

ABSTRACT
Staphylococcus aureus is a gram-positive pathogen that poses a major health concern, in part due to its large array of virulence factors that allow infection and evasion of the immune system. One of these virulence factors is the bicomponent pore-forming leukocidin LukAB. The regulation of lukAB expression is not completely understood, especially in the presence of immune cells such as human polymorphonuclear neutrophils (hPMNs). Here, we screened for transcriptional regulators of lukAB during the infection of primary hPMNs. We uncovered that PerR, a peroxide sensor, is vital for hPMN-mediated induction of lukAB and that PerR upregulates cytotoxicity during the infection of hPMNs. Exposure of S. aureus to hydrogen peroxide (H2O2) alone also results in increased lukAB promoter activity, a phenotype dependent on PerR. Collectively, our data suggest that S. aureus uses PerR to sense the H2O2 produced by hPMNs to stimulate the expression of lukAB, allowing the bacteria to withstand these critical innate immune cells.
IMPORTANCE
Staphylococcus aureus utilizes a diverse set of virulence factors, such as leukocidins, to subvert human neutrophils, but how these toxins are regulated is incompletely defined. Here, we identified the peroxide-sensitive repressor, PerR, as a required protein involved in the induction of lukAB in the presence of primary human neutrophils, a phenotype directly linked to the ability of PerR to sense H2O2. Thus, we show that S. aureus coordinates sensing and resistance to oxidative stress with toxin production to promote pathogen survival.
KEYWORDS: MRSA, cytotoxins, PerR, neutrophils, LukAB, pore-forming toxins, Staphylococcus aureus
INTRODUCTION
Staphylococcus aureus (S. aureus) can cause multiple illnesses including skin infections, pneumonia, and bacteremia (1). With the rise of antibiotic resistance, treating infections such as methicillin-resistant S. aureus (MRSA) has become increasingly difficult. S. aureus success as a pathogen is in part because it possesses a large collection of virulence factors (2, 3). These include the bicomponent pore-forming leukocidins, which target and rupture the membranes of immune cells that the host requires for protection from invasive pathogens (4). S. aureus strains associated with human infections, including community-associated MRSA strains from the USA300 lineage, produce up to five leukocidins that target human cells: leukocidin AB (LukAB, also known as LukGH), Panton–Valentine leukocidin (PVL, also known as LukSF-PV), leukocidin ED (LukED), gamma hemolysin HlgAB, and gamma hemolysin HlgCB (5). These toxins preferentially target leukocytes, using specific proteinaceous receptors to do so (6–9). S. aureus differentially activates the expression of leukocidin loci (10, 11), which is hypothesized to be important to combat the host-mediated attack during infection and promote pathogen survival. Most of the leukocidins share 60%–80% sequence similarity, aside from LukAB, which only shares 30%–40% sequence similarity to the other toxins (12, 13). Interestingly, among the leukocidins, the promoter activity of lukAB has been shown to be the most active during tissue culture infection of human polymorphonuclear neutrophils (hPMNs) (10). Additionally, LukAB is the predominant toxin that lyses hPMNs in these tissue culture models (9, 10, 12–14), both through intracellular and extracellular mechanisms, which aids in S. aureus escape of the immune system (10). These findings together with the observation that the lukAB operon is found in over 99% of S. aureus isolates have positioned LukAB as an attractive vaccine candidate (15, 16).
The network involved in the regulation of toxins in S. aureus includes two-component systems that activate expression, such as the S. aureus exoprotein (Sae) system and the accessory gene regulator (Agr) system. Other transcription factors such as the repressor of toxins (Rot) are also involved in the regulation of leukocidins (17, 18). The SaeRS system is a major activator of all the toxins (5, 19–21), but it is still unknown why lukAB specifically is more active in the presence of hPMNs compared to the other leukocidins. We hypothesized that these and/or other uncharacterized regulators may be involved in the upregulation of lukAB during infection of hPMNs.
This study aimed to identify transcriptional regulators involved in the hPMN-mediated activation of lukAB. We performed a high-throughput screen that utilized a luminescent transcriptional reporter to measure lukAB promoter activity in transposon mutants. The work revealed that the inactivation of perR reduces lukAB promoter activity and S. aureus-mediated cytotoxicity in neutrophils. PerR is a peroxide sensor that represses many genes involved in iron storage and oxidative stress response (22, 23). Our data demonstrate that H2O2, which is released by hPMNs (24, 25), induces lukAB through a PerR-mediated mechanism. Collectively, these data suggest that S. aureus uses PerR to upregulate the lukAB promoter when the bacterium encounters hPMNs, thus dually coordinating the protection against oxidative damage and a counterattack to kill hPMNs.
RESULTS
Identification of transcriptional regulators that alter promoter activity of lukAB during infection of hPMNs
We hypothesize that the lukAB promoter (PlukAB) may be regulated by various transcriptional regulators in different environmental conditions. To uncover transcription factors involved in the regulation of PlukAB, we utilized a sublibrary of the Nebraska Transposon Mutant Library (26) described by Balasubramanian et al. (11), which consists of strains with mutations in genes likely to be involved in transcription and translation. This mutant library was constructed in JE2, a S. aureus strain in the USA300 background (26). USA300 strains are associated with the current epidemic of community-associated MRSA infections (27). The regulatory sublibrary was transduced with a plasmid containing the lukAB promoter driving expression of the click beetle red luciferase (CBR-luc) (28). The reporter sublibrary was then subcultured for 3 hours, followed by a 3-hour infection of hPMNs. D-Luciferin was added, and the promoter activity was measured (Fig. 1A). We included two internal controls, rot::bursa (increased promoter activity) (29) and saeR::bursa (no to low promoter activity) (19). Indeed, we observed increased lukAB promoter activity in the absence of rot and decreased lukAB promoter activity in the absence of saeR, validating the screen. Altogether, the screen uncovered 72 mutants that had at least 1.75-fold less PlukAB activity compared to wild-type JE2, and these gene products were categorized as potential activators of PlukAB (Fig. 1B; see Table S1). Conversely, we identified 51 mutants with at least 1.75-fold more PlukAB activity than wild-type JE2, suggesting that they contained mutations in gene products that could act as repressors of PlukAB (Fig. 1C).
Fig 1.

Mutations in non-essential genes affect the regulation of the lukAB promoter. (A) A luminescence screen was conducted on a JE2 transposon library containing a reporter plasmid where the lukAB promoter (PlukAB) was fused to the luciferase gene. The library is composed of 250 mutants with mutations in non-essential genes that may have regulatory roles. The promoter activity was measured by the luminescence of the JE2 transposon library in RPMI + HEPES + 5% normal human serum (NHS) with hPMNs after 3 hours of infection. Created with BioRender.com. (B) Results show potential activators of PlukAB, which have an average luminescence less than wild-type JE2 (n = 4 donors; six independent colonies for controls) at a multiplicity of infection (MOI) of 8. Mutant strains shown have a minimum of a 1.75-fold difference compared to wild-type JE2. Error bars indicate the standard error of the mean (SEM). (C) Results show potential repressors of PlukAB, which have an average luminescence greater than wild-type JE2 (n = 4 donors; six independent colonies for controls, MOI = 8). Mutant strains shown have a minimum of a 1.75-fold difference compared to wild-type JE2. Error bars indicate SEM.
