

Abstract
Objective
Evaluation of the clinical utility of a genetic diagnosis in CP remains limited. We aimed to characterize the clinical utility of a genetic diagnosis by exome sequencing (ES) in patients with CP and related motor disorders.
Methods
We enrolled participants with CP and “CP masquerading” conditions in an institutional ES initiative. In those with genetic diagnoses who had clinical visits to discuss results, we retrospectively reviewed medical charts, evaluating recommendations based on the genetic diagnosis pertaining to medication intervention, surveillance initiation, variant‐specific testing, and patient education.
Results
We included 30 individuals with a molecular diagnosis and clinical follow‐up. Nearly all (28 out of 30) had clinical impact resulting from the genetic diagnosis. Medication interventions included recommendation of mitochondrial multivitamin supplementation (6.67%, n = 2), ketogenic diet (3.33%, n = 1), and fasting avoidance (3.33%, n = 1). Surveillance‐related actions included recommendations for investigating systemic complications (40%, n = 12); referral to new specialists to screen for systemic manifestations (33%, n = 10); continued follow‐up with established specialists to focus on specific manifestations (16.67%, n = 5); referral to clinical genetics (16.67%, n = 5) to oversee surveillance recommendations. Variant‐specific actions included carrier testing (10%, n = 3) and testing of potentially affected relatives (3.33%, n = 1). Patient education‐specific actions included referral to experts in the genetic disorder (30%, n = 9); and counseling about possible changes in prognosis, including recognition of disease progression and early mortality (36.67%, n = 11).
Interpretation
This study highlights the clinical utility of a genetic diagnosis for CP and “CP masquerading” conditions, evident by medication interventions, surveillance impact, family member testing, and patient education, including possible prognostic changes.
Introduction
Cerebral palsy (CP) refers to a “group of permanent disorders of the development of movement and posture, causing activity limitation, that are attributed to a non‐progressive disturbance that occurred in the developing fetal or infant brain.” 1 CP can be classified by the topographical distribution of motor impairment (monoplegia, diplegia, triplegia, hemiplegia, and quadriplegia) and by the predominant tone abnormality involved (spastic, dyskinetic, hypotonic‐ataxic, or mixed). 2 CP is a descriptive diagnosis that does not confer etiology. 3 Common acquired risk factors include prematurity, intrapartum asphyxia, infection, perinatal stroke, and intracranial hemorrhage. 4 , 5 Individuals with no risk factors underlying the diagnosis of CP are deemed to have “cryptogenic CP.” 6 In some cases, individuals have a diagnosis of, or symptoms that resemble, CP, yet they also have findings that are inconsistent with the definition, such as regression or progressive symptoms; these individuals may have CP‐masquerading conditions. 7
Recent studies have shown that a substantial portion of individuals with CP, particularly cryptogenic CP and CP masquerading conditions, may have an underlying monogenic disorder, which may require specific monitoring and treatment. One meta‐analysis determined that 35% of individuals with cryptogenic CP and 7% of individuals with non‐cryptogenic CP have a genetic diagnosis established by exome sequencing (ES). 8
Despite emerging literature about the contribution of genetics to CP and related motor disorders, evaluation of the clinical utility of a genetic diagnosis for individuals with CP and CP masquerading conditions remains limited. To address this gap, we characterized the clinical utility of a genetic diagnosis in individuals with CP and CP‐masquerading conditions, using retrospective analysis of clinical data.
Methods
Participant recruitment/selection and CP classification
Participants were part of the IRB‐approved Boston Children's Hospital (BCH) CP Sequencing Study, which falls under the Children's Rare Disease Cohorts (CRDC) initiative, an institutional effort facilitating ES for different disease cohorts. 9 The goal of the BCH CP Sequencing Study is to conduct research ES for patients with CP and CP masqueraders. We included individuals who fulfilled standard criteria for CP. 10 We also included individuals whom we termed “CP masqueraders”; we defined these as individuals with the following two core features: (1) abnormalities in the development of movement and posture, causing activity limitation, attributed to disturbances in the developing fetal or infant brain, often accompanied by disturbances of sensation, perception, cognition, communication, and behavior, by epilepsy, and by secondary musculoskeletal problems; (2) history of developmental regression, progressive symptoms, or other findings inconsistent with the definition of CP. In essence, CP masqueraders are individuals who might meet clinical criteria for CP apart from regression or progressive symptoms.
For individuals who fulfilled standard criteria for CP, we designated whether the CP was cryptogenic or non‐cryptogenic. Based on review of medical records, if the participant had any CP‐associated risk factors, as previously defined, 11 we described the CP as non‐cryptogenic. Otherwise, if the participant had no known risk factors, we described the CP as cryptogenic. The risk factors are as follows: (1) prematurity (≤32 weeks); (2) periventricular/intraventricular hemorrhage; (3) intracranial hemorrhage; (4) perinatal stroke; (5) evidence of other acute perinatal event (such as acute onset of decreased fetal movements); (6) hypoxic ischemic injury; (7) kernicterus; (8) fetal infection; (9) maternal infection at delivery leading to sepsis in the mother; (10) neonatal infection leading to sepsis; (11) neonatal respiratory arrest; (12) neonatal cardiac arrest; (13) hydrocephalus; (14) traumatic brain injury. For each participant, we designated a primary motor phenotype (spastic diplegic, spastic quadriplegic, spastic hemiplegic, dyskinetic, or hypotonic‐ataxic).
Participants could be any age or sex. We excluded participants with a known molecular diagnosis prior to study enrollment that completely explained their phenotype. Patients provided written informed patient consent for this study.
Exome sequencing and analysis
Participants underwent research ES, with parental DNA included for trio ES when possible. Full methods regarding DNA isolation, ES, and variant prioritization/interpretation are described in our initial cohort analysis. 11 We classified variants based on American College of Medical Genetics and Genomics (ACMG) criteria. 12 We clinically confirmed pathogenic/likely pathogenic (P/LP) variants through direct sequencing by GeneDx, which generated a Clinical Laboratory Improvement Amendments (CLIA) report. We considered P/LP variants in genes broadly associated with neurological or neurodevelopmental phenotypes, including genes not explicitly linked to CP. We have previously published the results of first 50 cases in this effort, 11 which now includes 193 probands.
