

Abstract
Background
Ferroptosis, an iron‐dependent form of regulated cell death, is a major cell death mode in myocardial ischemia reperfusion (I/R) injury, along with mitochondrial permeability transition‐driven necrosis, which is inhibited by cyclosporine A (CsA). However, therapeutics targeting ferroptosis during myocardial I/R injury have not yet been developed. Hence, we aimed to investigate the therapeutic efficacy of deferasirox, an iron chelator, against hypoxia/reoxygenation‐induced ferroptosis in cultured cardiomyocytes and myocardial I/R injury.
Methods and Results
The effects of deferasirox on hypoxia/reoxygenation‐induced iron overload in the endoplasmic reticulum, lipid peroxidation, and ferroptosis were examined in cultured cardiomyocytes. In a mouse model of I/R injury, the infarct size and adverse cardiac remodeling were examined after treatment with deferasirox, CsA, or both in combination. Deferasirox suppressed hypoxia‐ or hypoxia/reoxygenation‐induced iron overload in the endoplasmic reticulum, lipid peroxidation, and ferroptosis in cultured cardiomyocytes. Deferasirox treatment reduced iron levels in the endoplasmic reticulum and prevented increases in lipid peroxidation and ferroptosis in the I/R‐injured myocardium 24 hours after I/R. Deferasirox and CsA independently reduced the infarct size after I/R injury to a similar degree, and combination therapy with deferasirox and CsA synergistically reduced the infarct size (infarct area/area at risk; control treatment: 64±2%; deferasirox treatment: 48±3%; CsA treatment: 48±4%; deferasirox+CsA treatment: 37±3%), thereby ameliorating adverse cardiac remodeling on day 14 after I/R.
Conclusions
Combination therapy with deferasirox and CsA may be a clinically feasible and effective therapeutic approach for limiting I/R injury and ameliorating adverse cardiac remodeling after myocardial infarction.
Keywords: cyclosporin A, deferasirox, ferroptosis, ischemia reperfusion injury
Subject Categories: Basic Science Research, Ischemia, Translational Studies
Nonstandard Abbreviations and Acronyms
- 4‐HNE
4‐hydroxynonenal
- AMI
acute myocardial infarction
- CsA
cyclosporine A
- ER
endoplasmic reticulum
- HO‐1
heme oxygenase
- H/R
hypoxia‐reoxygenation
- I/R
ischemia/reperfusion
- MPT
mitochondrial permeability transition
Research Perspective.
What Is New?
Ferroptosis and mitochondrial permeability transition‐driven necrosis are major modes of cell death in myocardial ischemia reperfusion injury.
Therapeutics targeting ferroptosis are not yet developed, although cyclosporin A is an established inhibitor of mitochondrial permeability transition‐driven necrosis.
Deferasirox effectively chelates iron in the endoplasmic reticulum and suppresses ferroptosis induced by hypoxia‐reoxygenation; it reduces the infarct size in a mouse model of myocardial ischemia reperfusion injury, and combination therapy with deferasirox and cyclosporin A synergistically limits the infarct size during ischemia reperfusion injury, thereby ameliorating adverse cardiac remodeling after ischemia reperfusion injury.
What Question Should Be Addressed Next?
The clinical safety of deferasirox in patients with acute myocardial infarction should be confirmed.
Clinical studies will be necessary to determine the efficacy of deferasirox in reducing infarct size in patients with acute myocardial infarction undergoing percutaneous coronary intervention.
Ultimately, further investigation is required to confirm whether combination therapy with deferasirox and cyclosporin A can improve cardiac function and heart failure in the late phase in patients with acute myocardial infarction undergoing percutaneous coronary intervention.
Myocardial ischemia/reperfusion (I/R) injury has been considered a promising therapeutic target since cardiologists recognized that its inhibition can prevent myocardium loss after revascularization during acute myocardial infarction (AMI). 1 Revascularization therapy can improve clinical outcomes, including survival, during AMI. 2 , 3 , 4 , 5 , 6 However, the number of patients with heart failure after myocardial infarction (MI) has increased. 7 It is therefore imperative to develop new therapeutics targeting I/R injury to further reduce the infarct size and preserve cardiac function after MI.
Mitochondrial permeability transition (MPT)‐driven necrosis is recognized as a pivotal mode of regulated cell death in I/R injury. 8 Griffiths and Halestrap first reported that mitochondrial pores close during cardiac ischemia and open upon subsequent reperfusion. 9 Di Lisa et al and Hausenloy et al demonstrated that mitochondrial pore‐opening leads to MPT‐driven myocyte necrosis during the reperfusion phase, whereas cyclosporine A (CsA) inhibits mitochondrial pore‐opening and prevents cell death, 10 thus reducing the infarct size in rodent models of I/R. 11 , 12 Thereafter, Nakagawa et al and Baines et al independently demonstrated that cyclophilin D regulates MPT pore‐opening, while CsA inhibits cyclophilin D, thereby preventing MPT‐driven necrosis. 13 , 14 Thus, the roles of MPT‐driven necrosis and CsA in I/R injury were established. Indeed, a phase 2 clinical trial reported reductions in the infarct size after CsA treatment in patients receiving percutaneous coronary intervention for ST‐segment–elevation myocardial infarction. 15 However, a phase 3 clinical trial did not demonstrate a clinical benefit in cardiac remodeling at 1 year after CsA treatment directed against I/R. 16 Collectively, these data suggest that CsA may be effective in mitigating I/R injury; however, CsA monotherapy is not sufficient to clinically ameliorate adverse cardiac remodeling after percutaneous coronary intervention in patients with AMI. Thus, further investigations are necessary to discover novel therapeutic targets and develop clinically useful agents for I/R injury.
Ferroptosis is an iron‐dependent form of regulated cell death that is initiated by oxidative stress attributable to iron overload and subsequent lipid peroxidation. 17 Ferroptosis is endogenously controlled by glutathione peroxidase 4, a scavenger of lipid peroxides, and can be inhibited by iron chelators and lipophilic antioxidants. 10 , 18 We and others have shown that ferroptosis participates in myocardial I/R injury. 19 , 20 Of note, we also demonstrated that ferroptosis and MPT‐driven necrosis predominantly contribute to myocardial I/R injury, and iron overload in the endoplasmic reticulum (ER) is caused by the upregulation of heme oxygenase‐1 (HO‐1) expression in response to I/R insult, resulting in ferroptosis. 20 Thus, inhibiting ferroptosis in conjunction with CsA treatment that targets MPT‐driven necrosis may be an effective combinatorial strategy for maximally limiting myocardial I/R injury. 20 However, therapeutics targeting ferroptosis in I/R injury have not yet been established.
Lipophilic antioxidants can feasibly prevent ferroptosis in human diseases. Indeed, we showed that ethoxyquin, a lipophilic antioxidant, prevents ferroptosis in doxorubicin‐induced cardiotoxicity. 21 However, no lipophilic antioxidants with clinical safety are currently available. Alternatively, iron chelators represent a potential treatment modality against ferroptosis in I/R injury. As therapeutics for I/R injury should be initiated between ischemia and reperfusion during clinical AMI, rapid diffusion into myocytes during I/R injury is required for potential therapeutic agents to be effective against ferroptosis. Treatment before ischemia with dexrazoxane, an iron chelator against Fe3+ approved for suppressing doxorubicin cardiotoxicity, reduces the infarct size in rodent models of I/R. 22 , 23 However, treatment with dexrazoxane after the onset of ischemia, which is applicable for clinical AMI treatment, does not effectively reduce the infarct size in a porcine model of I/R. 24 This may be because of the need for dexrazoxane to be metabolized to ADR‐925 for full iron‐chelating activity after its administration. 25 Collectively, these findings indicate that further investigations are needed to identify an iron chelator that is effective against I/R injury, particularly in the context of clinical AMI and reperfusion therapy.
Deferasirox is a ferric iron (Fe3+) chelator that has been proven safe and effective in patients with transfusion‐dependent iron overload. 26 Deferasirox exhibits high cell‐membrane permeability and iron chelation activity in myocytes, 27 suggesting that it may be capable of rapidly diffusing into the I/R‐injured myocardium after reperfusion during AMI. Thus, deferasirox is a prospective therapeutic agent for ferroptosis in myocardial I/R injury. However, its efficacy against ferroptosis in myocardial I/R injury remains to be fully elucidated.