Activators differentially regulate lukAB in the presence of hPMNs
As we were interested in discovering transcriptional regulators that played a role in the upregulation of PlukAB in the presence of hPMNs, we analyzed the potential activators of PlukAB further. We selected regulators that were significantly different from wild-type JE2 and possessed features indicative of the direct regulation of downstream genes, such as genes that encoded for proteins that contained a helix-turn-helix motif or were part of two-component systems. These selection criteria narrowed our screen to 15 potential activators (Fig. 2). Utilizing the experimental design described in Fig. 1, we tested the activation of PlukAB luminescence by the potential activators in the presence and absence of hPMNs. In the secondary screen, only some of the mutants continued to show attenuated PlukAB activity, suggesting that the luciferase reporter is best suited to detect strains with exceedingly impacted gene regulation (Fig. 2A). In contrast, when the assay was repeated in media alone, the luminescence of most of these mutants was greater than wild-type JE2 (Fig. 2B). This suggests that the attenuation of PlukAB activity is dependent on the presence of hPMNs for these strains. We infer that the general luminescence is greater for all strains in media alone because S. aureus is phagocytosed during infection, which decreases the ability for D-luciferin to diffuse into the bacteria. Therefore, the difference in log2 fold change of the luminescence for the two conditions was compared (Fig. 2C). Mutants with a negative log2 fold change had a decreased activation of PlukAB compared to wild-type JE2, and mutants with a positive log2 fold change had an increased PlukAB activity. All the mutants tested had a shift in their regulation of PlukAB between the two conditions. Specifically, some mutants showed increased PlukAB activation in media alone and decreased activation in the presence of hPMNs (Fig. 2C). Therefore, these regulators seem to act as repressors in the absence of hPMNs and as activators in the presence of hPMNs. This suggests a switch in the regulation of lukAB that is dependent on the presence of hPMNs.
Fig 2.

Regulation of lukAB promoter activity in the presence or absence of hPMNs. (A) PlukAB luminescence values of selected PlukAB activators in the presence of hPMNs in media containing RPMI + HEPES + 5% NHS. The results shown are from two independent experiments each performed with three colonies of each strain repeated in four blood donors (n = 12, MOI = 8). The dotted line represents wild-type JE2. Statistical analysis was performed using one-way ANOVA with multiple comparisons to determine the statistical significance of mutants compared to wild-type JE2. Error bars indicate SEM. (B) Luminescence values of selected PlukAB activators grown as in panel (A) but in the absence of hPMNs. The results shown are from two independent experiments each performed with three colonies of each strain (n = 6). The dotted line represents wild-type JE2. Statistical analysis was performed using one-way ANOVA with multiple comparisons to determine the statistical significance of mutants compared to wild-type JE2. Error bars indicate SEM. (C) Log2 fold change of luminescence of mutants compared to wild-type JE2 in the presence or absence of hPMNs (n = 6–12). Statistical analysis was performed using unpaired t-tests with Welch’s correction to compare the log2 fold change of luminescence in the two conditions for each mutant strain. Log2 fold change of luminescence was used to compare the two conditions to account for the reduction in raw luminescence values because of phagocytosed bacteria, which reduces the efficacy of D-luciferin to cross the bacterial membrane. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. ns, not significant.
PerR regulates lukAB promoter activity and cytolytic activity in the presence of hPMNs
LukAB is responsible for the lysis of hPMNs in tissue culture models of S. aureus infection (10, 13). Thus, in addition to promoter activity, we analyzed the ability of the 15 potential activators to enhance the lysis of hPMNs. We observed that all the selected mutants were deficient in hPMN killing (Fig. 3A).
Fig 3.

Decreased hPMN cytotoxicity of potential lukAB activator mutants. (A) Cytotoxicity values of selected PlukAB activators. Cytotoxicity was measured as percent lactate dehydrogenase (LDH) release from lysed hPMNs. The results shown are from two independent experiments each performed with three colonies of each strain repeated in four blood donors (n = 12, MOI = 8). The dotted line represents wild-type JE2. Statistical analysis was performed using one-way ANOVA with multiple comparisons to determine the statistical significance of mutants compared to wild-type JE2. Error bars indicate SEM. (B) Cytotoxicity of strains wild-type JE2, perR::bursa, ΔperR, and the complement strain (ΔperR::perR). The values are averages of eight independent experiments with two colonies of each strain repeated in 23 blood donors (n = 18–46, MOI = 18). The increase in MOI was used to induce increased cytotoxicity. Statistical analysis was performed using one-way ANOVA with multiple comparisons to determine the statistical significance of mutants compared to wild-type JE2. Error bars indicate SEM. (C) Cytotoxicity of Newman wild-type and ΔperR. The results shown are averages from three independent experiments with two colonies of each strain repeated in nine blood donors (n = 18, MOI = 18). Statistical analysis was performed using an unpaired t-test with Welch’s correction to determine the statistical significance of mutants compared to wild-type JE2. Error bars indicate SEM. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
We then analyzed the transposon mutants for their ability to lyse hPMNs and their PlukAB activity in the presence of hPMNs. We identified eight mutants that had attenuated cytotoxicity and an average negative log2 fold change of PlukAB activity in the presence of hPMNs (Table 1). Among these, we observed that saeR::bursa and saeS::bursa, the response regulator and sensor histidine kinase of the SaeRS two-component system, were both hits as activators in our analysis, further validating the findings of our screen. The two other strains that displayed a pronounced attenuation of cytotoxicity and decreased luminescence in the presence of hPMNs were perR::bursa and arlR::bursa (Table 1).
TABLE 1.
List of mutants with low lukAB promoter activity and cytotoxicity
| Locus tag | NE number | Gene description | Gene name | Luciferase with hPMNs ± SEM | P-value hPMNs vs media only | LDH ± SEM | P-value LDH | DNA-binding domain |
|---|---|---|---|---|---|---|---|---|
| SAUSA300_0691 | NE1622 | DNA-binding response regulator SaeR | saeR | 25,790.9 ± 4,072.3 | <0.0001 | 3.7 ± 1.2 | <0.0001 | Helix-turn-helix |
| SAUSA300_1842 | NE665 | Transcriptional regulator, Fur family (repressor) | perR | 2,790,645.6 ± 509,781.5 | <0.0001 | 1.4 ± 0.8 | <0.0001 | Helix-turn-helix |
| SAUSA300_2558 | NE1116 | Nisin susceptibility-associated sensor histidine kinase | nsaS | 6,761,540.4 ± 947,821.9 | <0.0001 | 8.6 ± 2.6 | <0.0001 | |
| SAUSA300_0690 | NE1296 | Sensor histidine kinase SaeS | saeS | 62,617.4 ± 16,194.2 | 0.0001 | 3.9 ± 1.7 | <0.0001 | |
| SAUSA300_1308 | NE1684 | DNA-binding response regulator ArlR | arlR | 2,639,434.3 ± 456,560.1 | 0.0004 | 5.8 ± 1.8 | <0.0001 | Helix-turn-helix |
| SAUSA300_2599 | NE1132 | Intercellular adhesion transcription regulator (biofilm operon icaADBC repressor IcaR) | icaR | 7,348,020.1 ± 1,201,711.2 | 0.0005 | 8.3 ± 1.7 | <0.0001 | Helix-turn-helix |
| SAUSA300_0503 | NE354 | Transcriptional regulator, gntR family protein (PLP-dependent aminotransferase family protein) | pdxR | 8,444,604.6 ± 1,142,922.4 | 0.0052 | 7.7 ± 2.3 | <0.0001 | Helix-turn-helix |
| SAUSA300_1307 | NE1183 | Sensor histidine kinase protein ArlS | arlS | 9,235,594.9 ± 1,771,937.7 | 0.0018 | 12.4 ± 2.8 | 0.0004 | Helix-turn-helix |
Previous studies have established the importance of the ArlRS two-component system for S. aureus virulence (30–35), which further validated our study.
