

Abstract
Advanced reduction processes (ARP) have garnered increasing attention for the treatment of recalcitrant chemical contaminants, most notably per- and polyfluoroalkyl substances (PFAS). However, the impact of dissolved organic matter (DOM) on the availability of the hydrated electron (eaq–), the key reactive species formed in ARP, is not completely understood. Using electron pulse radiolysis and transient absorption spectroscopy, we measured bimolecular reaction rates constant for eaq– reaction with eight aquatic and terrestrial humic substance and natural organic matter isolates ( kDOM,e–aq), with the resulting values ranging from (0.51 ± 0.01) to (2.11 ± 0.04) × 108 MC–1 s–1. kDOM,e–aq measurements at varying temperature, pH, and ionic strength indicate that activation energies for diverse DOM isolates are ≈18 kJ mol–1 and that kDOM,e–aq could be expected to vary by less than a factor of 1.5 between pH 5 and 9 or from an ionic strength of 0.02 to 0.12 M. kDOM,e–aq exhibited a significant, positive correlation to % carbonyl carbon for the isolates studied, but relationships to other DOM physicochemical properties were surprisingly more scattered. A 24 h UV/sulfite experiment employing chloroacetate as an eaq– probe revealed that continued eaq– exposure abates DOM chromophores and eaq– scavenging capacity over a several hour time scale. Overall, these results indicate that DOM is an important eaq– scavenger that will reduce the rate of target contaminant degradation in ARP. These impacts are likely greater in waste streams like membrane concentrates, spent ion exchange resins, or regeneration brines that have elevated DOM concentrations.
Keywords: dissolved organic matter, hydrated electron, kinetics, electron pulse radiolysis, reducing moieties
Short abstract
Electron pulse radiolysis was used to measure the bimolecular reaction rate constant between the hydrated electron and eight dissolved organic matter isolates.
1. Introduction
Dissolved organic matter (DOM) is a complex, heterogeneous mixture of organic compounds naturally occurring in surface waters and groundwaters.1 DOM acts as a radical scavenger, thereby lowering the concentration of reactive species available for target contaminant degradation in advanced oxidation processes (AOP)2,3 and advanced reduction processes (ARP).4−6 Reactions between DOM and oxidizing radicals have been well characterized in the context of AOP, including hydroxyl radicals (•OH),7,8 sulfate radicals (SO4•–),9 carbonate radicals (CO3•–),10 chlorine radicals (Cl• and Cl2•–),11 and bromine radicals (Br• and Br2•–).12 In ultraviolet-advanced reduction processes (UV-ARP), the hydrated electron (eaq–) is considered to be the main reducing species4,5,13−15 with an aqueous reduction potential of −2.9 V.16 However, despite the growing interest in eaq–-mediated contaminant degradation, the reactivity of eaq– with DOM is not well understood.
The scavenging of eaq– by DOM represents an intrinsic limitation for the application of ARP in contaminated waters. ARP have been shown to degrade recalcitrant contaminants such as bromate,15,17−22 halogenated organic compounds,4,13,14,23−31 and per- and polyfluoroalkyl substances (PFAS),32−42 but the majority of these studies have been performed in relatively clean systems (e.g., lab-grade water). Some studies have tested ARP for treating concentrated waste streams produced from membrane filtration reject42 or regeneration of adsorbents,43 which have elevated DOM concentrations. Ren et al. also demonstrated that increasing the concentration of Aldrich humic acid inhibited the degradation of perfluorooctanoic acid in the UV/sulfite system.6 Another study conducted by Duan and Batchelor showed inhibited perchlorate degradation with increasing DOM concentration in the UV/sulfite ARP.44 As noted in these studies, and in parallel to the UV-AOP literature, this inhibition of target contaminant degradation can occur by DOM shielding the eaq– sensitizer from absorbing UV photons, by scavenging eaq–, or by a combination of both processes. While the impact of light screening can be predicted based on absorbance measurements,45 accurate predictions of eaq– scavenging by DOM are limited by the lack of reported bimolecular rate constants for this reaction.
The objectives of this study were to evaluate how the reactivity of eaq– with DOM depends on DOM physicochemical properties, environmental conditions, and the prolonged eaq– exposure typically encountered in ARP systems. These objectives were accomplished by quantifying bimolecular reaction rate constants (kDOM,e–aq) using electron pulse radiolysis for eight humic substance and natural organic matter (NOM) isolates in buffered solution at neutral pH and measuring kDOM,e–aq values as a function of pH, ionic strength, and temperature for selected DOM samples. The isolates employed represent a wide range of physiochemical properties and chemical composition, being derived from both terrestrial and aquatic sources.46 Insights into the variability of kDOM,e–aq among samples were gleaned by evaluating correlations to the physicochemical properties of DOM. Lastly, the impact of prolonged eaq– exposure on DOM-eaq– scavenging was evaluated in a 24 h UV/sulfite experiment conducted with Suwanee River natural organic matter II. Results from this study provide a means for estimating the eaq– scavenging capacity of DOM in ARP, informing how this scavenging capacity changes with environmental conditions, and indicate that eaq– scavenging by DOM can impact the efficacy of target contaminant degradation even after significant eaq– exposure.
2. Materials and Methods
2.1. DOM Isolates, Chemicals, and Sample Preparation
Chemicals were purchased from Sigma-Aldrich or VWR and are listed in Table S1 in the Supporting Information (SI). In addition, eight humic substance and natural organic matter (NOM) isolates were purchased from the International Humic Substances Society (IHSS) and used for electron pulse radiolysis experiments, including Elliott Soil humic acid IV (ESHA IV), Pahokee Peat fulvic acid II (PPFA II), Pahokee Peat humic acid I (PPHA I), Pony Lake fulvic acid (PLFA), Suwannee River fulvic acid II (SRFA II), Suwannee River humic acid II (SRHA II), Suwannee River natural organic matter II (SRNOM II), and Upper Mississippi River natural organic matter (MRNOM). The IHSS catalog number for each isolate is available in SI Table S2. Additional IHSS catalog numbers were used for optical measurements (see SI Table S3) and kDOM,e–aq comparison (SI Tables S5–S8), including Suwannee River fulvic acid III (SRFA III), Suwannee River humic acid III (SRHA III), and Elliott Soil humic acid V (ESHA V). All DOM stock solutions were prepared at a concentration of 200 mg L–1 in 10 mM dibasic phosphate that was adjusted to pH 5, 7, or 9 using HClO4, HCl, or NaOH. HClO4 was used for the pulse radiolysis studies. Ionic strength was varied using NaClO4. Ultrapure water (≥18.2 MΩ·cm) used for all experiments was obtained from either the Notre Dame University Radiation Laboratory reverse osmosis water treatment system or a Barnstead purification system (Thermo Fisher).
2.2. Analytical Measurements
Analytical measurements included pH, absorbance, dissolved organic carbon (DOC), and anion analysis. pH measurements were made with either an Orion Research pH/millivolt meter 811 (Notre Dame Radiation Laboratory) or a Thermo Scientific Orion Versa Star Pro combined with a micro Mettler Toledo LE422 pH probe (Texas A&M). A Cary-100 spectrophotometer (Agilent) with a 1 cm pathlength quartz cuvette was used to measure absorbance spectra, which were used to calculate specific ultraviolet absorbance at 254 nm (SUVA254) and spectral slope (S300–600) for the isolates. SI Text S1 provides additional measurement and calculation details for SUVA254 and S300–600. DOC measurements were performed by Hazen Huffman Laboratories in Golden, Colorado. Prior to DOC analysis, samples were acidified with trace-metal-grade nitric acid (70%) to pH ≤ 2.0 and stored at 4 °C. Anion analysis, except for sulfite, was conducted on a Dionex Integrion ion chromatography system equipped with a conductivity detector, a Dionex IonPac AS19 column (4 mm × 250 mm), a Dionex IonPac AG19 (4 mm × 50 mm) guard column, and a Dionex ADRS 600 (4 mm) suppressor. Anions were eluted with 20 mM KOH at a 1.0 mL min–1 flow rate and a 50 mA suppressor current. The column was temperature-controlled at 30 °C. Sulfite concentrations were quantified using the 5,5′-dithiobis(2-nitrobenzoic acid) assay and a thiol molar absorption coefficient of 14,000 M–1 cm–1, as described previously.47,48
2.3. Electron Pulse Radiolysis Techniques
kDOM,e–aq values were measured using the linear accelerator system at the University of Notre Dame Radiation Laboratory.49 Numerous studies have utilized this approach to quantify bimolecular rate constants for reactions between organic compounds and various radical species.50−54 Methods previously established and utilized in this study for bimolecular reaction rate determination are briefly discussed below.