Therefore, in this study, we aimed to examine the suppressive effect of deferasirox on ferroptosis in cultured cardiomyocytes and in a mouse model of I/R injury. We further assessed the efficacy of combinational deferasirox and CsA therapy on the infarct size and cardiac remodeling during the early and late phases of I/R in a mouse model, respectively.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Animals
All experimental protocols used in this study were approved by the Committee on Ethics of Animal Experiments at the Kyushu University Faculty of Medical and Pharmaceutical Sciences and were performed in accordance with the Guidelines for Animal Experiments of Kyushu University (A22‐009), Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, revised in 2011), and ARRIVE 2.0 guidelines. C57BL/6J mice and Sprague–Dawley rats (CLEA Japan) were housed in a temperature‐ and humidity‐controlled room, fed a commercial diet (CRF‐1, Oriental Yeast), and given free access to water.
Murine Myocardial I/R Model
Myocardial I/R was induced in mice as described previously. 20 , 28 Briefly, 9‐ to 12‐week‐old male mice were anesthetized via 1% to 2% isoflurane inhalation. The intercostal space was then opened under mechanical ventilation, and myocardial ischemia was induced by ligating the left anterior descending artery for 30 minutes, followed by reperfusion. The left anterior descending arteries of the animals in the sham group were sutured without ligation. Mice were orally administered deferasirox (200 mg/kg diluted in polyethylene glycol [28214‐05, Nacalai Tesque, Kyoto, Japan] to 40 mg/mL; D5905, Tokyo Chemical Industry) using a disposable feeding needle for mice (5202; Fuchigami, Kyoto, Tokyo) 10 minutes before reperfusion. CsA was injected via the femoral vein (2.5 mg/kg diluted in saline solution to 7.5 mg/mL; 3999406A1032, Novartis International) 10 minutes before reperfusion. Mouse hearts were excised 24 hours after reperfusion for histological and biochemical analyses.
Echocardiography
Echocardiography was performed as described previously. 29 , 30 Briefly, the mice were anesthetized using 1% to 2% isoflurane, and M‐mode images were obtained from the short‐axis view at the level of the papillary muscles using a Vevo1100 ultrasonography system (FUJIFILM VisualSonics).
Infarct Size Measurements
Infarct sizes were measured as described previously. 20 The left anterior descending arteries were reoccluded, and 0.3 mL of 2% Evans Blue dye (E2129, Sigma) was injected into the inferior vena cava to identify the area at risk (AAR) 24 hours after reperfusion. Once the peripheral limbs turned blue, the heart was rapidly excised and rinsed in normal saline solution, and the left ventricle (LV) was frozen in liquid nitrogen. The LV was then cut into five 1 mm‐thick slices and incubated in 1% 2,3,5‐TTC (T0520, Tokyo Chemical Industry) for 15 minutes at 37 °C. The infarct area (IFA, white area) and AAR (red and white areas) of each segment were measured using ImageJ software v1.44 (National Institutes of Health). Thereafter, the AAR/LV, IFA/LV, and IFA/AAR ratios were calculated.
Iron Measurements in Total Myocardium and ER Fraction
Total myocardial iron was examined 24 hours after a single administration of deferasirox (200 mg/kg) as described previously. 31 Briefly, the myocardium was lysed in radioimmunoprecipitation buffer, and the iron concentrations in the lysates were measured using a Metallo Assay Kit Iron LS (Metallogenics, FE31M) according to the manufacturer's instructions. The data are shown as the ratio of iron concentration per protein concentration (μg/mg). Iron in the ER fraction was examined as described previously. 20 The ER fraction was extracted from the myocardium using an Endoplasmic Reticulum Enrichment Extraction Kit (NBP2‐29482, Novus Biologicals). The ER fraction pellets were lysed with isosmotic homogenization buffer, and the suspensions were used for iron measurements.
Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling Staining
Terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) staining was performed using an In Situ Apoptosis Detection Kit (MK500, Takara) as described previously. 20 , 32 , 33 Briefly, hearts were retrieved 24 hours after reperfusion and stored in 10% formalin. Next, they were cut along the short axis into base, middle, and apex pieces. Each piece was embedded in paraffin, cut into 3‐μm‐thick slices, and stained according to the manufacturer's instructions. The sliced samples were deparaffinized and washed with PBS. The deparaffinized samples were processed with proteinase K (20 mg/mL, 161‐28 701, FUJIFILM Wako Pure Chemical Corp) for 15 minutes and washed 3 times with PBS. The samples were then labeled with an anti‐cardiac troponin T antibody (ab209813, Abcam) and placed in an incubator at 37 °C for 60 minutes. The samples were then washed again with PBS, and fragmented DNA within the samples was labeled with Labeling Safe Buffer containing terminal deoxynucleotide transferase (MK501, Takara). The samples were then incubated with an appropriate secondary antibody (DI‐1594, Vector Laboratories) for 90 minutes before being washed with PBS, sealed with a mounting agent (VECTASHIELD H1800, Vector Laboratories), and observed under a fluorescence microscope (BZ‐X800, Keyence). TUNEL‐positive and cardiac troponin T‐positive cells were counted as TUNEL‐positive cardiomyocytes.
Electron Microscopy for I/R‐Injured Myocardium
Electron microscopic images of the I/R‐injured myocardium were obtained as described previously. 34 Briefly, the myocardium at ischemic regions were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (PB, pH 7.4) at 4 °C overnight. After fixation, the samples were washed 3 times with 0.1% PB at 4 °C for 2 hours. The samples were dehydrated in an ethanol series, infiltrated with propylene oxide (PO) twice for 30 minutes, and placed in a 70:30 mixture of PO and resin (Quetol‐812, Nisshin EM, Tokyo, Japan) for 1 hour. The samples were then transferred to fresh 100% resin and polymerized at 60 °C for 48 hours. The polymerized resins were sliced into ultrathin (70‐nm) sections using an ultramicrotome (Ultracut UCT, Leica, Vienna, Austria) with a diamond knife, and the sections were mounted on copper grids. Before imaging, the sections were stained with 2% uranyl acetate at room temperature for 15 minutes and then washed with distilled water, followed by secondary staining with a lead‐stain solution (Sigma‐Aldrich, Tokyo, Japan) at room temperature for 3 minutes. Electron microscopic images were obtained by blinded technicians upon focusing on the mitochondria. Damaged mitochondria (as a feature of ferroptosis) were defined as mitochondria with reduced mitochondrial cristae or ruptured outer mitochondrial membranes as reported previously. 35 The damaged mitochondria were quantified relative to the total number of mitochondria.
Wheat Germ Agglutinin and Masson Trichrome Staining
Wheat germ agglutinin staining was performed using wheat germ agglutinin Alexa Fluor 488 (W11261, Thermo Fisher Scientific) to measure the cross‐sectional area. The deparaffinized samples were autoclaved in 10 mM sodium citrate (pH 6.0) for antigen retrieval. Wheat germ agglutinin solutions (5 μg/mL) were then mounted on the samples before they were incubated overnight at 4 °C in a dark room. The samples were washed with PBS, sealed with a mounting agent (VECTASHIELD H1200), and examined under a fluorescence microscope (BZ‐X800). Cardiomyocytes (100 cells per sample) cross‐sectioned at the nuclear level were selected, and their cross‐sectional area was measured using ImageJ software (v1.44). The average of the measured cross‐sectional areas was used as the cross‐sectional area of each sample. To evaluate interstitial fibrosis, the sliced samples were subjected to Masson Trichome staining. After excluding the infarct area, the interstitial fibrosis area and total myocardial area were measured using ImageJ software (v1.44) by capturing a tiling image of the entire section at the middle of the left ventricle. Thereafter, the collagen volume fraction (%) was determined by calculating the ratio of the interstitial fibrosis area to the total myocardial area.
