

Abstract
Severe dengue (SD) is a major cause of morbidity and mortality. To define DENV target cells and immunological hallmarks of SD progression in children’s blood, we integrated two single cell approaches capturing cellular and viral elements: virus-inclusive single cell RNA-Seq (viscRNA-Seq 2) and targeted proteomics with secretome analysis and functional assays. Beyond myeloid cells, in natural infection, B cells harbor replicating DENV capable of infecting permissive cells. Alterations in cell type abundance, gene and protein expression and secretion, and cell-cell communications point towards increased immune cell migration and inflammation in SD progressors (SDp). Concurrently, antigen presenting cells from SDp demonstrate intact uptake, yet impaired interferon response and antigen processing and presentation signatures, partly DENV-modulated. Increased activation, regulation, and exhaustion of effector responses and expansion of HLA-DR-expressing, adaptive-like NK cells also characterize SDp. These findings reveal DENV target cells in human blood and provide insight into SD pathogenesis beyond antibody-mediated enhancement.
Introduction
Dengue virus (DENV), a mosquito-borne, positive-sense single-strand RNA flavivirus infects approximately 400 million people annually globally1. Most DENV-infected individuals are asymptomatic or experience dengue fever (D), an acute febrile illness. However, approximately 5% of symptomatic patients progress within several days of symptom onset to severe dengue (SD), manifesting by hemorrhage, shock, organ failure and sometimes death2, 3.
The identification of SD progressors (SDp) early during infection is ineffective, relying on broadly-defined and nonspecific clinical warning signs that often develop late during the disease course and whose global implementation has increased hospitalization rates, challenging resource allocation and not eliminating mortality4, 5. Moreover, the development of effective dengue vaccines and antivirals to prevent SD progression has been hindered6.
A major risk factor for SD is the presence of non-neutralizing anti-DENV antibodies, mediating antibody-dependent enhancement (ADE) upon secondary infection with a heterologous DENV serotype (of four circulating)7, 8. Nevertheless, reliance on bulk tissue assays (e.g. whole blood microarrays) and immunocompromised mouse models have somewhat limited characterization of the cellular targets of DENV and the roles of other immune responses in SD pathogenesis in humans. Better understanding SD pathogenesis may aid with the discovery of candidate biomarkers and preventive therapies.
We have previously developed virus-inclusive single-cell RNA-Seq (viscRNA-Seq) to simultaneously detect host and viral transcripts, and used it to investigate molecular signatures of SD progression in peripheral blood mononuclear cell (PBMC) samples from adults9, 10. Recently, we have reported altered immune responses, some of which were more pronounced in children vs. adults, in PBMCs of SDp relative to D patients via mass cytometry (CyTOF)11. That analysis was limited to 15 functional markers and did not detect DENV-harboring cells. To overcome these limitations and comprehensively investigate the cellular and molecular determinants accompanying SD progression in children at a genome scale, here we used a systems immunology approach. We profiled the viral and host transcriptomes in PBMCs derived from 19 DENV-infected children and 4 healthy controls using an optimized viscRNA-Seq 2 approach and computed the abundance, gene expression and communication network of multiple distinct cell types across disease categories. We then validated key observations by single-cell proteomics, bulk secretomics, and functional assays in patient-derived samples. Our findings reveal DENV target cells in the human blood and point to a coordinated but aberrant immune response accompanying SD progression.
Results
viscRNA-Seq reveals changes in cell subtype abundance in SD
To characterize the immune response accompanying SD progression during natural infection in children, we profiled the transcriptome of PBMCs collected from individuals enrolled in our Colombia dengue cohort presenting within 1 to 7 days from symptom onset and testing DENV positive5, 10. Disease severity was defined as dengue (D), dengue with warning signs (DWS) and severe dengue (SD) at presentation and discharge. Here, we studied samples from four healthy donors (H, n = 4) and 19 DENV-infected 4–17-year-old children, of whom seven met criteria of SD within less than a day to two days following enrollment (SDp, n = 7), and twelve had an uncomplicated disease course (DWS, n = 4; D, n = 8) (Fig. 1a, Table 1, Supplementary Table 1, see Methods for sample selection).
Fig.1: viscRNA-Seq 2 analysis of human-derived PBMCs defines 21 immune cell subtypes and alterations in their abundance accompanying SD progression.
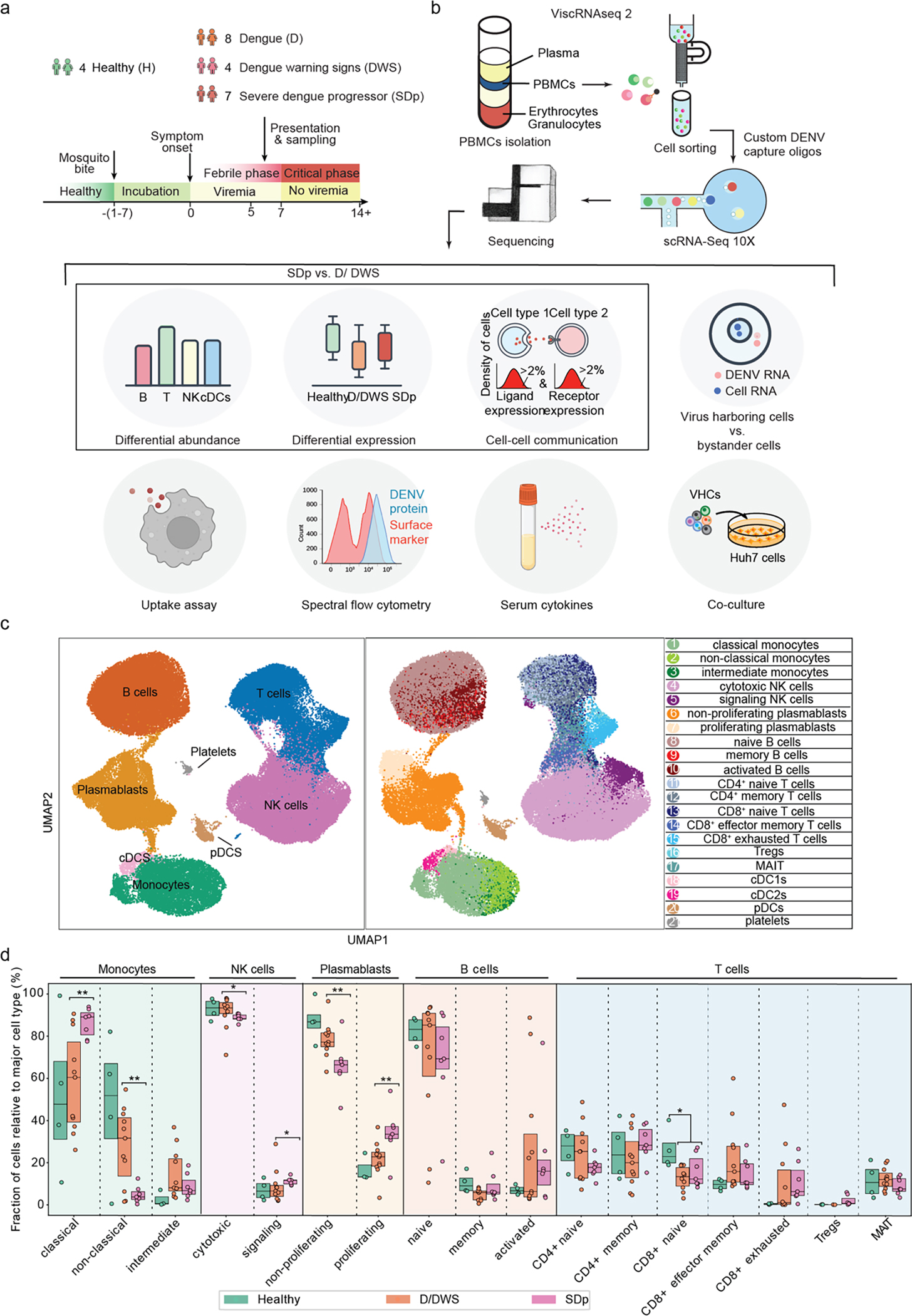
a-b, Schematic of the study design. a, PBMC and serum samples were collected from children enrolled in the Colombia dengue cohort upon presentation to the clinic (D, DWS, SD) and from healthy children (H). b, Schematic of the experimental and computational components of viscRNA-Seq 2 and outline of the performed functional and validation assays on patient-derived PBMCs. c, UMAP embedding of the viscRNA-Seq 2 dataset indicating broad cell types (left) and subtypes (right). d, Box plots showing the fractions (%) of cell subtypes within each major immune cell type. The Box horizontal lines indicate the first, second (median) and third quartiles. Each dot represents a participant, color-coded by disease severity: H (green, n = 4); D/DWS (orange, n = 8/3); SDp (pink, n = 7). p values by two-tailed KS test are shown.
Table 1. Human samples used in this study.
General information and specific assays samples were used for. UD, undetectable; NA, non-applicable; ND, not determined; UA, unavailable; equiv, equivocal; FC, flow cytometry; 1°, primary; 2°, secondary.
| General information |
Assays samples were used
for |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Diagnosis | Age | Gender | Dengue exposure | Viral load (copies/ml) | Day of fever | Days before SD | viscRNA-Seq 2 | Cytokines | Co-culture | Uptake | FC target cells | FC functional |
|
| |||||||||||||
| 1–140 | SD | 16 | F | 1° | UD | 5 | <1 | v | v | ||||
| 5–030 | SD | 4 | F | 2° | 5.84 E5 | 6 | 2 | v | v | ||||
| 5–041 | SD | 13 | F | 1° | 4.91 E4 | 5 | 1 | v | v | ||||
| 5–044 | SD | 11 | F | 2° | 5.11 E7 | 5 | 1 | v | v | ||||
| 5–193 | SD | 5 | M | 2° | UD | 5 | 2 | v | v | ||||
| 1–144 | SD | 17 | M | 2° | 9.12 E3 | 7 | 1 | v | v | ||||
| 1–002 | SD | 16 | F | 2° | 5.57 E4 | 4 | <1 | v | v | ||||
|
| |||||||||||||
| 6–023 | D | 11 | M | 2° | UD | 6 | - | v | v | ||||
| 6–029 | D | 11 | F | 2° | 1.78 E2 | 7 | - | v | v | ||||
| 1–019 | D | 13 | F | 1° | UD | 2 | - | v | v | ||||
| 5–154 | D | 5 | F | ND | 2.69 E8 | 4 | - | v | v | ||||
| 5–130 | D | 12 | M | 2° | UD | 7 | - | v | v | ||||
| 5–010 | D | 10 | F | 2° | 3.61 E6 | 7 | - | v | v | ||||
| 6–001 | D | 15 | M | 1° | 9.02 E8 | 3 | - | v | v | ||||
| 1–113 | D | 17 | M | 2° | 8.13 E3 | 1 | - | v | v | ||||
|
| |||||||||||||
| 5–089 | DWS | 11 | M | 1° | 2.46 E9 | 3 | - | v | |||||
| 6–020 | DWS | 13 | F | 1° | 2.00 E10 | 2 | - | v | |||||
| 6–025 | DWS | 6 | M | equiv | 1.23 E10 | 3 | - | v | |||||
| 6–028 | DWS | 5 | M | 1° | 8.07 E9 | 6 | - | v | |||||
|
| |||||||||||||
| 3–073 | H | 13 | F | NA | NA | NA | - | v | |||||
| 3–074 | H | 8 | F | NA | NA | NA | - | v | |||||
| 3–012 | H | 12 | M | NA | NA | NA | - | v | |||||
| 3–047 | H | 5 | M | NA | NA | NA | - | v | |||||
|
| |||||||||||||
| 6–031 | DWS | 23 | F | 2° | 5.17 E5 | 5 | - | v | v | v | |||
| 6–007 | DWS | 14 | M | 1° | 1.62 E9 | 2 | - | v | v | v | |||
| 5–055 | DWS | 12 | M | 2° | 1.99 E6 | 5 | - | v | v | v | |||
| 5–011 | DWS | 17 | F | 2° | 4.66 E8 | 4 | - | v | v | v | |||
| 1–151 | DWS | 29 | M | 1° | 4.64 E9 | 2 | - | v | v | v | |||
| 5–216 | DWS | 32 | M | 2° | 2.54 E5 | 3 | - | v | v | ||||
| 1–087 | DWS | 9 | F | 2° | UD | 4 | - | v | v | ||||
| 1–095 | DWS | 6 | M | 2° | UD | 7 | - | v | v | ||||
| 1–116 | DWS | 10 | F | 2° | UD | 5 | - | v | v | ||||
| 1–118 | DWS | 20 | M | 2° | 4.79 E2 | 2 | - | v | v | ||||
| 1–129 | DWS | 53 | F | 2° | 2.56 E4 | 7 | - | v | v | ||||
|
| |||||||||||||
| #1 | H (bank) | 36 | M | NA | NA | NA | - | v | v | ||||
| #2 | H (bank) | 27 | M | NA | NA | NA | - | v | v | ||||
| #3 | H (bank) | 33 | F | NA | NA | NA | - | v | v | ||||
| #4 | H (bank) | 41 | M | NA | NA | NA | - | v | v | ||||
| #5 | H (bank) | 29 | F | NA | NA | NA | - | v | v | ||||
Improvements incorporated in the viscRNA-Seq 2 platform included magnetic enrichment of rare cell types, redesigned oligos for DENV viral RNA (vRNA) capture, and switch to 5’-droplet-based microfluidics (10X Genomics) to increase sequenced cell number (see methods, Fig. 1b). This approach enabled simultaneous coverage of transcriptomes from 181,221 PBMCs, of which 105,698 were high quality (see methods, Supplementary Table 2). Because of large patient-to-patient sample variability, we annotated cell types and subtypes within each patient following Leiden clustering12. Overall, 8 cell types and 21 subtypes were identified, embedded (Fig. 1c), and verified by expression of unique marker genes (Extended Data Fig. 1a).
First, we assessed whether SD progression is accompanied by alterations in cell abundance. Since samples were enriched for less abundant cell populations (see Methods), we compared the composition of cell subtypes within the major immune cell populations (rather than cell type frequencies) in SDp, symptomatic non-progressors (D, DWS), and healthy controls (Fig. 1d). The fractions of classical monocytes, signaling NK cells, proliferating plasmablasts and regulatory T cells (Tregs) were expanded in SDp vs. non-progressors, albeit Treg expansion did not meet statistical significance (Fig. 1d, Extended Data Fig. 1b). CD4+ memory and CD8+ exhausted T cell fractions were larger in SDp than non-progressors, albeit these differences were not statistically significant. In contrast, smaller fractions of non-classical monocytes, cytotoxic NK cells and non-proliferating plasmablasts were found in SDp relative to non-progressors (Fig. 1d). Differences in cell subtype abundance were in agreement with those measured in our CyTOF dataset11 (see methods, Extended Data Fig. 1c,d), did not correlate with serum viral load (Extended Data Fig. 1e), were independent of age (<10, 10–13 and 14–17 years), not affected by pregnancy status (1 SDp patient was pregnant) (Extended Data Fig. 2a,b), and only partially associated with prior DENV exposure (Extended text and Extended Data Fig. 2c), indicating a potential biological association with SD progression.
A machine learning classifier trained only on signaling NK cell and non-classical monocyte fractions distinguished SDp in 16 out of 18 leave-one-out comparisons (Extended Data Fig. 2d), rising to 17 out of 18 upon addition of Treg fraction (Extended Data Fig. 2e).
SD progression is thus accompanied by alterations in immune cell subtypes, characterized by increased pro-inflammatory CD14+ monocytes and proliferating plasmablasts, yet suppressed effector cells (decreased cytotoxic NK cells, Treg emergence).
Reduced antigen presentation signatures but not uptake in SD
We profiled the transcriptional landscape of APCs between individual SDp and D patients via pairwise differential expression analysis, which enables visualization of effect size and noise levels in the data, compensating for the unknown empirical distribution of gene expression and the small sample size (see Methods, Extended Data Fig. 3a, Supplementary Table 3). Gene ontology (GO) analysis of differentially expressed genes (DEGs) in monocytes revealed upregulation of pathways associated with pro-inflammation and activation and downregulation of pathways associated with cytokine-mediated signaling, type I and type II interferon response, and antigen processing and presentation in SDp vs. D (Extended Data Fig. 3b).
DEGs between SDp and D were also identified in monocyte subtypes and other APCs: cDCs and B cells. Among the upregulated genes in monocytes were pro-inflammatory genes, FCγ-receptor signaling genes (FCGR1A, FCGR2A, PRKCD, BTK, ITPR2), scavenger receptor (CD163), and genes involved in cell adhesion and migration, with greatest upregulation in intermediate monocytes (Fig. 2a). Pro-inflammatory and cell adhesion and migration genes were also upregulated in cDCs from SDp vs. D, with variable expression patterns in cDC1s and cDC2s (Fig. 2b). B cells from SDp showed upregulation of genes related to host and viral translation, RNA binding, and differentiation (Fig. 2c).
Fig.2: Immunological determinants of SD progression in patient-derived antigen presenting cells (APCs).