Assessment of clinical utility
Assessment of clinical utility was not part of the research protocol but rather based on retrospective review of clinical notes pertaining to return of results visits. Participants with a clinically confirmed P/LP variant in a human disease gene had a clinical visit or phone call with S.S. to discuss results. We retrospectively reviewed how the molecular diagnosis impacted clinical care in this group of individuals. Specifically, we assessed whether an action occurred in any the following nine domains across four major themes.
Medication intervention
Suggested treatment changes. Recommendations for changes in the treatment plan including medications and diet.
Surveillance initiation
-
2
Recommendations for surveillance studies. Recommendations for new tests to investigate neurological or non‐neurological manifestations of the genetic disorder.
-
3
Referrals to new specialists. Referrals to new specialists to screen for specific systemic manifestations of the genetic disorder. For example, this category may entail referral to nephrology to screen for nephropathy in a patient with a genetic condition associated with this finding.
-
4
Recommendations for continued follow‐up with existing specialists. Recommendations for continued follow‐up with established specialists to screen for, or continue to manage, manifestations of the genetic disorder, based on knowledge of specific systemic manifestations of the disorder. For example, this category may entail referral to screen for optic atrophy in a patient already seeing ophthalmology for general vision assessment.
-
5
Referrals to clinical genetics for facilitation of surveillance. Referrals to clinical genetics for help with overseeing surveillance plan regarding genetic disorder‐related features, ensuring appropriate existing surveillance is underway or facilitating additional surveillance as needed.
Variant‐specific testing/genetic counseling needs
-
6
Recommendations for carrier testing. Recommendations for testing of unaffected relatives to see if they are carriers (heterozygous for variant in a gene implicated in an autosomal recessive or X‐linked disorder).
-
7
Recommendations for targeted testing. Recommendations for targeted testing of potentially affected relatives (individuals who may have symptoms or conditions possibly related to the proband's gene but no molecular diagnosis).
Patient education
-
8
Referrals to experts in the specific genetic disorder for education. Referrals to expert in the genetic disorder or category of genetic disorder who have expertise with the pathophysiology, natural history, or related research studies of the specific genetic disorder. For example, this category may entail referral a specialist who is known for conducting research on a specific genetic diagnosis.
-
9
Counseling about possible prognosis changes. Counseling about possible changes in prognosis, including discussion about the possibility that the clinical course may be progressive rather than static and the possibility of early death.
Conceptually, there may be some overlap among these categories; however, practically, these categories may mirror clinical practice (e.g., a patient may have discussion with the provider about sending specific tests while also receiving a referral to a specialist who would assume ownership over that aspect of surveillance). For each of the four themes, the theme was “positive” if an action occurred in any of the domains within that theme.
Statistics
We used descriptive statistics to describe percentage of the cohort impacted in each of these themes and domains.
Results
Demographics, genetic results
As of 24 April 2023, 193 probands in the BCH CP Sequencing Study had undergone research ES analysis. There were 10 CP masqueraders, 81 with cryptogenic CP, 96 with non‐cryptogenic CP, and 6 participants with classification that was unknown due to limited details about perinatal or early developmental history. We identified a P/LP variant in a human disease gene in 40 individuals: 6/10 (60%) in the CP masquerader category, 25 out of 81 (31%) in the cryptogenic CP category, 7 out of 96 (7%) in the non‐cryptogenic CP category, and 2 out of 6 (33%) in the unknown category.
For this analysis, we focused on those probands who had undergone clinical evaluation (n = 25) or phone visit (n = 5) to discuss study results, amounting to a total of 30 probands out of the 40 participants with P/LP variants. Regarding the other 10 individuals with P/LP not included in this analysis, two had been seen after the cutoff date for the retrospective chart review. One had not been seen for return of results due to the patient being out of region. Three had not been seen due to scheduling difficulties. For four patients, the eventually assigned P/LP variants had been noted before (the patients had been enrolled due to concerns for additional features not entirely explained by the variants), but these variants were still felt to be the most likely causative ones; these participants were de‐prioritized for return of results.
The 30 individuals in this current analysis included all 13 individuals with P/LP variants reported in our initial cohort analysis, 11 plus an additional 17 individuals. Detailed information about the demographics, motor phenotype, and genetic variants in these 30 individuals is shown in Table 1. Among these 30 individuals, 5 were in the CP masquerader category, 18 were in the cryptogenic CP category, 5 were in the non‐cryptogenic CP category, and 2 had classification that was unknown due to limited details about perinatal or early developmental history. The most common genes affected were ATL1, COL4A1, and SPAST (n = 2 participants with a P/LP variant in that gene), and all other genes were associated with one participant with a variant in that gene.
Table 1.
Demographics and variant characteristics of individuals with a genetic diagnosis in the cohort.
