-associated toxicities.17,18 Expression of FXN protein by more than 20-fold relative to WT expression levels was shown to impair electron transport chain complexes I and II and alter mitochondrial ultrastructure, leading to cardiomyocyte cell death and cardiac deficits.17 Huichalaf et al. confirmed that toxic levels of FXN following the administration of an AAV9-CAG-FXN construct, which affected mouse livers and hearts, caused a reduction in the iron-sulfur cluster biosynthesis by affecting the equilibrium between different complexes involving the cysteine desulfurase and the iron-sulfur cluster scaffold protein.18,33
To avoid potential toxicity associated with AAV constructs that contain strong promoters, we selected a constitutive and relatively lower-strength human PGK promoter to drive FXN expression.20 The goal was to transduce more cells using a relatively higher AAV dose, while maintaining FXN expression per cell under a certain threshold to avoid toxicity. Tissue transduction as measured by VCN per diploid genome was detected in the hearts of all AAV-treated mice, with a trend toward reflecting dose levels. No significant differences in VCN were observed with an extension of the postdose duration from 4 to 12 weeks, regardless of AAV-hFXN dosing at 1E+14 vg/kg (p = 0.9622), or AAV-mFXN dosing at either 3E+13 (p = 0.635) or 1E+14 vg/kg (p = 0.4955), suggesting no degradation of the transgene DNA during the additional 8 weeks. PGK was selected as a ubiquitous promoter because FXN is a highly conserved mitochondrial protein expressed throughout the body. In the present study, the total percentage of cells exhibiting FXN expression as assessed by IHC staining and quantified with HALO image analysis was similar in the lower dose of 3E+13 vg/kg AAV-treated FXN-MCK mice, with 58.6% of the tissue displaying positive stain for FXN compared to vehicle-treated WT mice at 54.4% in the heart. In addition, there were no significant differences between the lower dose of 3E+13 vg/kg AAV-treated FXN-MCK mice and the vehicle-treated WT mice in the FXN total protein expression level as measured by ELISA from the homogenized heart tissues at 4 weeks postdose. This suggests that the level and pattern of FXN expression following a single AAV-hFXN intravenous dose of 3E+13 vg/kg is comparable to that of WT. At the higher dose of 1E+14 vg/kg, consistent with the ELISA results, the total percentage of cells exhibiting FXN expression in the heart increased to a similar level at 79% (AAV-mFXN) or 85% (AAV-hFXN), and the FXN expression level reached 21.2-fold compared to WT endogenous level. The strongly positive and moderately positive signals likely contribute to the observed increase in protein expression in the ELISA, showing a 7- to 12-fold increase at 4 weeks postdose. Incorporating regulatory elements, further optimizing the promoter, or identifying and testing the endogenous FXN promoter could yield an expression pattern and level closer to FXN expression in WT animals. Although the highest dose in this study reached slightly over a 20-fold FXN expression compared to WT levels, no toxicity was observed in the heart for a duration up to 12 weeks postdose, and all of the higher-dose AAV-treated FXN-MCK mice survived until the scheduled necropsy at 18 WOA, with no sign of declining health. However, a longer-duration study may be beneficial to rule out possible delayed cardiac toxicity resulting from the chronic overexpression of protein accumulation over time, perhaps in a WT rodent toxicity study. No test article-related toxicities were detected in any selected tissues at both time points, except for the vacuolation observed in the DRG, which was observed only in the higher-dose groups (1E+14 vg/kg). No overt toxicity such as neuronal degeneration was observed in the DRGs of AAV-treated mice. Furthermore, since this effect is not well understood and is subclinical in this study, the severity remains unclear at this time. Conducting a dose-finding study and a 6-month nonhuman primate toxicity study in the future will be beneficial to define the minimum effective dose and provide a comprehensive safety assessment.
Although it is important to avoid expression FXN levels that may trigger toxicity, it is also critical to achieve minimally efficacious levels. Although homozygous deletion of FXN is embryonically lethal, heterozygous FXN-knockout mice are indistinguishable from their WT littermates in physical and behavioral phenotype.34 Human heterozygous carriers with an estimated 50% of normal FXN protein expression manifest no clinical symptoms.16,35 These observations suggest that FXN levels at or above 50% of WT levels can have meaningful therapeutic effects, in particular, in preventing or reversing FRDA-associated cardiomyopathy. A recent study by Munoz-Zuluaga and collaborators36 showed that achieving a minimum therapeutic level of expression may require reaching at least 30% of the healthy human FXN level (>17.7 ng/mg), considering that FRDA heterozygotes have FXN levels ranging from 30% to 80% of normal. Given that PGK is a lower-strength promoter compared to CAG and other ubiquitous promoters, it is anticipated that a dose level 3–4 times higher would be necessary, likely in the range of E+12 vg/kg, to reach approximately 30% of the endogenous FXN level. Correspondingly, the PGK promoter-driven FXN expression resulted in the improvement of cardiomyopathy and extension of survival in all AAV-treated FXN-MCK mice, in contrast to vehicle-treated FXN-MCK mice.