DOM stock solutions (200 mg L–1) were diluted with phosphate buffer in one of two dilution series (series 1: 160, 120, 80, and 40 mg L–1; series 2: 150, 100, and 50 mg L–1) in quartz cuvettes, purged with argon gas for at least 2 min, and sealed with glass stoppers. Water radiolysis via 7 ns electron pulses yielded 0.27 μmol eaq– per J of energy absorbed.16 The transient eaq– decays were monitored at 720 nm on a microsecond time scale for the dilution series as well as a phosphate buffer blank (DOM at 0 mg L–1) also purged with argon. Transient decay traces were averaged (∼30 traces) and fit with a first-order exponential decay plus baseline model to extract the pseudo-first-order decay constants of eaq– (k′), which were then plotted vs the DOM concentration to obtain kDOM,e–aq. In this analysis, the change in k′ is due solely to the change in DOM concentration because the scavenging capacity of the background solvent is constant. Bimolecular rate constants were normalized to carbon concentration using the carbon mass % provided by the IHSS.55 Additional details involving pulse radiolysis techniques are discussed in SI Text S2.
2.4. Photochemical Irradiation Experiments
Irradiation experiments were conducted in duplicate immersion well reactors (Ace Glass) with an exterior glass body and interior quartz sleeve. Reactors were filled with ultrapure water (∼590 mL) and 1.0 mM borate buffer (pH 10.0) and purged with nitrogen gas for at least 45 min prior to and during experiments. The temperature in the reactors was controlled at 20 °C using a recirculating chiller. A low-pressure Hg, non-ozone forming lamp (10 W LSE Lighting GPH212T5L/4P) was powered on for at least 15 min and briefly turned off before concentrated stock solutions of sulfite, SRNOM II, and monochloroacetic acid (MCAA) were added to the reactor. UV/sulfite experiments were performed at a pH at least 2 pH units above the pKa of HSO3– (pKa = 7.2) and under anerobic conditions to minimize eaq– scavenging impacts of HSO3–, H+, and O2.16,45,56,57 After spiking, the solution was mixed for at least 30 s and stirring was maintained throughout experiments using a magnetic stir bar at 400 rpm. Experiments were initiated by turning on the lamps and collecting aliquots of solution using a stainless-steel needle and syringe. Samples were collected in either falcon tubes or 1.5 mL polypropylene vials and stored at 4 °C before analysis. UV irradiance was measured as 1.26 × 10–8 Es cm–2 s–1 using uridine actinometry58 and the previously measured average reactor pathlength was determined as 2.23 cm using the H2O2 method.47,59,60
3. Results and Discussion
3.1. DOM Isolate and eaq– Bimolecular Rate Constant Measurements
kDOM,e–aq values at pH 7.0 and 22 ± 2 °C varied by approximately a factor of 4, ranging from (0.51 ± 0.01) to (2.11 ± 0.04) × 108 MC–1 s–1 (Figure 1A). kDOM,e–aq values were determined by finding the pseudo-first-order rate constant from the transient eaq– decay data (Figure 1B) and plotting the pseudo-first-order rate constant against the DOM concentration (Figure 1C). A linear fit to the data in Figure 1C yields kDOM,e–aq as the slope with the y-intercept representing any eaq– scavengers other than DOM (e.g., H+ in acidic conditions) present in the background solvent (see SI Text S2 for additional discussion). SI Figure S1 contains similar pseudo-first-order plots for the other DOM isolates and SI Text S3 discusses the minimal impact of IHSS catalog number on DOM-specific kDOM,e–aq values.
Figure 1.

Bimolecular rate constant measurements between eaq– and DOM isolates (kDOM,e–aq). Isolates include Elliott Soil IV humic acid (ESHA IV), Pahokee Peat II fulvic acid (PPFA II), Pahokee Peat I humic acid (PPHA I), Upper Mississippi River natural organic matter (MRNOM), Pony Lake fulvic acid (PLFA), Suwannee River II humic acid (SRHA II), Suwannee River II natural organic matter (SRNOM II), and Suwannee River II fulvic acid (SRFA II). kDOM,e–aq in (A) were determined by measuring (B) transient absorption decay kinetics of eaq– at 720 nm for various [DOM] and plotting (C) pseudo-first-order rate constant as a function of [DOM]. (B, C) Data for SRFA II only. The solid line in (C) represents a linear fit to the data using the least squares method with the slope reported as the kDOM,e–aq. Similar (C) plots for other DOM isolates are found in SI Figure S1. Error bars in (A) represent the standard error of the slope in (C). kDOM,e–aq were compared to bimolecular rate constants between other radicals7−11 and DOM isolates in (D). DOM-eaq– experiments conducted at pH 7.0 ± 0.1, 22 ± 2 °C, and 10.0 mM phosphate buffer. All other radical experiments in (D) were conducted at pH 7.0, room temperature, and varying concentrations of phosphate buffer.
kDOM,e–aq values for soil humic substance isolates ranged from (0.66 ± 0.02) × 108 MC–1 s–1 (PPFA II) to (1.14 ± 0.04) × 108 MC–1 s–1 (PPHA I) and largely overlap with those of aquatic isolates, which ranged from (0.51 ± 0.01) × 108 MC–1 s–1 (MRNOM) to (2.11 ± 0.04) × 108 MC–1 s–1 (SRFA II). Soil humic acid kDOM,e–aq values were lower than that for SRHA II, an aquatic humic acid. The kDOM,e–aq value for PPFA II, a terrestrial fulvic acid, was also lower than the kDOM,e–aq for SRFA II, an aquatic fulvic acid. However, not all kDOM,e–aq values for aquatic isolates were higher than isolate terrestrial kDOM,e–aq values. For example, MRNOM and PFLA (aquatic origin) had lower kDOM,e–aq values than PPHA I (soil origin). Isolates from the Suwannee River had the largest kDOM,e–aq values, with SRFA II and SRNOM II exhibiting similar reactivity and SRHA II being ∼20% lower. Overall, while kDOM,e–aq is variable among these DOM samples, there is no clear trend between isolation procedure (humic substance vs natural organic matter) or source (aquatic vs soil).
The range of kDOM,e–aq values reported on an MC–1 s–1 basis falls within the range of bimolecular reaction rate constants reported in the literature for oxidizing radicals’ reaction with DOM (Figure 1D and SI Table S4).7−11 On average, kDOM,e–aq values were exceeded only by •OH and Cl• values. DOM is a primary sink for oxidizing radicals in sunlit waters and advanced oxidation processes. In anaerobic systems, such as electron transfer in anaerobic bottom waters and sediments or engineered systems like ARP, DOM will be an important eaq– scavenger. Based on an average of the values measured for humic substance and NOM isolates, we recommend a kDOM,e–aq value of 1.2 × 108 MC–1s–1 (1.0 × 104 L mgC–1 s–1). Employing this value yields an eaq– scavenging capacity of 1.0 × 105 s–1 at a dissolved organic carbon concentration of 10 mgC L–1.
3.2. Impact of pH, Temperature, and Ionic Strength on kDOM,e–aq
We evaluated the impact of pH, temperature, and ionic strength on kDOM,e–aq for two isolates, SRFA II and ESHA IV (Figure 2). Increasing pH from 7 and 9 caused small but significant increases in kDOM,e–aq for both ESHA IV and SRFA II (Figure 2A, between 1.1- and 1.3-fold). Conversely, increasing pH from 5 and 7 caused a 1.4-fold decrease in kDOM,e–aq for SRFA II.
Figure 2.

Influence of (A) pH, (B) ionic strength, and (C) temperature on bimolecular rate constants for SRFA II and ESHA IV. Experiments conducted at 22 ± 2 °C, pH 7.0 ± 0.1, and 10.0 mM dibasic phosphate buffer unless otherwise specified. The ZB in (B) was calculated from the Brønsted-Bjerrum equation (eq 3.1) using the charge of eaq– (i.e., ZA = −1). Error bars represent the standard error of the bimolecular rate constant (majority of error bars are within markers). Additional plots of the pseudo-first-order rate constant against the DOM concentration for each pH, temperature, and ionic strength condition are provided in SI Figures S3 and S4.