| Family type | Sex | Classification | Primary motor phenotype | Gene | Variant | Inheritance | Zygosity | ACGME classification |
|---|---|---|---|---|---|---|---|---|
| Trio | F | Spastic diplegic | B4GALNT1 | NM_001478.4: c.1149_1156del (p.Gly384AlafsTer51); NM_001478.4: c.1072G>A (p.Asp358Asn) | Paternally inherited, maternally inherited | Compound heterozygous | L/P, VOUS | |
| Proband | F | Spastic diplegic | SPTAN1 | NM_001130438.3: c.55C>T (p.Arg19Trp) | Unknown | Heterozygous | L/P | |
| Trio | F | CP masquerader | Dyskinetic | ECHS1 | NM_004092.3: c.458A>G (p.Tyr153Cys); NM_004092.3: c.161G>A (p.Arg54His) | Maternally inherited, paternally inherited | Compound heterozygous | L/P, P |
| Trio | F | CP masquerader | Spastic quadriplegic | RARB | NM_000965.4: c.638 T>C (p.Leu213Pro) | De novo | Heterozygous | L/P |
| Trio | F | CP masquerader | Spastic diplegic | HEXA | NM 000520.4: c.754C>T (p.Arg252Cys); NM 000520.4: c.1274 1277dup (p.Tyr427IlefsTer5) | Paternally inherited, maternally inherited | Compound heterozygous | L/P, P |
| Duo | M | CP masquerader | Spastic diplegic | SPAST | NM_014946.3: c.1168A>G (p.Met390Val) | Not maternally inherited | Heterozygous | P |
| Proband | F | CP masquerader | Spastic quadriplegic | SPAST | NM_014946.3: c.1085C>T (p.Ser362Phe) | Unknown | Heterozygous | L/P |
| Trio | M | Cryptogenic CP | Spastic diplegic | ADAT3 | NM_138422.2: c.430G>A (p.Val144Met) | Maternally and paternally inherited | Homozygous | P |
| Duo | F | Cryptogenic CP | Spastic quadriplegic | THOC2 | NM_001081550.1: c.1550A>G (p.Tyr517Cys) | De novo | Heterozygous | P |
| Duo | F | Cryptogenic CP | Spastic diplegic | MAPK8IP3 | NM 001040439.1: c.60C>A (p.Cys20Ter) | Not maternally inherited | Heterozygous | P |
| Trio | M | Cryptogenic CP | Spastic quadriplegic | ATL1 | NM 015915.4:c.1220A>T (p.Lys407Met) | De novo | Heterozygous | L/P |
| Duo | M | Cryptogenic CP | Spastic quadriplegic | SLC16A2 | NM_006517.4: c.148G>T (p.Glu50Ter) | De novo | Hemizygous | P |
| Trio | F | Cryptogenic CP | Spastic diplegic | GNB1 | NM_002074.5: c.239 T>C (p.Ile80Thr) | De novo | Heterozygous | P |
| Trio | F | Cryptogenic CP | Spastic quadriplegic | SLC2A1 | NM 006516.2: c.1030dup (p.Met344AsnfsTer37) | De novo | Heterozygous | L/P |
| Trio | M | Cryptogenic CP | Hypotonic‐ataxic | POLR2A | NM_000937.4: c.3922 T>A (p.Tyr1308Asn) | De novo | Heterozygous | L/P |
| Trio | F | Cryptogenic CP | Spastic diplegic | GNAO1 | NM_020988.2: c.625C>T (p.Arg209Cys) | De novo | Heterozygous | P |
| Trio | M | Cryptogenic CP | Dyskinetic | CTNNB1 | NM_001904.4: c.725_731dup (p.Gly245AsnfsTer28) | De novo | Heterozygous | P |
| Trio | F | Cryptogenic CP | Hypotonic‐ataxic | PDHX | NM_003477.2: c.1345del (p.Leu449Ter) | Maternally and paternally inherited | Homozygous | L/P |
| ACADM | NM_000016.4: c.799G>A (p.Gly267Arg) | Maternally and paternally inherited | Homozygous | P | ||||
| Trio | M | Cryptogenic CP | Spastic diplegic | KCNB1 | NM_004975.4: c.934C>T (p.Arg312Cys) | De novo | Heterozygous | L/P |
| Trio | M | Cryptogenic CP | Spastic diplegic | ATL1 | NM_015915.4: c.756C>A (p.Asn252Lys) | De novo | Heterozygous | P |
| Trio | M | Cryptogenic CP | Spastic hemiplegic | CHD3 | NM_001005271.3: c.3689A>G (p.His1230Arg) | De novo | Heterozygous | L/P |
| Trio | F | Cryptogenic CP | Spastic hemiplegic | ASXL3 | NM_030632.3: c.4034_4035dup (p.Ile1346ProfsTer15) | De novo | Heterozygous | P |
| Trio | M | Cryptogenic CP | Hypotonic‐ataxic | CHD8 | NM_001170629.2: c.4645dup (p.Ala1549GlyfsTer8) | De novo | Heterozygous | L/P |
| Trio | M | Cryptogenic CP | Spastic quadriplegic | FOXG1 | NM_005249.5: c.256del (p.Gln86ArgfsTer106) | Not maternally inherited | Heterozygous | P |
| Trio | F | Cryptogenic CP | Hypotonic‐ataxic | ABHD16A | NM_021160.3: c.1402C>T (p.Arg468Ter) | Maternally and paternally inherited | Homozygous | L/P |
| Trio | M | Non‐cryptogenic CP | Spastic hemiplegic | SATB2 | NM_015265.3: c.715C>T (p.Arg239Ter) | De novo | Heterozygous | P |
| Trio | F | non‐cryptogenic CP | spastic quadriplegic | ZMYM2 | NM_003453.4: c.2843dup (p.Glu949ArgfsTer11) | De novo | Heterozygous | P |
| Trio | M | Non‐cryptogenic CP | Spastic quadriplegic | COL4A1 | NM_001845.4: c.443G>A (p.Gly148Glu) | De novo | Heterozygous | L/P |
| Duo | F | Non‐cryptogenic CP | Spastic quadriplegic | COL4A1 | NM 001845.4: c.3592G>A (p.Gly1198Arg) | Not maternally inherited | Heterozygous | P |
| Trio | F | Non‐cryptogenic CP | Spastic diplegic | RASA1 | NM_002890.3: c.1494_1495insCTAC (p.Gly499LeufsTer3) | Maternally inherited | Heterozygous | L/P |
There were two participants with classification (cryptogenic CP, non‐cryptogenic CP, or CP masquerader) that was unknown due to limited details about perinatal or early developmental history.
F, female; L/P, likely pathogenic; M, male; P, pathogenic; VOUS, variant of uncertain significance.