PerR is the main peroxide sensor in gram-positive bacteria such as Bacillus subtilis and S. aureus (36, 37) and is necessary for S. aureus pathogenesis in various animal models including a murine skin abscess, Caenorhabditis elegans, fruit fly, and zebrafish (22, 23, 38–40). PerR is an oxidation-sensing transcriptional regulator, mainly functioning in iron storage and oxidative stress resistance pathways (22, 36, 41–50). The PerR regulon includes katA (catalase), ahpCF (alkyl hydroperoxide reductase), bcp (bacterioferritin comigratory protein), trxB (thioredoxin-disulfide reductase), fur (ferric uptake regulator), ftn (ferritin), and mrgA (ferritin-like Dps homolog) (22). PerR is a metal-dependent regulator that binds to Zn2+ in combination with either Fe2+ or Mn2+, and its function is altered depending on the metal to which it is bound (22, 23, 43, 51). PerR senses low levels of H2O2 when bound to Fe2+ in S. aureus resulting in the derepression of its regulon (22, 23, 52).
To further validate the role of PerR in the regulation of S. aureus toxins, we tested the cytotoxicity of isogenic perR deletion and complementation strains. The ΔperR strain was constructed by phage transducing a perR::ermC mutation into JE2. The complement ΔperR::perR strain was made using pIMAY* (53) to replace the erm cassette with a wild-type perR. We observed that both the ΔperR and perR::bursa mutants exhibit attenuated cytotoxicity toward hPMNs (Fig. 3B). Of note, the phenotype was restored to wild-type levels in the complementation strain. We next tested if the deletion of perR resulting in altered cytotoxicity was a USA300-specific phenotype. We tested cytotoxicity in the strain Newman, a methicillin-sensitive S. aureus strain (54), and observed that the ΔperR strain also exhibits decreased cytotoxicity of hPMNs as compared to the wild-type strain (Fig. 3C). Together, these data demonstrate that PerR is required for the full lytic activity of S. aureus when the bacteria are exposed to hPMNs.
Hydrogen peroxide induces lukAB promoter activity
One of the major roles of PerR is sensing H2O2 in the environment (23, 51, 55, 56). hPMNs generate H2O2 by first synthesizing superoxide via NADPH oxidase (NOX2), which then undergoes dismutation to form H2O2 (24, 25) to attack pathogens (46, 57). We next tested if H2O2 alone could induce lukAB promoter activity. Using the luciferase reporter strains described above, we observed that H2O2 indeed induces PlukAB in wild-type JE2 but not in perR::bursa. We also tested the role of SaeR in this H2O2-mediated induction of PlukAB and observed that saeR::bursa, like perR::bursa, exhibited very little induction of lukAB regardless of H2O2 (Fig. 4A). These data suggest that H2O2-mediated PlukAB activity is dependent on both PerR and SaeRS. Of note, the impact of 0.1 mM H2O2 exposure on PlukAB induction was independent of H2O2 antimicrobial activity as we detected no significant difference in colony-forming units between any of the strains and treatments (Fig. 4B). We also observed that H2O2 induces PlukAB in a dose-dependent manner (Fig. 4C). Collectively, these data suggest that PerR plays a role in regulating cytotoxicity through the upregulation of lukAB in response to H2O2 produced by hPMNs.
Fig 4.
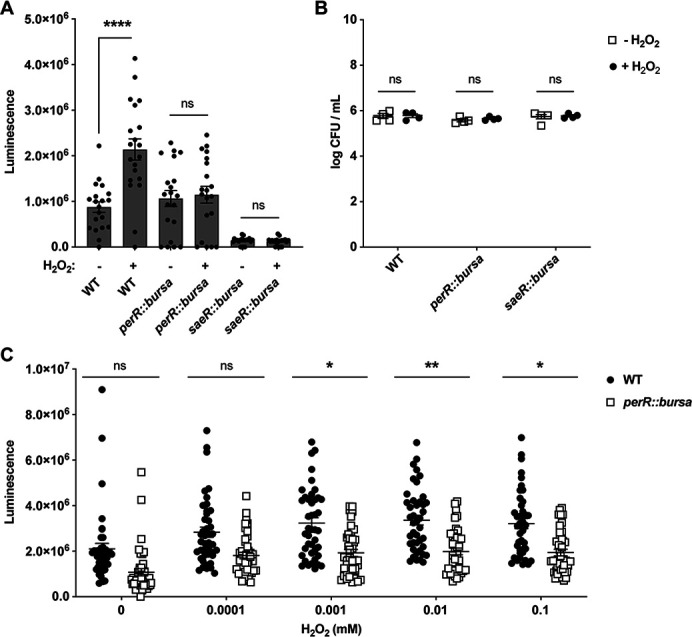
H2O2 treatment increases the promoter activity of lukAB. (A) PlukAB luminescence values after a 1-hour treatment with 0 or 0.1 mM of H2O2 in wild-type JE2, perR::bursa, and saeR::bursa. The results shown are from two independent experiments each performed with 10 colonies of each strain (n = 20). Statistical analysis was performed using unpaired t-tests with Welch’s correction to determine the statistical significance of 0.1 mM H2O2 treatment. Error bars indicate SEM. (B) CFUs per milliliter were calculated by plating the bacteria after exposure to H2O2 for 1 hour. The results shown are from two independent experiments each performed with two colonies for each strain (n = 4). Statistical analysis was performed using unpaired t-tests with Welch’s correction to determine the statistical significance. Error bars indicate SEM. (C) PlukAB luminescence values after treating bacteria with various concentrations of H2O2 for 1 hour. The results shown are from eight independent experiments each performed with six colonies of each strain (n = 48). Statistical analysis was performed on average luminescence per experiment using unpaired t-tests with Welch’s correction to determine the statistical significance of mutants compared to wild-type JE2 at each concentration. Error bars indicate SEM. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
PerR binding sites are found throughout the S. aureus chromosome
To gain insight into how PerR controls the activation of the PlukAB, we performed a bioinformatic analysis where the sequences of the PerR binding site (22, 45), whose consensus is ATTATAATTATTATAAT, were used to query the chromosome of the USA300 strain LAC. Of note, the S. aureus PerR consensus sequence was initially identified in strain 8325-4 (22, 45). For this purpose, the sequence motif scanning software FIMO (58) was used. The hits were mapped to the reference annotation of the LAC genome, and the distance of the putative PerR binding site sequence, with respect to the genes in which it occurred, was calculated. Genes were considered to have a putative PerR binding site if an alignment occurred at most 100 bp upstream of the start codon of the gene or within the coding sequence. Figure 5 summarizes our findings. Figure 5A depicts the sequence motif of the PerR binding sites identified in strain 8325-4, while Fig. 5B shows the alignment positions and scores for the PerR binding site motif. Among the loci containing putative PerR binding sites, we observe that binding sites for ahpC, katA, ftnA, dps, fur, and perR were identified, as expected. Some of these genes possessed more than one predicted PerR binding site. In addition, some interesting hits included SAUSA300_1202 and SAUSA300_1203, which are conserved hypothetical proteins that were not tested in our screen, and SAUSA300_0084 and SAUSA300_1200, which are transcriptional regulators. In our screen, SAUSA300_0084 behaved like an activator of lukAB. SAUSA300_1200 was not tested in our screen. Altogether, these data suggest that PerR may directly regulate many loci in S. aureus and that PerR may impact gene expression by acting directly on regulated operators or indirectly by controlling the expression of other master regulators.