Primary Culture of Isolated Cardiomyocytes
Neonatal rat ventricular myocytes were prepared as described previously. 36 , 37 Briefly, Sprague–Dawley rats were euthanized via isoflurane (5%) overdose, after which their hearts were rapidly excised. After digesting the myocardial tissues with trypsin (25300‐062, Thermo Fisher Scientific) and type‐2 collagenase (LS004176, Worthington Biochemical), the isolated cardiomyocytes were suspended in DMEM (D5796, Sigma‐Aldrich) containing 10% fetal bovine serum (SH30910.03, HyClone Laboratories) and 1% penicillin/streptomycin (26253‐84, Nacalai Tesque). The cells were plated twice in 100‐mm culture dishes for 70 minutes each to reduce the number of nonmyocytes. The nonadherent cells were plated in culture dishes (Primaria, Corning) at a density of approximately 2.5×105 cells/mL and cultured at 37 °C in humidified air with 5% CO2.
Hypoxia‐Reoxygenation Experiments with Cultured Cardiomyocytes
Cultured cardiomyocytes were subjected to H/R treatment as described previously. 20 , 28 Briefly, hypoxia was induced by replacing the standard medium with conditioned medium that was kept in a hypoxic chamber (APM‐50D, Astec) overnight (5% CO2, 94.5% N2, 0.5% O2 at 37 °C); the cultured cardiomyocytes were then incubated in the hypoxic chamber for 24 hours. Reoxygenation was induced by replacing the hypoxic medium with normoxic medium. Fer‐1 (50 μmol/L, SML0583, Abcam) or deferasirox (1, 10, 100, or 200 μmol/L) was added 1 hour before H/R treatment.
Western Blotting Analysis
Western blotting was performed as described previously 34 , 38 with the following primary antibodies against: 4‐hydroxy‐2‐nonenal (4‐HNE; MHN‐020P, JaICA; Shizuoka), acrolein (MAR‐020n, JaICA), cyclooxygenase‐2 (COX2; #12282, Cell Signaling Technology [CST]), heme oxygenase‐1 (HO‐1; #43966, CST), cleaved caspase substrate motif (#8698, CST), BAX (#14796, CST), and BCL2 (ab182858, Abcam); the antibody against GAPDH (internal standard; Santa Cruz Biotechnology, Inc., sc‐32 233) was used as an internal control to standardize the signal intensities.
Reverse Transcription‐Quantitative Polymerase Chain Reaction
Reverse transcription‐quantitative polymerase chain reaction was performed as described previously with slight modifications. 39 Briefly, total RNA was extracted from cultured cardiomyocytes using an ISOSPIN Cell & Tissue RNA kit (314‐08211; NIPPON GENE CO., LTD., Tokyo, Japan). cDNA was generated via reverse transcription using a ReverTra Ace qPCR RT kit (FSQ‐201; TOYOBO, Osaka, Japan). Reverse transcription‐quantitative polymerase chain reaction was performed on an Applied Biosystems QuantStudio3 system (A28567; Thermo Fisher Scientific) with the THUNDERBIRD SYBR qPCR Mix (#QPS‐101; TOYOBO). The forward (F) and reverse (R) primer sequences were as follows: Rps18: F 5′‐AAGTTTCAGCACATCCTGCGAGTA‐3′, R 5′‐TTGGTGAGGTCAATGTCTGCTTTC‐3′; Ptgs2: F 5′‐TGAACACGGACTTGCTCACTTTG‐3′, R 5′‐AGGCCTTTGCCACTGCTTGTA‐3′.
Boron‐Dipyrromethene Probing for Lipid Peroxides
Boron‐dipyrromethene (BODIPY) was used to probe for lipid peroxides as described previously. 20 Briefly, lipid peroxides in cultured cardiomyocytes were probed with C11 BODIPY 581/591 (D3861, Thermo Fisher Scientific) after 24 hours of hypoxia and 1 hour of reoxygenation. The cells were harvested by trypsinization and resuspended in PBS. They were then incubated with PBS containing C11 BODIPY 581/591 (2 mmol/L) for 15 minutes at 37 °C and strained through a 40‐mm cell strainer (43–10 040, pluriSelect Life Science). The strained cells were analyzed using a BD FACSLyric flow cytometer (BD Biosciences) equipped with a 488 nm laser for excitation. At least 10 000 cells were analyzed per condition. Histograms were prepared using the FlowJo software (v10).
Cell Viability Assay Using Calcein AM
Cell viability was evaluated using a Cell Counting Kit F (CK‐06, Dojindo) as previously described. 31 , 40 Briefly, cultured cardiomyocytes were subjected to 24 hours of hypoxia and 24 hours of reoxygenation, washed with PBS, and then incubated in calcein AM solution diluted in PBS (1:500 ratio of calcein AM to PBS) for 15 minutes at 37 °C. Thereafter, cell viability was measured fluorometrically (excitation wavelength of 490 nm and emission wavelength of 520 nm) with a Varioskan LUX Multimode Microplate Reader (Thermo Fisher Scientific).
Iron Measurements Using FerroFarRed, FerroOrange, and Mito‐FerroGreen
Iron concentrations in the ER, cytoplasm, and mitochondria were measured using FerroFarRed (GC903, Goryo Chemical), FerroOrange (F374, Dojindo), and Mito‐FerroGreen (M489, Dojindo) as previously described. 20 Briefly, cultured cardiomyocytes were seeded on 35‐mm glass‐bottomed dishes (AGC Techno Glass). Hypoxia was induced using conditioned media in the presence or absence of 200 μmol/L deferasirox (kept in a hypoxic chamber), after which the cells were incubated in the hypoxic chamber for 4 hours. The cells were then washed 3 times with PBS and incubated for 1 hour with serum‐free medium containing 5 μmol/L FerroFarRed and 0.1 μmol/L ERseeing (FDV‐0038; Funakoshi). FerroOrange and Mito‐FerroGreen were used at the final concentrations of 1 and 5 μmol/L, respectively. Thereafter, the cells were washed 3 times with PBS and fixed with 4% paraformaldehyde for 10 minutes at 4 °C. After washing the cells 3 times again with PBS, they were mounted on microscopic slides using a mounting medium containing DAPI (VECTASHIELD H1800, Vector Laboratories) and observed under a BZ‐X800 fluorescence microscope (Keyence). Image analysis was performed using ImageJ software (v1.44).
Statistical Analysis
All data are presented as the mean±SEM. To assume a normal distribution of data in principle, the ratiometric data in Figures 1C, 2B and 2C, 4B and 4D, 5B, Figures S1B and S1D, and S2B–E were log‐transformed using the log2X data transformation. The assumption of normal distribution and equal variance was applied to both the nonratiometric data and log‐transformed data because this study is exploratory in nature. Differences between 2 groups were statistically compared using unpaired t‐tests, and those between ≥3 groups were statistically compared using 1‐way ANOVA with Tukey honestly significant difference test for multiple pairwise comparisons. P values <0.05 indicated statistical significance. GraphPad Prism (v9.4.1, GraphPad Software) was used for preparing quantification panels, and JMP software (v16, SAS Institute) was used for all statistical analyses.
Figure 1. Deferasirox (DFX) reduces hypoxia (Hx)‐induced iron accumulation in the endoplasmic reticulum (ER) in cultured cardiomyocytes.
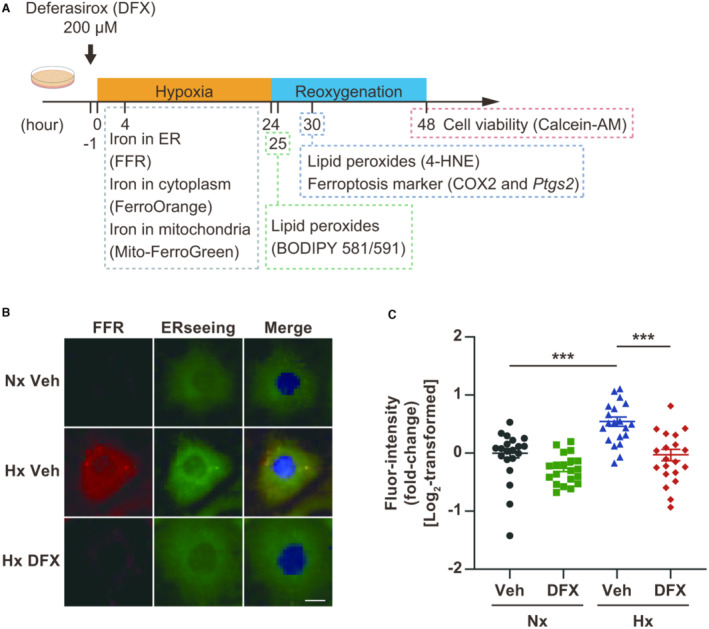
A, Experimental protocol of hypoxia‐reoxygenation (H/R) for inducing ferroptosis in cultured cardiomyocytes. B, Representative images of cardiomyocytes treated with FerroFarRed (FFR; red) to detect iron in the ER. The ER and nuclei were co‐stained with ERseeing (green) and DAPI (blue), respectively. Scale bar: 10 μm. C, Quantification of the fluorescence intensity of FFR in FFR‐stained cardiomyocytes (n=20, each group). Data are presented as the mean±SEM. Statistical significance was determined using 1‐way ANOVA with Tukey post hoc test. ***P<0.001. 4‐HNE indicates 4‐hydroxynonenal; BODIPY, boron‐dipyrromethene; COX2, cyclooxygenase 2; Nx, normoxia; and Veh, vehicle.