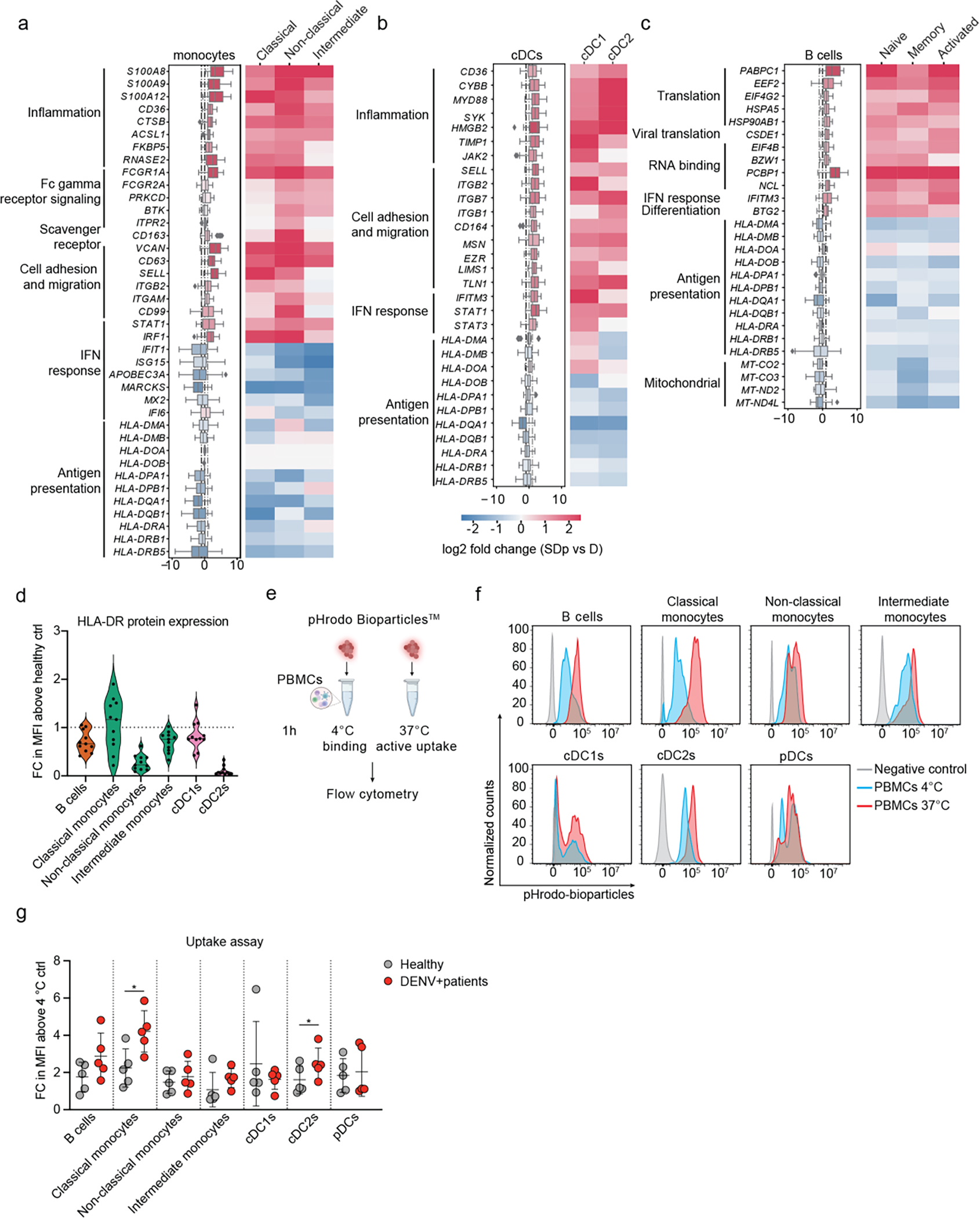
a-c, Differentially expressed genes (DEGs) between D and SDp detected via pairwise comparison of patients’ average (see Methods) in monocyte (a), cDC (b) and B cell (c) populations (Box plots, left) and the corresponding distinct cell subtypes (heatmaps, right). Data are color-coded based on median log2 fold change of pairwise comparisons. Box plots’ horizontal lines indicate the first, second (median) and third quartiles. Whiskers extend to ±1.5 × IQR. n = 15 participants: D (n = 8); SDp (n = 7). d, Violin plot showing HLA-DR cell surface expression measured via spectral flow cytometry in patient-derived PBMCs expressed as fold change (FC) of mean fluorescence intensity (MFI) above healthy control. Dotted line represents mean value of healthy controls normalized at value 1. Horizontal lines in violin plots represent median. n =11 paticipants from 2 independent experiments. e, Schematic of the uptake experiments shown in panels f and g. Patient-derived PBMCs were incubated for 1 hour either at 4 °C or 37 °C with pHrodo Bioparticles followed by fluorescence measurement via spectral flow cytometry. f, Representative histograms showing the percentage of pHrodo Bioparticles positive cells in distinct cell subtypes derived from a healthy individual incubated with no bioparticles (grey) or with bioparticles at 4 °C (cyan) or 37 °C (red). g, Bioparticle uptake quantification in the indicated cell subtypes shown as FC of mean MFI above the corresponding 4 °C control. Each dot represents a participant, color-coded by disease severity: H (gray, n = 5); DENV-infected (red, n = 5), from two independent experiments. Error bars represent mean values ± SD. p values by two-tailed Mann-Whitney test are shown.
Whereas interferon signaling genes, such as IRF1 and STAT1, were upregulated in all APC subtypes in SDp vs. D, downstream interferon stimulated genes (ISGs), including IFIT1, ISG15, MARCKS and APOBEC3A, where upregulated in cDCs and B cells but downregulated in monocytes (particularly non-classical) (Fig. 2a–c).
Multiple MHC-II genes were universally downregulated in all APC subtypes in SDp vs. D (Fig. 2a–c). Except for CD14+ monocytes, showing large patient-to-patient variability, lower expression of HLA-DR protein was also detected on APCs by flow cytometry of PBMC samples from six DWS patients than healthy individuals (Fig. 2d). HLA-DR transcript level correlated with HLA-DR protein expression previously measured on APCs by CyTOF11 (Extended Data Fig. 3c), proposing decreased antigen presentation capacity by select immune types as another phenotype of SD progression. Interestingly, this finding was associated with upregulation of FCGR1A (CD64), FCGR2A (CD32) and CD163 in monocytes from SDp vs. D (Fig. 2a) and in the combined DENV-infected population vs. healthy controls (Extended Data Fig. 3d). Flow cytometry analysis confirmed the latter at the protein level, showing increased expression levels of CD64 (in agreement with our existing CyTOF dataset11) and CD163, yet variable expression of CD32, on APC subsets in the six DWS patients vs. healthy individuals (Extended Data Fig. 3e). To functionally profile the phagocytic ability of APCs, we measured uptake activity of DENV-infected (DWS) patient-derived APCs using a pHrodo bioparticles assay either at 4 °C (binding control) or 37 °C (active uptake) followed by flow cytometry (Fig. 2e). A 2–4-fold increase in mean fluorescence intensity (MFI) above control, indicative of increased uptake, was measured in five DENV-infected vs. five healthy individuals (Fig. 2f,g) in classical (CD14+CD16−) monocytes, cDC2s (CD1c+), and B cells (CD19+CD20+), albeit the latter was not statistically significant in this sample size.
Comparison of the DEGs described above between primary and secondary infections showed possible association of prior DENV exposure with increased uptake genes in APCs (associated with ADE), but not with alterations observed in genes involved in antigen presentation, inflammation, migration, adhesion, and IFN response (Supplemental Text and Extended Data Fig. 4). These findings provide evidence that SD progression is associated with an inflammatory phenotype accompanied by impaired interferon response, possibly associated with prior DENV exposure, and impaired antigen presentation signatures in APCs despite intact uptake activity.
Effector cell activation and exhaustion accompany SD
We extended the differential expression analysis between SDp and D to effector cells (Supplementary Table 3). In NK cells, GO analysis of DEGs revealed upregulation of T cell proliferation or activation and adaptive immune response and downregulation of chemotaxis, migration and cytokine signaling pathways (Extended Data Fig. 5a). Cytotoxic and signaling NK cells demonstrated upregulation of genes involved in cytoskeleton rearrangement and protein translation and folding in SDp (Fig. 3a). Cytotoxic NK cells upregulated CD2 (cytotoxicity activator), MHC-II (HLA-DRB1, HLA-DPA, HLA-DPB1), pro-inflammatory (CCL5 and IL32), and cell migration genes and downregulated KLRB1 (cytotoxicity suppressor) (Fig. 3a). This signature, suggestive of cytotoxic NK cell activation, was accompanied by upregulation of LAG3, an exhaustion marker, and downregulation of ISGs (LY6E, ISG15, IFI6) (Fig. 3a).
Fig.3: Phenotypic alterations in effector cells associated with SD progression.
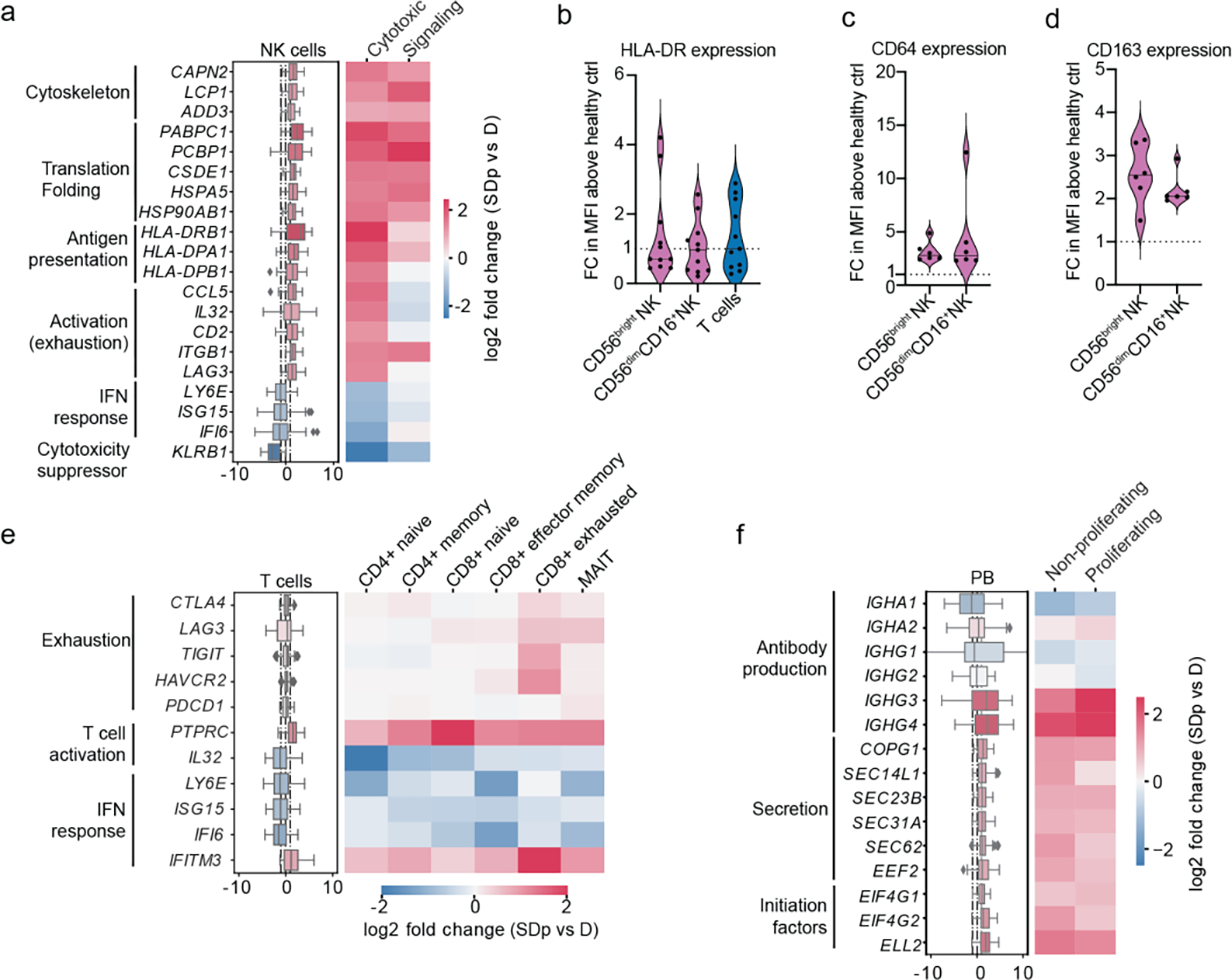
a, e, f, DEGs between D and SDp detected via pairwise comparison of patients average in NK cell (a) T cell (e) and plasmablast (f) populations (Box plots, left) and distinct corresponding cell subtypes (heatmaps, right). Data are color-coded based on median log2 fold change of pairwise comparisons. Box plots’ horizontal lines indicate the first, second (median) and third quartiles. Whiskers extend to ±1.5 × IQR. The heatmap for Tregs (e, starred) was computed using group averages instead of patient averages due to their low abundance. n = 15 participants: D (n = 8); SDp (n = 7). b-d, Violin plots showing cell surface expression of HLA-DR (b) CD64 (c) and CD163 (d) measured in patient-derived NK (b-d) and T cells (b) via spectral flow cytometry expressed as fold change (FC) of mean fluorescence intensity (MFI) above healthy control. Each dot represents a participant [n = 11 (b); n = 6 (c, d)] from two independent experiments. Dotted lines represent mean values of healthy controls normalized at value 1; horizontal lines in violin plots represent median. These measurements were conducted side-by-side with those shown in Fig. 2d.
In contrast with APCs, the expression of HLA-DR protein measured by flow cytometry in PBMC samples was higher in DWS patient-derived CD56bright and CD56dimCD16+ NK cells than healthy controls (Fig. 3b). HLA-DR transcript level correlated with HLA-DR protein expression measured on NK cells from the same patients via CyTOF (Extended Data Fig. 3c). Moreover, HLA-DRA positive NK cell abundance was greater in SDp than D (Extended Data Fig. 5b). These findings were associated with higher expression of CD64, CD32, and CD163 proteins measured by flow cytometry in patient-derived NK cells than healthy, and no alterations in uptake activity (Fig. 3c,d; Extended Data Fig. 5c,d).
Evidence for exhaustion and regulation in SDp was identified also in T cells, particularly exhausted CD8+ T cells and Tregs, whose abundance increased in SDp (Fig. 1d). These cells demonstrated upregulation of CTLA4, LAG3, TIGIT, HAVCR2, and PDCD1, albeit Treg cell numbers were low (Fig. 3e and Extended Data Fig. 5e). These findings were associated with signatures suggestive of attenuated effector functions across most T cell subtypes (Fig. 3e). For example, IL32, whose expression typically increases upon T cell activation to induce pro-inflammatory cytokine production in myeloid cells, was downregulated in SDp. Moreover, multiple ISGs, including LY6E, ISG15, and IFI6, but not IFITM3, were downregulated in SDp vs. D.
Proliferating and non-proliferating plasmablasts in SDp demonstrated upregulation of specific immunoglobulin constant region genes (IGHG3 and IGHG4), in correlation with IgG3 and IgG4 expression measured via CyTOF, and genes involved in protein translation and secretion (Fig. 3f and Extended Data Fig. 3c).
Whereas upregulation of IGHG and antibody secretion genes in plasmablasts and of exhaustion genes in T and NK cells were associated with prior DENV exposure, the majority of the observed responses in effector cell subtypes were not or only partially associated with prior DENV exposure (Supplemental Text and Extended Data Fig. 5f). These findings provide evidence that SD progression is associated with activation and regulation of effector lymphocyte responses and the presence of cytotoxic NK cells that may be taking on an antigen-presenting role.
DENV replicates in B cells and modulates their transcriptome
To determine whether some of the detected transcriptomic signatures are induced by DENV, we first sought to define the cell types that harbor vRNA. We used oligonucleotides complementary to the positive strand of the viral genome that matched the patients’ serotypes (DENV1 and DENV3). Viral reads were detected in the samples described above, yet their numbers were small. To improve vRNA detection, samples from four DWS patients with high serum viral load were included among the 19 processed by viscRNA-Seq 2. We detected over 3,000 vRNA molecules across 1,368 cells from 10 DENV-infected samples, majority of which were from these DWS patients (Extended Data Fig. 6a and Supplementary Table 4). vRNA molecule number positively correlated with the corresponding serum viral load but not with disease severity (Fig. 4a). Focusing on three DWS samples with the highest number of vRNA-harboring cells (VHCs) (and viremia), we observed that B cells were the predominant VHCs; over 40% of B cells (vs. less than 5% of monocytes and NK cells) harbored vRNA and showed highest number of DENV reads per cell (Fig. 4a–c; Extended Data Fig. 6a,b).
Fig.4: DENV actively replicates in patient-derived B cells and alters their transcriptional profile.