Fig 5.
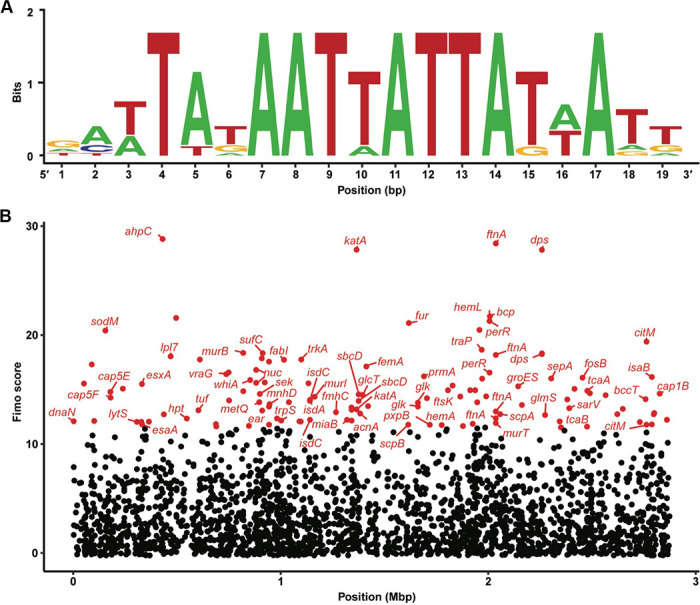
PerR binding site predicted in many potential genes. (A) PerR binding site sequence motif based on binding site sequences from strain S. aureus 8325-4. (B) Genes predicted to have a PerR binding site. Each dot represents a predicted binding site that lies within a gene or at most 100 bp upstream of it. The horizontal axis represents the coordinates in the assembly at which the binding site occurs, while the vertical axis represents the alignment score; that is how closely the predicted binding site resembles the sequence motif in (A). Genes in red have a FIMO alignment score in the upper 5% and are labeled with their symbol, unless undefined in the reference assembly.
DISCUSSION
Neutrophils, a crucial part of the innate immune response, are one of the first immune cells to respond to an infection (59). To better understand S. aureus pathogenesis, we were interested in the regulation of lukAB during infection of hPMNs. LukAB plays a vital role during tissue culture infection of hPMNs (10, 12, 13), and it is a promising vaccine candidate (16). In this study, we conducted a screen of a transposon mutant library to identify potential new regulators of lukAB during infection of hPMNs. We identified PerR, a peroxide regulator, as a key protein for hPMN-mediated induction of PlukAB. Our data illustrate that PerR influences lukAB regulation to increase S. aureus cytotoxicity (Fig. 2 and 3). We observed that lukAB promoter activity is induced by H2O2 (Fig. 4), suggesting that H2O2 released by hPMNs may play a role in the observed regulation (Fig. 6). Collectively, our data show that S. aureus can sense H2O2 via PerR to increase toxin production in response to hPMNs in an attempt to evade these critical innate immune leukocytes.
Fig 6.
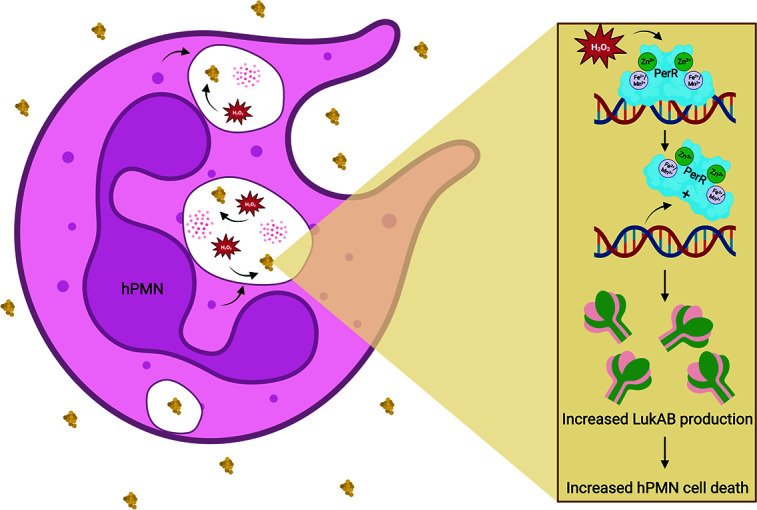
Model of the role of PerR in LukAB-mediated S. aureus virulence. hPMNs release H2O2 in the presence of S. aureus in the phagolysosome. In response, PerR, a dimeric peroxide sensor, stimulates the production of LukAB, which increases hPMN cell death. Figure made with BioRender.
Although we found genes that may play a regulatory role in toxin production, we do not know whether this regulation occurs directly or indirectly. Our PerR in silico binding site mapping experiment suggests that the impact on PlukAB might be indirect as no binding site was identified in the promoter or lukAB coding sequence. PerR may instead be regulating one or more regulators that directly bind to the promoter of lukAB. Such indirect regulation is seen in S. aureus by several different regulators, including RNAIII and Rot. RNAIII, the effector molecule of agr, regulates a number of different virulence factors through the activation and repression of downstream regulators such as Rot and SarT (17, 30, 60–67). Rot also indirectly regulates toxins through the repression of the SaeRS system (17, 21, 30). Our data suggest that PerR does not directly regulate lukAB nor global regulators such as sae, agr, and rot. Instead, PerR binding sites were found on the promoters of fur, sarV, a lysR-like (SAUSA300_0093), and upstream of the histidine kinases lytS and airS. Future studies are needed to elucidate the molecular mechanism of PerR-mediated regulation of PlukAB and the contribution of these genes.
