

Abstract
Objective
Progression prediction is a significant unmet need in people with progressive multiple sclerosis (pwPMS). Studies on glial fibrillary acidic protein (GFAP) have either been limited to single center with relapsing MS or were based solely on Expanded Disability Status Scale (EDSS), which limits its generalizability to state‐of‐the‐art clinical settings and trials applying combined outcome parameters.
Methods
Serum GFAP and NfL (neurofilament light chain) were investigated in EmBioProMS participants with primary (PP) or secondary progressive MS. Six months confirmed disability progression (CDP) was defined using combined outcome parameters (EDSS, timed‐25‐foot walk test (T25FW), and nine‐hole‐peg‐test (9HPT)).
Results
243 subjects (135 PPMS, 108 SPMS, age 55.5, IQR [49.7–61.2], 135 female, median follow‐up: 29.3 months [17.9–40.9]) were included. NfL (age‐) and GFAP (age‐ and sex‐) adjusted Z scores were higher in pwPMS compared to HC (p < 0.001 for both). 111 (32.8%) CDP events were diagnosed in participants with ≥3 visits (n = 169). GFAP Z score >3 was associated with higher risk for CDP in participants with low NfL Z score (i.e., ≤1.0) (HR: 2.38 [1.12–5.08], p = 0.025). In PPMS, GFAP Z score >3 was associated with higher risk for CDP (HR: 2.88 [1.21–6.84], p = 0.016). Risk was further increased in PPMS subjects with high GFAP when NfL is low (HR: 4.31 [1.53–12.13], p = 0.006).
Interpretation
Blood GFAP may help identify pwPPMS at risk of progression. Combination of high GFAP and low NfL levels could distinguish non‐active pwPMS with particularly high progression risk.
Introduction
Despite the advances in diagnosing and managing multiple sclerosis (MS), care for people with progressive multiple sclerosis (pwPMS) remains challenging. Limited treatment options for the progressive disease phase, lack of reparative treatment to restore function, and insufficient tools to evaluate the disease course are some of the critical limitations. Indeed, reliable methods to predict progression in pwPMS are still a significant unmet need. Numerous clinical algorithms and imaging‐based approaches revealed modest applicability in clinical settings.
Body‐fluid biomarkers offer a unique possibility to investigate the pathophysiology of PMS at a “single protein” resolution. Indeed, biomarkers of neuroaxonal injury, such as neurofilament light chain (NfL), reflect disease activity and are associated with long‐term disease progression. 1 Nevertheless, their potentials were mainly demonstrated in relapsing–remitting MS (RRMS), while results in PMS, especially for individual application, have been conflicting. 2 , 3 , 4 , 5 On the other hand, glial fibrillary acidic protein (GFAP), an astrocyte marker, was consistently associated with markers of disease severity, 2 , 6 , 7 , 8 , 9 , 10 , 11 and, recently, progression in MS. 2 However, previous studies were restricted to single‐center studies 12 or predominantly relapsing MS. 2 More extensive studies reporting GFAP levels in PwPMS either utilized definitions that are not routinely applied in the current clinical setting 12 and/or were solely based on an EDSS‐based definition of ongoing progression, which limits its generalization to current state‐of‐the‐art clinical settings and trials in PMS.
EMerging blood BIOmarkers in PROgressive Multiple Sclerosis (EmBioProMS) is a prospective multicenter observational study initiated in 2018, aiming to define the association between novel blood biomarkers and disease progression in a deeply phenotyped PMS population. Here, we measured serum concentrations of NfL and GFAP, aiming to (1) define their association with clinical diagnosis of confirmed disability progression (CDP) using combined outcome parameters and (2) investigate their predictive potential regarding future diagnosis of CDP.