The impact of pH on kDOM,e–aq may be attributed to changes in the reactivity of DOM moieties at different protonation states, the impact of ionization on DOM molecular size, and the accessibility of reducible moieties to eaq–. Protonation of carboxylic acids generally increases the eaq– bimolecular rate constant (e.g., acetic acid, k = 2 × 108 M–1 s–1; acetate, k < 1 × 106 M–1 s–1).16 This could explain the decrease in kDOM,e–aq for SRFA II between pH 5 and 7 (we were unable to measure kDOM,e–aq at pH 5 for ESHA IV). Increasing pH from 7 to 9 results in a greater fraction of ionized phenolic moieties, which have a lower reactivity than their corresponding protonated species. However, phenol is much less reactive with eaq– (k = 2 × 107 M–1 s–1)16 compared to carboxylic acids. Increasing protonation of phenols and carboxylic acids with decreasing pH lowers the DOM charge density,61 making the reaction of eaq– with DOM more favorable from an electrostatic perspective. A competing effect is the impact of ionization state on DOM size. As the pH increases, electrostatic repulsion between negatively charged DOM moieties intensifies, resulting in molecular expansion62 and easier access to the reducible DOM moieties (eaq– is formed in the aqueous phase upon absorption of radiation). Thus, the slight increase in kDOM,e–aq between pH 7 and 9 is consistent with an increase in accessibility of eaq– to reducible DOM moieties.
The ionic strength trend observed for both SRFA II and ESHA IV at pH 7 behaved according to the Brønsted-Bjerrum equation, eq 3.1, (i.e., the rate constant for like-charged reactants increases with increasing ionic strength).
| 3.1 |
In eq 3.1, k2,I represents the bimolecular rate constant at ionic strength I, k2,I = 0 represents the bimolecular rate constant at infinite dilution, and ZAZB is the product of the charges of the reactants. We approximated k2,I=0 with kDOM,e–aq values measured in 10 mM phosphate buffer (I = 0.02 M at pH 7). Due to the negative charge of eaq–, a higher ionic strength results in a shielding of the like-charged reactants, directly decreasing the coulombic repulsion forces and increasing reactivity with anionic species. This shielding effect was observed for both SRFA II and ESHA IV when ionic strength was increased (Figure 2B) but to slightly different extents, with kDOM,e–aq increasing by 1.3-fold for ESHA IV and 1.5-fold for SRFA II. One possible explanation is that, at pH 7 and high ionic strength, DOM structures have expanded such that the reducible moieties are more accessible to eaq– and some negatively charged DOM moieties have been shielded. This explanation is consistent with the abovementioned impact of increasing pH from 7 to 9 for these same isolates. Furthermore, using the Brønsted-Bjerrum equation, we calculated the ZB value, using a (−1) charge for eaq–.16 The calculated ZB values for SRFA II and ESHA IV were −0.68 and −0.44, respectively, which is much less negative than DOM charge density values reported based on other methods.63 One possibility is that negatively charged moieties are spatially distant from the site of eaq– reaction. Another explanation is that increasing DOM charge impacts DOM’s three-dimensional structure and that the subsequent effect on kDOM,e–aq is not fully captured by eq 3.1. Future research measuring kDOM,e–aq under a greater range of pH values and ionic strength conditions could help discern among these possibilities.
Activation energies (Ea) for the reaction of eaq– with SRFA II and ESHA IV at pH 7 were calculated using the measured temperature-dependent kDOM,e–aq values and the Arrhenius equation (eq 3.2)
| 3.2 |
where A is the Arrhenius pre-factor, R is the gas constant, and T is the temperature (K). Plotting ln (kDOM,e–aq) against 1000/T yields a slope −Ea/R from which Ea was determined (Figure 2C). The activation energies for SRFA II and ESHA IV were the same within error (18.5 ± 1.4 and 17.7 ± 2.2 kJ mol–1, respectively), suggesting that an average Ea of 18 kJ mol–1 can generally be applied to assess the temperature dependence of eaq– scavenging by DOM in ARP systems.
3.3. Relationships between Bimolecular Rate Constants and DOM Composition
We investigated correlations between the measured kDOM,e–aq values and DOM physiochemical properties to provide clues to the factors governing the reactivity of DOM with eaq–. Physicochemical properties included elemental ratios (H/C and O/C), SUVA254, S300–600, carbon distribution from 13C NMR, and number-average molecular charge (MnQ). These physicochemical properties for each DOM isolate along with the respective kDOM,e–aq values are shown in Table 1 for standard experimental conditions (pH 7.0, 10 mM phosphate buffer, 22 ± 2 °C). Elemental ratios and carbon distributions were taken from the IHSS website for each isolate’s catalog number, MnQ was taken from previous studies,64,65 and SUVA254 and S300–600 were measured in this study. Additional information about measurement and calculation of the DOM physicochemical properties and kDOM,e–aq values under nonstandard conditions are provided in the Supporting Information (Texts S4 and S5 and Tables S7–S9).
Table 1. kDOM,e–aq Values and Characterization Data for Humic Substance and NOM Isolates.
| samplea | SUVA254b,c (L mgC–1 m–1) | S300–600b (nm–1) | H/Cd | O/Cd | % aromatice | % carbonyle | MnQd (charge molecule–1) | kDOM,e–aqf (108 MC–1 s–1) | kDOM,e–aqf (104 L mgC–1 s–1) |
|---|---|---|---|---|---|---|---|---|---|
| ESHA IVb | 7.4 | 0.0074 | 0.05 | 0.54 | 41 | 1 | 10.8 | 0.85 ± 0.02 | 0.71 ± 0.02 |
| PPFA II | 5.9 | 0.0134 | 0.07 | 0.84 | 39 | 3.6 | 14.7 | 0.66 ± 0.02 | 0.55 ± 0.01 |
| PPHA I | 6.1 | 0.0090 | 0.07 | 0.66 | 47 | 5 | 12.4 | 1.14 ± 0.04 | 0.95 ± 0.03 |
| PLFA | 1.2 | 0.0170 | 0.10 | 0.60 | 12 | 1.2 | 3.20 | 0.83 ± 0.01 | 0.69 ± 0.01 |
| SRFA IIb | 4.3 | 0.0158 | 0.08 | 0.82 | 22 | 5 | 7.98 | 2.11 ± 0.04 | 1.76 ± 0.03 |
| SRHA IIb | 5.1 | 0.0124 | 0.08 | 0.80 | 31 | 6 | 10.4 | 1.60 ± 0.02 | 1.33 ± 0.02 |
| SRNOM II | 3.2 | 0.0146 | 0.08 | 0.82 | 23 | 8 | 8.64 | 2.07 ± 0.05 | 1.73 ± 0.04 |
| MRNOM | 2.8 | 0.0147 | 0.09 | 0.83 | 19 | 3 | 9.61 | 0.51 ± 0.01 | 0.43 ± 0.01 |
Experiments conducted at standard conditions of 22 ± 2 °C, pH 7.0 ± 0.1, and 10.0 mM dibasic phosphate buffer unless otherwise specified.
IHSS catalog numbers vary for SUVA254 and S300–600 values. See SI Table S3.
Values based on [DOC] calculated from isolate mass per volume normalized to IHSS percent carbon.
Values unavailable for DOM isolates prepared in nonstandard conditions.
Values listed here are significant figures reported on the IHSS website.66
Average kDOM,e–aq value is 1.22 ± 0.63 × 108 MC–1 s–1 or 1.02 ± 0.53 × 104 L mgC–1 s–1.
Of the physicochemical properties examined, kDOM,e–aq had the strongest relationship with the % carbonyl carbon as determined by 13C NMR. The positive linear correlation between kDOM,e–aq and % carbonyl carbon (ρ = 0.809) was statistically significant (p = 0.0046) (Figure 3A) and is consistent with the known high reactivity of eaq– with carbonyl compounds.16 For example, a sampling of literature bimolecular eaq– rate constants for model organic compounds shows that carbonyl-containing compounds exhibit consistently higher reactivity than other functional groups (Figure 3B).
Figure 3.