Figure 2. Deferasirox (DFX) suppresses lipid peroxidation caused by hypoxia/reoxygenation (H/R) in cultured cardiomyocytes.
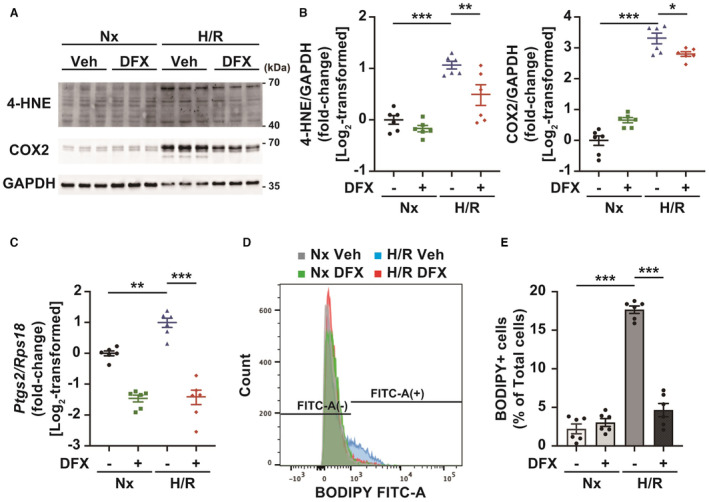
A, Western blotting for proteins modified by 4‐hydroxynonenal (4‐HNE) and cyclooxygenase 2 (COX2). GAPDH represents the internal control. B, Quantification of 4‐HNE protein modifications (left panel) and COX2 expression (right panel; n=6, each group). C, Gene expression of Ptgs2, measured by reverse transcription‐quantitative polymerase chain reaction (n=6, each group). D, Representative flow cytometric analysis of cultured cardiomyocytes probed with C11 boron‐dipyrromethene (BODIPY) after H/R. E, Quantification of C11‐BODIPY–positive cells (n=6, each group). Data are presented as the mean±SEM. Statistical significance was determined using 1‐way ANOVA with Tukey post hoc test. *P<0.05, **P<0.01, ***P<0.001.
Figure 4. Treatment with deferasirox (DFX) suppresses total myocardial and endoplasmic reticulum (ER) fraction iron contents in I/R‐injured myocardium.
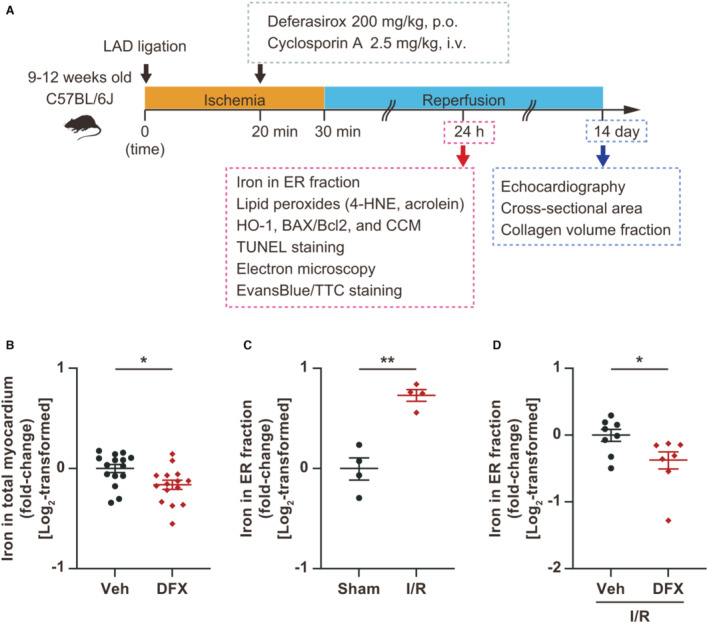
A, Experimental protocol for inducing ischemia reperfusion (I/R) in mice. B, Measurement of total myocardial iron levels 24 hours after DFX administration (200 mg/kg, per os [p.o.]; n=15, each group). C, Measurement of iron levels in the ER fraction of I/R‐injured myocardium 24 hours after I/R (n=4, each group). D, Measurement of iron levels in the ER fraction of I/R‐injured myocardium treated with vehicle or DFX (200 mg/kg, per os [p.o.]; n=8 in vehicle group and 7 in DFX group). Data are presented as the mean±SEM. Statistical significance was determined using unpaired t‐test. *P<0.05, **P<0.01. 4‐HNE indicates 4‐hydroxynonenal; CCM, cleaved caspase substrate motif; HO‐1, heme oxygenase‐1; Hx, hypoxia; LAD, left anterior descending artery; Nx, normoxia; TUNEL, terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling; and Veh, vehicle.
Figure 5. Treatment with deferasirox (DFX) reduces lipid peroxidation in ischemia reperfusion (I/R)‐injured myocardium.
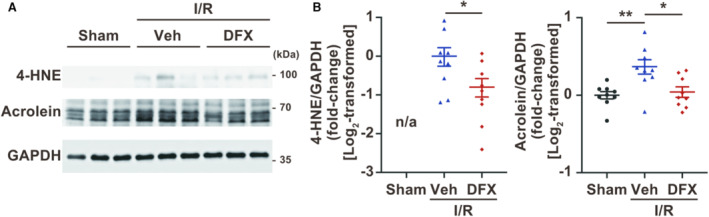
A, Western blotting for proteins modified by 4‐HNE and acrolein. GAPDH represents the internal control. B, Quantification of 4‐HNE protein modifications (left panel; n=9, each group) and acrolein protein modifications (right panel; n=9, each group). Data are presented as the mean±SEM. Statistical significance was determined using unpaired t‐tests (B, left panel) and 1‐way ANOVA with Tukey post hoc test (B, right panel). 4‐HNE indicates 4‐hydroxynonenal; n/a, not applicable; and Veh, vehicle. *P<0.05, **P<0.01.
Results
Deferasirox Suppresses H/R‐Induced Iron Overload, Lipid Peroxidation, and Ferroptosis in Cultured Cardiomyocytes
Ferroptosis was induced in cultured cardiomyocytes by H/R (Figure 1A), after which iron levels in the ER increased (Figure 1B and 1C) as previously reported. 20 Deferasirox treatment abolished hypoxia‐induced increases in the iron level within the ER (Figure 1B and 1C). Hypoxia did not increase iron levels in the cytoplasm or mitochondria, although deferasirox treatment reduced basal iron levels in both (Figure S1A through S1D). Consistent with these findings, deferasirox reduced H/R‐induced 4‐HNE modification of cardiac proteins in cultured cardiomyocytes, resulting in the suppression of cyclooxygenase 2 (COX2) expression, which was used as a potential marker for ferroptosis (Figure 2A and 2B). 18 The expression of Ptgs2, which encodes COX2, was upregulated by H/R; this upregulation was suppressed by deferasirox (Figure 2C). Deferasirox treatment also reduced the number of BODIPY–positive cells in cultured cardiomyocytes after H/R (Figure 2D and 2E). Furthermore, deferasirox significantly prevented H/R‐induced ferroptosis in cultured cardiomyocytes at concentrations of 10 to 200 μM (Figure 3A and 3B). Collectively, deferasirox effectively prevented H/R‐induced ferroptosis by chelating iron in cultured cardiomyocytes.
Figure 3. Deferasirox (DFX) prevents ferroptosis caused by hypoxia/reoxygenation (H/R) in cultured cardiomyocytes.