The FXN-MCK mice showed abnormal cardiac phenotypes, including left ventricular hypertrophy, functional impairment as measured by echocardiogram, and body weight reduction before AAV treatment at 6 WOA, indicating that they were treated at a symptomatic stage, when presumably the FXN insufficiency had already caused structural and functional deficits. At the second terminal time point of 18–19 WOA, when all of the vehicle-treated control FXN-MCK mice had been previously euthanized, the ejection fraction and left ventricular mass normalized to body weight in the AAV-treated FXN-MCK mice were indistinguishable from the vehicle-treated WT control mice. This shows that the AAV-mediated transfer of the FXN gene and de novo expression of the protein are capable of not only preventing the progression of cardiac FRDA-associated pathology but also of reversing cardiac deficits of the disease. Relatively lower levels of the cardiac injury biomarker, myosin light chain, detected when comparing AAV-treated FXN-MCK to vehicle-treated FXN-MCK mice further confirmed the efficacy of the AAV-mFXN and AAV-hFXN vectors. At the cellular level, FXN deficiency leads to abnormalities in mitochondrial function, particularly in the regulation of the iron-sulfur cluster that interacts with proteins in the electron transport chain complexes, presumably due to a defect in electron entry and transfer, which ultimately results in the excess generation of reactive oxygen species and reduction of ATP production.3 Furthermore, a decrease in the expression level of electron transport chain complexes II has been reported in the FXN-MCK mouse model.13,37 As a surrogate measure of mitochondrial health, it was shown that immunoreactive levels of complexes I and II from the FXN-MCK heart tissues were restored to WT levels following AAV treatment.
FXN deficiency has been shown in previous reports to cause a range of disturbances that are reflected in this FXN-MCK mouse model by deficient iron clearance and consequent myocardial tissue damage.13,37,38 Iron accumulation and myocardial fibrosis in the hearts of FXN-MCK mice were observed in this study. Consistent with the rest of the data, iron accumulation and fibrosis were diminished in the hearts of AAV-treated FXN-MCK mice at all dose levels compared to vehicle-treated FXN-MCK mice. Histological examination also showed that AAV-treated FXN-MCK mice showed an absence or lessening of cardiomyocyte degeneration, myocardial fibrosis, and atrial thrombi that were commonly observed in vehicle-treated FXN-MCK mice.21,26 It is interesting that improvement of the paled and vacuolated centrilobular hepatocytes in the livers of FXN-MCK mice were also alleviated with AAV treatment, possibly due to secondary relief from ischemia resulting from cardiac compromise. A less likely but possible explanation is that the apparent histological improvement in the liver is an artifact of tissue collection that caused cytoplasmic clearance and vacuolation. In general, there was no evidence of an AAV8-FXN-induced toxic effect in the heart that would be distinguishable from what was observed in vehicle-treated FXN-MCK mice.
In conclusion, progression of the cardiomyopathy-like phenotype that develops in FXN-MCK mice was prevented and/or reversed with a single dose of an AAV8-vector containing an FXN transgene, as evidenced by an increase in median survival and improved cardiac function and morphology. These results support the potential of this well-tolerated and efficacious AAV8 gene therapy to treat patients with FRDA that develop severe, and often lethal, cardiovascular complications.