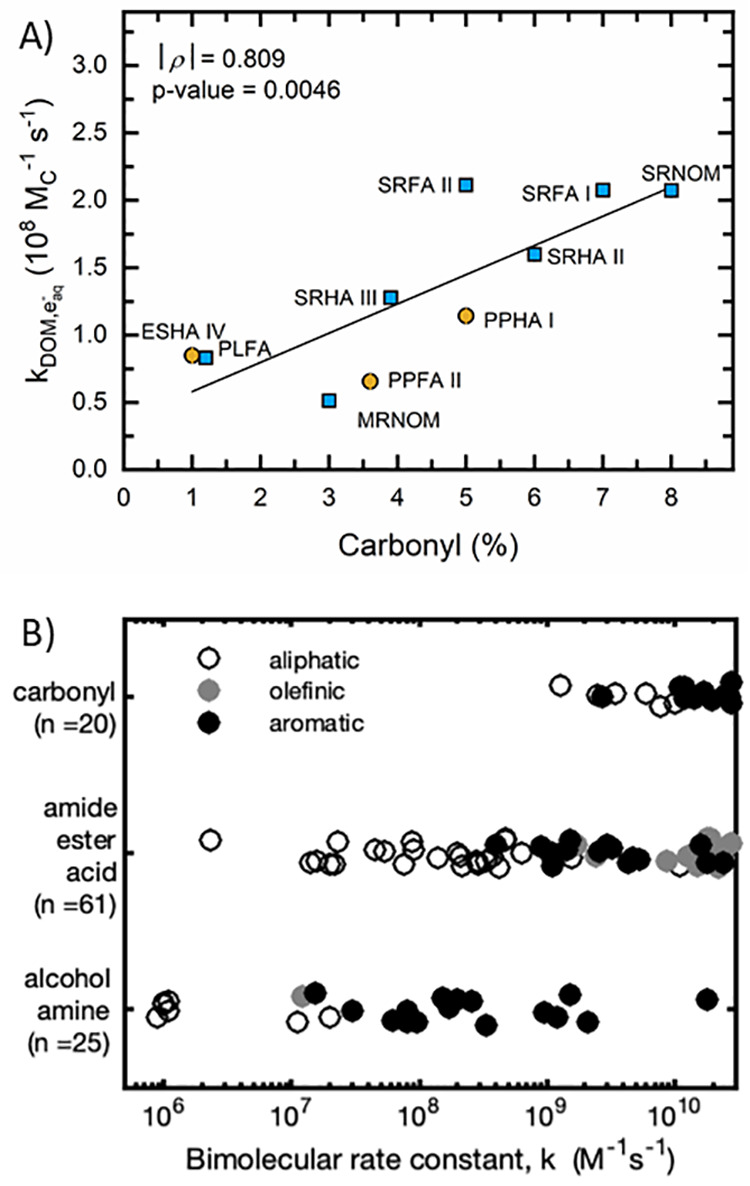
Relationships between DOM composition and eaq– bimolecular rate constant. (A) Correlation kDOM,e–aq and % carbonyl carbon as determined by 13C NMR and reported by the IHSS. Markers refer to values derived from the slope of first-order rate constants vs [DOM] (e.g., Figure 1C), and error bars refer to the standard error of the slope (majority of error bars are within markers). Marker color represents terrestrial (brown) and aquatic isolates (blue). SRNOM data represent SRNOM I for carbonyl % and SRNOM II for kDOM,e–aq. All other IHSS catalog numbers match exactly. Experiments conducted at 22 ± 2 °C, pH 7.0 ± 0.1, and 10.0 mM dibasic phosphate buffer. (B) Bimolecular rate constants between model organic compounds and eaq– from literature sources (accessed via https://kinetics.nist.gov/solution/).67
The lack of strong correlations between kDOM,e–aq and other DOM physicochemical properties (e.g., % aromaticity, MnQ) was surprising given the known impact of charge and functional group on the reactivity of model organic compounds with eaq–. For example, we hypothesized that kDOM,e–aq would tend to decrease with increasing DOM negative charge (MnQ), but this was not observed (see SI Figure S5). Similarly, we expected kDOM,e–aq to be positively correlated to the electron accepting capacity,68 but this was also not observed (see SI Figure S5). In comparison, recent reports of bimolecular rate constants for oxidizing radicals such as SO4•– and halogen radicals (X• and X2•–) with DOM have yielded significant correlations with DOM physicochemical properties such as SUVA254 and electron donating capacity.9,11,12 Bimolecular rate constants for DOM with •OH have not been described by a single parameter; rather multiple linear regression models or principal component analysis has been employed.51,69 Preliminary attempts were made in this study to correlate groups of 7–9 parameters, but these attempts only confirmed that % carbonyl carbon was the most significant predictor of kDOM,e–aq. These statistical analyses may prove useful in future studies on the reactivity of DOM with eaq– but will require a larger sample set than analyzed here.
Overall, the lack of correlations between kDOM,e–aq and DOM physicochemical properties observed herein indicates that kDOM,e–aq is not governed by a single aspect of DOM’s composition. It is likely, however, that aquatic-based DOM will have a larger impact on eaq– scavenging in ARP treatment due to the presence of a higher % carbonyl carbon.
3.4. Comparison of Organic Model Compounds and DOM Reaction with eaq–
Prior studies of radical reactions (•OH, SO4•–, X• and X2•–; X = Cl–, Br–) with DOM have shown that measured rate constants can be reasonably well predicted using an average value of individual reacting components chosen to represent DOM composition.7,9,11,12 To test this hypothesis for eaq– reaction, a set of model compounds with known eaq– bimolecular rate constants were selected based on prior compilations for oxidizing radicals,7,9,11,12 and the below equation was applied (eq 3.3)
| 3.3 |
where αi and ki are the fractional contribution and bimolecular rate constant (units of MC–1s–1) of model organic compound i. αi was varied across the three scenarios listed below to evaluate the range of possible % aromatic and % carbonyl carbon present in these isolates.
Scenario 1: Each model compound is set to an equal concentration resulting in 49.2% aromatic carbon and 5.3% carbonyl carbon. A 49.2% aromatic carbon is higher than aquatic isolates but only slightly higher than soil humic acids.
Scenario 2: The % aromatic carbon was chosen to be 20% and is partitioned equally between the aromatic compounds in the model compound set. The αi for acetone is set to 0.07 and the remaining αi’s are distributed equally among methyl acetate, tert-butanol, and alanine.
Scenario 3: The % aromatic carbon was set to 40% and αi for acetone is set to 0. The high aromatic % and low carbonyl % for this scenario are representative of characterization data for ESHA IV and V.
Contrary to the good agreement observed in previous studies for DOM reactions with oxidizing radicals,7,9,11,12 the three scenarios tested all resulted in kDOM,e–aq values that were either at the upper end or exceeded kDOM,e–aq values measured by pulse radiolysis (Table 2). The lower measured kDOM,e–aq values could be the result of geometric effects (reactive eaq– moieties are not accessible to radiolytically formed eaq–), charge impacts (DOM typically exhibits a large negative charge, which may slow down kDOM,e–aq relative to singly charged organic compounds), or a combination of these factors. Future research is needed to discern among these possibilities.