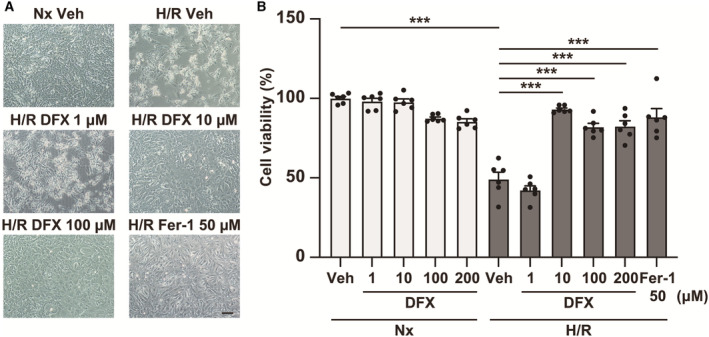
A, Representative images of cardiomyocytes subjected to H/R, treated with vehicle or 1–200 μM DFX. B, Cell viability of cultured cardiomyocytes subjected to H/R, treated with vehicle or 1 to 200 μM DFX (n=6, each group). Ferrostatin‐1 (Fer‐1) represents the ferroptosis inhibition positive control. Statistical significance was determined using 1‐way ANOVA with Tukey post hoc test. ***P<0.001. Hx indicates hypoxia; Nx, normoxia; and Veh, vehicle.
Deferasirox Reduces Total Myocardial and ER Fraction Iron Levels in I/R‐Injured Myocardium
We then examined the effect of deferasirox on the I/R‐injured myocardium following a protocol that was designed in accordance with the clinical scenario of AMI treatment (Figure 4A). A single 200 mg/kg (per os) dose of deferasirox suppressed total myocardial iron levels within 24 hours of treatment (Figure 4B). Moreover, iron levels in the ER fraction were increased in the I/R‐injured myocardium (Figure 4C), while deferasirox treatment significantly decreased them (Figure 4D).
Deferasirox Suppresses Lipid Peroxide Levels in a Murine Model of I/R
Deferasirox treatment also reduced lipid peroxide levels, represented by the modification of proteins with 4‐HNE and acrolein (Figure 5A and 5B) in the I/R‐injured myocardium, although deferasirox did not affect HO‐1 expression (Figure S2A and S2B). In contrast, apoptosis, represented by the BAX/BCL2 ratio and caspase cleavage, was also induced during I/R injury but was not inhibited by deferasirox treatment (Figure S2A and S2C through S2E). Collectively, deferasirox specifically suppressed ferroptosis in the I/R‐injured myocardium.
Deferasirox Reduces Myocardial Injury, Cell Death, and Infarct Size in a Mouse Model of I/R
Deferasirox treatment decreased the number of TUNEL‐positive cells in the I/R‐injured myocardium (Figure 6A and 6B). Given that deferasirox did not inhibit apoptosis in the I/R‐injured myocardium (Figure S2A and S2C through S2E), the reductions in the number of TUNEL‐positive cells were assumed to be attributed to reductions in ferroptosis. Mitochondrial abnormalities such as reduced cristae and ruptured outer mitochondrial membranes, which were examined by electron microscopy, are characteristic features of ferroptosis, 35 and electron microscopic analyses revealed that mitochondrial abnormalities in the I/R‐injured myocardium were reduced by deferasirox treatment (Figure 6C and 6D). Consistently, deferasirox treatment also significantly reduced the infarct size 24 hours after I/R (Figure 6E and 6F).
Figure 6. Treatment with deferasirox (DFX) reduces myocardial injury and infarct size in a murine model of ischemia reperfusion (I/R) injury.
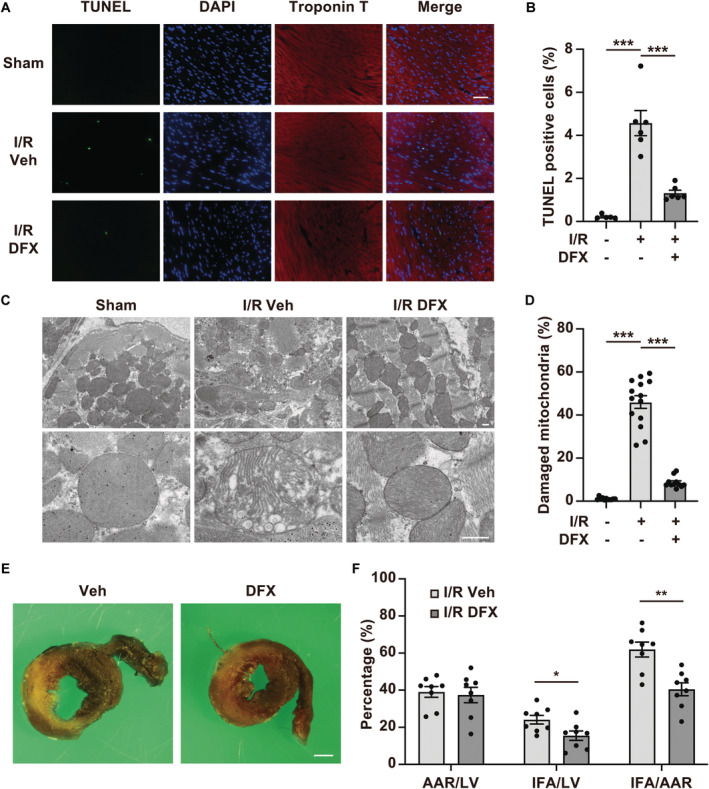
A, Representative terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) staining images of I/R‐injured myocardial tissues from mice treated with vehicle control (Veh) or DFX. Scale bar: 50 μm. B, Quantification of TUNEL‐positive cells 24 hours after reperfusion (n=5 in sham group, n=6 in I/R+Veh, n=6 in I/R+DFX). C, Representative images of I/R‐injured myocardium obtained by electron microscopy. Scale bar: 500 nm. D, Quantification of the number of damaged mitochondria (n=11 in sham group, n=14 in I/R+Veh, n=11 in I/R+DFX). The damaged mitochondria are presented as the proportion of damaged mitochondria to the total number of mitochondria. E, Representative images showing double staining with TTC and Evans Blue in hearts from mice with I/R after treatment with Veh or DFX. Scale bar: 1 mm. F, AAR per LV, IFA per LV, and IFA per L percentages in mice with I/R (n=8, each group). AAR indicates area at risk; IFA, infarct area; and LV, left ventricle. Data are presented as the mean±SEM. Statistical significance was determined using unpaired t‐test. *<0.05, **P<0.01, ***P<0.001.
Concomitant Treatment With Deferasirox and CsA Synergistically Reduces Infarct Size in a Mouse Model of I/R
CsA is an established therapeutic agent that prevents MPT‐driven necrosis in myocardial I/R injury by inhibiting cyclophilin D and MPT pore‐opening. 12 , 13 , 14 CsA treatment during ischemia significantly reduced the infarct size after I/R by approximately the same degree as deferasirox treatment did (Figure 7A and 7B). Meanwhile, administration of deferasirox with CsA further reduced the infarct size in I/R, suggesting that combination therapy exhibited an additive effect (Figure 7A and 7B).
Figure 7. Combination therapy with deferasirox (DFX) and cyclosporin A (CsA) synergistically reduces infarct size in a murine ischemia reperfusion (I/R) model.