As previously reported, 11 retrospective review showed that the ECHS1 variant and ADAT3 variant had been identified prior to, but not known at the time of, enrollment; these variants were independently identified using our pipeline. Additionally, the THOC2 variant had been identified by our pipeline but not prioritized as our research analysis was with a proband‐parent duo; this variant was, however, identified on clinical ES for that individual as part of trio analysis. One of the two participants with an ATL1 variant was known to have this variant (as detected on a gene panel) prior to enrollment, but there was uncertainty about whether it explained the full severity of the individual's motor phenotype; after enrollment and research ES analysis, we determined that this variant was indeed the full explanation for the individual's presentation. Finally, the individual with the HEXA variant had undergone biochemical testing in parallel to research ES, and biochemical testing suggested the diagnosis in advance of genetic sequencing.
Clinical utility
The majority of probands with return of genetics results visits (93%, 28 out of 30) had clinical impact due to the genetic diagnosis as defined by an action occurring in any of domains #1–9, summarized by the four themes of Medication Intervention, Surveillance Initiation, Variant‐Specific Testing, and Patient Education (Fig. 1). Ten percent (3 out of 10) had possible medication intervention (domain #1); 70% (21/30) underwent surveillance initiation (domains #2, #3, #4, and/or #5); 13% (4 out of 30) had variant testing (domains #6 and/or #7); and 43% (13 out of 30) had an action pertaining to patient education (domains #8 and/or #9). The domains with relatively high percentage (≥30%) of associated action were Recommendations for surveillance studies (40%, 12 out of 30), Referrals to new specialists (33.33%, 10 out of 30), Referrals to experts in the specific genetic disorder for education (30%, 9 out of 30), and Counseling about possible prognosis changes (36.67%, 11 out of 30).
Figure 1.
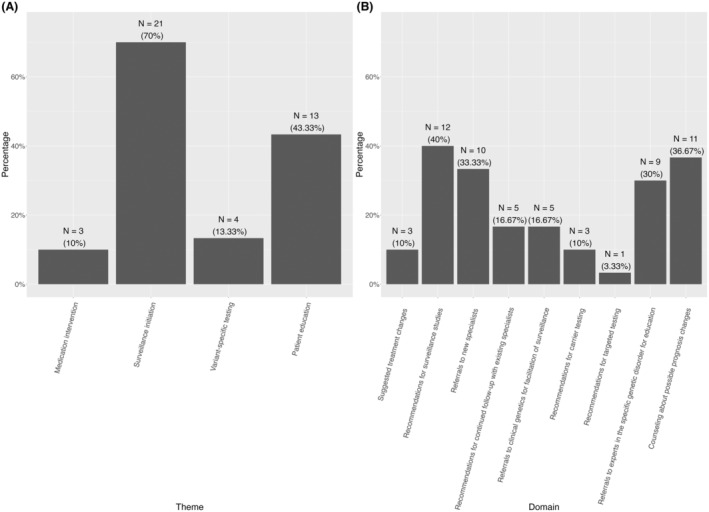
Clinical impact for individuals with a genetic diagnosis in the cohort pertaining to (A) 4 major themes (B) across nine domains. For each of the four themes, the theme was “positive” if an action occurred in any of the domains within that theme. The Medication Intervention theme corresponds to the domains of Suggested treatment changes. The Surveillance Initiation theme corresponds to the domains of Recommendations for surveillance studies; Referrals to new specialists; Recommendations for continued follow‐up with existing specialists; and Referrals to clinical genetics for facilitation of surveillance. The Variant‐Specific Testing theme corresponds to the domains of Recommendations for carrier testing and Recommendations for targeted testing. The Patient Education theme corresponds to the domains of Referrals to experts in the specific genetic disorder for education and Counseling about possible prognosis changes.
Medication intervention
Three (10%) participants had suggestions for treatment changes (Table 2). Two (6.67%) participants received recommendations to consider mitochondrial multivitamin supplementation. One participant (3.33%) received a recommendation to consider the ketogenic diet. One participant (3.33%) received a recommendation to avoid fasting.
Table 2.
Suggestions for medication/dietary interventions recommended to individuals with a genetic diagnosis in the cohort.
| Number of individuals who received this recommendation | Percentage of cohort | Gene involved (number of individuals) | |
|---|---|---|---|
| Mitochondrial multivitamin supplementation | 2 | 6.67 | ECHS1 (1) |
| PDHX (1) | |||
| Ketogenic diet | 1 | 3.33 | SLC2A1 (1) |
| Fasting avoidance | 1 | 3.33 | ACADM (1) |
There was one patient with dual diagnoses (PDHX‐related disorder and ACADM‐related disorder); the screening recommendations correspond to the respective genetic disorder as specified.
Surveillance initiation
Forty percent (12 out of 30) had recommendations for surveillance studies as a result of the genetic diagnosis (Table 3). Recommended imaging studies included brain MRI (3.33%, 1 out of 30); brain magnetic resonance angiography (MRA) (6.67%, 2 out of 30); kidney ultrasound (10%, 3 out of 30); and liver ultrasound (6.67%, 2 out of 30). Neurophysiological studies included electromyography/nerve conduction studies (EMG/NCS) (13.33%, 4 out of 30) to evaluate for possibility of neuropathy even if suggestive features were not present and EEG (6.67%, 2 out of 13) to evaluate background/interictal activity even if clinical suspicion for seizures were absent. Laboratory studies included creatine kinase (CK) (10%, 3 out of 30) and serum creatinine (6.67%, 2 out of 30). In one individual with dual diagnoses (PDHX‐ and ACADM‐related disorders), recommendations for laboratory testing included basic chemistry panel, liver function studies, plasma amino acids, urine organic acids, lactate, pyruvate, acylcarnitine profile, and free and total carnitine level. One individual with SPAST‐related disorder received suggestion for urodynamics studies due to enhancement of pretest suspicion by the genetic diagnosis.
Table 3.