Methods
Study design
EmBioProMS protocol is detailed elsewhere. 10 , 13 In summary, EmBioProMS is an observational, prospective, multicenter study 10 that recruited 247 PwPMS in eight centers in Germany. PPMS was defined according to the 2017 McDonald criteria, 14 while SPMS was defined as participants with previous RRMS (fulfilling the 2017 McDonald criteria) who developed worsening of neurological symptoms independent of relapses for at least 1 year before study inclusion. Exclusion criteria were RRMS or other known major inflammatory or non‐inflammatory nervous system diseases. All PwPMS who were eligible to participate and did not have any exclusion criteria were screened and included until the targeted recruitment numbers were fulfilled. PwPMS in acute relapse were not considered eligible for participation but could be included at a later time point. In the first phase, three visits were planned: baseline, Month 6 (±3 months), and Month 18 (±6 months). In the following extension phase, yearly visits (±6 months) were added. Additional unscheduled visits were allowed to increase retention following the restrictions imposed by the COVID‐19 pandemic (starting 03.2020) (Table S1). The date and symptoms of the first manifestation, date of the diagnosis, number of documented relapses, date of the most recent relapse, duration of the progressive phase, and concomitant diseases were recorded at the baseline visit. At baseline and at the following visits, medical history, EDSS, Nine‐Hole Peg Test (9‐HPT), and Timed 25‐Foot Walk Test (T25FW) were evaluated through a certified EDSS rater. Visits of participants who were treated with an immunotherapy/disease‐modifying treatment (DMT) up to the day of the visit or in a predefined period before the visit were assigned to the treated group, depending on the therapy: corticosteroids in the last 30 days, any interferon preparation, glatiramer acetate, natalizumab, dimethyl fumarate, teriflunomide, fingolimod, methotrexate, or azathioprine, siponimod, cyclophosphamide, ofatumumab in the previous 3 months; rituximab, ocrelizumab, or mitoxantrone in the previous 12 months; or cladribine or alemtuzumab in the previous 24 months. Otherwise, visits/ participants were considered untreated. Data management was conducted within the platform of the German MS registry. 15 In this analysis, participants with at least one biomarker value were included.
Evaluation of disability progression
The cutoff date for the current analysis in EmBioProMS was 13 March 2023. Disability progression was defined prospectively using combined outcome parameters as increase of EDSS, 9‐HPT, or T25FW, confirmed at ≥6 months. The combined outcome parameter applied the EDSS step increase thresholds of ≥1 and ≥0.5 points for reference EDSS of <5.5 and ≥5.5, respectively, or an increase in the 9‐HPT score or the T25FW score by 20% or more 16 , 17 using a roving reference EDSS/T25FW/9‐HPT approach to identify recurrent events. Here, each EDSS/T25FW/9‐HPT served as a reference for the following assessment. Progression‐independent of relapse activity (PIRA) was defined in case of no evidence of clinical activity (i.e., relapse) between reference EDSS/T25FW/9‐HPT visit and time of diagnosing the CDP event.
Biomarker measurements
Serum samples were collected at each visit and stored at −80°C. NfL and GFAP levels were measured using the Neurology 2‐Plex B kit from Quanterix on a Simoa HD‐X analyzer® according to the manufacturer's instructions. All samples were measured in duplicates, with the same kit lot, at the same machine by a lab technician blinded to the clinical characteristics. The intra‐assay coefficient of variation (CV) of all samples was <20% (few measurements had to be replicated). Quality control samples were included at each run with inter‐assay CV <20%. Age‐adjusted z scores for NfL and GFAP were generated using large reference datasets as described before. 1 , 2 Here, Z scores measure deviation from healthy controls: for example, a biomarker Z score of 1 means that the concentration deviates by 1 standard deviation from values in the reference database adjusted for relevant physiological factors.
Statistical analysis
Summary statistics were applied to describe the different variables, that is, median with interquartile range for continuous variables and frequencies with percentages for categorical variables. Marginal means were used when summary statistics spanned multiple visits. Group differences between SPMS and PPMS were tested with Mann–Whitney U‐tests for age at disease onset and baseline values of disease duration, EDSS, and biomarkers. Tests on biomarkers were always based on reference‐population‐standardized Z scores, as described above. One sample t‐test was used to compare baseline biomarker mean values to 0. Log‐transformed clinical severity metrics (EDSS, T25FW, and 9HPT time as dependent variable) were analyzed in relation to biomarker concentrations using univariable and multivariable linear mixed models. Continuous or dichotomized biomarker levels were modeled for lesion counts, treatment categories, or clinical disability outcomes with univariable and multivariable general or logistic linear mixed models with individual ID as random intercept, correcting for age at onset, sex, EDSS, center effect, disease duration, and treatment status (monoclonal antibodies, orals, platform, or others). Linear mixed models used a first‐order autoregressive covariance structure. Fisher's least significant difference was used as a post hoc test in linear mixed models. The Kaplan–Meier method was used to estimate survival curves. Cox regression was used to model the time from the participant's inclusion to the first CDP based on dichotomized biomarker Z scores cutoff adjusting for age at onset, sex, EDSS, center effect, disease duration, and treatment status at baseline (monoclonal antibodies, orals, platform, or others). Separate analyses were run for each disease phenotype (PPMS and SPMS). No adjustment for multiple testing was carried out in this hypothesis‐driven analysis; descriptive p‐values (two‐sided) are reported and referred to as statistically significant if smaller than 5%. SPSS (version 29.0) and R version 4.2.2 were used for all computations.