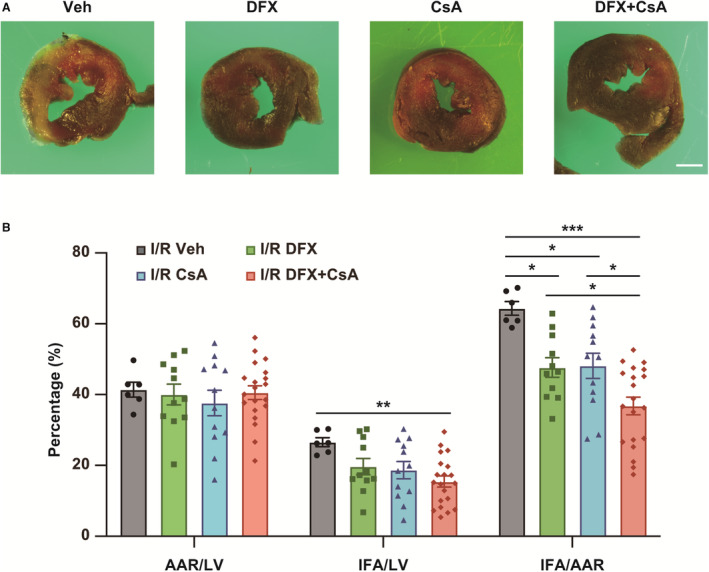
A, Representative images showing double staining with TTC and Evans Blue in I/R mice treated with vehicle control (Veh), DFX, CsA, or DFX+CsA. Scale bar: 1 mm. B, AAR per LV, IFA per LV, and IFA per AAR percentages in I/R mice. AAR indicates area at risk; IFA, infarct area; and LV, left ventricle. I/R Veh, n=6; I/R DFX, n=11; I/R CsA, n=12; I/R DFX+CsA, n=20. Data are presented as the mean±SEM. Statistical significance was determined using 1‐way ANOVA with Tukey post‐hoc test. *P<0.05, **P<0.01, ***P<0.001.
Combinational Deferasirox and CsA Therapy Ameliorates Adverse Cardiac Remodeling After I/R Injury
Finally, we examined the impact of combination therapy with deferasirox and CsA on adverse cardiac remodeling during the late phase of I/R, following the protocol shown in Figure 4A. Combination therapy significantly preserved left ventricular ejection fraction and left ventricular end‐systolic diameter while preventing LV dilation, represented by left ventricular end‐diastolic diameter, on day 14 after I/R (Figure 8A and Table). Moreover, combination therapy also alleviated cardiac hypertrophy, represented by the heart weight/tibial length (Figure 8B). Consistent with these findings, the cross‐sectional area and interstitial fibrosis were suppressed in I/R mice treated with deferasirox and CsA when compared with those of I/R mice treated with vehicle (Figure 8C and 8D).
Figure 8. Combination therapy with deferasirox (DFX) and cyclosporin A (CsA) prevents adverse cardiac remodeling in the late phase of ischemia reperfusion (I/R).
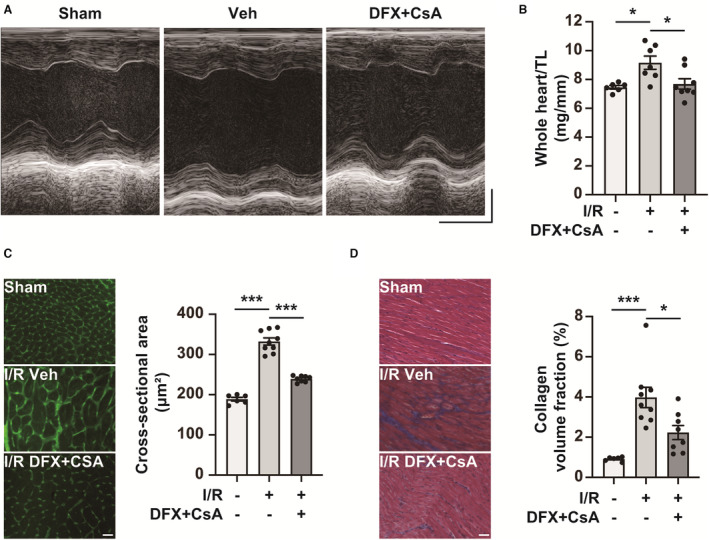
A, Representative M‐mode echocardiogram images of sham mice and I/R mice treated with vehicle control (Veh) or DFX+CsA on day 14 after I/R. Horizontal scale bar: 100 msec. Vertical scale bar: 1 mm. B, Whole heart weight per tibial length (TL) of sham mice (n=6) and I/R mice treated with Veh (n=7) or DFX+CsA (n=8) on day 14 after I/R. Two of the I/R mice treated with Veh were excluded because of their death after echocardiography. C, Cross‐sectional area on day 14 after I/R. Representative wheat germ agglutinin staining images of the left ventricle in sham mice and I/R mice treated with Veh or DFX+CsA (left panel). Scale bar: 50 μm. Quantification of cross‐sectional area (right panel; n=6 in sham group, n=9 in Veh group, n=8 in DFX+CsA group). D, Collagen volume fraction (interstitial fibrosis) on day 14 after I/R. Representative Masson trichrome staining of the left ventricle in mice with Veh or DFX+CsA (left panel). Scale bar: 50 μm. Quantification of interstitial fibrosis as the collagen volume fraction (right panel; n=6 in sham group, n=9 in Veh group, n=8 in DFX+CsA group). Data are presented as the mean±SEM. Statistical significance was determined using 1‐way ANOVA with Tukey post hoc test. *P<0.05, ***P<0.001.
Table .
Echocardiographic Parameters in a Murine Model of Ischemia/Reperfusion (I/R) on Day 14 After I/R Injury
| Variables | Sham | I/R | |
|---|---|---|---|
| Vehicle | Vehicle | Deferasirox+CsA | |
| n | 6 | 9 | 8 |
| LVEDD, mm | 3.4±0.3 | 4.4±0.4* | 3.9±0.4† |
| LVESD, mm | 2.1±0.3 | 3.6±0.5* | 2.8±0.4‡ |
| LVEF, % | 70±4 | 39±9* | 53±5§ |
Data are presented as the mean±SEM. Statistical significance was determined using 1‐way ANOVA with Tukey post hoc test. CsA indicates cyclosporin A; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; and LVESD, left ventricular end‐systolic diameter.
P<0.001 vs. sham+vehicle.
P<0.05
P<0.01
P<0.001 vs I/R+vehicle.
Discussion
I/R injury is recognized as a therapeutic target for further reducing the infarct size and preserving cardiac function after AMI. However, CsA monotherapy for I/R injury does not provide significant clinical benefits in terms of cardiac function during the late phase of MI. 16 As such, effective therapies for I/R injury remain an unmet medical need. 41 Here, we examined the suppressive effect of deferasirox, an Fe3+ chelator, on ferroptosis in cultured cardiomyocytes and a murine model of I/R injury and found 3 major findings as follows. First, deferasirox effectively suppressed H/R‐induced iron overload in the ER, lipid peroxidation, and ferroptosis in cultured cardiomyocytes. Second, deferasirox treatment during ischemia effectively reduced ferroptosis in the I/R‐injured myocardium; monotherapy with deferasirox or CsA during ischemia reduced the infarct size by similar extents after I/R injury. Third, combinational deferasirox and CsA therapy synergistically reduced the infarct size during I/R injury and ameliorated adverse cardiac remodeling in the late phase.
Ferroptosis is an emerging concept of iron‐dependent regulated cell death that is reportedly involved in physiological homeostasis and various pathological conditions. 42 Previously, we used glutathione peroxidase 4‐transgenic mice to demonstrate that ferroptosis and MPT‐driven necrosis are major modes of cell death under I/R conditions and that their dual inhibition represents a potentially viable therapeutic strategy for limiting I/R injury. 20 However, identification of an appropriate ferroptosis inhibitor that demonstrates cell permeability, capacity for diffusion into the heart, and safety in humans has proven challenging. Moreover, under the current clinical AMI treatment scenario, therapeutics against reperfusion injury should be initiated during ischemia, which further limits the scope of potential therapeutic candidates. We identified various potential ferroptosis inhibitors in drug screening, 21 some of which prevented H/R‐induced ferroptosis in cultured cardiomyocytes. However, most of the candidate inhibitors did not reduce the infarct size of a murine I/R model when initiated between coronary occlusion and recanalization in preliminary experiments. These results are consistent with the evidence that DXZ, when administered during ischemia, did not confer cardioprotection in a porcine I/R model, 24 whereas its initiation before ischemia has been shown to reduce the infarct size. 19 , 22 , 23 Deferasirox was developed to treat patients with iron overload caused by repeated transfusions. 27 Iron overload is referred to as hemochromatosis under these conditions, in which iron likely accumulates in multiple organs, including the liver and heart, resulting in cardiomyopathy. 27 Therefore, considering that deferasirox was developed to prevent iron accumulation in the heart and liver, it has a high affinity and rapidly diffusing capacity for the heart. Additionally, deferasirox reportedly exhibits superior cell permeability than other iron chelators such as deferoxamine and deferiprone. Furthermore, the safety of deferasirox in humans has been confirmed. Collectively, these characteristics support the protective potential of deferasirox over other iron chelators against myocardial I/R injury via ferroptosis inhibition.