Surveillance studies recommended to individuals with a genetic diagnosis in the cohort.
| Study | Number of individuals who received this recommendation | Percentage of cohort | Gene involved (number of individuals) | Potential complication/manifestation |
|---|---|---|---|---|
| Imaging | ||||
| Kidney ultrasound | 3 | 10.00 | COL4A1 (2) | Congenital renal anomalies |
| GNB1 (1) | Congenital renal anomalies | |||
| Brain MRI | 1 | 3.33 | RASA1 (1) | Vascular malformations |
| Brain MR angiography (MRA) | 2 | 6.67 | COL4A1 (1) | Aneurysms |
| RASA1 (1) | Vascular malformations | |||
| Liver ultrasound | 2 | 6.67 | COL4A1 (2) | Liver cysts |
| Neurophysiology | ||||
| Electromyography (EMG)/nerve conduction studies (NCS) | 4 | 13.33 | SPAST (2) | Neuropathy given possibility of polyneuropathy |
| SPTAN1 (1) | Neuropathy given possibility of polyneuropathy | |||
| B4GALNT1 (1) | Neuropathy given possibility of polyneuropathy | |||
| EEG | 2 | 6.67 | HEXA (1) | Evaluate background activity given elevated risk for seizures |
| KCNB1 (1) | Evaluate background activity given elevated risk for seizures | |||
| Laboratory studies | ||||
| Creatine kinase | 3 | 10.00 | COL4A1 (2) | Myopathy |
| PDHX (1) | Mitochondrial disorder, myopathy | |||
| Serum creatinine | 2 | 6.67 | COL4A1 (2) | Nephropathy |
| Estimation of glomerular filtration rate | 2 | 6.67 | COL4A1 (2) | Nephropathy |
| Urinalysis | 2 | 6.67 | COL4A1 (2) | Nephropathy |
| Thyroid function tests | 1 | 3.33 | GNB1 (1) | Hypothyroidism |
| Vitamin D levels | 1 | 3.33 | SATB2 (1) | Osteopenia |
| Complete blood cell count | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Serum chemistry | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Liver function tests | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Plasma amino acids | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Urine organic acids | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Serum lactate | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Serum pyruvate | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Acylcarnitine profile | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Free and total carnitine | 1 | 3.33 | PDHX (1) | Metabolic dysfunction |
| Other | ||||
| Urodynamic studies | 1 | 3.33 | SPAST (1) | Urological dysfunction |
One of the two individuals with COL4A1‐related disorder had already undergone MRA. There was one patient with dual diagnoses (PDHX‐related disorder and ACADM‐related disorder); the screening recommendations correspond to the respective genetic disorder as specified.
Thirty three percent (10 out of 30) of participants received recommendations to see a new specialist (Table 4). These specialties included Ophthalmology (20%, 6 out of 30) and Cardiology (16.67% 5 out of 30), and less commonly, Endocrinology (6.67%, 2 out of 30), Orthopedics (6.67%, 2 out of 30), Audiology (3.33%, 1 out of 30), Dentistry (3.33%, 1 out of 30), Nephrology (3.33%, 1 out of 30), Oncology (3.33%, 1 out of 30), and Vascular Anomalies (3.33%, 1 out of 30).
Table 4.
Specialists recommended to individuals with a genetic diagnosis in the cohort for the purpose of surveillance.
| Specialist | Number of individuals who received this recommendation | Percentage of cohort | Gene involved (number of individuals) | Potential complication/manifestation |
|---|---|---|---|---|
| Referral to a new specialist to screen for manifestations of genetic disorder | ||||
| Ophthalmology | 6 | 20.00 | ASXL3 (1) | Strabismus, decreased visual acuity |
| COL4A1 (2) | Cataracts, retinal artery tortuosity, Axenfeld‐Rieger anomaly | |||
| CTNNB1 (1) | Vitreoretinopathy, strabismus, refractive errors | |||
| POLR2A (1) | Strabismus and delayed visual maturation | |||
| SATB2 (1) | Strabismus and refractive errors | |||
| Cardiology | 5 | 16.67 | COL4A1 (2) | Mitral valve prolapse, supraventricular arrhythmias |
| CTNNB1 (1) | Congenital heart disease | |||
| GNB1 (1) | Congenital heart disease | |||
| RASA1 (1) | Heart failure from high‐flow vascular lesions | |||
| Endocrinology | 2 | 6.67 | ADAT3 (1) | Growth hormone deficiency |
| MAPK8IP3 (1) | Obesity, short stature, precocious puberty | |||
| Orthopedics | 2 | 6.67 | SATB2 (1) | Scoliosis, kyphosis |
| MAPK8IP3 (1) | Scoliosis | |||
| Audiology | 1 | 3.33 | POLR2A (1) | Sensorineural hearing loss |
| Dentistry | 1 | 3.33 | SATB2 (1) | Dental anomalies |
| Nephrology | 1 | 3.33 | COL4A1 (1) | Renal cysts, nephropathy |
| Oncology | 1 | 3.33 | GNB1 (1) | Hematological malignancies |
| Vascular Anomalies Specialist | 1 | 3.33 | RASA1 (1) | Arteriovenous malformations, capillary malformations |
| Continued follow‐up with an established specialist to evaluate for specific manifestations of the genetic disorder | ||||
| Ophthalmology | 2 | 6.67 | HEXA (1) | Macular degeneration, cherry‐red spot, visual loss |
| SPTAN1 (1) | Optic nerve atrophy | |||
| Orthopedics | 2 | 6.67 | ABHD16A (1) | Equinovarus/equinovalgus foot deformity, scoliosis |
| SLC16A2 (1) | Scoliosis, kyphosis | |||
| Endocrinology | 1 | 3.33 | SLC16A2 (1) | Abnormal thyroid tests with high serum T3 and low reverse T3 concentrations |
| Gastroenterology | 1 | 3.33 | SLC16A2 (1) | Nutritional difficulties |
| Nephrology | 1 | 3.33 | COL4A1 (1) | Renal cysts, nephropathy |
| Referral to clinical genetics | ||||
| Clinical Genetics | 5 | 16.67 | ADAT3 (1) | Facilitation of surveillance recommendations |
| CHD8 (1) | Facilitation of surveillance recommendations | |||
| COL4A1 (1) | Facilitation of surveillance recommendations | |||
| RASA1 (1) | Facilitation of surveillance recommendations | |||
| ZMYM2 (1) | Facilitation of surveillance recommendations | |||
There was one patient with dual diagnoses (PDHX‐related disorder and ACADM‐related disorder); the screening recommendations correspond to the respective genetic disorder as specified.