Materials and methods
AAV8-PGK-FXN vector construction and production
Both hFXNco and mFXNco coding sequences were subcloned into the proprietary plasmid pAAVAud2, which included the viral ITR22 from AAV2 and the human 3-PGK promoter (hPGK), an intron, and a polyadenylation signal, both from SV40. The AAV8 vectors used in these studies have been rendered replication defective by virtue of deletion of the rep and cap genes. The whole genome between these ITRs is fully synthetic. The lack of rep gene confirms that current recombinant AAV is unable to replicate alone or with helper viruses. The AAV8-hPGK-hFXNco (AAV-hFXN) and AAV8-hPGK-mFXNco (AAV-mFXN) vectors were produced by cotransfecting Expi-293F cells (Thermo Scientific, catalog no. A14527) with the human or mouse hPGK-FXNco plasmid and a proprietary AAV8 helper plasmid using polyethyleneimine. After 72 h of incubation, the vector was purified from cleared lysates by affinity (POROS GoPure AAV8 Prepacked Column, 5 mL, Thermo Scientific, catalog no. A36647) and anion exchange chromatography (POROS GoPure 50 HQ Ion-Exchange column, Thermo Scientific, catalog no. 4481315) on an AKTA pure high-performance liquid chromatography system, then buffer exchanged into Ringer’s lactate solution with 0.01% Pluronic (pH 7.4). All of the vectors were manufactured by Astellas Gene Therapies. Dosing solutions were formulated in Ringer’s lactate with 0.01% Pluronic. Lots X01-190226 (AAV-hFXN) and X02-190226 (AAV-mFXN) were produced for each test article. The titer of AAV-hFXN was 2.67E+13 vg/mL and AAV-mFXN was 2.99E+13 vg/mL. The endotoxin of AAV-hFXN was 5.96 endotoxin units (EU)/mL and AAV-mFXN was 3.28 EU/mL (Figure 1).
AAV-hFXN in vitro analysis
Cell lines and culture conditions
C2C12 cells were obtained from the American Type Culture Collection and cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% Anti-Anti. AC16 cells were obtained from Sigma and cultured in F12-DMEM supplemented with 12.5% heat-inactivated FBS and 1% antibiotic-antimycotic. Both cell lines were grown at 37°C in an atmosphere with 5% CO2.
Quantitative Immunofluorescence
Cells were seeded at 15,000 cells/well in black-walled, clear-bottom tissue culture (TC)-treated 96-well plates; 24 h later, the medium was removed and replaced with fresh medium containing the desired MOI of AAV-hFXN (calculated as vg/cell). After 4 h of incubation with AAV-hFXN-containing media, etoposide was added to a final concentration of 3 μM. At 72 h later, cells were fixed with 4% formaldehyde and analyzed by IF for FXN expression. Cells were incubated in Intercept Blocking Buffer (LI-COR 927-70001) for 30 min, then with rabbit anti-FXN primary antibody (Abcam 175402) at 2 μg/mL for 2 h, then with Alexa Fluor 488-tagged anti-rabbit secondary antibody (Invitrogen A11008) for 1 h. Plates were read on a SpectraMax M5 with excitation/emission of 490/525 and data were plotted on GraphPad Prism. Hoechst stain (Invitrogen R37605) was added for nuclear visualization in representative imaging. Images were taken on an Olympus IX73.
RNA extraction and droplet digital PCR (ddPCR) analysis
Cells were seeded at 15,000 cells/well in TC-treated 96-well plates; 24 h later, the medium was removed and replaced with fresh medium containing the desired MOI of AAV-hFXN (calculated as vg/cell). After 4 h of incubation with AAV-hFXN-containing media, etoposide was added to a final concentration of 3 μM. At 72 h later, cells were harvested in RLT buffer and RNA was extracted using the RNeasy 96 QIAcube HT kit (Qiagen 74171). Following RNA extraction, cDNA was synthesized using the SuperScript IV VILO Kit (Invitrogen 11766500). FXN mRNA was quantified using Bio-Rad’s ddPCR system with TaqMan primers and probe specifically designed to recognize coFXN expressed by AAV-hFXN.