In this study, we treated I/R model mice orally with deferasirox as this is the standard route for deferasirox administration in humans. 26 We previously demonstrated that the upregulation of HO‐1 expression in response to I/R causes iron overload in the ER after reperfusion and that the upregulation of HO‐1 expression occurs ≈ 6 hours after reperfusion. 20 These findings indicate that deferasirox should be delivered into the heart within 6 hours after reperfusion. Moreover, given that the time‐to‐maximum (Tmax) of deferasirox is 1.5 to 4 hours in humans, which may depend on the deferasirox dose (https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021882s019lbl.pdf), we deduced that oral deferasirox administration during ischemia could be effective in preventing ferroptosis in I/R. Furthermore, as administration of antiplatelet agents is required before percutaneous coronary intervention, concomitant oral administration of deferasirox with antiplatelet drugs may be clinically reasonable when treating AMI. Nevertheless, it may be necessary to reconsider the optimal route for deferasirox administration when using it clinically in this context in the future.
In this study, 200 mg/kg of deferasirox was used as the treatment dose in a murine I/R model. This dose is equivalent to 16 mg/kg in humans based on the human‐equivalent dose conversion factor (12.3, mice to humans), which is below the maximum deferasirox dose for humans (20 mg/kg) (https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021882s019lbl.pdf). The maximum plasma concentration (Cmax) and half‐life (T1/2) of deferasirox are approximately 50 μM and 17 hours in humans, respectively, after a single oral dose of 10 mg/kg deferasirox. Importantly, a plasma concentration of 10 μM, which prevents H/R‐induced ferroptosis in vitro, would be achieved in mice by administering a deferasirox dose of 200 mg/kg. However, further investigations are needed to optimize the dose of deferasirox against ferroptosis in patients with AMI.
Combination therapy with deferasirox and CsA is a reasonable strategy to improve their therapeutic efficacy against I/R injury, given that CsA treatment reduced the infarct size in an experimental I/R model and patients with AMI after reperfusion therapy, although no significant improvement in cardiac function in patients with late‐phase MI was observed in a phase 3 clinical trial. 16 Our previous and present findings show that dual therapy targeting ferroptosis and MPT‐driven necrosis markedly reduces the infarct size 24 hours after I/R injury in a murine model and preserves the left ventricular ejection fraction in the late phase of MI. This evidence supports the clinical application of combinational deferasirox and CsA therapy as a novel therapeutic approach for treating I/R injury in AMI.
Finally, it is important to note that ferroptosis in the I/R‐injured myocardium is initiated at the ER, although we observed damaged mitochondria through electron microscopy to be a characteristic feature of ferroptosis. Lipid radicals, which cause ferroptosis, initially arise in specific organelles, ultimately propagating to lipid bilayers in other organelles including the mitochondria and cell membrane, finally resulting in ferroptosis. 43 The mitochondria, possessing one of the largest membrane structures within the cell, may exhibit remarkable membrane damage under electron microscopy, which can be recognized as a feature of ferroptosis. Nevertheless, considering previous studies indicating the significance of Fe2+ in the ER (rather than in the mitochondria) in H/R‐induced ferroptosis, 20 the ER serves as the initial site for iron accumulation and generation of lipid radicals in I/R.
In conclusion, deferasirox is an effective inhibitor against I/R‐induced ferroptosis even when applied in accordance with clinical scenarios of AMI. Combination therapy with deferasirox and CsA may be a feasible approach for minimizing I/R injury during AMI and preserving cardiac function in the late phase after MI.
Sources of Funding
This work was supported by JSPS KAKENHI (grant numbers JP21K16090 to MI and JP20K08426 and JP23H02908 to TI), the Japan Foundation for Applied Enzymology (Vascular Biology of Innovation [VBIC] and Cardiovascular Innovative Conference [CVIC], to MI), the MSD Life Science Foundation, the Public Interest Incorporated Foundation (to MI), Novartis Pharma Grants for Basic Research 2020 (to MI), Kowa Life Science Foundation (to MI), SENSHIN Medical Research Foundation (to MI) and the Cardiovascular Research Fund (Tokyo, Japan) (to MI).
Disclosures
Tomomi Ide received research funding from SBI Pharmaceuticals and Pfizer Japan Co., Ltd. Hiroyuki Tsutsui received remunerations from Kowa, Teijin Pharma, Nippon Boehringer Ingelheim, Mitsubishi Tanabe Pharma, Pfizer Japan, Ono Pharmaceutical, Daiichi Sankyo, Novartis Pharma, Bayer Yakuhin, Otsuka Pharmaceutical, and AstraZeneca. Manuscript fees were received from Nippon Rinsho. Research funding was received from Mitsubishi Tanabe Pharma, Nippon Boehringer Ingelheim, IQVIA Services Japan, MEDINET, Medical Innovation Kyushu, Kowa, Daiichi Sankyo, Johnson & Johnson, and NEC Corporation. Scholarship funds or donations were received from Abbott Medical Japan, Otsuka Pharmaceutical, Boston Scientific Japan, Ono Pharmaceutical, Bayer Yakuhin, Nippon Boehringer Ingelheim, St. Mary's Hospital, Teijin Pharma, Daiichi Sankyo, and Mitsubishi Tanabe Pharma. A patent pertaining to the data presented in this paper is pending. This patent application has been submitted in collaboration with Sawai Pharmaceutical Co., LTD. (Osaka, Japan), and the inventors on the patent are K.I., M.I., and T.I. The remaining authors have no disclosures to report.
Supporting information
Figures S1–S2
Acknowledgments
We thank Midori Sato for providing excellent experimental technical support.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.031219
This manuscript was sent to Julie K. Freed, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 14.