A total of 16.67% (5 out of 30) of participants received recommendations to continue to follow‐up with established specialists to focus on related manifestations of the genetic disorder. Such specialties included Ophthalmology (6.67%, 2 out of 30), Orthopedics (6.67%, 2 out of 30), Endocrinology (3.33%, 1 out of 30), Gastroenterology (3.33%, 1 out of 30), and Nephrology (3.33%, 1 out of 30).
Five participants (16.67%) received referrals to clinical genetics to oversee surveillance recommendations and provide input into whether additional surveillance studies were needed.
Variant‐specific testing/genetic counseling needs
As noted in Table 5, three (10%) families received recommendations for carrier testing for unaffected relatives. One family (3.33%) with a child with RASA1‐related disorder received recommendations for targeted testing of potentially affected siblings of the proband.
Table 5.
Recommendations for carrier/targeted testing provided to individuals with a genetic diagnosis in the cohort.
| Number of individuals who received this recommendation | Percentage of cohort | Gene involved (number of individuals) | Rationale or indication | |
|---|---|---|---|---|
| Recommendations for carrier testing of unaffected relatives | 3 | 10.00 | ADAT3 (1) | Carrier status could impact the relative's reproduction decisions |
| B4GALNT1 (1) | Carrier status could impact the relative's reproduction decisions | |||
| PDHX (1) | Carrier status could impact the relative's reproduction decisions | |||
| Recommendations for testing of potentially affected relatives | 1 | 3.33 | RASA1 (1) | Molecular testing in family members with known vascular malformations but no prior genetic testing |
There was one patient with dual diagnoses (PDHX‐related disorder and ACADM‐related disorder); the screening recommendations correspond to the specific genetic disorder as specified.
Patient education
Thirty percent (9 out of 30) of the participants received a referral to an expert in the genetic disorder or category of genetic disorder (Table 6). Specifically, participants received referrals to a specialist in hereditary spastic paraplegias (HSPs) (20% 6 out of 30), a specialist in their specific genetic disorder (6.67%, 2 out of 30), and a specialist in mitochondrial disorders (3.33%, 1 out of 30).
Table 6.
Referrals to experts in genetic disorder or category of genetic disorder, and potential changes in prognosis for individuals with a genetic diagnosis in the cohort.
| Specialist | Number of individuals who received this recommendation | Percentage of cohort | Gene involved (number of individuals) |
|---|---|---|---|
| Referral to expert in genetic disorder or category of genetic disorder | |||
| Expert in HSP | 6 | 20.00 | ABHD16A (1) |
| ATL1 (1) | |||
| B4GALNT1 (1) | |||
| SPAST (2) | |||
| SPTAN1 (1) | |||
| Expert in specific genetic disorder | 2 | 6.67 | FOXG1 (1) |
| HEXA (1) | |||
| Expert in mitochondrial disorders | 1 | 3.33 | PDHX (1) |
| Number of individuals who received this prognosis | Percentage of cohort | Gene involved (number of individuals) | |
|---|---|---|---|
| Potential changes in prognosis | |||
| Static/unknown to progressive | 11 | 36.67 | ABHD16A (1) |
| ATL1 (2) | |||
| B4GALNT1 (1) | |||
| GNAO1 (1) | |||
| HEXA (1) | |||
| RARB (1) | |||
| SLC16A2 (1) | |||
| SPAST (2) | |||
| SPTAN1 (1) | |||
| Risk for early mortality | 2 | 6.66 | HEXA (1) |
| RARB (1) | |||
More than one‐third (36.67%, 11 out of 30) of the participants had counseling about changes in prognosis, including the genetic disorder possibly being associated with a progressive course and risk of early death (Table 6). Seven participants had variants in five different genes (ABHD16A, ATL1, B4GALNT1, SPAST, SPTAN1) associated with HSP, which is progressive. One participant received a diagnosis of a movement disorder that is possibly progressive (GNAO1‐related disorder). One participant received a diagnosis of a genetic condition, not an HSP subtype, associated with progressive spasticity (SLC16A2‐related disorder). One participant received a diagnosis of a genetic disorder associated with progressive motor impairment and early death (RARB‐related disorder). One participant received a diagnosis of Tay Sachs disease, a neurodegenerative disorder associated with variants in HEXA and early mortality.
Of these 11 participants with a potential prognosis change, 5 were in the cryptogenic CP category (ABHD16A‐related disorder, ATL1‐related disorder × 2, GNAO1‐related disorder, SCL16A2‐related disorder). In these individuals, should there be evidence of progressive symptoms (either in the future or presently after more careful evaluation of prior trajectory), a diagnosis of CP may no longer be applicable.
Discussion
This study extends the new field of CP genetics by focusing on those with a molecular diagnosis and characterizing how the diagnosis impacted management for each individual. Out of 30 participants with a molecular diagnosis, the majority (93%) had changes in their clinical management, including suggestions for targeted medications/dietary interventions, recommendations for surveillance studies, referrals to specialists, referrals to clinical genetics, recommendations for carrier/targeted testing, and changes in prognosis.
Thirty‐seven percent (11 out of 30) of individuals in our cohort had molecular diagnoses of disorders that may be progressive, most commonly different genetic subtypes of HSP. Some of these individuals were in the CP masquerader category, having received a label of or considered to have CP at one point, which is not an uncommon clinical scenario. For example, in one study, out of 20 families affected by HSP with onset <3 years of age, 14 out of 20 (70%) had received an initial diagnosis of CP. 13 Once a patient has a diagnosis of CP, this label may carry over through the course of encounters with child neurologists and other clinicians, unless there are explicit attempts to reevaluate the diagnosis based on review of longitudinal trajectory of symptoms.