In vitro western blot analysis
A total of 200,000 C2C12 cells/well were plated in 6-well TC-treated plates; 24 h later, the medium was removed and replaced with fresh medium containing the desired MOI of AAV-hFXN (calculated as vector genomes per cell). After 4 h of incubation with AAV-hFXN-containing media, etoposide was added to a final concentration of 3 μM. At 72 h later, cells were harvested in radioimmunoprecipitation assay lysis buffer (Sigma 20-188) containing complete protease inhibitor tablets (Roche 4693124001). Cells were incubated with light agitation at 4°C for 30 min, then spun down at 18,000 × g for 10 min. The supernatant was harvested and analyzed by bicinchoninic acid (BCA) for total protein content using the Pierce Microplate BCA Protein Assay Kit-Reducing Agent Compatible (Thermo Fisher Scientific 23252). Lithium dodecyl sulfate sample buffer (Invitrogen NP0007) was added to 1× final concentration and samples were incubated at 95°C for 10 min. Total protein, 30 μg, was loaded per well of a NuPAGE 4%–12% Bis-Tris gel (Invitrogen NP0322), as well as one lane of the Odyssey One-Color Protein Molecular Weight Marker (LI-COR 928-40000). The gel was run for 45 min at 180 V in NuPAGE MES SDS Running Buffer supplemented with NuPAGE antioxidant (Invitrogen NP0002 and NP0005, respectively). The separated proteins were then transferred to a polyvinylidene fluoride membrane using the Trans-Blot Turbo instrument from Bio-Rad, blocked in Intercept Blocking Buffer (LI-COR 927-70001) for 1 h under gentle agitation and then incubated with mouse anti-FXN monoclonal antibody (Invitrogen 45-6300) and rabbit anti- glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal antibody (Abcam 9485) for 2 h at room temperature under gentle agitation. The membrane was washed and then incubated with IRDye 800CW Donkey anti-mouse and IRDye 680CW goat anti-rabbit secondary antibodies (LI-COR 926-32212 and 926-68071, respectively) for 1 h at room temperature under gentle agitation. Membranes were washed and imaged on the LI-COR Odyssey imaging system.
AAV-hFXN and AAV-mFXN in vivo study
Animal model of FRDA
The FXN-MCK mice (B6.Cg-Fxnem2Lutzy Fxnem2.1Lutzy Tg(Ckmm-cre)5 Khn/J; The Jackson Laboratory stock no. 029720) contain a Cre-conditional FXN allele floxing exon 2, a global exon 2 deleted knockout FXN allele, and a Cre recombinase transgene driven by muscle creatine kinase promoter (MCK-Cre, B6.FVB(129S4)-Tg(Ckmm-cre)5 Khn/J; The Jackson Laboratory stock no. 006475) resulting in a specific ablation of FXN expression in cardiac and skeletal muscle. This mouse model is similar to the one developed by Puccio and collaborators, in which exon 4 was deleted.21 The phenotypes in the FXN-MCK mice include progressive weight loss associated with functional cardiac deficits leading to morbidity at 10 ± 1 WOA. The Jackson Laboratory has characterized the cardiomyopathic phenotypes from this mouse model as decreased heart rate, ejection fraction, and fractional shortening, as well as a significantly increased left ventricular mass distinguishable from nonmutant littermates (https://www.jax.org/strain/029720).
Animal study design
Animal procedures and experiments were performed by The Jackson Laboratory in compliance with applicable animal welfare acts and were approved by the local Institutional Animal Care and Use Committee and in accordance with Institutional Biosafety Committee protocols.
We evaluated the efficacy of AAV8-hPGK-hFXNco (AAV-hFXN) and AAV8-hPGK-mFXNco (AAV-mFXN) vectors each administered by a single intravenous dose to rescue survival and cardiomyopathy in an FXN-MCK mouse model of FRDA (Figure 1), using WT mice (C57BL/6J, The Jackson Laboratory stock no. 000664) and knockout Fxnflox/null::MCK-Cre conditional knockout mice (FXN-MCK) supplied by The Jackson Laboratory. At 6 WOA, mice were given a single dose of vehicle (Ringer’s lactate with 0.01% Pluronic) as control, or AAV-hFXN administered at 1E+14 vg/kg, or AAV-mFXN administered at 1E+14 vg/kg or 3E+13 vg/kg, respectively, via intravenous injection in the tail vein.
Two cohorts were included in this study (Figure 1). Cohort 1 animals were harvested at 10 WOA to provide the first postdose time point at 4 weeks’ duration. Two separate groups of animals were designated for bioanalytical analysis (BAN) and histology evaluation in cohort 1. Animals in cohort 2 continued to be monitored beyond 10 WOA until one animal reached a humane endpoint, at which time the entire cohort was euthanized (groups 3, 9, 12, and 15; group 6 terminated at 10 WOA). Since one animal in the AAV-mFXN lower-dose group (3E+13 vg/kg) reached the humane endpoint at 18 WOA, all of the animals in cohort 2 were euthanized for tissue collection at 12 weeks postdosing. There was only one group of animals designated for both BAN and histology evaluation in cohort 2; therefore, tissues were limited. Animal housing facilities were controlled for temperature and humidity, with a 12-h light/dark cycle and free access to water and standard rodent chow (Certified Rodent Diet no. 2014C, Envigo RMS). Safety assessments included clinical observations (BCS),27 body weight, analysis of clinical pathology parameters (including cardiac injury markers), histopathological evaluations, and echocardiography. These comprehensive evaluations aimed to provide a thorough assessment of the safety profile.