Table 2. Summary of Results from Applying Eq 3.3 to Predict Hydrated Electron Rate Constants for DOM Using Model Compounds.
| model compound dataa |
scenario 1 |
scenario 2 |
scenario 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| compound | formula | k (107 M–1 s–1) | k (107 MC–1 s–1) | αi | αiki | αi | αiki | αi | αiki |
| phenol | C6H6O | 2.00 | 0.33 | 0.11 | 0.037 | 0.04 | 0.01 | 0.08 | 0.03 |
| 2-hydroxybenzoate | C7H6O3 | 1000 | 143 | 0.11 | 15.873 | 0.04 | 5.71 | 0.08 | 11.43 |
| benzoate | C7H6O2 | 300 | 42.9 | 0.11 | 4.762 | 0.04 | 1.71 | 0.08 | 3.43 |
| benzyl alcohol | C7H8O | 20 | 2.86 | 0.11 | 0.317 | 0.04 | 0.11 | 0.16 | 0.46 |
| benzaldehyde | C7H6O | 2400 | 343 | 0.11 | 38.095 | 0.04 | 13.71 | 0.00 | 0.00 |
| acetone | C3H6O | 770 | 257 | 0.11 | 28.519 | 0.07 | 18.74 | 0.02 | 5.13 |
| methyl acetate | C3H6O2 | 870 | 2.9 | 0.11 | 0.322 | 0.24 | 0.70 | 0.19 | 0.56 |
| tert-butanol | C4H10O | 0.04 | 0.01 | 0.11 | 0.001 | 0.24 | 0.00 | 0.19 | 0.00 |
| alanine | C3H7O2N | 1.2 | 0.40 | 0.11 | 0.044 | 0.24 | 0.10 | 0.19 | 0.08 |
| Σiαi or Σiαiki | 1.00 | 8.8 × 108 MC–1s–1 | 1.00 | 4.08 × 108 MC–1s–1 | 1.00 | 2.11 × 108 MC–1s–1 | |||
Rate constants obtained from the NDRL/NIST solution kinetics database (kinetics.nist.gov/solution/).67
3.5. Impact of DOM on eaq– Exposure during the UV/Sulfite ARP
The measured kDOM,e–aq values indicate that DOM will be an important scavenger of eaq– in ARP. In treatment technologies in which eaq– is formed photochemically, DOM will also screen incoming UV photons from being absorbed by the eaq– sensitizer (e.g., sulfite), thereby decreasing the rate of eaq– formation. Both eaq– scavenging and light screening by DOM will decrease the rate of eaq–-mediated target contaminant degradation.45,47 In ARP, where DOM is continuously exposed to eaq–, the light screening characteristics and eaq– scavenging likely change over time as eaq– reactions modify DOM structure. The kDOM,e–aq values presented in Table 1, however, represent initial conditions before each DOM isolate has undergone transformation by eaq–.
To evaluate the impact of eaq– exposure on DOM light screening and eaq– scavenging, we performed an experiment in which 10 mM sodium sulfite was irradiated with low-pressure Hg vapor lamps (emitting at 254 nm) in the presence of 10 mgC L–1 SRNOM II. UV/sulfite experiments were conducted under anerobic conditions with a reactor pH ≥ 9.5 to minimize DOM’s reaction with radical species other than eaq–.16,45,56,57 Even though sulfite and sulfite radicals may directly react with DOM moieties, for simplicity it was assumed that DOM reacted predominantly with eaq– in the UV/sulfite system. However, it is not possible to categorically exclude DOM transformations by sulfite radicals. During a 24 h irradiation experiment, 20 μM chloroacetate (MCAA) was spiked at various time points to serve as an eaq– probe as demonstrated in our previous study.47 The resulting first-order degradation rate constants for chloroacetate transformation were used to calculate the eaq– concentration ([eaq–]t) and eaq– scavenging capacity (k′S,t) for each chloroacetate spike time, t.
Results from chloroacetate spikes over a 24 h UV/sulfite experiment indicate that the light screening and eaq– scavenging of 10 mgC L–1 SRNOM II dissipate after ∼4 h, resulting in an [eaq–]t that peaks at ∼7 h (Figure 4, see SI Text S6 for additional details on calculations). The rate of eaq– formation (Reaq–f,t), which is a function of the fraction of light absorbed by sulfite, increases from 2.3 × 10–7 M s–1 at 0 h to 3.8 × 10–7 M s–1 at 4 h. At 0 h, the solution absorbance at 254 nm was 0.49 cm–1, the calculated absorbance due to sulfite was 0.19 cm–1, and the fraction of light absorbed by sulfite was 38%. At 4 h, the fraction of light absorbed by sulfite was nearly 100%. This indicates that by ∼4 h eaq– reactions have completely attenuated the absorbance of SRNOM II at 254 nm and that the remaining absorbance is completely attributable to sulfite (see SI Figure S6). Furthermore, the k′S,tvalue decreased rapidly during the first ∼4 h of the UV/sulfite experiment. At 0 h, the measured k′S,t was 1.5 × 105 s–1, which agrees well with the value calculated the kDOM,e–aq value for SRNOM II in Table 1 [(10 mgC L–1) × (1.73 × 104 L mgC–1 s–1) = 1.73 × 105 s–1]. By ∼4 h, the measured k′S,t is consistent with the calculated k′S,t of 20 μM chloroacetate (2 × 104 s–1), indicating that the k′S,t from SRNOM II has been completely abated. Taken together, the results indicate that both light screening and eaq– scavenging decrease the [eaq–]t in the UV/sulfite system available for contaminant abatement. These impacts are anticipated to be greater at higher DOM concentrations. For example, Ren et al. demonstrated sustained light screening in the UV/sulfite system over 24 h at an Aldrich humic acid concentration of 50 mgC L–1.6 This may present a challenge for eaq–-based treatment of waste streams like ion exchange resin, regeneration brine, and reverse osmosis concentrate where DOM concentrations are elevated.
Figure 4.
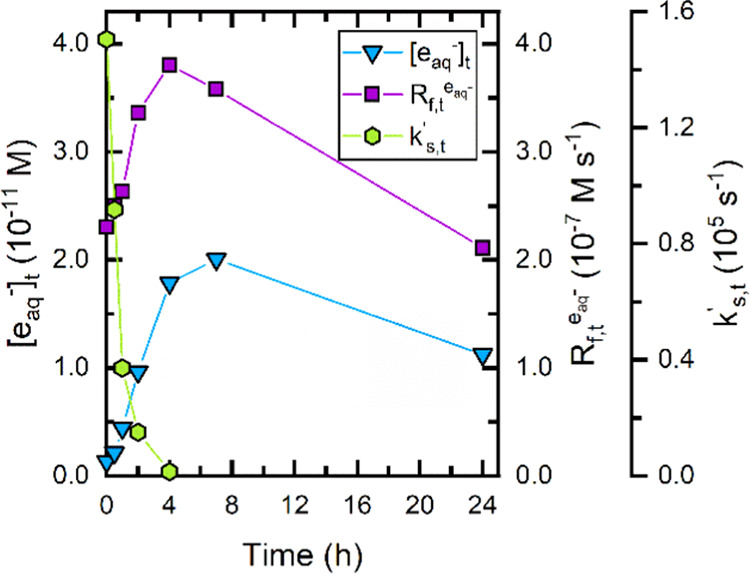
Photochemical parameters measured by chloroacetate for the UV/sulfite system in the presence of Suwanee River natural organic matter II (SRNOM II), including eaq– concentration ([eaq–]t), formation rate (Reaq–f,t), and scavenging capacity (k′S,t). SI Text S6 explains how these parameters were calculated. Experimental conditions: 10 W low-pressure Hg lamp, pH0 = 9.5, 20 °C, 10 mgC L–1 [SRNOM II]0, 10.4 mM [sulfite]0, 20 μM [MCAA]0 spikes, and 1.0 mM borate buffer in ultrapure water.
4. Significance for Hydrated Electron-Based Contaminant Degradation
ARP have received increasing attention for the destruction of recalcitrant chemical contaminants, most notably PFAS.45,57,70,71 However, the role of DOM in these treatment technologies has not been adequately addressed.45,57 Results from this study demonstrate that eaq– scavenging by DOM will significantly impact the rate of target contaminant degradation in ARP. We recommend that a kDOM,e–aq value of 1.2 × 108 MC–1 s–1 (1.0 × 104 L mgC–1 s–1) be used to evaluate the eaq– scavenging impact of DOM in future studies. Given that kDOM,e–aq values vary by a factor of 4, additional research is needed to develop structure-reactivity relationships to predict eaq– scavenging by DOM in different contexts.
Another implication of this research is that the increases in kDOM,e–aq resulting from high ionic strength or alkaline pH, as observed in treating concentrated waste streams, are unlikely to significantly impact the efficiency of eaq–-based treatment. We showed that increasing ionic strength from 0.02 to 0.12 M or increasing pH from 5 to 9 results in only a 1.5-fold increase in kDOM,e–aq. On the other hand, increasing the DOM concentration from 10 to 100 mgC L–1 results in a 10-fold increase in the eaq– scavenging capacity in addition to a significant increase in UV photon screening. Thus, increases in DOM concentration in these waste streams will likely outweigh any increase in kDOM,e–aq values that come from varying pH and ionic strength.