Five of the individuals with variants in genes associated with progressive genetic disorders in our cohort were in the cryptogenic CP category. This scenario emphasizes the notion that progressive genetic motor disorders can sometimes present insidiously, such that rate of progression may not be appreciable until years or sometimes decades have lapsed. In some cases, patients may not show evidence of progression at all. It is especially striking when an individual with CP (considered a static motor encephalopathy) obtains a genetic diagnosis associated with possible disease progression, signaling that the clinical canonical/strict diagnosis of CP may no longer be applicable. This scenario is particularly relevant if clinical disease progression occurs subsequently, or if upon review of the individual's past trajectory it becomes apparent there has indeed been clinical progression. This is in contrast to other major neurodevelopmental disorders (NDDs) (autism spectrum disorder [ASD], intellectual disability, epilepsy), in which symptomatic progression in the context of a genetic diagnosis does not take away the clinical NDD diagnosis. For example, a child with ASD based on gold‐standard clinical diagnostic criteria who develops progressive neurological signs and is found to have a genetic diagnosis still meets criteria for ASD.
Collectively there was a wide range of laboratory, imaging, and electrophysiological studies recommended based on molecular diagnoses, despite a relatively small number of participants in this analysis. With larger cohort sizes, we might expect recommendations for an even broader array of tests for surveillance purposes.
While 10 out of 30 (33.3%) received referrals to new specialists to screen for manifestations of the genetic disorder, a smaller, but still substantial, percentage [5 out of 30 (16.67%)] received recommendations to continue to follow‐up with established specialists to screen for new, or manage ongoing, manifestations of the disorder. From the perspective of the specialist, a specific molecular diagnosis may help contextualize findings and raise/lower threshold for considering certain treatments. Although we did not note referrals to research studies, this domain will become increasingly important to consider, as the number of natural history studies and clinical trials grows for different genetic NDDs.
Our study expands the limited literature surrounding the clinical utility of genetic testing in individuals with CP and related motor disorders. In one sequencing study of 150 individuals with CP, 37 out of 150 (24.7%) had a P/LP variant discovered by whole genome sequencing (WGS). Out of these 37 individuals, based on the genetic disorders, 20 out of 37 (54.1%) may have benefited from changes in clinical management, access to an approved medication, and/or eligibility for a clinical trial. 14 Similarly, in a cohort of 50 individuals from 49 different families with CP masqueraders or presentations of CP enriched for a genetic cause (what the authors termed “atypical CP”), 32 out of 49 (65%) families had a molecular diagnosis using next‐generation sequencing (WES and/or WGS). 15 Twenty‐five percent (8 out of 32) of families with a molecular diagnosis underwent targeted interventions, including deep brain stimulation (GNAO1‐related disorder, targeting dystonic/hyperkinetic crises); carbidopa/levodopa and 5‐hydroxytryptophan (CSTB‐related disorder, targeting myoclonus and dystonia); dopamine (TUBB4A‐related disorder, targeting dystonia); carbidopa/levodopa and 5‐hydroxytryptophan (PAK3‐related disorder, targeting self‐injurious behavior); lysine‐ and tryptophan‐restricted diet, sick day protocols, and carnitine and thiamine supplementation (GCDH‐related disorder, targeting prevention of metabolic decompensation); and immunological assessment (RANBP2‐related disorder, targeting prevention of immune dysregulation). 15
Compared to our current study, both of these investigations have reported an overall lower percentage of individuals with CP or CP‐like phenotypes and P/LP variants who experienced changes in clinical management. However, our study, with clinical utility as its primary focus, has defined clinical utility more broadly to include domains beyond medication/dietary interventions. Outside of targeted interventions, there is benefit in identifying, preventing, and treating medical comorbidities in genetic NDDs, 16 including genetic counseling, clarification of recurrence risk, restoration of reproductive confidence, and alleviation of guilt.
Studies evaluating other genetic NDDs also support the clinical utility of genetic testing. For example, one study showed that out of 38 patients with isolated NDDs (ID, ASD, learning disabilities, behavioral or psychiatric disorders, and/or seizures) with both P/LP chromosomal copy number variants and clinical follow‐up notes available for review, 16 out of 38 (42.1%) had management implications due to the genetic findings. 17 Another study found that out of 36 toddlers with ASD who had pathogenic findings on genetic testing, 26 out of 36 (72.2%) received medical recommendations based on the genetic findings. 18 Out of 100 patients with GDD/ID and prior non‐diagnostic genetic testing, WGS identified a cause in 21 out of 100 (21%), and 9 out of 21 (42.8%) underwent a change in clinical management (as assessed via telephone follow‐up), including initiation of targeted treatments, discontinuation of unnecessary interventions, and reproductive counseling. 19 In a cross‐sectional study of 418 patients with epilepsy with a genetic diagnosis, clinical management changes occurred in 208 out of 418 (49.8%), including addition of new medications, referral to a specialist, surveillance of systemic, non‐neurological manifestations, and discontinuation of existing medications. 20 Similarly, in a cohort of 53 patients with unexplained epilepsy found to have a genetic diagnosis, 22 out of 53 (41.5%) received at least one recommendation based on the diagnosis, pertaining to the areas of medication, systemic surveillance, referrals to specialists, referrals to clinical trials and research, and cascade testing. 21
Limitations
Our retrospective study had several limitations. The overall sample size of patients with a molecular diagnosis was small, limited to an initial cohort of 30 patients with intragenic variants. However, there is a limited number of existing studies about clinical utility of genetic testing for CP, making our findings noteworthy to report even with a small sample size.
Our study involved a retrospective analysis of medical charts. Accordingly, there may have been bias in ascertainment of clinical utility, given counseling of results was by a single provider at a single site. Notably, though, the majority of participants (n = 25) had clinical visits to discuss results, with recommendations discussed in accordance with best clinical practices. Given that there some can be some variability in discussing clinical recommendations, a prospective standardized approach may perhaps even increase evidence of actionability of findings.