Results
Cohort description
Out of 274, 243 participants from the EmBioProMS study had at least one NfL and GFAP value and were included in the analysis, comprising 108 patients with SPMS and 135 patients with PPMS; detailed clinical characteristics are shown in Table 1. At baseline, SPMS participants had longer disease duration and had higher EDSS (p < 0.001 for all) than PPMS patients. Seventy six out of 243 (31.3%) participants did not receive any DMT prior to study inclusion. In participants receiving DMT at baseline, ocrelizumab (n = 28) and 3‐monthly methylprednisolone (n = 17) represent the most common treatments in PPMS and SPMS, respectively. MRI of the brain or spinal cord was documented in 442 visits with a median offset to the visit of −162 [−516 to 30] and −9 [−497 to 51] days, respectively. 176 MRIs were conducted within 120 days of the visit, of which contrast‐enhancing lesions (CEL) were documented in only 11 scans.
Table 1.
Clinical characteristics of included EmBioProMS participants.
| PPMS (n = 135) | SPMS (n = 108) | Total (n = 243) | |
|---|---|---|---|
| At baseline | |||
| Baseline age [IQR] | 55.6 [49.7–60.9] | 55.3 [49.6–61.6] | 55.5 [49.7–61.2] |
| Sex (female:male) | 70:65 | 65:43 | 135:108 |
| Baseline disease duration [IQR] | 8.0 [4.0–13.0] | 19.5 [13.0–30.0] | 12.0 [6.0–21.0] |
| Baseline EDSS [IQR] | 4.0 [3.5–6.0] | 6.0 [4.0–6.0] | 4.5 [3.5–6.0] |
| Baseline immunotherapy (n, %) | |||
| Monoclonal antibodies | 29 (21.5) | 3 (2.8) | 32 (13.2) |
| Orals | 1 (0.7) | 16 (14.8) | 17 (7.0) |
| Injectables and others | 20 (14.8) | 29 (26.9) | 49 (20.2) |
| Untreated | 85 (63.0) | 60 (55.6) | 145 (59.7) |
| GFAP (pg/mL) a [IQR] | 131.6 [91.4–173.6] | 128.7 [97.9–174.8] | 131.2 [94.7–173.0] |
| GFAP Z score a [IQR] | 1.6 [0.8–2.7] | 1.7 [1.1–2.6] | 1.7 [0.9–2.7] |
| NfL (pg/mL) a [IQR] | 14.4 [10.9–18.8] | 15.5 [11.1–23.8] | 14.8 [10.9–19.5] |
| NfL Z score a [IQR] | 0.6 [−0.4–1.2] | 0.8 [−0.2–1.7] | 0.6 [−0.4–1.4] |
| At last follow‐up | |||
| Follow‐up duration in months | 29.7 [18.1–41.1] | 29.1 [16.4–39.6] | 29.3 [17.9–40.9] |
| EDSS [IQR] | 4.5 [3.5–6.0] | 6.0 [4.0–6.5] | 5.5 [4.0–6.0] |
| Immunotherapy (n, %) | |||
| Monoclonal antibodies | 67 (55.4%) | 19 (19.8%) | 86 (39.6%) |
| Orals | 1 (0.8%) | 16 (16.7%) | 17 (7.8%) |
| Injectables and others | 1 (0.8%) | 10 (10.4%) | 11 (5.1%) |
| Untreated | 52 (43.0%) | 51 (53.1%) | 103 (47.5%) |
| GFAP (pg/mL) b [IQR] | 140.3 [109.9–192.2] | 145.0 [97.5–188.8] | 141.8 [99.9–191.7] |
| GFAP Z score b [IQR] | 1.8 [1.0–2.7] | 1.7 [0.9–2.5] | 1.8 [1.0–2.6] |
| NfL (pg/mL) b [IQR] | 14.4 [10.7–19.2] | 15.7 [11.4–21.1] | 14.8 [11.2–20.3] |
| NfL Z score b [IQR] | 0.4 [−0.3–1.2] | 0.6 [−0.3–1.3] | 0.5 [−0.3–1.2] |
EDSS, Expanded Disability Status Scale; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; PPMS, primary progressive multiple sclerosis; SPMS, secondary progressive multiple sclerosis.
First available biomarker value; 97.1% from baseline visit.
Last available biomarker value, baseline value in subjects with no follow‐up visits. Data from follow‐ups were reported from the 218 subjects with at least two visits.