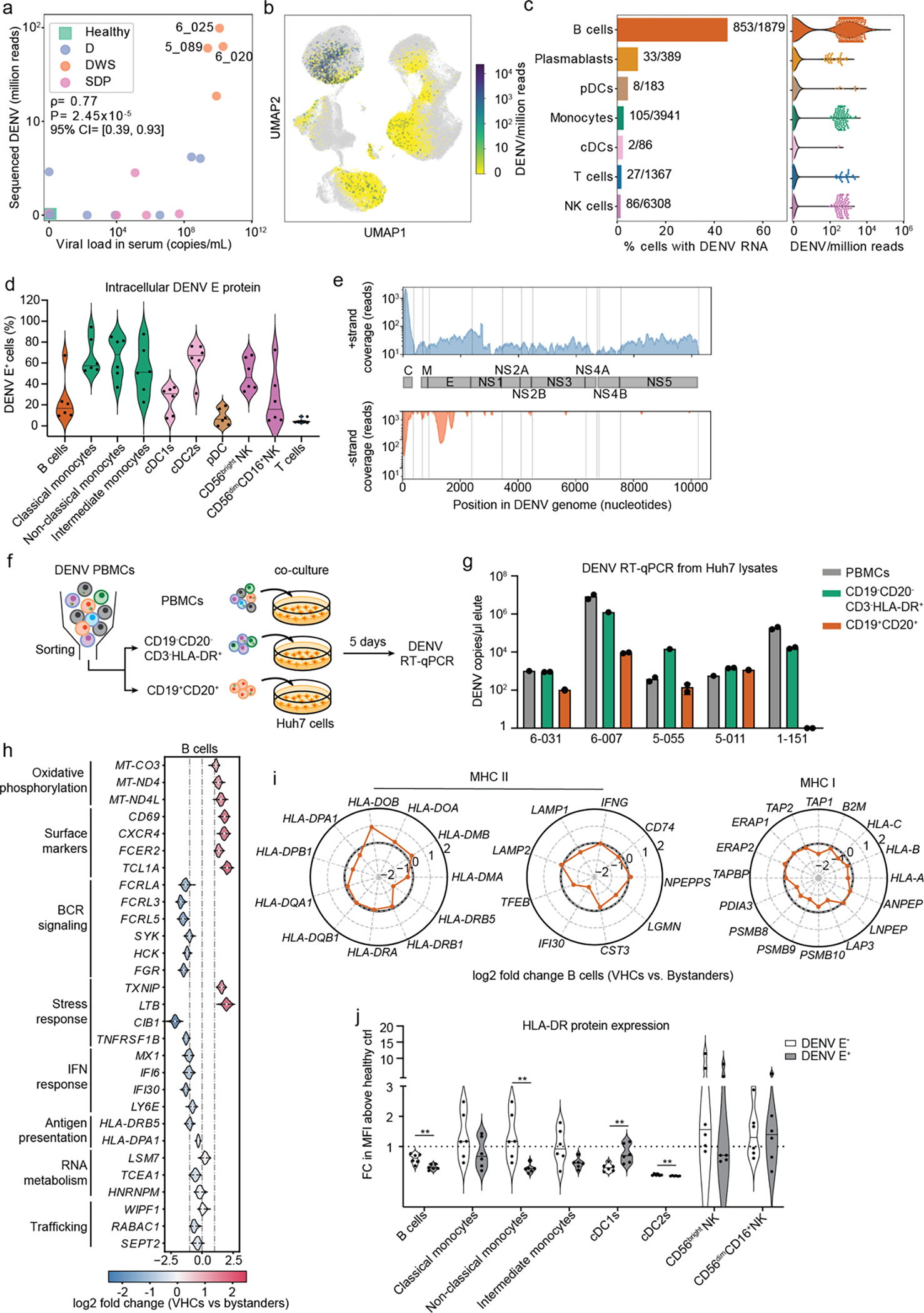
a, Scatter plot of normalized DENV reads measured in PBMCs by viscRNA-Seq 2 vs. serum viral load measured by RT-qPCR (two-tailed Spearman correlation coefficient ρ=0.76, p value=1.86e-5; 95% confidence interval were computed using bootstrapping strategy). Each dot represents a participant, color-coded by disease severity: H - green; D - blue; DWS - orange; SDp - pink. Sample IDs are shown for the three DWS patients with highest DENV read counts. b, UMAP color-coded by DENV reads per million reads (cpm) of the three patients labeled in a. c, Fraction of VHCs from the total (bar plot, left panel) and distribution of DENV read per million reads (violin plots, right panel) in the indicated immune cell types from the three patients labeled in a. d, Violin plot depicting the percentage of DENV E positive cells in B cells (CD19+), classical monocytes (CD14+CD16−), non-classical monocytes (CD14−CD16+), intermediate monocytes (CD14+CD16+), cDC1s (CD141+), cDC2s (CD1c+), pDCs (CD123+CD303+), CD56bright NK cells, CD56dimCD16+ NK cells, and T cells from the indicated DENV-infected DWS patients with viremia ranging from 105 to 109 viral copies/mL measured via spectral flow cytometry. e, Coverage of detected positive (+ strand) and negative (− strand) vRNA strands along the DENV genome. f, Schematic of the co-culture experiments shown in g. g, Bar plot showing DENV RNA copies per μL measured via RT-qPCR in Huh7 lysates following a 5-day co-culture with patient-derived total PBMCs (gray), or HLA-DR+ (CD19−CD20− CD3−HLA-DR+ cells, green) and B cell (CD19+CD20+, orange) fractions isolated from the same five DWS patients). Data are combined from two independent experiments (n = 5 participants). Dots represent technical replicates. h, Violin plots showing log2 fold change of DEGs between VHCs and corresponding bystander B cells in three DWS patients with highest vRNA reads shown in Fig.4a. DEGs were identified by the median log2 fold change of 100 comparisons between VHCs and equal numbers of random corresponding bystander cells. i, Radar plot showing log2 fold change of selected genes associated with MHC II and MHC I pathways between VHCs and corresponding bystander cells in B cells. j, Violin plot showing cell surface expression of HLA-DR in DENV+ and DENV− fractions, identified via DENV envelope immunolabelling of B cells, monocytes, DCs and NK cells measured via spectral flow cytometry and displayed as fold change (FC) of mean fluorescence intensity (MFI) above healthy control. Data combined from two independent experiments are shown in panels d [n = 11: DWS (n = 6); healthy controls (n = 5)] and j (n = 6). P values by two-tailed Mann-Whitney test are shown in panel J.
Since myeloid cells were reported to be the primary DENV targets in blood13, 14, yet vRNA was abundant in B cells, we measured the intracellular expression of DENV envelope (E) protein via spectral flow cytometry in PBMCs from six viremic DWS patients (Extended Data Fig. 6c, Table 1). Patient-derived B cells (CD19+CD20+) expressed moderate, albeit variable, E levels vs. healthy control (Fig. 4d; Extended Data Fig. 6d). High E expression levels were measured in classical (CD14+CD16−), non-classical (CD16+) and intermediate (CD14dimCD16+) monocytes, type II conventional dendritic cells (cDC2) (CD1c+), and CD56bright NK cells. E protein expression was low in cDC1s (CD141+) and practically absent in pDCs and T cells (Fig. 4d; Extended Data Fig. 6d).
To determine whether DENV actively replicates in B cells during natural infection, we leveraged the ability of viscRNA-Seq 2 to ascertain for each vRNA molecule which genomic strand it originated from. We detected 419 DENV reads across 255 cells that originated from the DENV negative strand (presumably synthesized during vRNA replication) (Extended Data Fig. 6e). These molecules were detected primarily in B cells and distributed at the 5’ end of the genome, similarly to positive-strand reads, and at position ~1700 bases—a region that does not contain tight RNA hairpins15, possibly providing easier template access for the poly-T oligonucleotide (Fig. 4e; Extended Data Fig. 6e). The negative-strand to total vRNA ratio was 14.1%, an order of magnitude higher than the ratio of antisense molecules for representative housekeeping genes (Extended Data Fig. 6f), favoring active DENV replication over an artifact of library preparation.
To assess the ability of patient-derived B cells to produce infectious virus particles and infect naive cells, we co-cultured DENV-permissive human hepatoma (Huh7) cells for five days with PBMCs from five DWS viremic patients or with two cell fractions isolated from the same samples via flow cytometry: i) B (CD19+CD20+) cells; and ii) HLA-DR+ non-B cells (CD45+CD3−CD19−CD20−HLA-DR+), followed by RT-qPCR measurement of intracellular DENV copies number (Fig. 4f, Extended Data Fig. 7a, Table 1). High DENV copy numbers were measured in co-cultures of PBMCs from all five patients, indicating effective infectious virus production (Fig. 4g). Isolated HLA-DR+ cells from five out of five patients effectively infected Huh7 cells with DENV copy numbers that were comparable or slightly lower than PBMCs. Remarkably, sorted B cells from four of the five patients infected Huh7 cells, with DENV copy numbers that were comparable or up to 2-fold lower than the PBMC and HLA-DR+ fractions (Fig. 4g). DENV copy numbers in the various co-cultures positively correlated with viremia level (Extended Data Fig. 7b).
To determine whether DENV alters the transcriptome of VHCs, we identified DEGs between vRNA-harboring and corresponding bystander B cells using a bootstrapping strategy (see Methods) (Fig. 4h). vRNA-harboring B cells showed upregulation of oxidative phosphorylation and surface marker (CD69, CXCR4) genes and downregulation of genes involved in BCR signaling (FCRLA, FCRL3, FCLR5, HCK, SYK, FGR) and ISGs (MX1, IFI6, IFI30, LY6E) relative to bystander B cells. MHC-II genes (HLA-DPA1, HLA-DRB5) and IFI30 (GILT, thiol reductase mediating antigen unfolding) were downregulated, and HLA-DOB (negative regulator of HLA-DM-mediated peptide loading16) was upregulated in virus-harboring vs. bystander B cells (Fig. 4i), proposing DENV-induced downregulation of antigen processing, loading and presentation capacity. Whereas MHC-I transcripts were not altered, TAP1 (antigen transporter) and the cytoplasmic peptidases PSMB8/9/10, and LAP3 were mildly downregulated in vRNA-harboring vs. bystander B cells (Fig. 4i). In agreement with the transcriptomic data, flow cytometry analysis of patient-derived PBMCs stained intracellularly for DENV envelope (E) protein revealed significantly reduced expression of HLA-DR on E+ vs. bystander B cells (Fig. 4j). DEG analysis between vRNA-harboring monocytes and NK cells was limited by noise (given small number of viral reads in these cells) (Extended Data Fig. 7c). Nevertheless, HLA-DR protein expression was significantly lower on E+ non-classical monocytes and cDC2s (Fig. 4j)—the myeloid cell subtypes in which we measured highest mean percentage of E+ cells (Fig. 4d)—yet, comparable between E+ and bystander NK cells (Fig. 4j).
These results reveal infectious DENV production in natural infection in circulating B cells, in addition to monocytes and cDCs13, 14 in a dynamic balance with bloodstream virions that is associated with a naïve B cell state and altered interferon signaling and antigen processing and presentation signatures. Some of the phenotypes observed in cells harboring vRNA vs. bystanders are also observed in SDp vs. D patients.
Altered cell-cell interactions and cytokine production in SD
At a systems level, orchestration of the immune response is facilitated by interactions between different cell types via recognition of ligands by membrane-bound receptors. By computationally analyzing cell-cell communications in SDp and D children we identified 1164 genes encoding proteins involved in known interactions (interacting genes)17, 18. The number of candidate interactions, most commonly involving monocytes and cDCs, was larger in SDp than D (Fig. 5a; Extended Data Fig. 8a,b).
Fig.5: Cell-cell communication and cytokine production are increased in SDp.
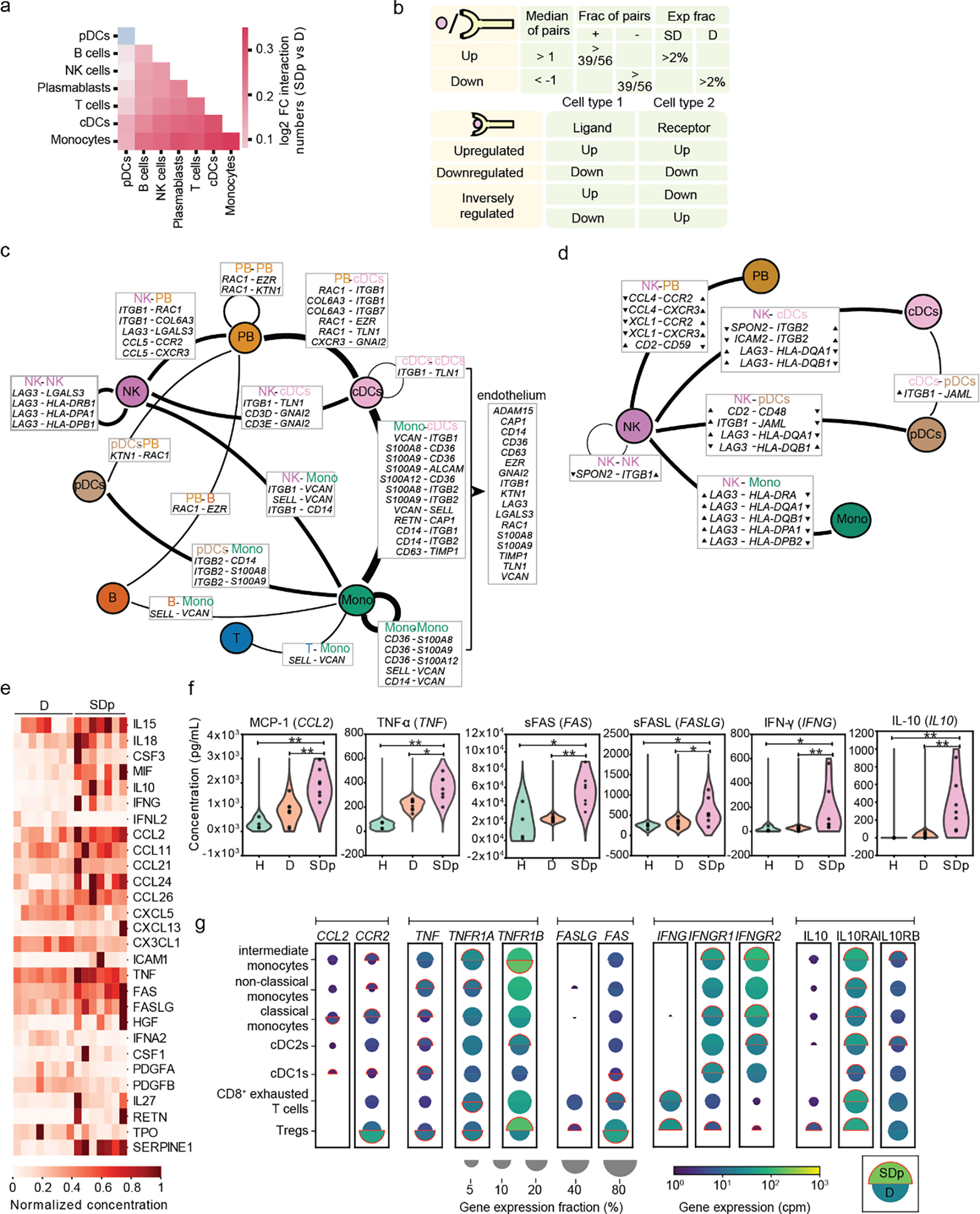
a, Heat map showing the log2 fold change in number of candidate interactions between SDp and D children defined by expression of both the ligand and receptor in at least 2% of cells within each cell type. b, Schematic showing the parameters for identifying interacting partners and (top), and the classification of DEIs (bottom). c, d, Interaction networks of upregulated (c) and inversely regulated (d) DEIs in SDp vs. D. Circles indicate cell types; lines indicate interaction partners with thickness representing the number of interactions; text boxes specify genes involved in identified candidate interactions and the cell types expressing them, including candidate interactions with endothelium based on Tabula Sapiens data (right panel in c); and arrowheads (in d) depict up- and down-regulation of the indicated genes. e, Heatmap showing serum concentrations of 27 differentially secreted cytokines (out of 80 tested) between SDp vs. D normalized to the highest corresponding concentration. f, Violin plots of serum concentration (pg/mL) of top differentially secreted cytokines in SDp vs. D. g, Split dot plots showing the expression level of transcripts of cytokines (corresponding to those in (f)), and cognate receptors measured via viscRNA-Seq 2. Top half of each split dot plot depicts the expression level in SDp; bottom half depicts the expression level in D; size depicts the percentage of gene expressing cells in the cell types indicated on the left; and color depicts gene expression level in counts per million (cpm). Red outline indicates the condition with the higher gene expression. Data in panels e and f are from n = 19 participants: H (n = 4), f only; D (n = 8); SDp (n = 7). p values by two-tailed Mann-Whitney test are shown in panel f.
We defined interacting DEGs (iDEGs) as ligand- or receptor-encoding genes that were also differentially expressed, and differentially expressed interactions (DEIs) as interactions whose ligand and receptor were iDEGs and passed a strict statistical randomization test (Fig. 5b; Extended Data Fig. 8c–d). DEIs were classified as upregulated, downregulated or inversely regulated according to the ligand and receptor regulation. No strongly downregulated DEIs were identified, and upregulated and inversely regulated DEIs were largely physical (Supplementary Table 5). iDEGs were abundant and largely upregulated in cDCs and monocytes, and abundant, yet variably regulated in NK cells (Extended Data Fig. 8e). Concurrently, cDCs and monocytes formed the largest hubs of the upregulated DEI network (Fig. 5c; Extended Data Fig. 8c). Candidate upregulated DEIs were predominantly involved in adhesion and migration (e.g. ITGB1-VCAN; SELL-VCAN) and pro-inflammation (e.g. S100s-CD36; RETN-CAP) (Fig. 5c; Extended Data Fig. 8f). Eighteen of 31 upregulated iDEGs involved in DEIs in SDp were also expressed in >5% of endothelial cells from the human cell atlas Tabula Sapiens19, suggesting potential roles in immune-endothelium interaction (Fig. 1c).