We did not include the family perspectives about the benefits/disadvantages of a genetic diagnosis for CP and related motor disorders. We did not differentiate between disorders linked to neurological/neurodevelopmental phenotypes broadly versus CP/motor phenotypes specifically, as there is no canonical list of “CP” genes. We did not determine if individuals followed through with recommendations. We did not investigate potential impact of a negative result for individuals, such as relief at the diagnostic odyssey, or clinical management changes, if any.
We did not evaluate chromosomal copy number variants, which in addition to single‐gene disorders have also been implicated in CP. 6 These limitations could be addressed by future prospective studies.
Conclusions
Limitations aside, our study findings should encourage clinicians taking care of individuals with CP and related motor disorders to periodically reevaluate the clinical diagnosis and pursue genetic testing for individuals with no clear perinatal risk factors or imaging findings firmly establishing the cause of the CP. For individuals with CP and related motor disorders, establishing a genetic diagnosis may lead to significant impact in their overall health, with targeted treatments, adequate surveillance of associated complications, appropriate genetic testing and reproductive guidance to family members, and accurate prognosis.
Conflict of Interest Statement
Siddharth Srivastava has received grants from NIH/NINDS; consulting fees from GLG, Guidepoint (which connected to a client, Fortress Biotech), Novartis, ExpertConnect, Orchard Therapeutics, Stoke Therapeutics. Michael Kruer has received grant support from PTC Therapeutics; consulting fees from PTC Therapeutics, Aeglea, Merz; participated on board for Aeglea and Merz.
Acknowledgments
We would like to thank the families for their participation in this study. SS receives support from the National Institute of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) (K23NS119666).
Funding Statement
This work was funded by National Institute of Health/National Institute of Neurological Disorders and Stroke ; NIH/NINDS grant K23NS119666.
References
- 1. Bax M, Goldstein M, Rosenbaum P, et al. Proposed definition and classification of cerebral palsy, April 2005. Dev Med Child Neurol. 2005;47(8):571‐576. [DOI] [PubMed] [Google Scholar]
- 2. Surveillance of Cerebral Palsy in Europe . Surveillance of cerebral palsy in Europe: a collaboration of cerebral palsy surveys and registers. Surveillance of Cerebral Palsy in Europe (SCPE). Dev Med Child Neurol. 2000;42(12):816‐824. [DOI] [PubMed] [Google Scholar]
- 3. Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic mimics of cerebral palsy. Mov Disord. 2019;34(5):625‐636. [DOI] [PubMed] [Google Scholar]
- 4. Colver A, Fairhurst C, Pharoah POD. Cerebral palsy. Lancet. 2014;383(9924):1240‐1249. [DOI] [PubMed] [Google Scholar]
- 5. Graham HK, Rosenbaum P, Paneth N, et al. Cerebral palsy. Nat Rev Dis Primers. 2016;2:15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Segel R, Ben‐Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84(16):1660‐1668. [DOI] [PubMed] [Google Scholar]
- 7. Lee RW, Poretti A, Cohen JS, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. 2014;16(4):821‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Srivastava S, Lewis SA, Cohen JS, et al. Molecular diagnostic yield of exome sequencing and chromosomal microarray in cerebral palsy: a systematic review and meta‐analysis. JAMA Neurol. 2022;79(12):1287‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rockowitz S, LeCompte N, Carmack M, et al. Children's rare disease cohorts: an integrative research and clinical genomics initiative. NPJ Genom Med. 2020;5(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenbaum P, Paneth N, Leviton A, et al. The definition and classification of cerebral palsy. Dev Med Child Neurol. 2007;109:8‐14. [PubMed] [Google Scholar]
- 11. Chopra M, Gable DL, Love‐Nichols J, et al. Mendelian etiologies identified with whole exome sequencing in cerebral palsy. Ann Clin Transl Neurol. 2022;9(2):193‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suchowersky O, Ashtiani S, Au P‐YB, et al. Hereditary spastic paraplegia initially diagnosed as cerebral palsy. Clin Park Relat Disord. 2021;5:100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Eyk CL, Webber DL, Minoche AE, et al. Yield of clinically reportable genetic variants in unselected cerebral palsy by whole genome sequencing. NPJ Genom Med. 2021;6(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matthews AM, Blydt‐Hansen I, Al‐Jabri B, et al. Atypical cerebral palsy: genomics analysis enables precision medicine. Genet Med. 2019;21(7):1621‐1628. [DOI] [PubMed] [Google Scholar]
- 16. Savatt JM, Myers SM. Genetic testing in neurodevelopmental disorders. Front Pediatr. 2021;9:526779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henderson LB, Applegate CD, Wohler E, Sheridan MB, Hoover‐Fong J, Batista DAS. The impact of chromosomal microarray on clinical management: a retrospective analysis. Genet Med. 2014;16(9):657‐664. [DOI] [PubMed] [Google Scholar]
- 18. Harris HK, Sideridis GD, Barbaresi WJ, Harstad E. Pathogenic yield of genetic testing in autism spectrum disorder. Pediatrics. 2020;146(4):e20193211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun Y, Peng J, Liang D, et al. Genome sequencing demonstrates high diagnostic yield in children with undiagnosed global developmental delay/intellectual disability: a prospective study. Hum Mutat. 2022;43(5):568‐581. [DOI] [PubMed] [Google Scholar]
- 20. McKnight D, Morales A, Hatchell KE, et al. Genetic testing to inform epilepsy treatment management from an international study of clinical practice. JAMA Neurol. 2022;79(12):1267‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Graifman JL, Lippa NC, Mulhern MS, Bergner AL, Sands TT. Clinical utility of exome sequencing in a pediatric epilepsy cohort. Epilepsia. 2023;64(4):986‐997. [DOI] [PubMed] [Google Scholar]