GFAP and NfL concentrations were available from 668/809 (82.6%) visits (including 236 visits at baseline). GFAP and NfL Z scores were elevated at baseline compared to the established reference mean (i.e., Z score of 0, p < 0.001 for both, Fig. 1). Most pwPMS had GFAP Z scores >1 (171/236, 72.5%, above the 84.1th percentile of the healthy population). 93/236 (39.4%) and 41/236 (17.4%) had GFAP Z scores >2 (97.7th percentile) and >3 (99.9th percentile), respectively. On the other hand, 84/236 (35.6%) of NfL values were >1, 53/236 (22.5%) of NfL values were >1.5 (93.3 percentile), and 30/236 (12.7%) >2.
Figure 1.
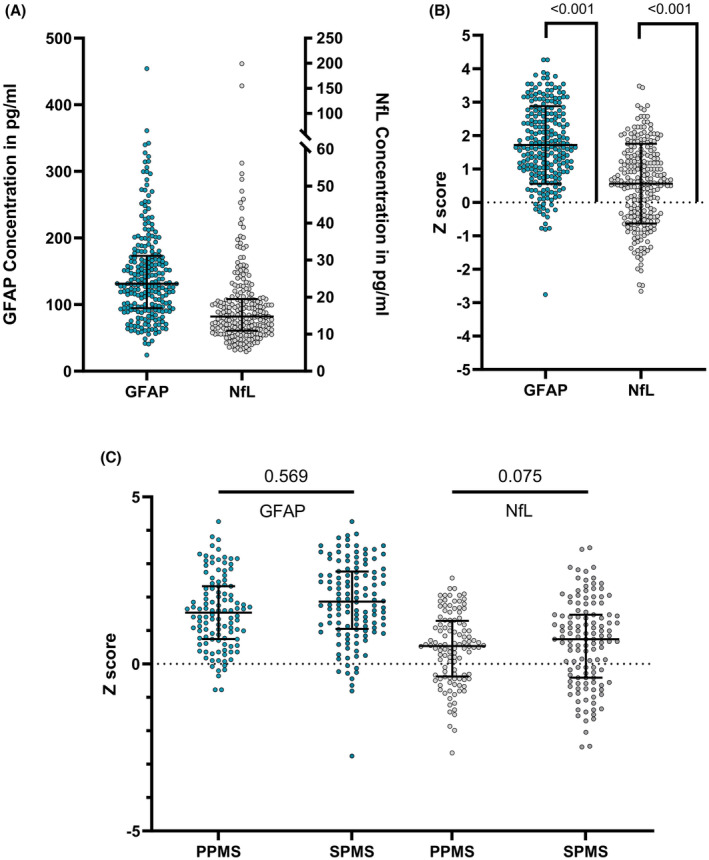
Baseline neurofilament light chain and glial fibrillary acidic protein in EmBioProMS population. Neurofilament light chain (NfL) and glial fibrillary acidic protein (GFAP) absolute values in EmBioProMS participants (A). Age‐adjusted Z scores at baseline were significantly elevated compared to levels in the healthy reference range (i.e., Z score of 0) (B). No statistically significant difference could be detected between participants with primary (PPMS) or secondary progressive multiple sclerosis (SPMS) regarding GFAP, while there was a trend for higher NfL Z scores in SPMS (C).
Higher GFAP Z scores were numerically positively associated with EDSS (mean difference per unit increase = 0.005 [−0.003 to 0.012], p = 0.201), T25FW time (0.010 [−0.005 to 0.025], p = 0.176) and 9‐HPT time (0.012 [0.000–0.025], p = 0.058). Similarly, higher NfL Z scores were associated with higher disease severity metrics (0.004 [−0.002 to 0.011], 0.010 [−0.003 to 0.023], and 0.017 [0.005–0.028], p = 0.225, 0.148, and 0.006 for EDSS, T25FW, and 9HPT time, respectively). In multivariable mixed models correcting for age at onset, sex, disease duration, treatment effect, and center effect, GFAP, and NfL association with 9HPT time was significant (Estimates: 0.015 and 0.016, p = 0.024 and 0.006, respectively).
GFAP Z scores did not differ significantly between different treatment categories (Table S2 ). For NfL, lower concentrations were found in PwPMS treated with monoclonal antibodies (marginal mean 0.35 [0.17–0.54]) compared to untreated participants (0.60 [0.45–0.75], p = 0.010), while no statistically significant difference could be seen under oral or other treatments (marginal mean 0.52 for both, p = 0. 636 and 0.548, respectively). In the available MRI dataset, a higher number of T2 lesion count (>8 lesions) was associated with higher GFAP and NfL Z scores (p = 0.002 and 0.046, respectively). Recent CEL lesions were associated with higher NfL (p = 0.016) but not significantly with GFAP Z scores (p = 0.961, Table S3). We documented only eight relapses during the FU period; therefore, we could not reliably assess the effect of relapses on the included biomarker concentrations.