Beyond involvement in variably-regulated iDEGs, NK cells were the hub of the network of inversely-regulated iDEGs—interactions likely involved in regulatory or compensatory mechanisms (Fig. 5d,e; Extended Data Fig. 8d). Candidate communications included: downregulated SPON2 in NK cells with upregulated ITGB1 and ITGB2 in myeloid subtypes; upregulated LAG3 in NK cells with downregulated HLA-DQA1/HLA-DQB1 in myeloid subtypes, and downregulated CCL4 and XCL1 in NK cells (Fig. 5d; Extended Data Fig. 8f–h), supporting inhibition of NK cell function and tissue migration.
To strengthen our understanding of paracrine communications, we measured the levels of 80 secreted factors in the sera of the same patients and identified 27 differentially secreted cytokines between SDp and D (Fig. 5e, Supplementary Table 6). Computational analyses revealed specific myeloid and T cell subtypes that may contribute to their secretion and/or interact with these cytokines (Fig. 5f,g). Among the differentially secreted cytokines, increased CCL2 concentration in SDp was associated with upregulation of transcripts for both CCL2 in cDC1s and its receptor, CCR2, on myeloid subtypes, yet downregulation of CCR2 on Tregs. Increased TNFα, sFAS, and sFASLG concentration in SDp’s sera suggested apoptosis induction, yet the viscRNA-Seq data revealed distinct responses, with proapoptotic signatures (upregulation of TNF and TNFRSF1A) in monocytes and cDCs vs. antiapoptotic signatures (downregulation of TNF, TNFRSF1A and upregulation of the anti-apoptotic TNFRSF1B) in Tregs. Increased IFNγ concentration was associated with IFNG upregulation on Tregs and exhausted T cells accompanied by IFNGR1 and IFNGR2 upregulation on APCs. Furthermore, increased concentration of IL-10 appeared to be partially driven by Tregs and was accompanied by upregulation of IL10RA/B on myeloid and regulatory and exhausted T cells.
SD progression is thus linked to increased coordinated interactions involving myeloid cells associated with migration and pro-inflammation, yet aberrant orchestration of the innate-adaptive immune junction by poorly coordinated NK cell interactions.
A systems immunology model of SD progression
Integrating viscRNA-Seq 2 and functional data, we propose a systems immunology model of SD progression centered around a disjointed immune response (Fig. 6). Uncomplicated D/DWS is characterized by a multifaceted immune response that balances antigen recognition, uptake, processing and presentation with initiation of DENV-specific responses driven by T and B lymphocytes. Contrastingly, SD progression is linked to increased plasmablasts and coordinated myeloid cell interactions promoting migration and pro-inflammation, yet aberrant orchestration of the innate-adaptive immune junction, mediated in part by DENV replication in B cells and myeloid cells, and in part by suppressive Tregs and uncoordinated NK cell interactions, promoting tissue injury and reducing viral clearance.
Fig. 6: Immune hallmarks of early SD progression.
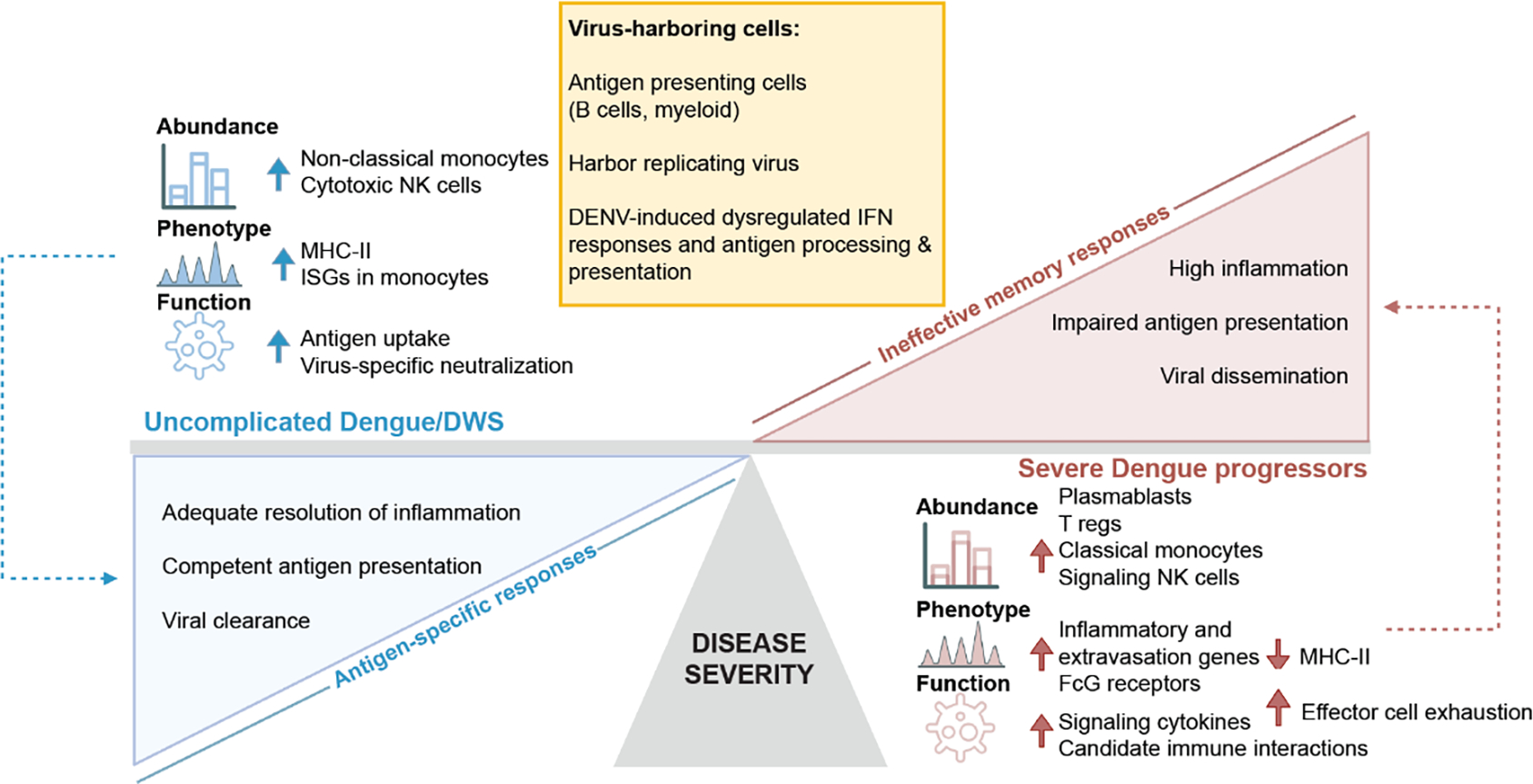
Schematic representation depicting the differences in cell type abundance, phenotype, and immunological function of distinct cell subtypes in SDp (red) vs. uncomplicated D/DWS (blue), and specific attributes of virus-harboring cells (VHCs) (yellow). We propose that ineffective memory responses, accompanied by defective antigen presentation and inflammation resolution mechanisms, in part mediated by active DENV replication in B cells and myeloid cells in human blood, prevail in SDp, whereas adequate presentation of antigens and effective antigen-specific responses characterize D/DWS patients.
Discussion
The pathogenesis of SD infection in humans remains poorly understood. Here, we integrate two single cell approaches, to capture cellular and viral elements in children’s PBMCs, with functional assays and secretome analysis. We discover the cellular targets of DENV in the human blood and immunological determinants of SD progression that are in part DENV-induced, including alterations in cell subtype abundance, DEGs, cell-cell communications, candidate immune blood cells driving cytokine secretion, and potential immunological pathways induced in natural dengue infection.
Myeloid cells were previously identified as DENV target cells in natural infection: DENV proteins were detected in cDCs, monocytes, and macrophages in patients’ blood via flow cytometry20,21 and in cadaver tissues via immunohistochemistry22,23. These cells were also found to be susceptible to DENV infection in vitro3,20. Our results showing high-level replication in monocytes via E protein staining and co-culture experiments, albeit low number of vRNA molecules, support these findings. Extending prior findings showing that B cells harbor the majority of positive-strand vRNA in children’s blood in adult patients10, 24, we reveal active replication in patients’ B cells via three orthogonal experimental methods: measuring negative-strand vRNA, E protein expression via flow cytometry, and viral spread upon co-culture with permissive cells. Previously, evidence for infectious DENV production was demonstrated only in healthy donor-derived B cells in vitro-infected at a high multiplicity of infection (MOI=20)25, but not in patient-derived B cells.
Subgenomic flavivirus RNAs (sfRNAs) might account for the greater vRNA abundance in B cells yet greater E protein expression in myeloid cells, since sfRNAs can (i) suppress antiviral IFN response20, 21 consistent with our finding that vRNA-harboring B cells downregulate IFN response genes relative to bystander cells; and (ii) compete for packaging with DENV genomic RNA22, increasing intracellular vRNA. Patient-to-patient variability and differences in sensitivity may also account for the former and for the discrepancy in magnitude between VHCs and E+ monocytes and NK cells. Nevertheless, since the fraction of detected vRNA molecules resulting from active replication vs. passive internalization and/or adsorption onto the cell surface is unknown, the comparison across cell types is hardly statistically unbiased. Interestingly, vRNA-harboring B cells upregulate markers and transcripts for CD69 and CXCR4—markers for antigen internalization and homing to germinal centers for antigen presentation, respectively. We speculate that by internalizing vRNA, these B cells might differentiate in absence of specific BCR signaling and prematurely home to secondary lymphoid organs, thereby expanding the pool of low-affinity plasma cells and antibodies, the hallmark of ADE.
While monocyte subtypes demonstrate comparable level of DENV E protein and SDp-specific upregulation of genes and interactions promoting inflammation and migration, their abundances are distinctly altered. Classical monocytes are expanded and activated, possibly contributing to SD pathogenesis by promoting migration to lymphoid organs, conferring sustained inflammation and forming a niche for DENV replication, as suggested by others23. Contrastingly, the fraction of non-classical monocytes, patrolling the endothelium and protecting from protease-mediated damage23, is drastically reduced, potentially impairing immune surveillance and inducing endothelial damage24.
We reveal downregulation of antigen processing and presentation genes in APCs in SDp vs. D, yet intact uptake. Reduced HLA-DR expression, particularly on non-classical monocytes, cDC2s, and B cells (primary DENV targets), is partly driven by DENV, expanding previous findings of suppression of cell maturation and HLA-DR expression in blood donor-derived DCs upon DENV infection25. Our results validate this finding in natural infection in several DENV target cell subtypes and propose that this viral-mediated immune modulation may contribute to reduced antigen presentation in SD progression. Interestingly, no such drop in HLA-DR expression was detected in E+ vs. bystander NK cells, supporting a hypothesis that the viral elements detected in NK cells represent adherence of infected cell debris engaged by NK cells to the NK cell membrane26 rather than active replication.
We reveal downregulation of additional antigen processing machinery components in vRNA-harboring B cells. It is tempting to hypothesize that since DENV E, Pre M and NS1 proteins harbor disulfide bridges27, the downregulation of IFI30 (GILT)--critical for unfolding antigens with disulfide bridges16, 28--could impact peptidome generation. While Epstein-Barr viral microRNA downregulates IFI30 ex vivo in primary B cells, no such modulation has been reported in natural infection29. HLA-DOB upregulation may impair HLA-DM-mediated antigen uploading on conventional MHC-II, as reported in other disease models16. Similarly, albeit mild, the downregulation of TAP1, key for antigen transport, and cytoplasmic peptidases in vRNA-harboring B cells proposes possible DENV-induced modulation of cross presentation, described in DNA viruses16.
We provide evidence for dysregulated IFN responses in SDp, including increased serum IFNγ levels (as reported30), largely derived from T and NK cells, and increased IFN signaling genes (e.g. STAT1, IRF) in multiple cell types. Nevertheless, signatures of antigen presentation, effector responses, and ISGs—functions typically stimulated by IFNγ31—are suppressed in APCs of SDp. These findings suggest that DENV infection, and in part active replication in APC subtypes, might disrupt their ability to adequately respond to IFNs and trigger antigen-specific T cell-responses, pointing to impaired coordination between innate and adaptive immunity during SD progression. Conversely, antigen uptake is greater in all APC subsets from DENV-infected patients than healthy donors, possibly compensating for the deficient antigen presentation.
Our findings point to NK cells as a key correlate of SD progression. Cytotoxic NK cells in SDp exhibit activation signatures, including upregulation of CD2 and downregulation of the inhibitory receptor KLRB1. They also demonstrate increased exhaustion in SDp, with higher LAG3 and PD-L1 expression detected by CyTOF11, and lower abundance relative to all NK cells, suggesting reduced killing of DENV-infected cells. Concurrently, signaling NK cells with pro-inflammatory and migratory phenotypes are exapanded, possibly contributing to tissue injury and vascular permeability, more commonly reported in children32. We also observe expansion of “adaptive-like” NK cells with increased expression of MHC-II transcripts and proteins, IL32, CD2 and LAG3 in SDp. These NK cells might activate antigen-specific T cell responses, as shown in cytomegalovirus infection33, or be active in T cell killing34, 35. These findings combined with our cell-cell communication analysis propose that imbalanced NK cell responses with reduced cell killing yet increased inflammation and migration may be implicated in SD progression.
We provide evidence for emergence of Tregs characterized by increased expression of CTLA4, LAG3, TIGIT, HAVCR2, and PDCD in SDp. These findings contrast a study showing reduced suppression36, yet agree with CTLA4 and PD-1 overexpression detected on Tregs by CyTOF11. Notably, the abundance of Tregs measured longitudinally via CyTOF declined prior to SD onset, supporting protection11, possibly mediated in part by inhibition of vasoactive cytokine production37. However, Treg activation can also suppress immune responses, such as via IL-10 secretion38, thereby promoting pathogenesis. While IL-10 has been implicated in SD39, 40 and shown to suppress DENV-specific responses upon restimulation of patient-derived T cells41, the specific cell subtypes orchestrating this response remained unknown. Based on the increased serum IL-10 level, upregulation of IL10 transcripts particularly in Tregs, and upregulation of IL10RA/B transcripts in myeloid cells, it is intriguing to hypothesize that this pathway is activated by Treg-secreted IL-10 and contributes to the downregulation of antigen presentation and IFN response signatures in APCs of SDp, as shown in other disease models42.
In addition to IL-10 and IFNγ, among the 27 differentially secreted cytokines we identify, MCP-1 and TNF have been implicated in increased plasma leakage and hepatic dysfunction in an independent pediatric dengue cohort39. The increased serum MCP-1 level and CCL2 (MCP-1) and CCR2 transcripts we measure in myeloid cells from SDp may be associated with disease pathogenesis in humans, similarly to mice43. Increased serum TNFα and TNF and TNFRSF1A-B transcripts in myeloid cells of SDp reveal activation of the TNFα pathway, previously shown to mediate tissue injury via caspase-3-mediated cell death44 and correlate with SD45.
While non-neutralizing antibodies are key mediators of SD immunopathogenesis, we provide evidence that beyond ADE, functional and phenotypic alterations within cell subtypes affect the collective coordination and effectiveness of the immune system. Our findings support that prior DENV exposure is associated with expansion of memory B and Treg cell fractions and pathways involved in ADE and possibly effector cell exhaustion. Nevertheless, other hallmarks of SD progression including downregulation of MHC-II genes may not or only partially be associated with DENV exposure. Moreover, we reveal that some alterations, including in IFN responses and antigen presentation signatures, may result from DENV-mediated immunomodulation. Patient-specific attributes that interfere with antigen presentation, e.g. genetic or epigenetic determinants, may also predispose individuals to disease progression.
Our systems immunology approach establishes a broad, yet granular, picture of the immune response to DENV accompanying severe progression, highlighting cell-cell and virus-cell interactions and identifying B cells as a major DENV target in the bloodstream. While this study has several limitations (see extended text), it provides a template to gain detailed biomedical insight into an infectious disease and could be extended to other human pathogens.
Methods
Colombia Cohort
Ethics Statement.
All work with human subjects was approved by the Stanford University Administrative Panel on Human Subjects in Medical Research (protocols #35460 and #50513) and the ethics committees in biomedical research of the Fundación Valle del Lili (FVL, Cali, Colombia) and Centro de Atención y Diagnóstico de Enfermedades Infecciosas (CDI, Bucaramanga, Colombia). The parents or legal guardians of subjects provided written informed consent, and subjects between 2 to 17 years of age provided assent. Subjects were not involved in previous procedures and were all test-naïve. The demographics and clinical parameters of the subjects are summarized in Supplementary Table 1. Participants were reimbursed for their travel expenses and for meals during their clinic visit when relevant, yet no compensation was provided.
Study Population and Sample Collection.
The Colombia cohort participants involved in this study presented to the emergency room or clinics of FVL or CDI between March 2016 and Jan 2020. Enrollment criteria consisted of: i) age equal to or greater than 2 years; ii) presentation with an acute febrile illness of less than 7 day duration associated with one or more of the following symptoms or signs: headache, rash, arthralgia, myalgia, retroorbital pain, abdominal pain, positive tourniquet test, petechiae, and bleeding; and iiia) a positive dengue IgM antibody and/or NS1 antigen by the SD BIOLINE Dengue Duo combo device (Standard Diagnostic Inc., Korea) test or iiib) clinical presentation highly consistent with dengue and subsequent confirmation of diagnosis via serological testing and rRT-qPCR at Stanford. Patients were classified by infectious diseases specialists, both upon presentation and at recovery, as having dengue (D), dengue with warning signs (DWS), or severe dengue (SDp) according to 2009 WHO criteria3 (Supplementary Table 1). Patients who progressed to SD prior to enrollment were excluded from this study.