The data presented herein establish that PerR functions not only in oxidative stress resistance and iron storage pathways but also in lukAB regulation. This highlights a trend seen in S. aureus, where metabolic or stress response regulators are additionally involved in the regulation of virulence factors. RpiRc, PurR, and CodY are all metabolic regulators that have also been shown to control toxin expression (11, 68–70). RpiRc is involved in the catabolism of sugar and has been established to repress the rnaIII promoter, resulting in increased Rot levels and, therefore, decreased toxin expression (11). PurR functions as a repressor of purine biosynthesis and also participates in directly regulating virulence factors and master regulators of virulence (68). The canonical role of CodY enables S. aureus to adapt to environments with nutrient limitations and metabolic stress (71). In addition, CodY also functions as a repressor of virulence factors and regulators such as α-toxin and RNAIII (69). We posit that it is advantageous for the bacteria to have regulators that perform dual-functional roles, especially in the case of PerR; having a regulator that is able to sense and respond to H2O2 and upregulate a virulence factor that can kill PMNs that are producing H2O2 may promote bacterial survival and proliferation during infection.
Altogether, the findings presented here highlight the ability of S. aureus to sense host environments and respond in coordinated ways to both protect the bacterium and suppress the host immune system. Additional studies are needed to better understand how PerR is regulating the leukocidins and if other stress response regulators have dual functions. This knowledge will give us a better understanding of the complexities of virulence regulation during infection and may highlight key targets for future therapeutics.
MATERIALS AND METHODS
Purification of human neutrophils
Human PMNs were isolated by a Ficoll–Paque method as described before (72).
Bacterial cultures and growth conditions
All S. aureus strains were grown on tryptic soy agar (TSA) or TSA supplemented with appropriate antibiotics (chloramphenicol 10 µg/mL and erythromycin 2.5 µg/mL) at 37°C. Liquid cultures of S. aureus were grown in tryptic soy broth (TSB) and supplemented with antibiotics if needed. Liquid cultures were incubated at 37°C while shaking at 180 rpm. They were grown in 96-deep-well plates (Corning, 14-222-353) with 1 mL of growth medium. E. coli was grown in a flask containing 20 mL of Luria–Bertani broth. For subculturing S. aureus, a dilution of 1:100 was used from the overnight cultures into fresh media.
Construction of mutant strains
For all the strains, plasmids, and oligonucleotides used in this study, see Tables S2 and S3 in the supplemental materials. The PlukAB_luc strain was constructed as previously described in Anderson et al. (73). Briefly, the backbone of the PlukAB_luc plasmid originated from the plasmid pHC123 (kindly provided by Alex Horswill) (28) and was cut at the SalI and KpnI restriction sites before being ligated with the lukAB intergenic region and being transformed into DH5α and electroporated into AH-LAC. The primers pHC123_lukAB_F and pHC123_lukAB_R were used. The JE2 promoter–reporter library was generated by phage transduction using phage 80α lysate of the AH-LAC PlukAB_luc strain. The regulatory library was grown overnight in 400 µL of TSB in a round-bottomed deep-well plate. In the morning, 390 µL of fresh TSB was inoculated with 10 µL of the overnight culture and grown at 120 rpm until an optical density (OD600) of 1. Next, 5 µL of 1 M CaCl2 and 100 µL of phage lysate were added to each well, and this was left at room temperature for 20 minutes. We added 40 µL of 1 M Na citrate, and 10 µL of the mix was spot-platted onto TSA + Cm10 and grew overnight at 30°C. Colonies were picked from this plate and grown overnight, and then, 50 µL of the overnight culture was added to 50 µL of 20% glycerol and frozen down for further use.
The JE2 ΔperR strain was generated by phage transduction using phage φ11 lysate from Newman ΔperR::ermC (kindly provided by Anthony Richardson). Complementation of perR was performed with plasmid pIMAY* (kindly provided by Angelika Gründling via Addgene), which is used to stably integrate DNA into the natural site, resulting in a single-copy chromosomal insertion (53). JE2 ΔperR::perR was made by cloning the perR allele into the pIMAY* plasmid. pIMAY* was cut with XhoI and XmaI before ligation with the perR coding region. Primers PerR_PIMAY_F and PerR_PIMAY_R were used to amplify upstream and downstream regions of perR with base pair homology to pIMAY* for ligation. pIMAY*-perR plasmid was transformed into IM08B and electroporated in JE2ΔperR.
Ex vivo infection assay
Human PMNs were seeded on 96-well flat-bottom white tissue culture-treated plates (Corning, 3917) at a concentration of 2 × 105 cells/well at a final volume of 80 µL of phenol red-free Roswell Park Memorial Institute media (RPMI—Gibco, 11-835-055) supplemented with 10 mM HEPES (Corning, 25-060-CI) and 5% NHS (SeraCare, 1830-0003). One hundred and fifty microliters of the S. aureus subcultures was transferred to clear round-bottom plates (Corning, 3788) to measure OD600. The OD600 of the subcultures was obtained before infection using the PerkinElmer EnVision 2103 Multilabel Reader. For the primary and secondary screens, the cultures were grown overnight in 96-well plates and then subcultured for 3 hours. For the perR deletion and transposon cytotoxicity assay, cultures were grown overnight in 96-well plates and then subcultured for 3.5 hours. hPMNs were infected at an MOI of 8 (10 µL) or 18 (20 µL) and incubated at 37°C in 5% CO2 for 3 hours. MOI was confirmed by serial dilution and plating for CFU. Ten microliters of TSB was added to the media for a final volume of 100 µL in each well when experiments were conducted at an MOI of 8.
Luminescence reporter assay
The ex vivo infection assay described above was conducted on the Nebraska Transposon Mutant Library strains containing the pHC123 plasmid with the promoter of lukAB driving expression of the luciferase operon (26, 28). These strains were grown in 1 mL of TSB with 10 µg/mL of chloramphenicol (to retain reporter plasmid) in 96-deep-well plates overnight and subcultured for 3 hours the following day. After the 3-hour infection of hPMNs, background luminescence was measured by PerkinElmer EnVision 2103 Multilabel Reader before adding 15 mg/mL (10 µL/well) of D-luciferin (Thermo Fisher Scientific, 88293) resuspended in water to each well. Plates were stored in the dark for 30 minutes before luminescence was measured again. OD600 was accounted for in the analysis. The resulting OD600 was ~0.15 in the 96-well plate. The resulting raw luminescence values after infection were divided by the OD600 for each strain for the luminescence reporter assay.
Cytotoxicity assay
The ex vivo infection assay described above was performed before measuring the cytotoxicity of the strains. Subcultures were grown for 3 hours for the secondary screen and 3.5 hours for the perR deletion and transposon experiments to obtain a higher OD600. The resulting OD600 was ~0.2 in the 96-well plate. TSB was added to normalize the cultures to an OD600 of 0.19. After the 3-hour infection of hPMNs, plates were centrifuged at 1,500 rpm for 5 minutes at 4°C. Twenty-five microliters of supernatants was transferred to clear-bottom black 96-well plates (Corning, 3904). Cytotoxicity was measured by LDH release from hPMNs. Twenty-five microliters of the CytoTox‐One Homogeneous Membrane Integrity Assay (Promega, G7892) was also added to the black 96-well plates. LDH release was quantified per the manufacturer’s instructions using an EnVision 2103 Multilabel Reader.