The temporal nature of the eaq– formation rate, scavenging capacity, and concentration demonstrated in Figure 4 indicates that eaq– scavenging by DOM is long-lived and has the potential to significantly impact ARP performance. The results shown in Figure 4 also raise several questions to be addressed in future research. First, we observed that the absorbance at 254 nm and eaq– scavenging capacity of DOM were completely attenuated at ∼4 h of UV/sulfite treatment but [DOC] measured for samples collected at 2 and 24 h were the same within error of those measured at 0 h (SI Figure S9). This result indicates that the end products of eaq– reaction with DOM are not chromophoric (do not absorb at 254 nm) and do not volatilize in our system, which was continuously sparged with nitrogen gas. Future research is needed to elucidate the composition of these products to explain the lack of change in [DOC] during UV/sulfite treatment. Second, future research should also investigate the impact of DOM in the sequential oxidation–reduction system described by Liu et al.72 The oxidation step, which involves the formation of •OH from heat-activated persulfate, will likely be impacted by DOM given the known reactivity of •OH with DOM. While mineralization of DOM in this stage may alleviate eaq– scavenging by DOM in the subsequent reduction step, buildup of bicarbonate could negatively impact subsequent reductive treatment due to eaq– scavenging. Third, the temporal variation in eaq– photochemical parameters due to DOM modifications begs the question of how these parameters change in other photochemical treatment systems (i.e., UV-AOP). Although a prior study has evaluated this question and found minimal impacts under typical UV-AOP fluences (∼1000 mJ cm–2) using low-pressure Hg lamps,73 more studies are warranted.
Acknowledgments
The authors gratefully acknowledge support from the U.S. National Science Foundation (CBET #2050934, # 2050882). Thanks go to Dr. James Kiddle for help with conducting pulse radiolysis experiments. B.D.F. gratefully acknowledges financial support from the Texas A&M University Graduate Merit Fellowship, Stantec & AWWA Water Equation, and the Texas Engineering Foundation. The authors are thankful for the collaborative efforts at the Radiation Laboratory, which is supported by the Office of Basic Energy Sciences, U.S. Department of Energy.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.3c00909.
List of chemicals used in this study; SUVA254 and spectral slope calculations from DOM absorbance measurements; kinetic data for DOM-eaq– bimolecular rate constant determination; MnQ calculations; SRNOM II irradiation in UV/sulfite system; and measured DOC concentration during 24 h experiment (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Aiken G. R.; McKnight D. M.; Wershaw R. L.; MacCarthy P.. Humic Substances in Soil, Sediment, and Water; John Wiley & Sons, Inc., 1985. [Google Scholar]
- Ulliman S. L.; Miklos D. B.; Hübner U.; Drewes J. E.; Linden K. G. Improving UV/H2O2 Performance Following Tertiary Treatment of Municipal Wastewater. Environ. Sci.: Water Res. Technol. 2018, 4, 1321–1330. 10.1039/C8EW00233A. [DOI] [Google Scholar]
- Rosenfeldt E. J.; Linden K. G.; Canonica S.; von Gunten U. Comparison of the Efficiency of ·OH Radical Formation During Ozonation and the Advanced Oxidation Processes O3/H2O2 and UV/H2O2. Water Res. 2006, 40, 3695–3704. 10.1016/j.watres.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Li X.; Fang J.; Liu G.; Zhang S.; Pan B.; Ma J. Kinetics and Efficiency of the Hydrated Electron-induced Dehalogenation by the Sulfite/UV Process. Water Res. 2014, 62, 220–228. 10.1016/j.watres.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu H.; Shan C.; Zhang W.; Pan B. A Novel Combined Process for Efficient Removal of Se(VI) from Sulfate-rich Water: Sulfite/UV/Fe(III) Coagulation. Chemosphere 2018, 211, 867–874. 10.1016/j.chemosphere.2018.07.159. [DOI] [PubMed] [Google Scholar]
- Ren Z.; Bergmann U.; Leiviska T. Reductive Degradation of Perfluorooctanoic Acid in Complex Water Matrices by Using the UV/sulfite Process. Water Res. 2021, 205, 117676 10.1016/j.watres.2021.117676. [DOI] [PubMed] [Google Scholar]
- Westerhoff P.; Mezyk S. P.; Cooper W. J.; Minakata D. Electron Pulse Radiolysis Determination of Hydroxyl Radical Rate Constants with Suwannee River Fulvic Acid and Other Dissolved Organic Matter Isolates. Environ. Sci. Technol. 2007, 41, 4640–4646. 10.1021/es062529n. [DOI] [PubMed] [Google Scholar]
- McKay G.; Kleinman J. L.; Johnston K. M.; Dong M. M.; Rosario-Ortiz F. L.; Mezyk S. P. Kinetics of the Reaction Between the Hydroxyl Radical and Organic Matter Standards from the International Humic Substance Society. J. Soils Sediments 2014, 14, 298–304. 10.1007/s11368-013-0697-z. [DOI] [Google Scholar]
- Lei X.; Lei Y.; Guan J.; Westerhoff P.; Yang X. Kinetics and Transformations of Diverse Dissolved Organic Matter Fractions with Sulfate Radicals. Environ. Sci. Technol. 2022, 56, 4457–4466. 10.1021/acs.est.1c08388. [DOI] [PubMed] [Google Scholar]
- Yan S.; Liu Y.; Lian L.; Li R.; Ma J.; Zhou H.; Song W. Photochemical Formation of Carbonate Radical and its Reaction with Dissolved Organic Matters. Water Res. 2019, 161, 288–296. 10.1016/j.watres.2019.06.002. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Lei X.; Westerhoff P.; Zhang X.; Yang X. Reactivity of Chlorine Radicals (Cl• and Cl2•-) with Dissolved Organic Matter and the Formation of Chlorinated Byproducts. Environ. Sci. Technol. 2021, 55, 689–699. 10.1021/acs.est.0c05596. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Lei X.; Westerhoff P.; Tong X.; Ren J.; Zhou Y.; Cheng S.; Ouyang G.; Yang X. Bromine Radical (Br• and Br2•-) Reactivity with Dissolved Organic Matter and Brominated Organic Byproduct Formation. Environ. Sci. Technol. 2022, 56, 5189–5199. 10.1021/acs.est.2c00549. [DOI] [PubMed] [Google Scholar]
- Li X.; Ma J.; Liu G.; Fang J.; Yue S.; Guan Y.; Chen L.; Liu X. Efficient Reductive Dechlorination of Monochloroacetic Acid by Sulfite/UV Process. Environ. Sci. Technol. 2012, 46, 7342–7349. 10.1021/es3008535. [DOI] [PubMed] [Google Scholar]
- Xie B.; Li X.; Huang X.; Xu Z.; Zhang W.; Pan B. Enhanced Debromination of 4-Bromophenol by the UV/Sulfite Process: Efficiency and Mechanism. J. Environ. Sci. 2017, 54, 231–238. 10.1016/j.jes.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Wang T.; Yu S.; Yi P.; Li L. Influence of UV Lamp, Sulfur(IV) Concentration, and pH on Bromate Degradation in UV/Sulfite Systems: Mechanisms and Applications. Water Res. 2017, 111, 288–296. 10.1016/j.watres.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (·OH/·O–) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Botlaguduru V. S. V.; Batchelor B.; Abdel-Wahab A. Application of UV–Sulfite Advanced Reduction Process to Bromate Removal. J. Water Process Eng. 2015, 5, 76–82. 10.1016/j.jwpe.2015.01.001. [DOI] [Google Scholar]
- Liu X.; Wang L.; Sun Z.; Shao Y.; Yu T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts 2017, 7, 131 10.3390/catal7050131. [DOI] [Google Scholar]