The first day of fever (fever day 0) was defined by the patients or their relatives. Symptoms, warning signs, and laboratory parameters (including complete blood count, chemistry, and liver function test results) were documented by healthcare professionals.
10–40 mL of whole venous blood samples were collected upon enrollment and PBMCs and serum samples isolated. Sample transport, reception, and processing were strictly controlled using personal data assistants (PDAs) with barcode scanners.
For patients managed in an outpatient setting, follow up was conducted daily via phone calls, during which patients were provided information about the clinical warning signs and asked about their appearance, until full recovery when final diagnosis was determined. Organ damage was defined according to standard clinical endpoints for DENV infection46. Demographics and clinical information were collected at the time of presentation (Supplementary Table 1).
Healthy controls samples were obtained from children presenting to the clinic in FVL (Colombia) for routine checkups or prior to elective procedures.
Sample Selection.
For viscRNA-seq 2: PBMCs from 19 children infected with DENV (9 males and 10 females, age: 4–17 years) and 4 healthy controls (2 males and 2 females, age: 5–12 years) were selected (Table 1). Samples from 7 of 9 children who progressed to SD (out of approximately 200 enrolled) were selected based on: sufficient cell number and enrollment prior to meeting SD criteria. The eight dengue (D) patients were selected to largely match the age, gender, and prior exposure status of the SD patients. Four DWS samples were selected and used from patients with high viral load in serum to improve detection of viral reads, yet only three were used for cell subtype abundance determination due to small number of cells and B cell predominance in sample 6–028 (Supplementary Table 2). Four age- and gender-matched samples were also selected from healthy controls. Serum samples from the same SDp and D patients were used for the cytokine analysis (Table 1). For functional/validation assays: Due to the limited number of SDp and cell numbers in these pediatric samples, samples from 11 individuals with DWS, the most frequent diagnosis, were selected for these assays. For the co-culture assays, we chose DWS patients with various levels of viremia (ranging from 10^5 to 10^9 viral copies/mL). The healthy controls samples used for some of the functional assays were derived from adults from the Stanford blood bank.
Establishment of Dengue Diagnosis
Detection of DENV NS1 antigen and IgG/IgM.
The SD BIOLINE Dengue Duo combo test (Standard Diagnostic Inc., Korea) was used to identify dengue patients for enrollment to the study.
rRT-PCR assays for detection of DENV and other microbial pathogens.
To confirm the diagnosis of dengue and differentiate from infection with the co-circulating arboviruses, Zika virus and chikungunya virus, serum samples were screened via a qualitative, single-reaction, multiplex real-time reverse transcriptase PCR (rRT-PCR) that detects Zika, chikungunya, and DENV RNA. To identify the specific DENV serotype and quantify the viral load, samples positive for DENV in the screening assay were serotyped and quantitated using a separate DENV multiplex RT-qPCR47.
DENV serological assays.
Anti-DENV IgG were tested using DENV Detect™ IgG ELISA kit (InBios, Seattle, WA) as per manufacturer instructions. Briefly, serum samples were diluted 1:100 in sample dilution buffer and 50 μL was added per well. The top half (4 wells on each 8-well strip) of the ELISA plates were pre-coated with DENV derived recombinant antigen (DENRA) and the other half with normal cell antigen (NCA). Each sample went into two pairs of DENRA or NCA coated wells, one without and another containing 50 μL of 8 M Urea. Each plate contained a negative and a positive control provided in the kit. Plates were incubated in 37 °C for 1 hour and washed in a plate washer (Biotek 404 Select Microwasher). Next, we added 50 μL/well enzyme conjugate-HRP tagged goat anti-human IgG (included in the ELISA kit), incubated for 1 hour at 37 °C, and followed by wash. Next, 150 μL of EnWash was added, incubated for 5 min at 22 °C (room temperature) and washed. Finally, 75 μL of TMB solution was added, incubated at 22 °C in the dark for 10 min. The reaction was stopped by adding 50 μL stop solution. The plates were read at 450 nM by Spectra Max M2 (Molecular Devices) using SoftMax pro 7.0.3 software. Presence or absence of DENV IgG was interpreted from the ratio of readings from DENRA and NCA wells without urea. A ratio >= 2.84 was considered positive, 1.65–2.84 ‘equivocal’ and <= 1.65 as negative. The equivocal samples were repeated once. DENV IgG avidity was calculated by the ratio of reading in DENRA well without urea over the DENRA well with 8 M urea. High avidity (>0.6) was considered as secondary infection, moderate avidity (0.4–0.6), and low avidity (<0.6) as primary infection, in samples that showed a positive result by DENRA/NCA ratio.
PBMC isolation.
PBMCs were isolated using SepMate tubes (Stemcell Technologies) from 18 mL (>10 years), 9 mL (5–10 years) and 5 mL (2–5 years) of blood and stored at a cell concentration ranging from 1×10^6 to 5×10^6 cells/mL depending on the overall cell number. Briefly, whole blood was diluted 1:1 with phosphate-buffered saline (PBS) and added to a SepMate tube, which contained 15 mL of Ficoll. Tubes were then centrifuged for 10 minutes at 1,200g, after which the PBMC layer was poured off into a fresh tube and washed with PBS. Tubes were then centrifuged at 250 × g for 10 minutes and resuspended in freezing media. Cryovials containing PBMCs were then placed in a CoolCell at −80 ° C for 24 hours prior to being transferred to liquid nitrogen for storage.
Magnetic separation and rebalancing of PBMCs.
Samples containing ~1 mL of cryopreserved PBMCs were quickly thawed in a water bath at 37 °C in medium containing 10% DMSO. Nine mL of warm medium were added and cells were spun at 300 g for 8 minutes. The supernatant was discarded, and 2 mL of medium were added. The samples were filtered with 35 μm cell strainers to obtain a pure single-cell suspension. The samples were mixed with magnetic microbeads to label cell debris, dead cells, and dying cells (Miltenyi Biotec), and the mix was set on a magnet to elute only living cells. The flow through was spun, the supernatant was removed, and the cell pellet was resuspended in 150 μl cell resining buffer with 0.5% BSA (AutoMACS). To enrich for rare cell populations, the samples were split into 2 aliquots: a 30 μl aliquot was kept intact, and a second aliquot was labeled with anti-CD3- Phycoerythrin (PE) antibody (BioLegend) and mixed with anti-PE magnetic microbeads to reduce the number of T cells. Cell numbers in both aliquots were measured, and the two aliquots were remixed. The samples were then diluted to 1000~1400 cells/μl and processed via viscRNA-Seq 2.
Virus-inclusive single cell RNA-sequencing 2 (viscRNA-Seq 2).
The master mix containing 10,000–14,000 cells per sample was prepared following the standard 10X Genomics 5’ RNA-Seq protocol. A DENV capturing oligo was added to the master mix at a final concentration of 30 nM for capturing the specific DENV serotype identified via RT-qPCR in patient’s sera (in this work DENV 1 and DENV3) (DENV1: 5’-AAGCAGTGGTATCAACGCAGAGTACTTTCCCCAGCTTTTCCATGA-3’); DENV3: 5’-AAGCAGTGGTATCAACGCAGAGTACTTTCCCCACGTTTTCCATGA-3’). Samples were loaded into the droplet generator for partitioning single cells into an inverted emulsion. The Next GEM Single Cell 5’ reagent kit (10x Genomics) was used for reverse transcription, cDNA amplification and construction of gene expression libraries. Library quality was assessed using a 2100 Bioanalyzer High Sensitivity DNA kit (Agilent). Sequencing was performed in a paired-end mode with S1 and S4 flow cells (2 X 150 cycles) on a NovaSeq 6000 sequencer (Illumina).
Pre-processing of 10x Genomics Chromium scRNA-seq data.
CellRanger v3.1.0 (10x Genomics) was used to process the raw read pairs. To generate a gene expression matrix for each sample, we constructed a combined reference of (i) human genome (GRCh38) and (ii) DENV genome as an additional chromosome (GenBank GQ868569 for DENV 1 and MH544647 for DENV 3). We then aligned the reads to this reference and measured the number of UMIs for each host gene and vRNA in each cell. High quality cells were scored based on the following criteria: fewer than 15% mitochondrial reads; over 400 expressed genes; between 1,000 and 30,000 detected UMIs. We excluded genes that were expressed in less than three cells. The resulting dataset was normalized by the total UMI count, multiplied by 1,000,000 (cpm) and log2 transformed after addition of a pseudo count of 1.
Dimensional reduction, clustering and cell type annotation.
After normalization, principal component analysis (PCA) was performed on the 2,000 most variable features identified by dispersion-based methods. The normalized dispersion was obtained by scaling with the mean and standard deviation of the dispersions for genes falling into a given bin for mean expression of genes. The two-dimensional embedding was computed via UMAP based on the first 15 dimensions of the PCA reduction. The cells were clustered using the Leiden algorithm based on the first 15 PCA dimensions with modularity metric and a resolution parameter of 0.3. Clusters were annotated based on marker genes from public sources to identify immune cell types. The data from each annotated immune cell type were further embedded and clustered to annotate cell subtypes.
Detection of vRNA harboring cells (VHCs).
We counted the number of molecules aligning against the DENV genome in our composite reference. All cells with >= 1 detected viral molecules were considered VHCs, else they were considered bystanders. Since the number of viral molecules detected in a single cell was never above 24 and typically 1 or 2, the likelihood of vRNA spillover from separate droplets is low. Several analyses were carried out using slightly different thresholds for VHC vs. bystander and yielded similar results for the fraction of positive/negative strand reads. The analyses shown in Fig 4b,c,e,h,i; extended Fig. 6b,e,f and extended Fig. 7c were conducted on three DWS samples with the highest VHC numbers.
Differential expression via pairwise comparison of patient averages.
To identify differentially expressed genes across patient groups (e.g. SDp vs. D), a two-step procedure was used. First, gene expression was averaged within each cell type within each patient that had at least 5 cells of that type. For subtype-level analyses, at least 3 cells were required. Second, since the empirical distribution of gene expression is unknown and the sample size small, effect size and noise level were visualized separately. All pairs of patients (one in the first group, one in the second, and never two from the same group) were compared and the median log-fold change of all comparisons was used to estimate effect size, while the noise level was shown using box plots (Extended Data Fig 3.a). DEGs were defined via a custom threshold as those with a median log2 fold change greater than 1 or smaller than −1. The results of this pairwise comparison largely agreed with those obtained via a KS test on the same dataset (Supplementary Table 7). For DEGs between VHCs and bystanders, both VHC and non-VHC cells were subsampled to the number of VHCs from the same cell type, and 100 bootstraps of this population was computed to form an empirical distribution of differential expression.
Cell-cell communication analysis.
We extracted the list of known potential interaction partners from OmniPath and complemented it with a series of interactions mined from recent immunology literature (Supplementary Table 8). Given the shallow sequencing of droplet single cell platforms, interactions were counted if both the ligand and the receptor were expressed in more than 2 % of the cells within the respective cell type. Variations in this threshold did not affect our results. Interacting DEGs (iDEGs) were defined as ligands or receptors in our list of interactions which were clear DEGs (i.e. |log2-fold change| > 1 and an up- or -downregulation in at least 40 out of 54 pairwise patient comparisons) (Fig 5.b, top). Differential expressed interactions (DEIs) between D and SDp were classified as upregulated and downregulated if both the receptor and ligand were overexpressed or under-expressed, respectively, or as inversely regulated if one of the partner genes was overexpressed and the other under-expressed (Fig 5.b, bottom). A nonparametric label randomization test was utilized to define the significance of differential interactions. The p-value was calculated as the fraction of randomizations for which the distance from the origin was larger (i.e., differential expressions were more extreme) than in the actual data (Extended Data Fig 8.g). Violin plots were designed to show the expression of the interacting gene on the corresponding cell type in each patient to verify the robustness of DEIs passed the randomization test (Extended Data Fig 8.h).
Reanalysis of an existing CyTOF dataset.
To correlate the viscRNA-Seq findings at the transcript level with those we recently measured at the protein level via CyTOF11, we reanalyzed the (publicly available) CyTOF dataset using the published code at github.com/davidrglass/dengue, this time excluding patients diagnosed with SD at enrollment. Samples collected within 12 days of fever onset from 47 D patients and 21 patients who progressed to SD within several days of enrollment were included. Comparison between SDp and uncomplicated D for cell subtype abundance and protein expression in distinct cell subtypes was performed.
Spectral flow cytometry.
Cryopreserved PBMCs were thawed at 37 °C, washed, and incubated with the Zombie Aqua™ live/dead fixable dye (1:1000 dilution, Biolegend) and with 5 μl of FcR blocking reagent (Miltenyi) for 15 min at 22 °C (room temperature). Cells were labeled for 20 min at 4 °C with the following specific antibodies: BUV496 anti-CD19 (1:50 dilution, SJ25C1;BD Biosciences), BUV563 anti-CD56 (1:50 dilution MY31;BD Biosciences), BUV615 anti-HLA-DR (1:50 dilution,G46–6;BD Biosciences), BUV661 anti-CD20 (1:50 dilution, 2H7;BD Biosciences), BUV737 anti-CD16 (1:50 dilution, 3G8;BD Biosciences), BUV805 anti-CD3 (1:50 dilution, SK7;BD Biosciences), BV421 anti-CD1c (1:50 dilution, L161;Biolegend), BV605 anti CD11c (1:50 dilution, 3.9;Biolegend), BV711 anti CD163 (1:50 dilution, GHI/61;Biolegend), PeCy5 anti-CD123 (1:50 dilution, 6H6;Biolegend), PeCy7 anti-CD141 (1:50 dilution, M80’;Biolegend), APC anti CD14 (1:50 dilution, M5E2;Biolegend), AF700 anti CD303 (1:50 dilution, 201A;Biolegend), APC-Cy7 anti CD15 (1:50 dilution, W6D3;Biolegend) and PeCy5.5 anti-Flavivirus (1:25 dilution, 4G2;Novus Biologicals). Cells were fixed with 1% PFA for 24 hours, and data were acquired on an Aurora spectral analyzer (Cytek) and analyzed with the FlowJo v10.9 software (Tree Star). Flow cytometry experiments were performed on PBMCs from DWS patients and healthy controls given the limited availability of SDp samples.
Co-culture experiments assessing infectious DENV production.
Cryopreserved PBMCs were thawed at 37 °C and washed with cell culture media (RPMI (Gibco) supplemented with 10% FBS + 20 U/mL sodium heparin (Sigma) and 0.025 U/mL Benzonase (Sigma). Distinct cellular fractions were sorted via fluorescence activated cell sorting (FACS, BD ARIA) after staining with FITC anti-CD45 (1:100 dilution, HI30;Biolegend), BV605 anti-CD3 (1:50 dilution, OKT3;Biolegend), PE-Cy5.5 anti-CD19 (1:50 dilution, HIB19;BD Biosciences), PE anti-CD20 (1:50 dilution, 2H7;Biolegend), and APC-Cy7 anti-HLA-DR (1:50 dilution, L243;Biolegend) antibodies. 2×104 sorted cells were added to 4×104 human hepatoma cells (Huh7, Apath LLC, Brooklyn, NY ) and incubated for 96 hours at 37 °C. Co-cultured cells were then lysed and RNA extracted with RNeasy kit following manufacturer’s instructions. DENV RNA copies number were determined via RT-qPCR.
Uptake assay.
Cryopreserved PBMCs derived from DWS patients and healthy controls were thawed at 37 °C, washed with cold cell culture media (300 g, 4 °C) and pre-incubated for 30 minutes either at 4 °C or at 37 °C. 5×105 cells were then incubated with 1 mg/mL of pHrodo-labeled S. aureus bioparticles (ThermoFisher) or no bioparticles for 1 hour at 4 °C to determine non-specific binding on the cell surface (binding control), or at 37 °C for active uptake. Cells were harvested, labeled with fluorescently-labeled antibodies as described above, and pHrodo fluorescence was assessed by spectral flow cytometry.
Cytokine measurement.
The concentrations of cytokines and chemokines were measured in the serum of 15 DENV-infected patients’ (D = 8; SDp = 7) and 4 healthy controls (whose samples were studied by viscRNA-Seq 2) via Luminex Assay Human 80 (R&D System) following manufacturer’s instructions (Supplementary Table 6).
Cytokine-receptor communication.
The serum concentrations of cytokines of SDp and D patients were compared by Man-Whitney U test. Differentially secreted cytokines were defined as those with a p value < 0.05. The RNA expression of cytokine producing genes and corresponding receptors was then computed in distinct cell subtypes (Fig. 5g, Extended Data Fig. 8f).
Statistics, Reproducibility and data analysis.
Data were analyzed using Python 3 and Jupyter notebooks, R-studio and Flowjo v10.9. Statistical analysis were performed with Python 3, R-studio and Prism 9 (GraphPad). No statistical method was used to predetermine sample size, but our sample size are similar to those reported in previous publication (see examples in48). No data were excluded from the analysis. Data distribution was assumed to be normal, but this was not formally tested. Measurements of viral load, serological parameters and cytokine levels were blinded for the diagnosis of the patients. The remaining experiments and analyses were not performed blinded, as the same investigators processed samples, generated data and analyzed it.