Hydrogen peroxide treatment assay
Subcultures were grown for 3.5 hours. H2O2 30% (Sigma-Aldrich, 7722-84-1) was freshly diluted with H2O to varying concentrations (0.0001, 0.001, 0.01, 0.1, 1, and 5 mM). The final concentration of H2O2 added to the wells was 0.0001, 0.001, 0.01, and 0.1 mM. Fifty microliters of the subculture and 50 µL of H2O2 were added to 96-well flat-bottom white tissue culture-treated plates. Bacterial strains used in the experiment contained the pHC123_lukAB plasmid for luminescence readings. After a 1-hour H2O2 treatment, background luminescence was measured by the PerkinElmer EnVision 2103 Multilabel Reader before adding 15 mg/mL (10 µL/well) of D-luciferin. Plates were incubated in the dark for 30 minutes at room temperature before luminescence was measured again. To calculate CFU per milliliter, bacteria were plated after 1-hour H2O2 treatment.
Analysis of PerR binding sequence and regulon
PerR binding site sequences for S. aureus were obtained from the CollecTF database (http://www.collectf.org/browse/home/) and verified to be identical to those reported in Horsburgh et al. (22). A sequence logo for the aligned sequences was created using WEBLOGO (https://weblogo.berkeley.edu/logo.cgi). A S. aureus LAC reference assembly (NCBI ID: GCF_015475575.1) was scanned for PerR binding sites using FIMO v. 5.5.3 (https://meme-suite.org/meme/tools/fimo) (58) with the PerR binding site sequences and default parameters. Only FIMO hits falling within the first third of a coding sequence or up to 100 bp upstream of a coding sequence were retained; to this end, we filtered the FIMO hits using Python v. 3.9.2, the BioPython package v. 1.78, and GenBank LAC annotation files from NCBI (GCF_015475575.1). Locus tags corresponding to coding sequences in the annotation files of the LAC assembly (GCF_015475575.1) were translated to those of a USA300 FPR3757 assembly (GCF_000013465.1) via sequence similarity search with blastp v. 2.11.0, only retaining hits at 95% identity with 95% coverage. FIMO hits were plotted in R (74) v. 4.3.0 using ggplot2 v. 3.4.2 (75). Only hits with a FIMO score in the upper 5% were colored red, additionally labeling those with a gene symbol, if available.
Statistics
Statistical significance was determined using Prism 9.0 (GraphPad Software). One-way ANOVA with multiple comparisons was used to compare data sets with more than two strains. Unpaired t-tests with Welch’s correction were used for comparing data sets that only had two mutants.
ACKNOWLEDGMENTS
We thank members of the Torres laboratory for insightful feedback on this manuscript. We also thank Professor Angelika Gründling (Imperial College London) for gifting pIMAY* (53), Professor Alex Horswill (University of Colorado Anschutz School of Medicine) for gifting pHC123 (28), and Professor Anthony Richardson (University of Pittsburgh) for gifting Newman ΔperR strain.
This work was supported by the NIH–National Institute of Allergy and Infectious Diseases awards AI099394 (V.J.T.) and AI137336 (V.J.T. and B.S.). V.J.T. is also supported by ALSAC.
BioRender was used to create some images.
E.E.A. and V.J.T. designed the study. A.S., E.E.A., S.D., and M.P. generated strains, and A.S. performed the experiments. E.E.A. and S.D. trained A.S. A.P. performed the bioinformatic analyses, and B.S. and V.J.T. managed the project. A.S. wrote the first draft of the manuscript, and E.E.A., S.D., and V.J.T. worked on the final version of the manuscript. All authors commented on the manuscript.
AFTER EPUB
[This article was published on 18 January 2024 with errors in the Abstract and Importance. The errors were corrected in the current version, posted on 19 January 2024.]
Footnotes
This article is a direct contribution from Victor J. Torres, a member of the Infection and Immunity Editorial Board, who arranged for and secured reviews by Lindsey Shaw, University of South Florida, and Anthony Richardson, University of Pittsburgh.
Contributor Information
Victor J. Torres, Email: victor.torres@stjude.org.
Andreas J. Bäumler, University of California, Davis, Davis, California, USA
ETHICS APPROVAL
Buffy coats were obtained from anonymous healthy donors from the New York Blood Center. All experiments were performed according to NIH guidelines and U.S. federal law.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/iai.00526-23.
Strains and plasmids, as well as oligonucleotides, used in this study.
Primary screen measuring lukAB promoter activity in S. aureus JE2 regulatory transposon library containing pHC123 reporter plasmid.
PerR binding sites.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- 2. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. doi: 10.1038/nrmicro3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tam K, Torres VJ. 2019. Staphylococcus aureus secreted toxins and extracellular enzymes. Microbiol Spectr 7:7. doi: 10.1128/microbiolspec.GPP3-0039-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Otto M. 2014. Staphylococcus aureus toxins. Curr Opin Microbiol 17:32–37. doi: 10.1016/j.mib.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spaan AN, van Strijp JAG, Torres VJ. 2017. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15:435–447. doi: 10.1038/nrmicro.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alonzo F, Torres VJ. 2014. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev 78:199–230. doi: 10.1128/MMBR.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DuMont A.L, Torres VJ. 2014. Cell targeting by the Staphylococcus aureus pore-forming toxins: it’s not just about lipids. Trends Microbiol 22:21–27. doi: 10.1016/j.tim.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perelman SS, James DBA, Boguslawski KM, Nelson CW, Ilmain JK, Zwack EE, Prescott RA, Mohamed A, Tam K, Chan R, Narechania A, Pawline MB, Vozhilla N, Moustafa AM, Kim SY, Dittmann M, Ekiert DC, Bhabha G, Shopsin B, Planet PJ, Koralov SB, Torres VJ. 2021. Genetic variation of staphylococcal LukAB toxin determines receptor tropism. Nat Microbiol 6:731–745. doi: 10.1038/s41564-021-00890-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DuMont A.L, Yoong P, Day CJ, Alonzo F, McDonald WH, Jennings MP, Torres VJ. 2013. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A 110:10794–10799. doi: 10.1073/pnas.1305121110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DuMont Ashley L, Yoong P, Surewaard BGJ, Benson MA, Nijland R, van Strijp JAG, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841. doi: 10.1128/IAI.00095-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balasubramanian D, Ohneck EA, Chapman J, Weiss A, Kim MK, Reyes-Robles T, Zhong J, Shaw LN, Lun DS, Ueberheide B, Shopsin B, Torres VJ. 2016. Staphylococcus aureus coordinates leukocidin expression and pathogenesis by sensing metabolic fluxes via RpiRc. mBio 7:e00818-16. doi: 10.1128/mBio.00818-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, Torres VJ. 2011. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79:814–825. doi: 10.1111/j.1365-2958.2010.07490.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, DeLeo FR. 2010. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5:e11634. doi: 10.1371/journal.pone.0011634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malachowa N, Kobayashi SD, Braughton KR, Whitney AR, Parnell MJ, Gardner DJ, Deleo FR. 2012. Staphylococcus aureus leukotoxin GH promotes inflammation. J Infect Dis 206:1185–1193. doi: 10.1093/infdis/jis495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Copin R, Shopsin B, Torres VJ. 2018. After the deluge: mining Staphylococcus aureus genomic data for clinical associations and host pathogen interactions. Curr Opin Microbiol 41:43–50. doi: 10.1016/j.mib.2017.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez J, Sanders H, Henn J, Wilson JM, Malone D, Buoninfante A, Willms M, Chan R, DuMont AL, McLahan C, Grubb K, Romanello A, van den Dobbelsteen G, Torres VJ, Poolman JT. 2022. Vaccination with detoxified leukocidin AB reduces bacterial load in a Staphylococcus aureus minipig deep surgical wound infection model. J Infect Dis 225:1460–1470. doi: 10.1093/infdis/jiab219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guillet J, Hallier M, Felden B. 2013. Emerging functions for the Staphylococcus aureus RNome. PLoS Pathog 9:e1003767. doi: 10.1371/journal.ppat.1003767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balasubramanian D, Harper L, Shopsin B, Torres VJ. 2017. Staphylococcus aureus pathogenesis in diverse host environments. Pathog Dis 75:ftx005. doi: 10.1093/femspd/ftx005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flack CE, Zurek OW, Meishery DD, Pallister KB, Malone CL, Horswill AR, Voyich JM. 2014. Differential regulation of staphylococcal virulence by the sensor kinase SaeS in response to neutrophil-derived stimuli. Proc Natl Acad Sci U S A 111:E2037–E2045. doi: 10.1073/pnas.1322125111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zurek OW, Nygaard TK, Watkins RL, Pallister KB, Torres VJ, Horswill AR, Voyich JM. 2014. The role of innate immunity in promoting SaeR/S-mediated virulence in Staphylococcus aureus. J Innate Immun 6:21–30. doi: 10.1159/000351200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li D, Cheung A. 2008. Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect Immun 76:1068–1075. doi: 10.1128/IAI.01069-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69:3744–3754. doi: 10.1128/IAI.69.6.3744-3754.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ji C-J, Kim J-H, Won Y-B, Lee Y-E, Choi T-W, Ju S-Y, Youn H, Helmann JD, Lee J-W. 2015. Staphylococcus aureus PerR is a hypersensitive hydrogen peroxide sensor using iron-mediated histidine oxidation. J Biol Chem 290:20374–20386. doi: 10.1074/jbc.M115.664961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoffstein ST, Gennaro DE, Manzi RM. 1985. Neutrophils may directly synthesize both H2O2 and O2− since surface stimuli induce their release in stimulus-specific ratios. Inflammation 9:425–437. doi: 10.1007/BF00916342 [DOI] [PubMed] [Google Scholar]
- 25. Winterbourn CC, Kettle AJ, Hampton MB. 2016. Reactive oxygen species and neutrophil function. Annu Rev Biochem 85:765–792. doi: 10.1146/annurev-biochem-060815-014442 [DOI] [PubMed] [Google Scholar]
- 26. Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. The Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7 [DOI] [PubMed] [Google Scholar]
- 28. Miller RJ, Crosby HA, Schilcher K, Wang Y, Ortines RV, Mazhar M, Dikeman DA, Pinsker BL, Brown ID, Joyce DP, Zhang J, Archer NK, Liu H, Alphonse MP, Czupryna J, Anderson WR, Bernthal NM, Fortuno-Miranda L, Bulte JWM, Francis KP, Horswill AR, Miller LS. 2019. Development of a Staphylococcus aureus reporter strain with click beetle red luciferase for enhanced in vivo imaging of experimental bacteremia and mixed infections. Sci Rep 9:1. doi: 10.1038/s41598-019-52982-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benson MA, Ohneck EA, Ryan C, Alonzo F, Smith H, Narechania A, Kolokotronis S-O, Satola SW, Uhlemann A-C, Sebra R, Deikus G, Shopsin B, Planet PJ, Torres VJ. 2014. Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element. Mol Microbiol 93:664–681. doi: 10.1111/mmi.12682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jenul C, Horswill AR. 2018. Regulation of Staphylococcus aureus virulence. Microbiol Spectr 6. doi: 10.1128/microbiolspec.GPP3-0031-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, Buysse JM. 2004. Large-scale identification of genes required for full virulence of Staphylococcus aureus. J Bacteriol 186:8478–8489. doi: 10.1128/JB.186.24.8478-8489.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walker JN, Crosby HA, Spaulding AR, Salgado-Pabón W, Malone CL, Rosenthal CB, Schlievert PM, Boyd JM, Horswill AR. 2013. The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog 9:e1003819. doi: 10.1371/journal.ppat.1003819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fournier B, Hooper DC. 2000. A new two-component regulatory system involved in adhesion, autolysis, and extracellular proteolytic activity of Staphylococcus aureus. J Bacteriol 182:3955–3964. doi: 10.1128/JB.182.14.3955-3964.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Memmi G, Nair DR, Cheung A. 2012. Role of ArlRS in autolysis in methicillin-sensitive and methicillin-resistant Staphylococcus aureus strains. J Bacteriol 194:759–767. doi: 10.1128/JB.06261-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fournier B, Klier A, Rapoport G. 2001. The two-component system ArlS–ArlR is a regulator of virulence gene expression in Staphylococcus aureus. Mol Microbiol 41:247–261. doi: 10.1046/j.1365-2958.2001.02515.x [DOI] [PubMed] [Google Scholar]
- 36. Lee J-W, Helmann JD. 2007. Functional specialization within the Fur family of metalloregulators. Biometals 20:485–499. doi: 10.1007/s10534-006-9070-7 [DOI] [PubMed] [Google Scholar]
- 37. Zuber P. 2009. Management of oxidative stress in Bacillus. Annu Rev Microbiol 63:575–597. doi: 10.1146/annurev.micro.091208.073241 [DOI] [PubMed] [Google Scholar]
- 38. Cosgrove K, Coutts G, Jonsson I-M, Tarkowski A, Kokai-Kun JF, Mond JJ, Foster SJ. 2007. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol 189:1025–1035. doi: 10.1128/JB.01524-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SA. 2008. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol 10:2312–2325. doi: 10.1111/j.1462-5822.2008.01213.x [DOI] [PubMed] [Google Scholar]
- 40. Needham AJ, Kibart M, Crossley H, Ingham PW, Foster SJ. 2004. Drosophila melanogaster as a model host for Staphylococcus aureus infection. Microbiology (Reading) 150:2347–2355. doi: 10.1099/mic.0.27116-0 [DOI] [PubMed] [Google Scholar]