Disability progression and blood biomarkers
We documented 111 (33.2%) progression events in 86 subjects over 338 EmBioProMS visits. Most of the progression events were evident through worsening of T25FW (58/111, 52.3%), followed by 9HPT (40/111, 36.0%), while EDSS progression was evident in only 38/111 (34.2%). In 21 events, two criteria were fulfilled, while in seven cases, all three progression criteria showed relevant worsening. Recurrent events (two or three) were evident in 15/86 (17.4%) and 5/86 (5.8%) subjects, respectively. None of the CDP events was preceded by relapse activity, rendering all of them PIRA.
Time to first event analysis
The median time to first CDW event was 7.1 months [IQR 6.2–17.7]. Particularly elevated GFAP values at baseline (GFAP >3) were associated with a trend for higher risk for CDP (hazard ratio [HR]: 1.86 [0.97–3.58], p = 0.063) Figure 2A–C, Table S4. The predictive value of high GFAP (i.e., >3) was particularly evident in subjects with lowest NfL concentrations (i.e., ≤1.0) (2.38 [1.12–5.08], p = 0.025), NfL < 1.5 (2.06 [0.96–4.43], p = 0.065), and NfL ≤ 2.0 (1.90 [0.93–3.84], p = 0.079) compared to participants with who did not meet this criterion (Fig. 2D–F). At baseline, NfL Z scores >1.5 and 2 were associated with numerically, but not statistically significant, higher risk for CDP (aHR: 1.63 [0.91–2.92], and 1.59 [0.67–3.75], p = 0.100 and 0.290, respectively) Figure S1, Table S4.
Figure 2.
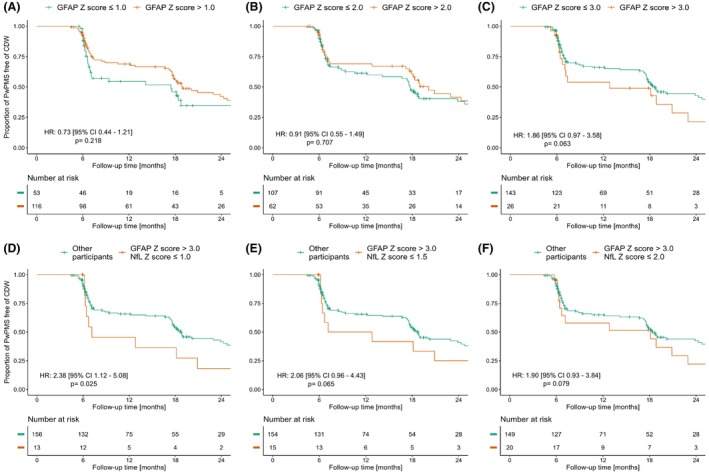
Predictive value of glial fibrillary acidic protein regarding first confirmed disability progression event. Kaplan–Meier curves for glial fibrillary acidic protein (GFAP) Z score and time to first confirmed disability progression events (CDP). Particularly high GFAP Z score (>3, C) was associated with a trend for a higher adjusted hazard ratio for confirmed disability progression (CDP), while GFAP Z scores cutoffs of 1 (A), or 2 (B) were not associated with statistically significant association with CDP. The predictive value of high GFAP Z score >3 (C) increased with lower neurofilament light chain (NfL) Z scores (i.e., ≤1.0, D), NfL ≤1.5 (E), and NfL ≤2.0 (F).
The predictive effect of the included biomarkers was distinct between the included PPMS and SPMS populations. In PPMS, GFAP >3 was associated with a 2.9‐fold higher risk for CDP (HR: 2.88 [1.21–6.84], p = 0.016), and the effect was particularly high in the subject with high GFAP (>3) and low NfL (≤1) with 4.3‐fold higher risk for CDP (4.31 [1.53–12.13], p = 0.006) Figure 3, Table S4. Higher GFAP values (>3) were not associated with statistically significant higher risk for CDP in the SPMS population (1.15 [0.37–3.61], p = 0.805) Table S4.
Figure 3.