The following open source software was used for this study: numpy (version 1.12.2)49, pandas (version 1.4.1)50, matplotlib (version 3.6.2)51, seaborn(version 0.11.1)52, anndataks 0.1.3 (version 0.1.3), pysam (version 0.18.0)53, scanpy (version 1.9.1)54, metascape (version 3.5)55 and GSEAPY (version 0.10.8)56.
Extended Data
Fig.1: Differences in cell subtype abundance in the viscRNA-seq 2 and CyTOF datasets are comparable and do not correlate with serum viral load.
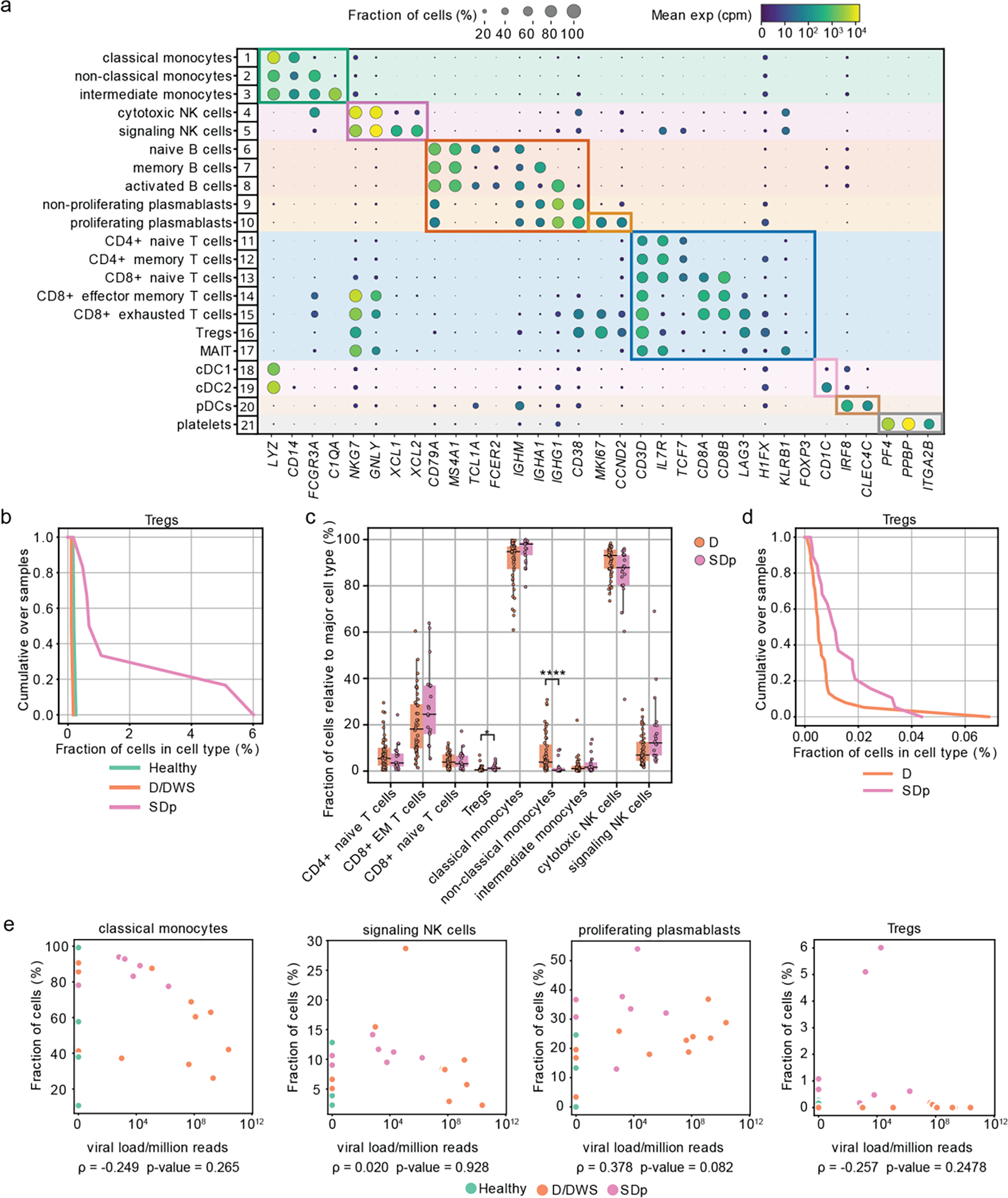
a, Dot plot depicting examples of marker genes used to annotate the indicated 21 immune cell subpopulations in the viscRNAseq 2 dataset (Fig.1). Dot size indicates the fraction of cells expressing the marker gene; color indicates expression level of the marker gene in cpm; and identification numbers of distinct cell populations refer to the UMAP in Fig.1a. b, Cumulative distribution of Treg fractions within T cells from all patients across disease severity in the viscRNAseq 2 dataset. c, Box plots showing the fractions of cell subtypes within T cells, monocytes, and NK cell populations in 21 SDp and 47 D patients computed from the (publicly available) CyTOF dataset of samples obtained from the same Colombia dengue cohort. Notably, in this reanalysis, samples from patients who presented with SD upon enrollment were excluded from the dataset, leaving only those from SD patients who progressed to SD within several days following enrollment. *p-value<0.05, ****p-value<0.0001 by Mann-Whitney-Wilcoxon test two-sided with Bonferroni correction. Each dot represents an individual patient, color-coded by disease severity. Horizontal lines signify the first, second (median) and third quartiles. d, Cumulative distribution of Treg fractions within T cells from all patients across disease severity measured via CyTOF. e, Scatter plots showing correlations between fractions of classical monocytes, signaling NK cells, proliferating plasmablasts, and Tregs and serum viral load measured by RT-qPCR. Each dot represents a single patient colored by disease severity (H= green; D/DWS= orange; SDp= pink). Spearman correlation coefficients and p-values are shown below each panel.
Fig.2: Alterations in cell type abundance are independent of age and pregnancy status and are partially associated with prior DENV exposure.
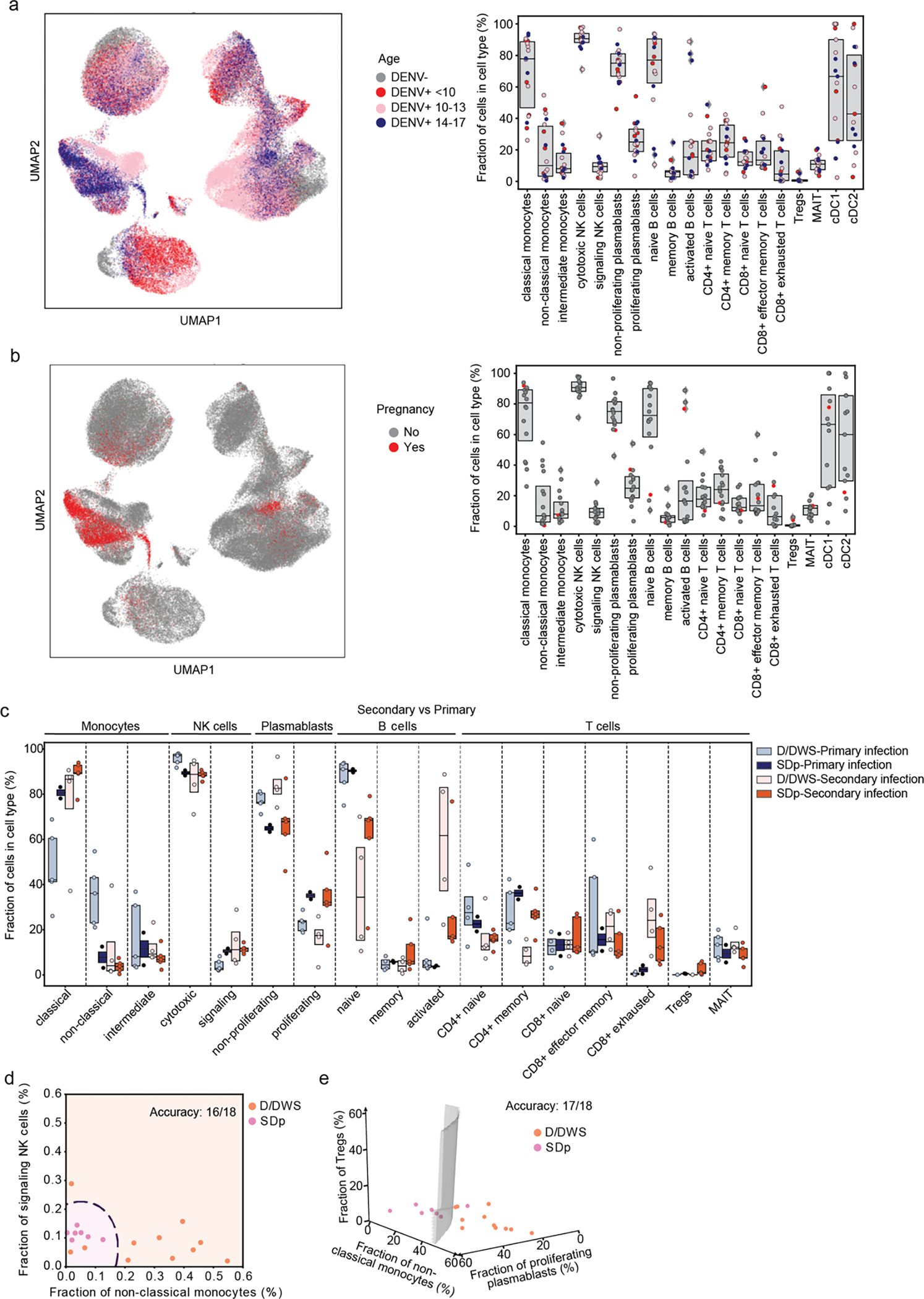
a, UMAP of the entire dataset (left panel) and box plots showing the fractions (%) of cell subtypes within each major immune cell type (right panel) color-coded by patients’ age (in years): <10 =red; 10–14 =pink; 14–17 =blue years old. Box plots’ horizontal lines indicate the first, second (median) and third quartiles, each dot Each dot represents an individual. b, UMAP (left panel) and box plots (right panel) showing distribution and fraction of cells (%) color-coded by pregnancy status, pregnant= red, non-pregnant= gray. c, Box plots showing the fractions (%) of cell subtypes within each major immune cell type by disease severity (SDp and D/DWS) and DENV exposure (primary and secondary). Each dot represents an individual, color-coded by disease severity and DENV exposure (D/DWS-primary =light blue; SDp-primary= dark blue; D/DWS-secondary= light orange; SDp-secondary= dark orange). Box plots’ horizontal lines indicate the first, second (median) and third quartiles. d,e Two (c)- and three (d) dimensional Support Vector Machine (SVM) classifiers for SDp versus D/DWS using the fraction of cells indicated on the axes. Accuracy is evaluated using leave-one-out cross-validation. For this prediction, we trained a support vector machine (SVM) regression model with a third-degree polynomial kernel using the class NuSVC in scikit-learn. We chose SVMs partly because they have a straightforward geometrical interpretation as one can directly plot the hypersurface with the nullcline of the decision function (black dashed curve in d and gray surface in e).
Fig.3: APCs from SDp show signatures of increased activation but decreased antigen presentation.
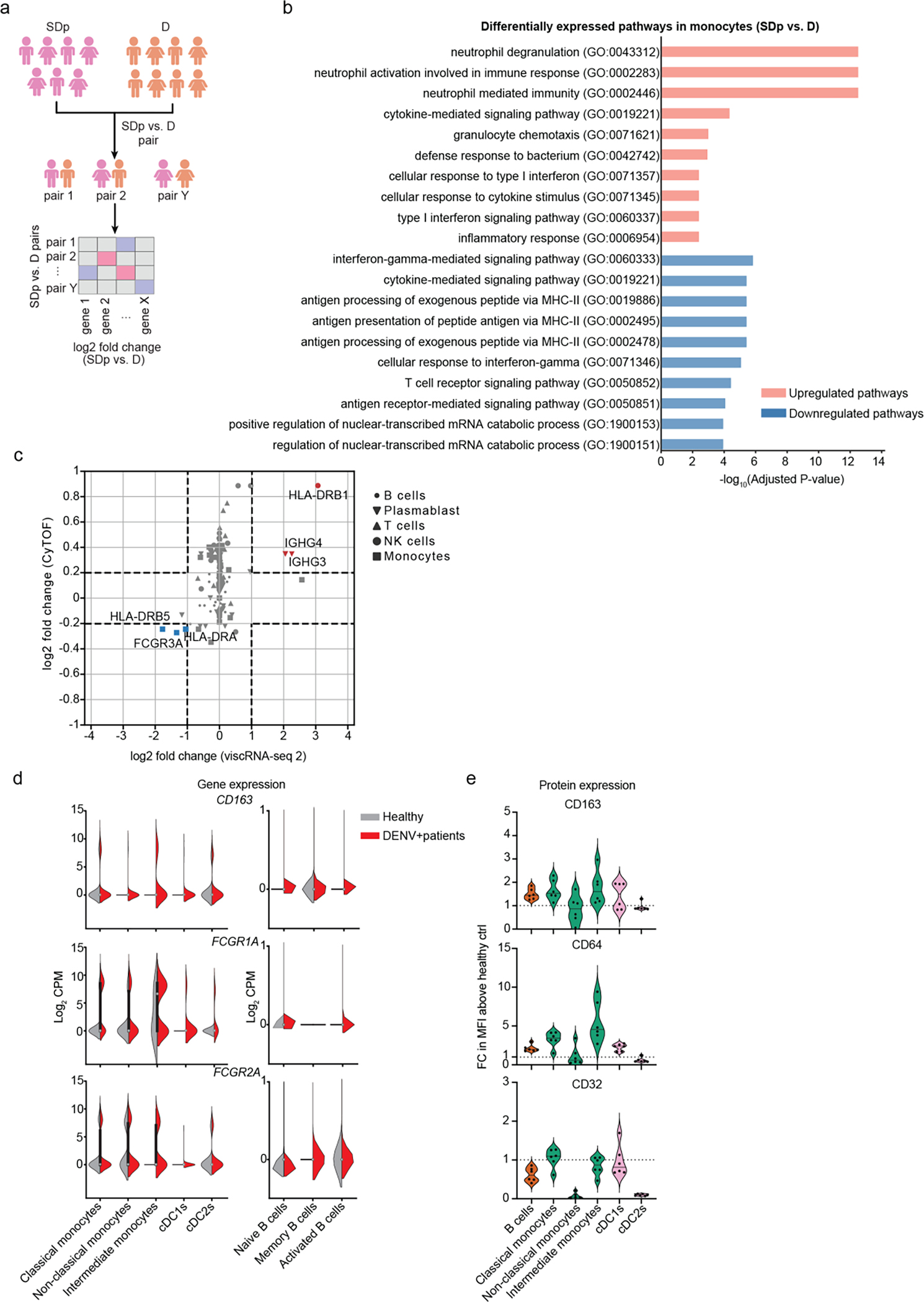
a, Schematic illustrating the pairwise comparison strategy used to identify DEGs between SDp and D patients. Pairs were generated considering only patients that showed n≥5 cells in a cell type or n≥3 in a cell subtype. For each gene, we calculated the geometric mean expression in various cell subtypes for each patient in the pair and the log2 fold change in expression between the two patients in the pair. A median log2 fold change was then obtained for each gene by analyzing all pair combinations. DEGs were defined as those with a median log2 fold change greater than 1 or smaller than −1. b, Pathway analysis showing top 10 upregulated and downregulated gene ontology (GO) terms between SDp and D in monocytes. DEGs for downstream pathway analysis were identified by a 2-sample Kolmogorov-Smirnov test using anndataks 0.1.3. Genes with >1 log2 fold change between the two groups and a p-value <=0.05 (after FDR correction for multiple hypotheses). Metascape58 and GSEAPY59 open source software. were used for pathway analysis of groups of up- or downregulated genes. c, Scatter plots depicting log2 fold change in expression between SDp and D measured at the transcript level via viscRNAseq 2 (x axis) and at the protein level via CyTOF (y axis). Dashed lines depict the log2 fold change cutoffs (|log2 fold change| <1 in the viscRNA-seq dataset; |log2 fold change| < 0.2 in the CyTOF dataset). Shapes represent specific cell types. Each symbol represents a single cellular factor color coded based on the expression pattern (red – upregulated in both datasets; blue – downregulated in both datasets; gray – unaltered in the two datasets). Notably, in this reanalysis of the CyTOF dataset, samples from patients who presented with SD upon enrollment were excluded. d, Violin plots showing expression (Log2 CPM) of CD163, FCGR1A and FCGR2A in monocytes, cDCs and B cell subtypes in healthy (gray) and DENV+ patients including SD and D (red). e, Violin plots showing expression of CD163, CD64 and CD32 proteins on B cells (CD19+), classical monocytes (CD14+CD16−), non-classical monocytes (CD14−CD16+), intermediate monocytes (CD14+CD16+), cDC1s (CD141+) and cDC2s (CD1c+) measured via spectral flow cytometry in DWS patient-derived PBMCs. Data are shown as fold change (FC) of mean fluorescence intensity (MFI) above healthy control (n=6, N=2). Dotted line represents mean value of healthy controls normalized at value 1, horizontal line in each violin plot represents median value.