- 41. Faulkner MJ, Helmann JD. 2011. Peroxide stress elicits adaptive changes in bacterial metal ion homeostasis. Antioxid Redox Signal 15:175–189. doi: 10.1089/ars.2010.3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L, Keramati L, Helmann JD. 1995. Coordinate regulation of Bacillus subtilis peroxide stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci U S A 92:8190–8194. doi: 10.1073/pnas.92.18.8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morrissey JA, Cockayne A, Brummell K, Williams P. 2004. The staphylococcal ferritins are differentially regulated in response to iron and manganese and via PerR and Fur. Infect Immun 72:972–979. doi: 10.1128/IAI.72.2.972-979.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peng W, Yang X, Wang N, Gao T, Liu Z, Liu W, Zhou D, Yang K, Guo R, Liang W, Chen H, Tian Y, Yuan F, Bei W, Dozois CM. 2022. PerR-regulated manganese import contributes to oxidative stress defense in Streptococcus suis. Appl Environ Microbiol 88:e0008622. doi: 10.1128/aem.00086-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Horsburgh MJ, Ingham E, Foster SJ. 2001. In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J Bacteriol 183:468–475. doi: 10.1128/JB.183.2.468-475.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beavers WN, Skaar EP. 2016. Neutrophil-generated oxidative stress and protein damage in Staphylococcus aureus. Pathog Dis 74:ftw060. doi: 10.1093/femspd/ftw060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duarte V, Latour J-M. 2010. PerR vs OhrR: selective peroxide sensing in Bacillus subtilis. Mol Biosyst 6:316–323. doi: 10.1039/b915042k [DOI] [PubMed] [Google Scholar]
- 48. Dubbs JM, Mongkolsuk S. 2012. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol 194:5495–5503. doi: 10.1128/JB.00304-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mongkolsuk S, Helmann JD. 2002. Regulation of inducible peroxide stress responses. Mol Microbiol 45:9–15. doi: 10.1046/j.1365-2958.2002.03015.x [DOI] [PubMed] [Google Scholar]
- 50. Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. 1998. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol 29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x [DOI] [PubMed] [Google Scholar]
- 51. Lee J-W, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537 [DOI] [PubMed] [Google Scholar]
- 52. Faulkner MJ, Ma Z, Fuangthong M, Helmann JD. 2012. Derepression of the Bacillus subtilis PerR peroxide stress response leads to iron deficiency. J Bacteriol 194:1226–1235. doi: 10.1128/JB.06566-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schuster CF, Howard SA, Gründling A. 2019. Use of the counter selectable marker PheS* for genome engineering in Staphylococcus aureus. Microbiology (Reading) 165:572–584. doi: 10.1099/mic.0.000791 [DOI] [PubMed] [Google Scholar]
- 54. Lorenz LL, Duthie ES. 1952. Staphylococcal coagulase: mode of action and antigenicity. Microbiology 6:95–107. doi: 10.1099/00221287-6-1-2-95 [DOI] [PubMed] [Google Scholar]
- 55. Helmann JD, Wu MFW, Gaballa A, Kobel PA, Morshedi MM, Fawcett P, Paddon C. 2003. The global transcriptional response of Bacillus subtilis to peroxide stress is coordinated by three transcription factors. J Bacteriol 185:243–253. doi: 10.1128/JB.185.1.243-253.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fuangthong M, Herbig AF, Bsat N, Helmann JD. 2002. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J Bacteriol 184:3276–3286. doi: 10.1128/JB.184.12.3276-3286.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gerber CE, Bruchelt G, Falk UB, Kimpfler A, Hauschild O, Kuçi S, Bächi T, Niethammer D, Schubert R. 2001. Reconstitution of bactericidal activity in chronic granulomatous disease cells by glucose-oxidase–containing liposomes. Blood 98:3097–3105. doi: 10.1182/blood.v98.10.3097 [DOI] [PubMed] [Google Scholar]
- 58. Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. doi: 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spaan AN, Surewaard BGJ, Nijland R, van Strijp JAG. 2013. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol 67:629–650. doi: 10.1146/annurev-micro-092412-155746 [DOI] [PubMed] [Google Scholar]
- 60. Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, Gaspin C, Vandenesch F, Romby P. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator rot by an antisense mechanism. Genes Dev 21:1353–1366. doi: 10.1101/gad.423507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mootz JM, Benson MA, Heim CE, Crosby HA, Kavanaugh JS, Dunman PM, Kielian T, Torres VJ, Horswill AR. 2015. Rot is a key regulator of Staphylococcus aureus biofilm formation. Mol Microbiol 96:388–404. doi: 10.1111/mmi.12943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP. 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol 61:1038–1048. doi: 10.1111/j.1365-2958.2006.05292.x [DOI] [PubMed] [Google Scholar]
- 63. Hsieh H-Y, Tseng CW, Stewart GC. 2008. Regulation of Rot expression in Staphylococcus aureus. J Bacteriol 190:546–554. doi: 10.1128/JB.00536-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bronesky D, Wu Z, Marzi S, Walter P, Geissmann T, Moreau K, Vandenesch F, Caldelari I, Romby P. 2016. Staphylococcus aureus RNAIII and its regulon link quorum sensing, stress responses, metabolic adaptation, and regulation of virulence gene expression. Annu Rev Microbiol 70:299–316. doi: 10.1146/annurev-micro-102215-095708 [DOI] [PubMed] [Google Scholar]
- 65. Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x [DOI] [PubMed] [Google Scholar]
- 66. Heinrichs JH, Bayer MG, Cheung AL. 1996. Characterization of the sar locus and its interaction with agr in Staphylococcus aureus. J Bacteriol 178:418–423. doi: 10.1128/jb.178.2.418-423.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schmidt KA, Manna AC, Gill S, Cheung AL. 2001. SarT, a repressor of α-hemolysin in Staphylococcus aureus. Infect Immun 69:4749–4758. doi: 10.1128/IAI.69.8.4749-4758.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sause WE, Balasubramanian D, Irnov I, Copin R, Sullivan MJ, Sommerfield A, Chan R, Dhabaria A, Askenazi M, Ueberheide B, Shopsin B, van Bakel H, Torres VJ. 2019. The purine biosynthesis regulator PurR moonlights as a virulence regulator in Staphylococcus aureus. Proc Natl Acad Sci U S A 116:13563–13572. doi: 10.1073/pnas.1904280116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol 190:2257–2265. doi: 10.1128/JB.01545-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seidl K, Stucki M, Ruegg M, Goerke C, Wolz C, Harris L, Berger-Bächi B, Bischoff M. 2006. Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrob Agents Chemother 50:1183–1194. doi: 10.1128/AAC.50.4.1183-1194.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sonenshein AL. 2005. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr Opin Microbiol 8:203–207. doi: 10.1016/j.mib.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 72. Reyes-Robles T, Lubkin A, Alonzo F 3rd, Lacy DB, Torres VJ. 2016. Exploiting dominant‐negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep 17:428–440. doi: 10.15252/embr.201670010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Anderson EE, Dyzenhaus S, Ilmain JK, Sullivan MJ, van Bakel H, Torres VJ. 2023. SarS is a repressor of Staphylococcus aureus bicomponent pore-forming leukocidins. Infect Immun 91:e0053222. doi: 10.1128/iai.00532-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. R Core Team . 2023. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 75. Wickham H. 2016. ggplot2: elegant graphics for data analysis. New York. doi: 10.1007/978-3-319-24277-4 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains and plasmids, as well as oligonucleotides, used in this study.
Primary screen measuring lukAB promoter activity in S. aureus JE2 regulatory transposon library containing pHC123 reporter plasmid.
PerR binding sites.