- Nawaz S.; Shah N. S.; Khan J. A.; Sayed M.; Al-Muhtaseb A. a. H.; Andersen H. R.; Muhammad N.; Murtaza B.; Khan H. M. Removal Efficiency and Economic Cost Comparison of Hydrated Electron-mediated Reductive Pathways for Treatment of Bromate. Chem. Eng. J. 2017, 320, 523–531. 10.1016/j.cej.2017.03.011. [DOI] [Google Scholar]
- Jung B.; Nicola R.; Batchelor B.; Abdel-Wahab A. Effect of Low- and Medium-pressure Hg UV Irradiation on Bromate Removal in Advanced Reduction Process. Chemosphere 2014, 117, 663–672. 10.1016/j.chemosphere.2014.09.086. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Yu S.; Li L.; Wang T.; Liao X.; Ye Y. An Overview of Advanced Reduction Processes for Bromate Removal from Drinking Water: Reducing Agents, Activation Methods, Applications and Mechanisms. J. Hazard. Mater. 2017, 324, 230–240. 10.1016/j.jhazmat.2016.10.053. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Yu S.; Li L.; Zhang Y.; Yi P. Degradation of Bromate by Fe(II)Ti(IV) Layered Double Hydroxides Nanoparticles Under Ultraviolet Light. Water Res. 2019, 150, 310–320. 10.1016/j.watres.2018.11.067. [DOI] [PubMed] [Google Scholar]
- Yu K.; Li X.; Chen L.; Fang J.; Chen H.; Li Q.; Chi N.; Ma J. Mechanism and Efficiency of Contaminant Reduction by Hydrated Electron in the Sulfite/Iodide/UV Process. Water Res. 2018, 129, 357–364. 10.1016/j.watres.2017.11.030. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhong J.; Fang L.; Wang L.; Ye M.; Shao Y.; Li J.; Zhang T. Trichloroacetic Acid Reduction by an Advanced Reduction Process Based on Carboxyl Anion Radical. Chem. Eng. J. 2016, 303, 56–63. 10.1016/j.cej.2016.05.130. [DOI] [Google Scholar]
- Liu X.; Yoon S.; Batchelor B.; Abdel-Wahab A. Degradation of Vinyl Chloride (VC) by the Sulfite/UV Advanced Reduction Process (ARP): Effects of Process Variables and a Kinetic Model. Sci. Total Environ. 2013, 454-455, 578–583. 10.1016/j.scitotenv.2013.03.060. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yoon S.; Batchelor B.; Abdel-Wahab A. Photochemical Degradation of Vinyl Chloride with an Advanced Reduction Process (ARP) – Effects of Reagents and pH. Chem. Eng. J. 2013, 215–216, 868–875. 10.1016/j.cej.2012.11.086. [DOI] [PubMed] [Google Scholar]
- Wentworth W. E.; Becker R. S.; Tung R. Thermal Electron Attachment to Some Aliphatic and Aromatic Chloro, Bromo, and Iodo Derivatives. J. Phys. Chem. A 1967, 71, 1652–1665. 10.1021/j100865a017. [DOI] [Google Scholar]
- Sun C.; Chang W.; Ma W.; Chen C.; Zhao J. Photoreductive Debromination of Decabromodiphenyl Ethers in the Presence of Carboxylates Under Visible Light Irradiation. Environ. Sci. Technol. 2013, 47, 2370–2377. 10.1021/es3045604. [DOI] [PubMed] [Google Scholar]
- Yu X.; Cabooter D.; Dewil R. Effects of Process Variables and Kinetics on the Degradation of 2,4-Dichlorophenol Using Advanced Reduction Processes (ARP). J. Hazard. Mater. 2018, 357, 81–88. 10.1016/j.jhazmat.2018.05.049. [DOI] [PubMed] [Google Scholar]
- Yazdanbakhsh A.; Eslami A.; Moussavi G.; Rafiee M.; Sheikhmohammadi A. Photo-assisted Degradation of 2, 4, 6-Trichlorophenol by an Advanced Reduction Process Based on Sulfite Anion Radical: Degradation, Dechlorination and Mineralization. Chemosphere 2018, 191, 156–165. 10.1016/j.chemosphere.2017.10.023. [DOI] [PubMed] [Google Scholar]
- Shoute L. C. T.; Mittal J. P.; Neta P. Reduction and Defluorination of Pentafluorophenol in Aqueous Solutions. J. Phys. Chem. B 1996, 100, 3016–3019. 10.1021/jp9513374. [DOI] [Google Scholar]
- Qu Y.; Zhang C.; Li F.; Chen J.; Zhou Q. Photo-reductive Defluorination of Perfluorooctanoic Acid in Water. Water Res. 2010, 44, 2939–2947. 10.1016/j.watres.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Song Z.; Tang H.; Wang N.; Zhu L. Reductive Defluorination of Perfluorooctanoic Acid by Hydrated Electrons in a Sulfite-mediated UV Photochemical System. J. Hazard. Mater. 2013, 262, 332–338. 10.1016/j.jhazmat.2013.08.059. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Liu T.; Wang H.; Han H.; Dong W. Hydrated Electron Based Decomposition of Perfluorooctane Sulfonate (PFOS) in the VUV/Sulfite System. Sci. Total Environ. 2017, 607-608, 541–548. 10.1016/j.scitotenv.2017.06.197. [DOI] [PubMed] [Google Scholar]
- Tian H.; Gao J.; Li H.; Boyd S. A.; Gu C. Complete Defluorination of Perfluorinated Compounds by Hydrated Electrons Generated from 3-Indole-Acetic-Acid in Organomodified Montmorillonite. Sci. Rep. 2016, 6, 32949 10.1038/srep32949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H.; Gu C. Effects of Different factors on Photodefluorination of Perfluorinated Compounds by Hydrated Electrons in Organo-montmorillonite System. Chemosphere 2018, 191, 280–287. 10.1016/j.chemosphere.2017.10.074. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Mi N.; Li C.; Teng Y.; Chen Y.; Gu C. Effects of Different Variables on Photodestruction of Perfluorooctanoic Acid in Self-assembled Micelle System. Sci. Total Environ. 2020, 742, 140438 10.1016/j.scitotenv.2020.140438. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Li C.; Gao J.; Dong H.; Chen Y.; Wu B.; Gu C. Efficient Reductive Destruction of Perfluoroalkyl Substances under Self-Assembled Micelle Confinement. Environ. Sci. Technol. 2020, 54, 5178–5185. 10.1021/acs.est.9b06599. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Teng Y.; Mi N.; Jin X.; Yang D.; Wang C.; Wu B.; Ren H.; Zeng G.; Gu C. Highly Efficient Hydrated Electron Utilization and Reductive Destruction of Perfluoroalkyl Substances Induced by Intermolecular Interaction. Environ. Sci. Technol. 2021, 55, 3996–4006. 10.1021/acs.est.0c07927. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Li Z.; Wong B. M.; Men Y.; Liu J. Defluorination of Per- and Polyfluoroalkyl Substances (PFASs) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management. Environ. Sci. Technol. 2019, 53, 3718–3728. 10.1021/acs.est.8b06648. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Kwon H.; Li Z.; Wong B. M.; Men Y.; Liu J. Degradation of Perfluoroalkyl Ether Carboxylic Acids with Hydrated Electrons: Structure-Reactivity Relationships and Environmental Implications. Environ. Sci. Technol. 2020, 54, 2489–2499. 10.1021/acs.est.9b05869. [DOI] [PubMed] [Google Scholar]
- Liu C. J.; McKay G.; Jiang D.; Tenorio R.; Cath J. T.; Amador C.; Murray C. C.; Brown J. B.; Wright H. B.; Schaefer C.; Higgins C. P.; Bellona C.; Strathmann T. J. Pilot-Scale Field Demonstration of a Hybrid Nanofiltration and UV-Sulfite Treatment Train for Groundwater Contaminated by Per- and Polyfluoroalkyl Substances (PFASs). Water Res. 2021, 205, 117677 10.1016/j.watres.2021.117677. [DOI] [PubMed] [Google Scholar]
- Cui J.; Deng Y. Hydrated Electron Degradation of PFOA Laden on Ion-Exchange Resins in the Presence of Natural Organic Matter. ACS ES&T Eng. 2022, 86–93. 10.1021/acsestengg.2c00253. [DOI] [Google Scholar]
- Duan Y.; Batchelor B. Impacts of Natural Organic Matter on Perchlorate Removal by an Advanced Reduction Process. J. Environ. Sci. Health, Part A 2014, 49, 731–740. 10.1080/10934529.2014.865462. [DOI] [PubMed] [Google Scholar]
- Fennell B. D.; Mezyk S. P.; McKay G. Critical Review of UV-Advanced Reduction Processes for the Treatment of Chemical Contaminants in Water. ACS Environ. Au 2022, 2, 178–205. 10.1021/acsenvironau.1c00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- What are Humic Substances?. https://humic-substances.org/. (accessed October 5, 2021).