Fig.4. DEGS between SDp and D in APCs are only partially associated with prior DENV exposure.
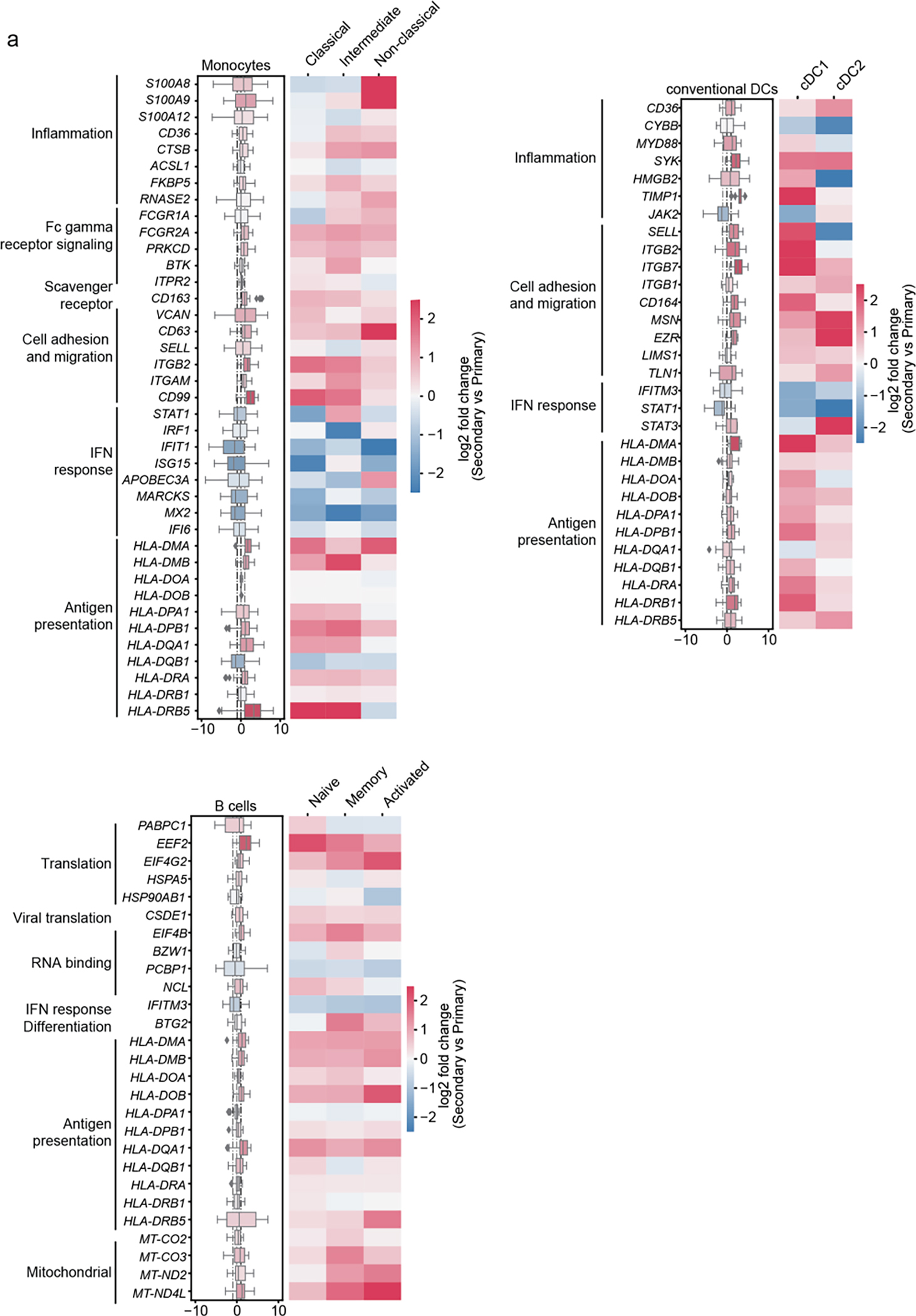
a, Differentially expressed genes (DEGs) between D and SDp (Fig. 2a-c) were analyzed based on DENV exposure (Secondary vs. Primary) in monocyte, cDC, and B cell populations (Box plots, left) and the corresponding distinct cell subtypes (heatmaps, right). Data are color-coded based on the median log2 fold change of pairwise comparisons. Box plots’ horizontal lines indicate the first, second (median) and third quartiles
Fig.5: NK cells in SDp show activation and adaptive-like signatures that may be linked to a prior DENV exposure.
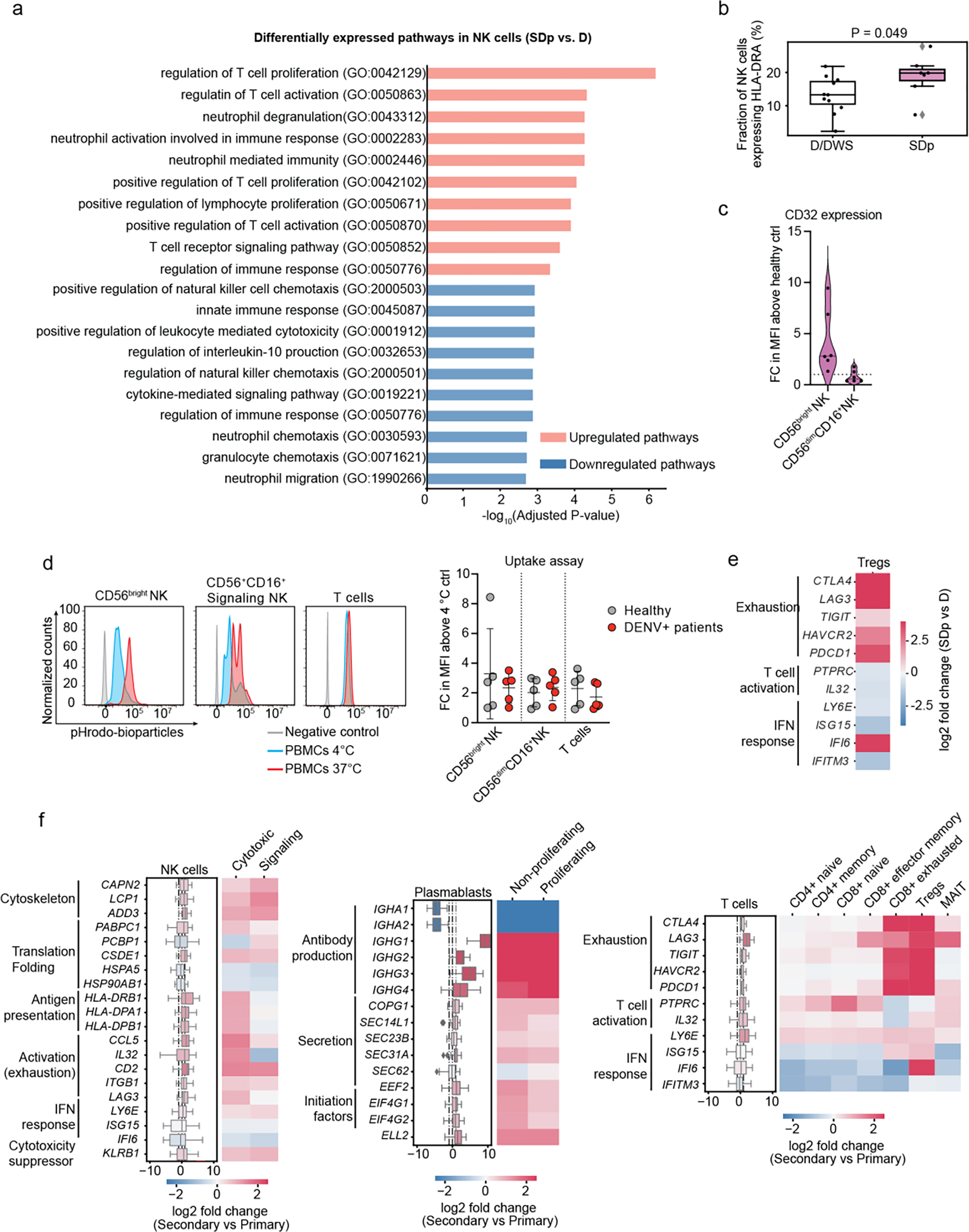
a, Pathway analysis showing top 10 upregulated and downregulated gene ontology (GO) terms between SDp and D in NK cells (see supplementary figure 3b for technical details). b, Fraction of NK cells expressing HLA-DRA gene out of the total NK cell population in D/DWS and SDp. c, Violin plots showing CD32 protein expression in CD56bright NK cells and CD56dimCD16+ NK cells measured via spectral flow cytometry in patient-derived PBMCs. Data are shown as fold change (FC) of mean fluorescence intensity (MFI) above healthy control (n=6, N=2). Dotted line represents mean value of healthy controls normalized to 1; horizontal line in each violin plot represents median value. d, Histograms showing the percentage of pHrodo Bioparticles positive cells in distinct cell subtypes derived from healthy (gray) and DENV-infected patients measured at 4 °C (cyan) or 37 °C (red). Dot plot showing bioparticle uptake quantification as FC of mean MFI above 4 °C control. Each dot represents an individual color-coded by disease status: healthy (gray) and DENV-infected (red) (n=5, N>2). e, DEGs between D and SDp detected in regulatory T cells (Tregs). Data are aggregated from all patients and color-coded based on log2 fold change. f, Differentially expressed genes (DEGs) between D and SDp (Fig. 3a–c) were analyzed based on DENV exposure (Secondary vs. Primary) for NK cell, plasmablast and T cell populations (Box plots, left) and the corresponding distinct cell subtypes (heatmaps, right). Data are color-coded based on median log2 fold change of pairwise comparisons. Box plots’ horizontal lines indicate the first, second (median) and third quartiles.
Fig.6: B cells from DENV-infected patients harbor replicating virus.
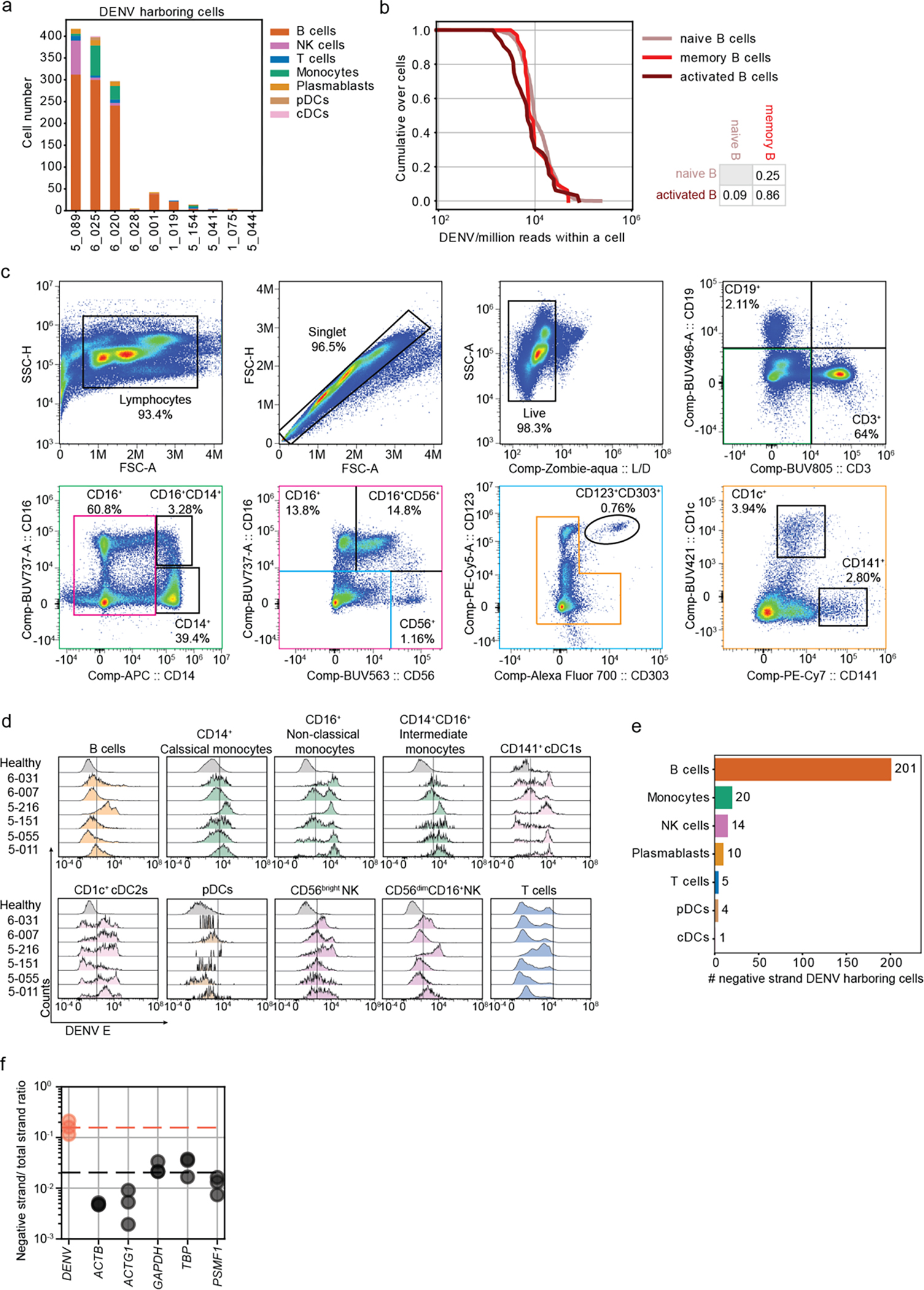
a, Stack bar plots showing the absolute numbers and distribution of VHCs across cell types in each of the 10 patients with detectable vRNA. b, Cumulative distributions of DENV reads/million reads per cell in B cell subtypes. p-values from KS test after Bonferroni correction between distinct B cell subtypes are indicated. c, Flow cytometry gating strategy used to define B cells (CD19+), T cells (CD3+), classical monocytes (CD14+CD16−), non-classical monocytes (CD14−CD16+), intermediate monocytes (CD14+CD16+), CD56bright NK cells, CD56dimCD16+ NK cells, pDCs (CD123+CD303+), cDC1s (CD141+) and cDC2s (CD1c+). d, Distributions of intracellular expression of DENV envelope (E) protein in B cells (CD19+), classical monocytes (CD14+CD16−), non-classical monocytes (CD14−CD16+), intermediate monocytes (CD14+CD16+), cDC1s (CD141+), cDC2s (CD1c+), pDCs (CD123+CD303+), CD56bright NK cells, CD56dimCD16+ NK cells, and T cells from the indicated DENV-infected patients with viremia ranging from 105 to 109 viral copies/mL (N=2,n=6 combined data) and healthy controls (N=2, n=6 combined data) measured via spectral flow cytometry. e, Number of negative strand RNA-harboring cells (VHCs) over total number of VHCs across immune cell types in three DWS patients with highest vRNA reads shown in Fig.4a. f, Dot plot showing the average ratio of negative strand over total strand of housekeeping genes (gray) and DENV RNA (orange) reads within the 20 single cells with the largest DENV read counts in 3 DWS patients with highest vRNA reads shown in Fig.4a.
Fig.7: Gating strategy and DEGs in VHCs vs. bystander cells.