Animal health and survival assessments
Physical examination of each animal to ensure that it was “bright, alert, responsive, and healthy,” and survival assessments were performed daily, and body weights were recorded weekly. Per protocol, animals reaching the following endpoint criteria (i.e., humane endpoints), regardless of age or time since dosing, were humanely euthanized and every attempt was made to collect tissues: >20% weight loss from maximum weight, rapid or sustained deterioration in health status resulting in a BCS (The Jackson Laboratory) of ≤2,27 unresponsive to meaningful stimuli, hunched posture, signs of respiratory distress, or any other condition that interferes with their ability to reach or consume adequate amounts of food or water. In cohort 2, the entire cohort was euthanized for tissue collection once one animal in the cohort reached a humane endpoint.
Cardiac assessments and analyses
Cardiac imaging was performed before dosing, before terminal sacrifice at 9–10 WOA for cohort 1, including both groups for bioanalytical endpoints and histological evaluation and at 18–19 WOA for the lifespan cohort. Animals were anesthetized with up to 5% isoflurane at a flow rate of 0.8–2.0 L/min in oxygen. Ultrasound echocardiography was performed by The Jackson Laboratory Center for Biometrics Core Service using Vevo 770/2100 high-frequency ultrasound with 30- and 40-MHz probes (VisualSonics, Toronto, ON, Canada). B-mode and M-mode images were recorded in parasternal short-axis view for measurement of left ventricular wall thickness and inner diameter. From these direct measurements, diastolic and systolic blood volumes, ejection fraction, fractional shortening, cardiac output, and stroke volume were calculated.
Serum muscle injury biomarkers analyses
Whole blood was collected and processed into plasma for the muscle injury biomarkers panel (cTnI , skeletal troponin, fatty acid binding protein, and myosin light chain) measured using a commercially available kit from MesoScale Discovery (catalog no. K15186C-1) and creatine kinase measured by The Jackson Laboratory Clinical Chemistry Core Service using the Beckman Coulter DxC 700 AU chemistry analyzer.
Tissue collection
Tissues were collected and either flash-frozen in liquid nitrogen and stored at −80°C for bioanalytical endpoints or fixed in 10% neutral buffered formalin for histological evaluation. In cohort 1, one group of animals was designated for bioanalytical endpoints, and heart, cerebellum, DRG, and spinal cord tissues were collected for both protein expression and VCN analysis. Another group of animals was designated for histological evaluation, and tissues including heart, liver, sciatic nerve, quadriceps muscle, spinal cord, diaphragm, DRG, and brain were collected and fixed. In cohort 2, heart tissues were collected for only bioanalytical endpoints including protein expression and VCN analysis, and other tissues including liver, sciatic nerve, quadriceps muscle, spinal cord, diaphragm, DRG, and brain were collected for histological evaluation.
Histological tissue staining
Tissues were sectioned to allow visualization of the four chambers of the heart (right/left atria and right/left ventricles). H&E staining was performed and evaluated by a board-certified veterinary pathologist to allow the assessment of general pathology and the size and shape of cardiac myocytes, whereas IHC analysis evaluated transgene FXN biodistribution, Masson’s trichrome staining evaluated fibrosis, and Perls’ Prussian blue staining evaluated iron accumulation within cardiac myocytes. Due to the blurry details of the cardiomyocytes, one female H&E-stained heart section was not evaluated in this study. For IHC, anti-FXN antibody (EPR21840, Abcam catalog no. ab219414) was used in conjunction with a 3,3′-diaminobenzidine-based, single chromogenic IHC approach. The images of FXN IHC staining then underwent a quantitative expression assessment using HALO image analysis software (version 3.2.1851.393e, Indica Labs, Albuquerque, New Mexico) to measure the percentage of positive cardiomyocyte (area) and expression levels (signal intensity). The expression levels of positive cardiomyocytes were defined as strong, moderate, and weak, and the whole-heart section was scanned, masked, and analyzed by an anatomic pathologist.
AAV transduction and FXN expression analysis
VCN analysis
Quantification of the vector biodistribution of the test article-specific DNA was performed using a qPCR analytical method. Quantification was performed using a linearized DNA plasmid quantification standard, vector AAVAud2-PGK-FXN, and a primer/probe set designed to specifically detect a coDNA amplicon in this linearized plasmid and in the test article (Figure 1; Table S9). genomic DNA (gDNA) samples were extracted from mouse tissue and run on the QuantStudio 6 Pro Real-Time PCR System to amplify test-article specific DNA and the endogenous housekeeping gene PCBP2 DNA. Quantification analysis was performed using Thermo Fisher Scientific Design & Analysis Software, version 2.3.3, to determine relative quantification of test article-specific DNA and PCBP2 DNA in each sample.