- Fennell B. D.; Odorisio A.; McKay G. Quantifying Hydrated Electron Transformation Kinetics in UV-Advanced Reduction Processes Using the Re-,UV Method. Environ. Sci. Technol. 2022, 56, 10329–10338. 10.1021/acs.est.2c02003. [DOI] [PubMed] [Google Scholar]
- Humphrey R. E.; Ward M. H.; Hinze W. Spectrophotometric Determination of Sulfite with 4,4′-Dithiodipyridine and 5,5′-Dithiobis-(2-Nitrobenzoic Acid). Anal. Chem. 1970, 42, 698–702. 10.1021/ac60289a021. [DOI] [Google Scholar]
- Whitham K.; Lyons S.; Miller R.; Nett D.; Treas P.; Zante A.; Fessenden R. W.; Thomas M. D.; Wang Y. In Linear Accelerator for Radiation Chemistry Research at Notre Dame, Proceedings Particle Accelerator Conference; IEEE: Dallas, TX, USA, 1995; pp 131–133.
- Jeong J.; Song W.; Cooper W. J.; Jung J.; Greaves J. Degradation of Tetracycline Antibiotics: Mechanisms and Kinetic Studies for Advanced Oxidation/Reduction Processes. Chemosphere 2010, 78, 533–540. 10.1016/j.chemosphere.2009.11.024. [DOI] [PubMed] [Google Scholar]
- Keen O. S.; McKay G.; Mezyk S. P.; Linden K. G.; Rosario-Ortiz F. L. Identifying the Factors that Influence the Reactivity of Effluent Organic Matter with Hydroxyl Radicals. Water Res. 2014, 50, 408–419. 10.1016/j.watres.2013.10.049. [DOI] [PubMed] [Google Scholar]
- Dong M. M.; Mezyk S. P.; Rosario-Ortiz F. L. Reactivity of Effluent Organic Matter (EfOM) with Hyroxyl Radical as a Function of Molecular Weight. Environ. Sci. Technol. 2010, 44, 5714–5720. 10.1021/es1004736. [DOI] [PubMed] [Google Scholar]
- Rickman K. A.; Mezyk S. P. Kinetics and Mechanisms of Sulfate Radical Oxidation of β-lactam Antibiotics in Water. Chemosphere 2010, 81, 359–365. 10.1016/j.chemosphere.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Cole S. K.; Cooper W. J.; Fox R. V.; Gardinali P. R.; Mezyk S. P.; Mincher B. J.; O’Shea K. E. Free Radical Chemistry of Disinfection Byproducts. 2. Rate Constants and Degradation Mechanisms of Trichloronitromethane (Chloropicrin). Environ. Sci. Technol. 2007, 41, 863–869. 10.1021/es061410b. [DOI] [PubMed] [Google Scholar]
- Elemental Compositions and Stable Isotopic Ratios of IHSS Samples. https://humic-substances.org/elemental-compositions-and-stable-isotopic-ratios-of-ihss-samples/. (accessed March 30, 2022).
- Maza W. A.; Breslin V. M.; Plymale N. T.; DeSario P. A.; Epshteyn A.; Owrutsky J. C.; Pate B. B. Nanosecond Transient Absorption Studies of the pH-dependent Hydrated Electron Quenching by HSO3-. Photochem. Photobiol. Sci. 2019, 18, 1526–1532. 10.1039/c9pp00063a. [DOI] [PubMed] [Google Scholar]
- Cui J.; Gao P.; Deng Y. Destruction of Per- and Polyfluoroalkyl Substances (PFAS) with Advanced Reduction Processes (ARPs): A Critical Review. Environ. Sci. Technol. 2020, 54, 3752–3766. 10.1021/acs.est.9b05565. [DOI] [PubMed] [Google Scholar]
- Jin S.; Mofidi A. A.; Linden K. G. Polychromatic UV Fluence Measurement Using Chemical Actinometry, Biodosimetry, and Mathematical Techniques. J. Environ. Eng. 2006, 132, 831–841. 10.1061/(ASCE)0733-9372(2006)132:8(831). [DOI] [Google Scholar]
- Beltran F. J.; Ovejero G.; Garcia-Araya J. F.; Rivas J. Oxidation of Polynuclear Aromatic Hydrocarbons in Water. 2. UV Radiation and Ozonation in the Presence of UV Radiation. Ind. Eng. Chem. Res. 1995, 34, 1607–1615. 10.1021/ie00044a013. [DOI] [Google Scholar]
- Crittenden J. C.; Hu S.; Hand D. W.; Green S. A. A Kinetic Model for H2O2/UV Process In a Completely Mixed Batch Reactor. Water Res. 1999, 33, 2315–2328. 10.1016/S0043-1354(98)00448-5. [DOI] [Google Scholar]
- Ritchie J. D.; Perdue E. M. Proton-binding Study of Standard and Reference Fulvic Acids, Humic Acids, and Natural Organic Matter. Geochim. Cosmochim. Acta 2003, 67, 85–96. 10.1016/S0016-7037(02)01044-X. [DOI] [Google Scholar]
- Avena M. J.; Vermeer A. W. P.; Koopal L. K. Volume and Structure of Humic Acids Studies by Viscometry pH and Electrolyte Concentration Effects. Colloids Surf., A 1999, 151, 213–224. 10.1016/S0927-7757(98)00504-4. [DOI] [Google Scholar]
- Green S. A.; Morel F. M. M.; Blough N. V. Investigation of Electrostatic Properties of Humic Substances by Fluorescence Quenching. Environ. Sci. Technol. 1992, 26, 294–302. 10.1021/es00026a008. [DOI] [Google Scholar]
- Li H.; McKay G. Relationships between the Physicochemical Properties of Dissolved Organic Matter and Its Reaction with Sodium Borohydride. Environ. Sci. Technol. 2021, 55, 10843–10851. 10.1021/acs.est.1c01973. [DOI] [PubMed] [Google Scholar]
- Acidic Functional Groups of IHSS Samples. https://humic-substances.org/acidic-functional-groups-of-ihss-samples/. (accessed March 30, 2022).
- 13C NMR Estimates of Carbon Distribution. https://humic-substances.org/13c-nmr-estimates-of-carbon-distribution-in-ihss-samples/. (accessed March 30, 2022).
- NDRL/NIST Solution Kinetics Database on the Web. 2002 ed.; National Institute of Standards and Technology: Notre Dame Radiation Laboratory, 2023.
- Aeschbacher M.; Graf C.; Schwarzenbach R. P.; Sander M. Antioxidant Properties of Humic Substances. Environ. Sci. Technol. 2012, 46, 4916–4925. 10.1021/es300039h. [DOI] [PubMed] [Google Scholar]
- Rosario-Ortiz F. L.; Mezyk S.; Doud D. F. R.; Snyder S. A. Quantitative Correlation of Absolute Hydroxyl Radical Rate Constants with Non-Isolated Effluent Organic Matter Bulk Properties in Water. Environ. Sci. Technol. 2008, 42, 5924–5930. 10.1021/es800349b. [DOI] [PubMed] [Google Scholar]
- Alalm M. G.; Boffito D. C. Mechanisms and Pathways of PFAS Degradation by Advanced Oxidation and Reduction Processes: A Critical Review. Chem. Eng. J. 2022, 450, 138352 10.1016/j.cej.2022.138352. [DOI] [Google Scholar]
- Liu F.; Guan X.; Xiao F. Photodegradation of Per- and Polyfluoroalkyl Substances in Water: A Review of Fundamentals and Applications. J. Hazard. Mater. 2022, 439, 129580 10.1016/j.jhazmat.2022.129580. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Bentel M. J.; Yu Y.; Ren C.; Gao J.; Pulikkal V. F.; Sun M.; Men Y.; Liu J. Near-Quantitative Defluorination of Perfluorinated and Fluorotelomer Carboxylates and Sulfonates with Integrated Oxidation and Reduction. Environ. Sci. Technol. 2021, 55, 7052–7062. 10.1021/acs.est.1c00353. [DOI] [PubMed] [Google Scholar]
- Vinge S. L.; Shaheen S. W.; Sharpless C. M.; Linden K. G. Nitrate with Benefits: Optimizing Radical Production During UV Water Treatment. Environ. Sci.: Water Res. Technol. 2020, 6, 1163–1175. 10.1039/C9EW01138B. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.