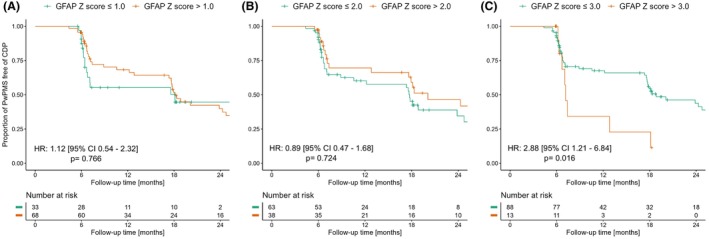
Prognostic value of glial fibrillary acidic protein in primary progressive multiple sclerosis participants. Glial fibrillary acidic protein (GFAP) Z scores >3 were associated with a higher risk of developing confirmed disability progression (CDP) in primary progressive multiple sclerosis (PPMS) (C). GFAP Z scores cutoffs of 1 (A) and 2 (B) were not significantly associated with higher risk for CDP in PPMS.
Discussion
Blood biomarkers of CNS pathology hold the promising potential to address a significant unmet need in PMS; reliable, reproducible tools to reflect and support the prediction of progression. In this study, we prospectively evaluate if GFAP and/or NfL are associated with CDW, assessed through state‐of‐the‐art outcome metrics in a deeply characterized prospective PMS cohort.
Pathological astrocyte activation has been demonstrated in MS brains. Indeed, extensive astrogliosis has been demonstrated inside and at the rim of mixed active lesions, 18 which has been attributed to progression in MS. 19 , 20 In line with that, a high GFAP level, an established marker of astrocyte involvement, was associated with more severe disease severity metrics and a higher risk of CDW. Our findings coincided with new reports demonstrating the predictive value of GFAP in relation to progression.
Besides GFAP, NfL levels predicted CDW in the included pwPMS. NfL predictive ability in regard to progression has been demonstrated in previous studies, 4 including in a recent large international collaboration with more than 12.000 NfL measurements. 21 Nevertheless, the predictive value of GFAP for CDP was stronger in our population compared to NfL. Moreover, this effect was particularly evident in subjects with low NfL concentrations.
Although not fully explained, this effect has recently been reported in other populations. 12 A number of hypotheses might help explain this effect, considering the variability of NfL and GFAP and the timing of assessment in relation to CDP. GFAP did not vary significantly within‐individual, compared to NfL, in pwMS. 2 NfL, on the other hand, has been shown to vary more significantly with significant elevation 1–2 years preceding the diagnosis of CDP. 21 In this study, NfL values decrease closer to the event and could reach a nadir at the time of CDP diagnosis. In studies with short FU duration, such as our current analysis, consistently high GFAP levels and declining NfL levels are expected to be seen closer to the event and could explain our findings (i.e., high GFAP and low NfL are associated with a higher hazard of progression). Indeed, in our study, the median time to first detection of the first CDP event was seven months, which is beyond the window of elevated NfL preceding CDP without relapses (~1–2 years preceding the event). Nevertheless, beyond the biomarker evolution related to disability, the combination of high GFAP and low NfL levels might recognize a particular subset of pwMS/pwPMS with distinct pathology and a high risk of progression.
Our analysis has some limitations, most notably the relatively short FU duration for PMS population. Nevertheless, the average duration of around 2.5 years is similar to Phase II and III studies in PMS, suggesting its potential applicability in clinical trials. Our SPMS population was relatively small, relatively old, and with advanced disease. Moreover, EDSS/T25/9HPT increase with no confirmation visit could not be included in the analysis, which relatively reduced the sample size. In addition, we generated the age‐ and sex‐adjusted GFAP Z scores from a comparably limited population, compared to NfL Z scores, and the proposed/generated cutoff values might differ compared to potential future broader reference datasets. Beyond that, our study has a limited MRI dataset, which did not allow the expansion of the definition of PIRA to include the MRI activity, which has been reported recently to be evident in up to 50% of PIRA events. 22
In summary, this study reports GFAP and NfL predictive value for combined outcome disability in a clinically defined PMS population. Our findings add to the promising, growing body of evidence regarding GFAP application regarding disability progression in MS.
Author Contributions
Conception and design: AA, KA, MCK, MS, JH, UKZ, IK, TS, AH, AS, AH, MK, TF, and HT. Data collection: AA, MCK, MS, JH, UKZ, IK, MH, TS, AH, SG, MK, and HT. Data analysis and interpretation: All authors; Manuscript drafting and review: All authors.
Funding Information
EmBioProMS was supported through a research grant from the German Multiple Sclerosis Society (DMSG) Federal Association, the German MS trust (Deutsche Multiple Sklerose‐Stiftung), the AMSEL Stiftung Ursula Späth, and the Bavarian MS Trust (Bayerische MS‐Stiftung).