ELISA analysis
Quantitation of mFXN and hFXN was performed with tissues from the heart, cerebellum, DRG (cervical), spinal cord, and liver using qualified ELISA kits from Abcam. The quantitated results are expressed as ng/mg of FXN in tissue, expressed as the quotient of each sample’s back-calculated concentration and its total protein result as determined by BCA assay. The size of mFXN and hFXN was assessed in heart tissues using western blot with antibodies from Abcam and Invitrogen (Thermo Fisher Scientific). For each analysis, the group and number of animals analyzed is reported in the respective figure legend.
Western blot analysis in mouse heart
mFXN and hFXN levels in mouse tissue lysate were analyzed by western blot using primary antibodies specific for mFXN (Abcam catalog no. ab219414), hFXN (Invitrogen catalog no. 45-6300), and GAPDH (Abcam catalog no. ab8245). The secondary antibodies used were IRDye 800CW donkey anti-rabbit immunoglobulin G (IgG) (LI-COR, 926-32213) and IRDye 680RD donkey anti-mouse IgG (LI-COR, 926-68072). Images were taken using an Odyssey DLx imaging system (LI-COR). For the oxidative phosphorylation (OXPHOS) complexes expression analysis, the primary antibodies used were anti-GAPDH (Novus Biologicals, 1D4, NB300-221) at 1:1,000 and anti-OXPHOS (Abcam/Mitosciences, MS601-360, MitoProfile Total antibody cocktail) at 1:500. The secondary antibodies were horseradish peroxidase-conjugated mouse or rabbit from Cell Signaling Technologies at 1:2,000. A total of 30 μg were loaded into a 4%–12% NuPAGE Bis-Tris Gel, SDS-PAGE (NP0336). Quantification of the bands was performed using the NIH ImageJ software and normalized to the loading control (GAPDH) (WT, N = 4; FXN-MCK, N = 5–6).
Statistical analyses
Survival was compared between treatment groups and age-matched WT controls using chi-square from the Mantel-Cox log rank test. The average body weight per sex and treatment group ± SD were compared between treatment groups and age-matched WT controls using the Student’s t test. Cardiac function parameters, cardiac biomarkers, heart weight normalized for body weight, and skeletal biomarkers were compared between treatment groups and age-matched WT controls with one-way ANOVA at each time point. Symbols indicate p values for significance designated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Data and code availability
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Acknowledgments
This research was funded by Astellas Gene Therapies. Medical writing support was provided by Laurie LaRusso, MS, ELS, of Chestnut Medical Communications, paid for by Astellas Gene Therapies. Additional editorial support and preparation of artwork files were provided by Joanne Fitz-Gerald, B.Pharm. and Jonathan A.C. Lee, Ph.D. of FourWave Medical Communications, funded by Astellas Gene Therapies.
Author contributions
J.C.C., M.R.R., C.D., F.U., B.M., and C.F. designed the study. J.C.C., M.R.R., M.C.S., S.L., P.P., F.B., C.A.J., M.C., H.M., M.W.L., S.H., L.V.N., and C.D. acquired and analyzed the data. H.M. and M.W.L. established the IHC assay, performed IHC, and analyzed the IHC results and the integrative analysis of the study data. C.L. developed and validated the mouse model and platforms for the FXN-MCK mouse. J.C.C., M.R.R., M.C.S., S.L., P.P., M.C., M.W.L., S.H., L.V.N., D.R.L., C.D., E.G.-G., F.U., B.M., and C.F. were involved in the data interpretation. All of the authors contributed to the drafting and critical review of the manuscript for important intellectual content.