REFERENCES
- 1. Braunwald E, Kloner RA. Myocardial reperfusion: a double‐edged sword? J Clin Invest. 1985;76:1713–1719. doi: 10.1172/JCI112160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simoons ML, Serruys PW, van den Brand M, Res J, Verheugt FW, Krauss XH, Remme WJ, Bar F, de Zwaan C, van der Laarse A, et al. Early thrombolysis in acute myocardial infarction: limitation of infarct size and improved survival. J Am Coll Cardiol. 1986;7:717–728. doi: 10.1016/s0735-1097(86)80329-1 [DOI] [PubMed] [Google Scholar]
- 3. White HD, Norris RM, Brown MA, Takayama M, Maslowski A, Bass NM, Ormiston JA, Whitlock T. Effect of intravenous streptokinase on left ventricular function and early survival after acute myocardial infarction. N Engl J Med. 1987;317:850–855. doi: 10.1056/NEJM198710013171402 [DOI] [PubMed] [Google Scholar]
- 4. Sheehan FH, Doerr R, Schmidt WG, Bolson EL, Uebis R, von Essen R, Effert S, Dodge HT. Early recovery of left ventricular function after thrombolytic therapy for acute myocardial infarction: an important determinant of survival. J Am Coll Cardiol. 1988;12:289–300. doi: 10.1016/0735-1097(88)90397-x [DOI] [PubMed] [Google Scholar]
- 5. Global Use of Strategies to Open Occluded Coronary Arteries in Acute Coronary Syndromes (GUSTO IIb) Angioplasty Substudy Investigators . A clinical trial comparing primary coronary angioplasty with tissue plasminogen activator for acute myocardial infarction. N Engl J Med. 1997;336:1621–1628. doi: 10.1056/NEJM199706053362301 [DOI] [PubMed] [Google Scholar]
- 6. Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20. doi: 10.1016/S0140-6736(03)12113-7 [DOI] [PubMed] [Google Scholar]
- 7. Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D'Agostino RB, Levy D, Kannel WB, Vasan RS. Long‐term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005 [DOI] [PubMed] [Google Scholar]
- 9. Griffiths EJ, Halestrap AP. Mitochondrial non‐specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–98. doi: 10.1042/bj3070093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200 [DOI] [PubMed] [Google Scholar]
- 12. Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia‐reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025 [DOI] [PubMed] [Google Scholar]
- 13. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317 [DOI] [PubMed] [Google Scholar]
- 14. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- 15. Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142 [DOI] [PubMed] [Google Scholar]
- 16. Cung TT, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D, Bonnefoy‐Cudraz E, Guerin P, Elbaz M, Delarche N, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489 [DOI] [PubMed] [Google Scholar]
- 17. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyamoto HD, Ikeda M, Ide T, Tadokoro T, Furusawa S, Abe K, Ishimaru K, Enzan N, Sada M, Yamamoto T, et al. Iron overload via heme degradation in the endoplasmic reticulum triggers ferroptosis in myocardial ischemia‐reperfusion injury. JACC Basic Transl Sci. 2022;7:800–819. doi: 10.1016/j.jacbts.2022.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tadokoro T, Ikeda M, Abe K, Ide T, Miyamoto HD, Furusawa S, Ishimaru K, Watanabe M, Ishikita A, Matsushima S, et al. Ethoxyquin is a competent radical‐trapping antioxidant for preventing ferroptosis in doxorubicin cardiotoxicity. J Cardiovasc Pharmacol. 2022;80:690–699. doi: 10.1097/FJC.0000000000001328 [DOI] [PubMed] [Google Scholar]
- 22. Ramu E, Korach A, Houminer E, Schneider A, Elami A, Schwalb H. Dexrazoxane prevents myocardial ischemia/reperfusion‐induced oxidative stress in the rat heart. Cardiovasc Drugs Ther. 2006;20:343–348. doi: 10.1007/s10557-006-0497-4 [DOI] [PubMed] [Google Scholar]
- 23. Neckář J, Boudíková A, Mandíková P, Stěrba M, Popelová O, Mikšik I, Dabrowská L, Mráz J, Geršl V, Kolář F. Protective effects of dexrazoxane against acute ischaemia/reperfusion injury of rat hearts. Can J Physiol Pharmacol. 2012;90:1303–1310. doi: 10.1139/y2012-096 [DOI] [PubMed] [Google Scholar]
- 24. Kamat P, Vandenberghe S, Christen S, Bongoni AK, Meier B, Rieben R, Khattab AA. Dexrazoxane shows no protective effect in the acute phase of reperfusion during myocardial infarction in pigs. PLoS One. 2016;11:e0168541. doi: 10.1371/journal.pone.0168541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jirkovský E, Jirkovská A, Bavlovič‐Piskáčková H, Skalická V, Pokorná Z, Karabanovich G, Kollárová‐Brázdová P, Kubeš J, Lenčová‐Popelová O, Mazurová Y, et al. Clinically translatable prevention of anthracycline cardiotoxicity by dexrazoxane is mediated by topoisomerase II beta and not metal chelation. Circ Heart Fail. 2021;14:e008209. doi: 10.1161/CIRCHEARTFAILURE.120.008209 [DOI] [PubMed] [Google Scholar]
- 26. Nolte F, Hochsmann B, Giagounidis A, Lubbert M, Platzbecker U, Haase D, Luck A, Gattermann N, Taupitz M, Baier M, et al. Results from a 1‐year, open‐label, single arm, multi‐center trial evaluating the efficacy and safety of oral deferasirox in patients diagnosed with low and int‐1 risk myelodysplastic syndrome (MDS) and transfusion‐dependent iron overload. Ann Hematol. 2013;92:191–198. doi: 10.1007/s00277-012-1594-z [DOI] [PubMed] [Google Scholar]
- 27. Glickstein H, El RB, Link G, Breuer W, Konijn AM, Hershko C, Nick H, Cabantchik ZI. Action of chelators in iron‐loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood. 2006;108:3195–3203. doi: 10.1182/blood-2006-05-020867 [DOI] [PubMed] [Google Scholar]
- 28. Deguchi H, Ikeda M, Ide T, Tadokoro T, Ikeda S, Okabe K, Ishikita A, Saku K, Matsushima S, Tsutsui H. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice. Circ J. 2020;84:1028–1033. doi: 10.1253/circj.CJ-19-1039 [DOI] [PubMed] [Google Scholar]
- 29. Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada K, Kang D, et al. Overexpression of TFAM or Twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10:e0119687. doi: 10.1371/journal.pone.0119687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, Kakino T, Oga Y, Nishizaki A, Sunagawa K. The Akt‐mTOR axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci Rep. 2015;5:15881. doi: 10.1038/srep15881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abe K, Ikeda M, Ide T, Tadokoro T, Miyamoto HD, Furusawa S, Tsutsui Y, Miyake R, Ishimaru K, Watanabe M, et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1‐dependent heme synthesis. Sci Signal. 2022;15:eabn8017. doi: 10.1126/scisignal.abn8017 [DOI] [PubMed] [Google Scholar]
- 32. Ikeda M, Ide T, Furusawa S, Ishimaru K, Tadokoro T, Miyamoto HD, Ikeda S, Okabe K, Ishikita A, Abe K, et al. Heart rate reduction with ivabradine prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Drugs Ther. 2022;36:257–262. doi: 10.1007/s10557-020-07123-5 [DOI] [PubMed] [Google Scholar]
- 33. Ikeda M, Ide T, Tadokoro T, Miyamoto HD, Ikeda S, Okabe K, Ishikita A, Sato M, Abe K, Furusawa S, et al. Excessive hypoxia‐inducible factor‐1α expression induces cardiac rupture via p53‐dependent apoptosis after myocardial infarction. J Am Heart Assoc. 2021;10:e020895. doi: 10.1161/JAHA.121.020895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inoue T, Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, Sunagawa K. Twinkle overexpression prevents cardiac rupture after myocardial infarction by alleviating impaired mitochondrial biogenesis. Am J Physiol Heart Circ Physiol. 2016;311:H509–H519. doi: 10.1152/ajpheart.00044.2016 [DOI] [PubMed] [Google Scholar]
- 35. Yu H, Yan J, Li Z, Yang L, Ju F, Sun Y. Recent trends in emerging strategies for ferroptosis‐based cancer therapy. Nanoscale Adv. 2023;5:1271–1290. doi: 10.1039/D2NA00719C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujino T, Ide T, Yoshida M, Onitsuka K, Tanaka A, Hata Y, Nishida M, Takehara T, Kanemaru T, Kitajima N, et al. Recombinant mitochondrial transcription factor A protein inhibits nuclear factor of activated T cells signaling and attenuates pathological hypertrophy of cardiac myocytes. Mitochondrion. 2012;12:449–458. doi: 10.1016/j.mito.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 37. Ikeda M, Ide T, Matsushima S, Ikeda S, Okabe K, Ishikita A, Tadokoro T, Sada M, Abe K, Sato M, et al. Immunomodulatory cell therapy using αGalCer‐pulsed dendritic cells ameliorates heart failure in a murine dilated cardiomyopathy model. Circ Heart Fail. 2022;15:e009366. doi: 10.1161/CIRCHEARTFAILURE.122.009366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arai S, Ikeda M, Ide T, Matsuo Y, Fujino T, Hirano K, Sunagawa K, Tsutsui H. Functional loss of DHRS7C induces intracellular Ca2+ overload and myotube enlargement in C2C12 cells via calpain activation. Am J Physiol Cell Physiol. 2017;312:C29–C39. doi: 10.1152/ajpcell.00090.2016 [DOI] [PubMed] [Google Scholar]
- 39. Furusawa S, Ikeda M, Ide T, Kanamura T, Miyamoto HD, Abe K, Ishimaru K, Watanabe M, Tsutsui Y, Miyake R, et al. Cardiac autoantibodies against cardiac troponin I in post‐myocardial infarction heart failure: evaluation in a novel murine model and applications in therapeutics. Circ Heart Fail. 2023;16:e010347. doi: 10.1161/CIRCHEARTFAILURE.122.010347 [DOI] [PubMed] [Google Scholar]
- 40. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI, et al. Mitochondria‐dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747. doi: 10.1172/jci.insight.132747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol. 2019;73:89–99. doi: 10.1016/j.jacc.2018.09.086 [DOI] [PubMed] [Google Scholar]
- 42. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–937. doi: 10.1016/j.cmet.2020.10.011 [DOI] [PubMed] [Google Scholar]
- 43. Henkel RR. Leukocytes and oxidative stress: dilemma for sperm function and male fertility. Asian J Androl. 2011;13:43–52. doi: 10.1038/aja.2010.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2