Conflict of Interest Statement
AA received research funding from Department of Defense, and UCSF Weill Institute for Neurosciences, all not related to that work. MK received travel funding, speaker honoraria, and research support from Bristol Myers Squibb, Merck, Novartis and Roche, all not related to this manuscript. MS received consulting and/or speaker honoraria from Alexion, Bayer, Biogen, Bristol‐Myers‐Squibb, Merck, Roche, and Sanofi Genzyme; none related to this work. JH reports a grant for OCT research from the Friedrich‐Baur‐Stiftung and Merck, personal fees and nonfinancial support from Merck, Alexion, Novartis, Roche, Celgene, Biogen, Bayer and Horizon and nonfinancial support of the Sumaira‐Foundation and Guthy‐Jackson Charitable Foundation, all outside the submitted work. UKZ has received speaking fees, travel support, and financial support for research activities from Alexion, Almirall, Bayer, Biogen, Celgene, Janssen, Merck Serono, Novartis, Octapharm, Roche, Sanofi Genzyme, Teva, as well as EU, BMBF, BMWi, and DFG. None resulted in a conflict of interest. IK has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities from Alexion, Almirall, Bayer, Biogen, Hexal, Horizon, Merck, Neuraxpharm, Roche/Chugai, and Sanofi, all outside the submitted work. AS has no personal pecuniary interests to disclose other than being the lead of the German MS Registry, which receives project funding from a range of public and corporate sponsors, recently including The German Innovation Fund (G‐BA), The German Retirement Insurance, The German MS Trust, The German MS Society, Biogen, BMS, Merck, Novartis, Roche, and Sanofi. All outside the submitted work. AH received travel funding, consulting and/or speaker honoraria from Alexion, Argenx, and Horizon; none related to this manuscript. MCK has served on advisory boards and received speaker fees/travel grants from Merck, Sanofi‐Genzyme, Novartis, Biogen, Jansen, Alexion, Celgene/Bristol‐Myers Squibb and Roche and received research grants from Merck, Sanofi‐Genzyme, and Celgene/Bristol‐Myers Squibb. SG reports research support from Alnylam Pharmaceuticals, CSL Behring, Else Kröner Fresenius Foundation, Deutsche Forschungsgemeinschaft, and Hannover Biomedical Research School (HBRS) and consulting and/or speaker honoraria from Alnylam Pharmaceuticals and Merck all outside the submitted work. TF reports personal fees for consultancies (including data monitoring committees) in the past three years from Bayer, Biosense, Webster, Cardialysis, CSL Behring, Enanta, Fresenius Kabi, Galapagos, IQVIA, Immunic, Janssen, Kyowa Kirin, Lilly, Liva Nova, Minoryx, Mylan, Novartis, Roche, Vifor; all outside the submitted work. UZ received grants from the European Research Council (ERC), German Ministry of Education and Research (BMBF), German Research Foundation (DFG), Takeda Pharmaceutical Company Ltd., and consulting fees from CorTec GmbH, all not related to this work. JK reported receiving grants from Swiss National Science Foundation, Swiss MS Society, Biogen, Celgene, Merck, Novartis, Roche, Sanofi, Progressive MS Alliance, University of Basel, Octave Bioscience. TK has received speaker honoraria and/or personal fees for advisory boards from Bayer Healthcare, Merck, Novartis Pharma, Sanofi‐Aventis/Genzyme, Roche Pharma, Alexion/Astra Zeneca and Biogen as well as grant support from Novartis and Chugai Pharma in the past. HT received consulting and/or speaker honoraria from Alexion, Bayer, Biogen, Celgene, GSK, Jannssen, Merck, Novartis, Roche, Sanofi Genzyme, and TEVA; none related to this work. All other authors report no conflict of interest to this work.
Supporting information
Table S1. Number of included visits and their intervals.
Table S2. Effect of treatment on NfL and GFAP Z scores in EmBioProMS.
Table S3. Association between MRI metrics and assessed biomarkers.
Table S4. Predictive value of NfL and GFAP for first disability progression event.
Figure S1. Predictive value of neurofilament light chain (NfL) regarding first confirmed disability progression (CDP) event.
Acknowledgments
We want to thank all participants, study nurses, technicians, and physicians for their engagement. We want to thank Prof. Martin Kerschensteiner and Prof. Reinhard Hohlfeld for their support. Moreover, we want to thank the German MS Society, Federal Association (DMSG), the German MS Trust (Deutsche Multiple Sklerose‐Stiftung), and the AMSEL Stiftung Ursula Späth and Bavarian MS Trust (Bayerische MS‐Stiftung) for their ongoing generous funding.
Funding Statement
This work was funded by Aktion Multiple Sklerose Erkrankter Landesverband (AMSEL); Bayerische MS‐Stiftung; Deutschen Multiple Sklerose Gesellschaft .
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, Hayrettin Tumani, upon reasonable request.