Declaration of interests
J.C.C., M.R.R., M.C.S., S.L., F.B., C.A.J., M.C., B.M., and C.F. are employees of and/or own equity in Astellas Gene Therapies. P.P. and F.U. are former employees of Astellas Gene Therapies. H.M. is an employee of Diverge Translational Science Laboratory. M.W.L. is the founder, owner, and chief executive officer of Diverge Translational Science Laboratory; participated in scientific advisory boards sponsored by Solid Biosciences, Vial, Astellas Gene Therapies, and Ichorion Therapeutics (formerly); received research support from Astellas Gene Therapies, Solid Biosciences, Taysha Gene Therapies, Kate Therapeutics, Prothelia, Ultragenyx, Dynacure, Rocket Pharmaceuticals, Carbon Biosciences, and Voyager Therapeutics; and a paid consultant to Astellas Gene Therapies, Encoded Therapeutics, Lacerta Therapeutics, Affinia Therapeutics, AGADA Biosciences, Modis Therapeutics, Dynacure, BioMarin, Vertex Pharmaceuticals, Locanabio, Rocket Pharmaceuticals, and Entrada Therapeutics. D.R.L. has received grant funding from Astellas, PTC, Reata, Design, and Novartis.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101193.
Supplemental information
References
- 1.Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 2.Lazaropoulos M., Dong Y., Clark E., Greeley N.R., Seyer L.A., Brigatti K.W., Christie C., Perlman S.L., Wilmot G.R., Gomez C.M., et al. Frataxin levels in peripheral tissue in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2015;2:831–842. doi: 10.1002/acn3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark E., Johnson J., Dong Y.N., Mercado-Ayon E., Warren N., Zhai M., McMillan E., Salovin A., Lin H., Lynch D.R. Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease. Neuronal Signal. 2018;2 doi: 10.1042/NS20180060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anzovino A., Lane D.J.R., Huang M.L.H., Richardson D.R. Fixing frataxin: ‘ironing out’ the metabolic defect in Friedreich's ataxia. Br. J. Pharmacol. 2014;171:2174–2190. doi: 10.1111/bph.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koeppen A.H. Friedreich's ataxia: pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011;303:1–12. doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cossée M., Schmitt M., Campuzano V., Reutenauer L., Moutou C., Mandel J.L., Koenig M. Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc. Natl. Acad. Sci. USA. 1997;94:7452–7457. doi: 10.1073/pnas.94.14.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delatycki M.B., Bidichandani S.I. Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 2019;132 doi: 10.1016/j.nbd.2019.104606. [DOI] [PubMed] [Google Scholar]
- 8.Harding A.E. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 9.Tsou A.Y., Paulsen E.K., Lagedrost S.J., Perlman S.L., Mathews K.D., Wilmot G.R., Ravina B., Koeppen A.H., Lynch D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011;307:46–49. doi: 10.1016/j.jns.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 10.Koeppen A.H., Ramirez R.L., Becker A.B., Bjork S.T., Levi S., Santambrogio P., Parsons P.J., Kruger P.C., Yang K.X., Feustel P.J., Mazurkiewicz J.E. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One. 2015;10 doi: 10.1371/journal.pone.0116396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weidemann F., Störk S., Liu D., Hu K., Herrmann S., Ertl G., Niemann M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013;126(Suppl 1):88–93. doi: 10.1111/jnc.12217. [DOI] [PubMed] [Google Scholar]
- 12.Ocana-Santero G., Díaz-Nido J., Herranz-Martín S. Future prospects of gene therapy for Friedreich's ataxia. Int. J. Mol. Sci. 2021;22:1815. doi: 10.3390/ijms22041815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perdomini M., Belbellaa B., Monassier L., Reutenauer L., Messaddeq N., Cartier N., Crystal R.G., Aubourg P., Puccio H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nat. Med. 2014;20:542–547. doi: 10.1038/nm.3510. [DOI] [PubMed] [Google Scholar]
- 14.Gérard C., Xiao X., Filali M., Coulombe Z., Arsenault M., Couet J., Li J., Drolet M.C., Chapdelaine P., Chikh A., Tremblay J.P. An AAV9 coding for frataxin clearly improved the symptoms and prolonged the life of Friedreich ataxia mouse models. Mol. Ther. Methods Clin. Dev. 2014;1 doi: 10.1038/mtm.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piguet F., de Montigny C., Vaucamps N., Reutenauer L., Eisenmann A., Puccio H. Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich ataxia. Mol. Ther. 2018;26:1940–1952. doi: 10.1016/j.ymthe.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plasterer H.L., Deutsch E.C., Belmonte M., Egan E., Lynch D.R., Rusche J.R. Development of frataxin gene expression measures for the evaluation of experimental treatments in Friedreich's ataxia. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belbellaa B., Reutenauer L., Messaddeq N., Monassier L., Puccio H. High levels of frataxin overexpression lead to mitochondrial and cardiac toxicity in mouse models. Mol. Ther. Methods Clin. Dev. 2020;19:120–138. doi: 10.1016/j.omtm.2020.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huichalaf C., Perfitt T.L., Kuperman A., Gooch R., Kovi R.C., Brenneman K.A., Chen X., Hirenallur-Shanthappa D., Ma T., Assaf B.T., et al. In vivo overexpression of frataxin causes toxicity mediated by iron-sulfur cluster deficiency. Mol. Ther. Methods Clin. Dev. 2022;24:367–378. doi: 10.1016/j.omtm.2022.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vannocci T., Notario Manzano R., Beccalli O., Bettegazzi B., Grohovaz F., Cinque G., de Riso A., Quaroni L., Codazzi F., Pastore A. Adding a temporal dimension to the study of Friedreich's ataxia: the effect of frataxin overexpression in a human cell model. Dis. Model. Mech. 2018;11 doi: 10.1242/dmm.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin J.Y., Zhang L., Clift K.L., Hulur I., Xiang A.P., Ren B.Z., Lahn B.T. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puccio H., Simon D., Cossée M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 22.Ling C., Wang Y., Lu Y., Wang L., Jayandharan G.R., Aslanidi G.V., Li B., Cheng B., Ma W., Lentz T., et al. The adeno-associated virus genome packaging puzzle. J. Mol. Genet. Med. 2015;9:175. doi: 10.4172/1747-0862.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weng L., Wang Q., Yu S., Yang X., Lynch D.R., Mesaros C., Blair I.A. Evaluation of antibodies for western blot analysis of frataxin protein isoforms. J. Immunol. Methods. 2019;474 doi: 10.1016/j.jim.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weng L., Laboureur L., Wang Q., Guo L., Xu P., Gottlieb L., Lynch D.R., Mesaros C., Blair I.A. Extra-mitochondrial mouse frataxin and its implications for mouse models of Friedreich's ataxia. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-72884-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmucker S., Argentini M., Carelle-Calmels N., Martelli A., Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008;17:3521–3531. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 26.Payne R.M. The heart in Friedreich's ataxia: basic findings and clinical implications. Prog. Pediatr. Cardiol. 2011;31:103–109. doi: 10.1016/j.ppedcard.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullman-Culleré M.H., Foltz C.J. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab. Anim. Sci. 1999;49:319–323. [PubMed] [Google Scholar]
- 28.Llorens J.V., Soriano S., Calap-Quintana P., Gonzalez-Cabo P., Moltó M.D. The role of iron in Friedreich's ataxia: insights from studies in human tissues and cellular and animal models. Front. Neurosci. 2019;13:75. doi: 10.3389/fnins.2019.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H., Zhan Q., Huang B., Wang Y., Wang X. AAV-mediated gene therapy: Advancing cardiovascular disease treatment. Front. Cardiovasc. Med. 2022;9 doi: 10.3389/fcvm.2022.952755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 32.Jensen M.K., Bundgaard H. Cardiomyopathy in Friedreich ataxia: exemplifying the challenges faced by cardiologists in the management of rare diseases. Circulation. 2012;125:1591–1593. doi: 10.1161/CIRCULATIONAHA.112.095364. [DOI] [PubMed] [Google Scholar]
- 33.Schmucker S., Martelli A., Colin F., Page A., Wattenhofer-Donzé M., Reutenauer L., Puccio H. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cossée M., Puccio H., Gansmuller A., Koutnikova H., Dierich A., LeMeur M., Fischbeck K., Dollé P., Koenig M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000;9:1219–1226. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- 35.Campuzano V., Montermini L., Lutz Y., Cova L., Hindelang C., Jiralerspong S., Trottier Y., Kish S.J., Faucheux B., Trouillas P., et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 36.Munoz-Zuluaga C., Gertz M., Yost-Bido M., Greco A., Gorman N., Chen A., Kooner V., Rosenberg J.B., De B.P., Kaminsky S.M., et al. Identification of safe and effective intravenous dose of AAVrh.10hFXN to treat the cardiac manifestations of Friedreich's ataxia. Hum. Gene Ther. 2023;34:605–615. doi: 10.1089/hum.2023.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitnall M., Suryo Rahmanto Y., Sutak R., Xu X., Becker E.M., Mikhael M.R., Ponka P., Richardson D.R. The MCK mouse heart model of Friedreich's ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. USA. 2008;105:9757–9762. doi: 10.1073/pnas.0804261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanson E., Sheldon M., Pacheco B., Alkubeysi M., Raizada V. Heart disease in Friedreich's ataxia. World J. Cardiol. 2019;11:1–12. doi: 10.4330/wjc.v11.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.