References
- 1. Benkert P, Meier S, Schaedelin S, et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: a retrospective modelling and validation study. Lancet Neurol. 2022;21(3):246‐257. [DOI] [PubMed] [Google Scholar]
- 2. Meier S, Willemse EAJ, Schaedelin S, et al. Serum glial fibrillary acidic protein compared with neurofilament light chain as a biomarker for disease progression in multiple sclerosis. JAMA Neurol. 2023;80(3):287‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gafson AR, Jiang X, Shen C, et al. Serum neurofilament light and multiple sclerosis progression independent of acute inflammation. JAMA Netw Open. 2022;5(2):e2147588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Leppert D, Kropshofer H, Haring DA, et al. Blood neurofilament light in progressive multiple sclerosis: post hoc analysis of 2 randomized controlled trials. Neurology. 2022;98(21):e2120‐e2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang X, Shen C, Teunissen CE, et al. Glial fibrillary acidic protein and multiple sclerosis progression independent of acute inflammation. Mult Scler. 2023;29(9):1070‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huss A, Otto M, Senel M, Ludolph AC, Abdelhak A, Tumani H. A score based on NfL and glial markers may differentiate between relapsing‐remitting and progressive MS course. Front Neurol. 2020;11:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abdelhak A, Hottenrott T, Morenas‐Rodriguez E, et al. Glial activation markers in CSF and serum from patients with primary progressive multiple sclerosis: potential of serum GFAP as disease severity marker? Front Neurol. 2019;10:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abdelhak A, Huss A, Kassubek J, Tumani H, Otto M. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep. 2018;8(1):14798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abdelhak A, Foschi M, Abu‐Rumeileh S, et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat Rev Neurol. 2022;18(3):158‐172. [DOI] [PubMed] [Google Scholar]
- 10. Abdelhak A, Huss A, Stahmann A, et al. Explorative study of emerging blood biomarkers in progressive multiple sclerosis (EmBioProMS): design of a prospective observational multicentre pilot study. Contemp Clin Trials Commun. 2020;18:100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robinson T, Abdelhak A, Bose T, et al. Cerebrospinal fluid biomarkers in relation to MRZ reaction status in primary progressive multiple sclerosis. Cell. 2020;9(12):2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barro C, Healy BC, Liu Y, et al. Serum GFAP and NfL levels differentiate subsequent progression and disease activity in patients with progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2023;10(1):e200052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abdelhak A, Krumbholz M, Senel M, et al. Patient‐centered approach might effectively tackle the definition of progression in chronic neurological diseases: results from the EmBioProMS trial in progressive multiple sclerosis. medRxiv. 2021;2021.2009.2007.21262777. [Google Scholar]
- 14. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162‐173. [DOI] [PubMed] [Google Scholar]
- 15. Ohle LM, Ellenberger D, Flachenecker P, et al. Chances and challenges of a long‐term data repository in multiple sclerosis: 20th birthday of the German MS registry. Sci Rep. 2021;11(1):13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koch MW, Cutter GR, Giovannoni G, et al. Comparative utility of disability progression measures in PPMS: analysis of the PROMiSe data set. Neurol Neuroimmunol Neuroinflamm. 2017;4(4):e358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209‐220. [DOI] [PubMed] [Google Scholar]
- 18. Healy LM, Stratton JA, Kuhlmann T, Antel J. The role of glial cells in multiple sclerosis disease progression. Nat Rev Neurol. 2022;18(4):237‐248. [DOI] [PubMed] [Google Scholar]
- 19. Cubas‐Nunez L, Gil‐Perotin S, Castillo‐Villalba J, et al. Potential role of CHI3L1+ astrocytes in progression in MS. Neurol Neuroimmunol Neuroinflamm. 2021;8(3):e972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol. 2015;6:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abdelhak A, Benkert P, Schaedelin S, et al. Neurofilament light chain elevation and disability progression in multiple sclerosis. JAMA Neurol. 2023;80:1317‐1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tur C, Carbonell‐Mirabent P, Cobo‐Calvo A, et al. Association of early progression independent of relapse activity with long‐term disability after a first demyelinating event in multiple sclerosis. JAMA Neurol. 2022;80:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Number of included visits and their intervals.
Table S2. Effect of treatment on NfL and GFAP Z scores in EmBioProMS.
Table S3. Association between MRI metrics and assessed biomarkers.
Table S4. Predictive value of NfL and GFAP for first disability progression event.
Figure S1. Predictive value of neurofilament light chain (NfL) regarding first confirmed disability progression (CDP) event.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, Hayrettin Tumani, upon reasonable request.