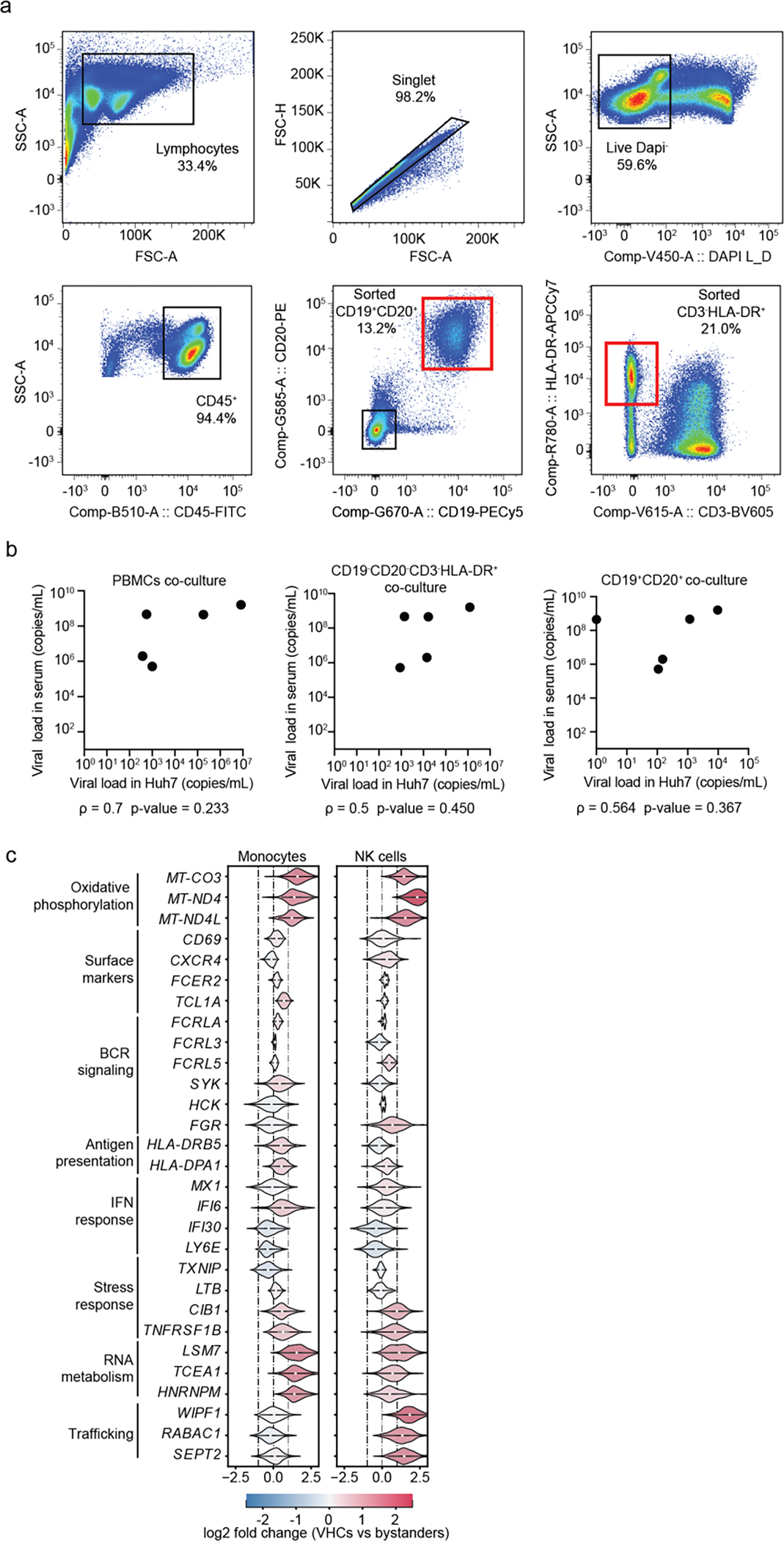
a, FACS gating strategy used to sort populations of B cells (CD19+, CD20+) and CD19−CD20− CD3−HLA-DR+ cells for co-culture experiments. b, Scatter plots showing correlations between viral load in serum and viral load in Huh7 cell lysates (DENV copies /mL) following 5-day co-culture either with PBMCs or with CD19−CD20− CD3−HLA-DR+ and B cell (CD19+, CD20+) fractions from the same patients. Spearman correlation coefficients and p-values are shown below each panel. c, Violin plots showing log2 fold change of DEGs between VHCs and corresponding bystander monocytes and NK cells in 3 DWS patients with highest vRNA reads shown in Fig.4a. DEGs were identified by the median log2 fold change of 100 bootstrapped comparisons between VHCs and equal numbers of subsampled bystander cells. Note large noise driven by the small number of viral reads in these cell subtypes.
Fig.8: Cell-cell communications are altered in SDp.
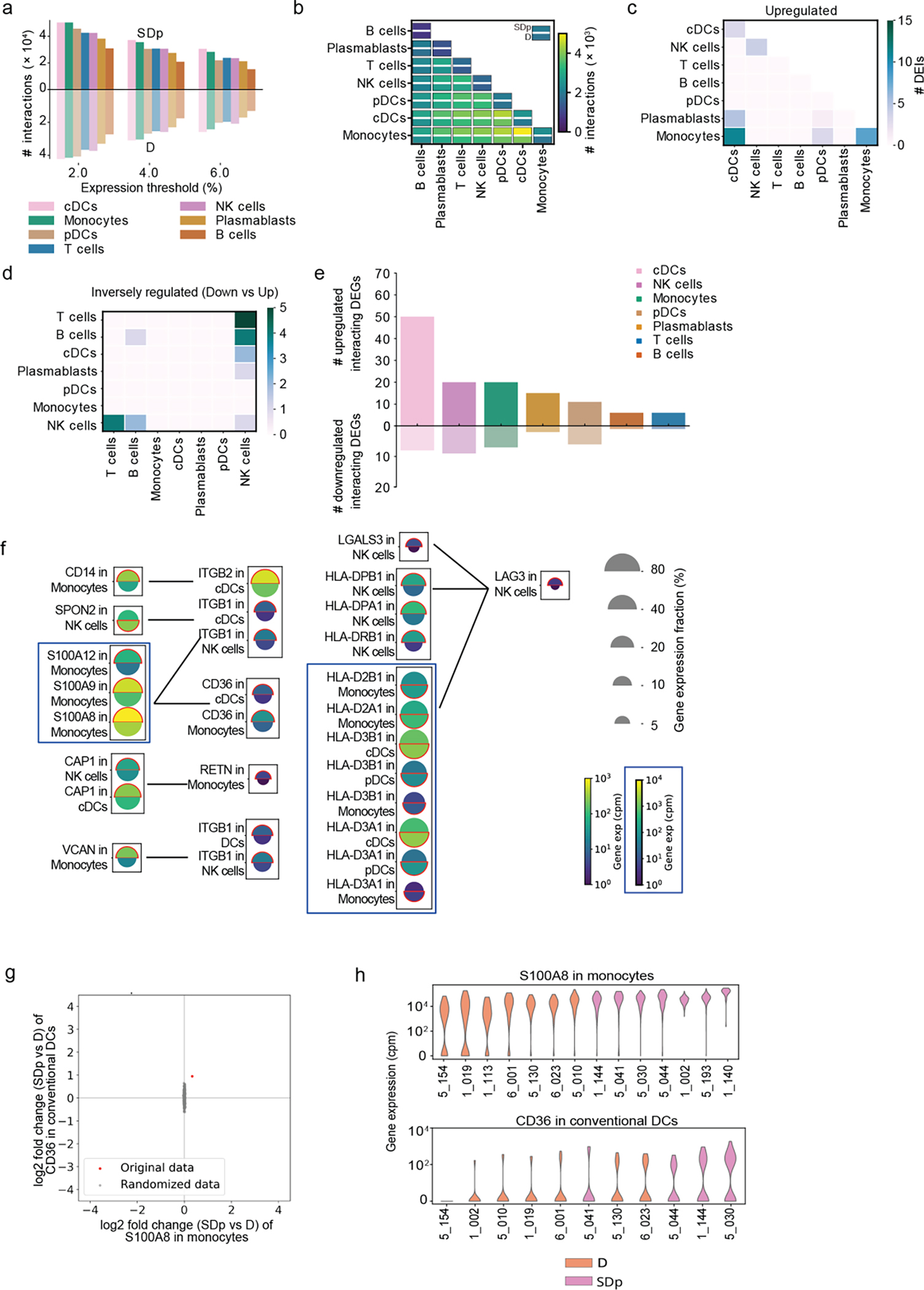
a, Bar plot showing the number of candidate interactions in each cell type in SDp (top) and D (bottom) based on different expression thresholds (2%, 4%, and 6%). b, Heatmap showing the number of candidate interactions between the indicated cell types in SDp (top rectangle) and D (bottom rectangle). c, d, Heatmaps showing the number of upregulated and inversely regulated DEIs shown in fig. 6c and 6d, respectively, across cell types. e, Bar plot showing the number of up- and down-regulated interacting DEGs across cell types in SDp vs. D. f, Split dot plots showing gene expression in representative DEIs. Top half of each spilt dot plot depicts the expression level in SDp; bottom half depicts the expression level in D; spilt dot plot size depicts the percentage of gene expressing cells in the cell types indicated; and color depicts gene expression level in counts per million (cpm). Red outline indicates the condition with the higher gene expression. g, Example of label randomization for the S100A8-CD36 interaction. The red dot indicates the log2 fold change of the expression of either gene in the corresponding cell type in SDp vs D. The 1000 gray dots indicate the log2 fold changes after randomly picking SDp and D cells following cell shuffling. The p-value was calculated as the fraction of randomizations for which the distance from the origin was larger than in the real data. h, Violin plots depicting ligand-receptor expression (S100A8-CD36 interaction) in each patient.
Supplementary Material
Supplementary Table 1: Patients ID and clinical data
Supplementary Table 2: Number of high-quality cells for each patient
Supplementary Table 3: Differentially expressed genes defined by median log2 fold change of ≥ 1 or ≤−1 between SDp and D patients by pairwise comparisons.
Supplementary Table 4: Total number of viral RNA harboring cells
Supplementary Table 5: Candidate cell-cell interactions in SDp vs. D patients
Supplementary Table 6: Cytokine concentration in patients serum
Supplementary Table 7: DEGs between SDp and D patients are comparably detected via KS test and pairwise comparison.
Supplementary Table 8: Additional cell-cell interactions - source table
Acknowledgements:
This work was supported by an Investigator Initiated Award number W81XWH1910235 from the Department of Defense (DoD) office of the Congressionally Directed Medical Research Programs (CDMRP)/Peer Reviewed Medical Research Program (PRMRP), Catalyst and Transformational Awards from Dr. Ralph & Marian Falk Medical Research Trust, a National Institute of Allergy and Infectious Diseases (NIAID) grant U19 AI057229 supplement to S.E. and funds from the Chan Zuckerberg Biohub to S.Q. and S.E.. S.E. is a Chan Zuckerberg Biohub – San Francisco Investigator, who is also supported by a NIAID grant RO1AI158569, an Investigator Initiated Award number W81XWH2210283 and an expansion award number W81XWH2110456 from the DoD office of the CDMRP/PRMRP, and a Defense Threat Reduction Fundamental Research to Counter Weapons of Mass Destruction grant HDTRA11810039. L.G. was supported by EMBO Postdoctoral Fellowship ALTF 584–2021. Z.Y. was supported by a Thrasher Research Fund early career award program grant and by a postdoctoral fellowship from the Maternal and Child Health Research Institute, Lucile Packard Foundation for Children’s Health. V.D. was supported by a Chan Zuckerberg Biohub Collaborative Postdoctoral Fellowship. M.L.R. was supported by the A.P. Giannini Foundation Postdoctoral Fellowship and the Harold Amos Medical Faculty Development Program. I.O. was supported by a Sue Merigan Student Scholar Fund in Infectious Diseases and Geographic Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr. Catherine Blish for her guidance in the interpretation of NK cell data. BioRender software was used for preparation of Figure 6. Co-first authors L.G., Z.Y., Y.X. and V.D. contributed equally to this manuscript and each reserves the right to list themselves as first authors in public outlets.
Footnotes
Competing Interest Statement:
The authors declare no competing interests.
Code availability. The code used in the present study is publicly available at: https://github.com/echosun77/dengue.
Data availability.
Sequencing data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO accession number GSE220969. Fcs file used for CyTOF data reanalysis are available at FlowRepositiry: https://flowrepository.org/id/FR-FCM-Z5MQ, IDFR-FCM-Z5MQ. Source data are provided with this manuscript.
References
- 1.Bhatt S et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khursheed M et al. A comparison of WHO guidelines issued in 1997 and 2009 for dengue fever - single centre experience. JPMA. The Journal of the Pakistan Medical Association 63, 670–674 (2013). [PubMed] [Google Scholar]
- 3.WHO Guidelines Approved by the Guidelines Review Committee. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control: New Edition. World Health Organization Copyright © 2009, World Health Organization.: Geneva, 2009. [Google Scholar]
- 4.Barniol J et al. Usefulness and applicability of the revised dengue case classification by disease: multi-centre study in 18 countries. BMC Infect Dis 11, 106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu YE et al. An 8-gene machine learning model improves clinical prediction of severe dengue progression. Genome Med 14, 33 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Meng Y, Halloran ME & Longini IM Jr. Dependency of Vaccine Efficacy on Preexposure and Age: A Closer Look at a Tetravalent Dengue Vaccine. Clinical Infectious Diseases 66, 178–184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang TT et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 355, 395–398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katzelnick LC et al. Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson M et al. A 20-Gene Set Predictive of Progression to Severe Dengue. Cell Rep 26, 1104–1111.e1104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanini F, Pu SY, Bekerman E, Einav S & Quake SR Single-cell transcriptional dynamics of flavivirus infection. Elife 7, e32942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson ML et al. Magnitude and kinetics of the human immune cell response associated with severe dengue progression by single-cell proteomics. Sci Adv 9, eade7702 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Scientific reports; 2019. p. 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyle JL, Beatty PR & Harris E Dengue Virus Infects Macrophages and Dendritic Cells in a Mouse Model of Infection. The Journal of infectious diseases 195, 1808–1817 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Kou Z et al. Monocytes, but not T or B cells, are the principal target cells for dengue virus (DV) infection among human peripheral blood mononuclear cells. Journal of medical virology 80, 134–146 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Dethoff EA et al. Pervasive tertiary structure in the dengue virus RNA genome. Proceedings of the National Academy of Sciences 115, 11513–11518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pishesha N, Harmand TJ & Ploegh HL A guide to antigen processing and presentation. Nature Reviews Immunology 22, 751–764 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Türei D, Korcsmáros T & Saez-Rodriguez J OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nature Methods 13, 966–967 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nature Protocols 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Jones RC et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slonchak A & Khromykh AA Subgenomic flaviviral RNAs: What do we know after the first decade of research. Antiviral Res 159, 13–25 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Barnard TR, Abram QH, Lin QF, Wang AB & Sagan SM Molecular Determinants of Flavivirus Virion Assembly. Trends Biochem Sci 46, 378–390 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Syenina A et al. Positive epistasis between viral polymerase and the 3’ untranslated region of its genome reveals the epidemiologic fitness of dengue virus. Proc Natl Acad Sci U S A 117, 11038–11047 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maheshwari D et al. Contrasting behavior between the three human monocyte subsets in dengue pathophysiology. iScience 25, 104384 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naranjo-Gómez JS et al. Different phenotypes of non-classical monocytes associated with systemic inflammation, endothelial alteration and hepatic compromise in patients with dengue. Immunology 156, 147–163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer DR et al. Differential effects of dengue virus on infected and bystander dendritic cells. J Virol 79, 2432–2439 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalbeth N et al. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol 173, 6418–6426 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Lai CY et al. Antibodies to envelope glycoprotein of dengue virus during the natural course of infection are predominantly cross-reactive and recognize epitopes containing highly conserved residues at the fusion loop of domain II. J Virol 82, 6631–6643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poluektov Y, Kim A & Sadegh-Nasseri S HLA-DO and Its Role in MHC Class II Antigen Presentation. Frontiers in Immunology 4, 260 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tagawa T et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J Exp Med 213, 2065–2080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandey N et al. Serum levels of IL-8, IFNγ, IL-10, and TGF β and their gene expression levels in severe and non-severe cases of dengue virus infection. Arch Virol 160, 1463–1475 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Schroder K, Hertzog PJ, Ravasi T & Hume DA Interferon-γ: an overview of signals, mechanisms and functions. Journal of Leukocyte Biology 75, 163–189 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Guzman MG et al. Effect of age on outcome of secondary dengue 2 infections. International journal of infectious diseases : IJID : official publication of the International Society for Infectious Diseases 6, 118–124 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Costa-García M et al. Human Cytomegalovirus Antigen Presentation by HLA-DR+ NKG2C+ Adaptive NK Cells Specifically Activates Polyfunctional Effector Memory CD4+ T Lymphocytes. Front Immunol 10, 687 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama M et al. Natural killer (NK)-dendritic cell interactions generate MHC class II-dressed NK cells that regulate CD4+ T cells. Proc Natl Acad Sci U S A 108, 18360–18365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reighard SD et al. Therapeutic Targeting of Follicular T Cells with Chimeric Antigen Receptor-Expressing Natural Killer Cells. Cell Rep Med 1, 100003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jayaratne HE et al. Regulatory T-cells in acute dengue viral infection. Immunology 154, 89–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lühn K et al. Increased frequencies of CD4+ CD25(high) regulatory T cells in acute dengue infection. J Exp Med 204, 979–985 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhry A et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira RA et al. Circulating cytokines and chemokines associated with plasma leakage and hepatic dysfunction in Brazilian children with dengue fever. Acta Trop 149, 138–147 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Chen LC et al. Correlation of serum levels of macrophage migration inhibitory factor with disease severity and clinical outcome in dengue patients. Am J Trop Med Hyg 74, 142–147 (2006). [PubMed] [Google Scholar]
- 41.Malavige GN & Ogg GS Pathogenesis of vascular leak in dengue virus infection. Immunology 151, 261–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couper KN, Blount DG & Riley EM IL-10: the master regulator of immunity to infection. J Immunol 180, 5771–5777 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Guabiraba R et al. Role of the chemokine receptors CCR1, CCR2 and CCR4 in the pathogenesis of experimental dengue infection in mice. PLoS One 5, e15680 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wati S et al. Tumour necrosis factor alpha (TNF-alpha) stimulation of cells with established dengue virus type 2 infection induces cell death that is accompanied by a reduced ability of TNF-alpha to activate nuclear factor kappaB and reduced sphingosine kinase-1 activity. J Gen Virol 92, 807–818 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Mangada MM et al. Dengue-specific T cell responses in peripheral blood mononuclear cells obtained prior to secondary dengue virus infections in Thai schoolchildren. The Journal of infectious diseases 185, 1697–1703 (2002). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 46.Tomashek KM et al. Development of standard clinical endpoints for use in dengue interventional trials. PLoS Negl Trop Dis 12, e0006497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waggoner JJ et al. Comparison of the FDA-approved CDC DENV-1–4 real-time reverse transcription-PCR with a laboratory-developed assay for dengue virus detection and serotyping. J Clin Microbiol 51, 3418–3420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waickman AT et al. Temporally integrated single cell RNA sequencing analysis of PBMC from experimental and natural primary human DENV-1 infections. PLoS Pathog 17, e1009240 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris CR et al. Array programming with NumPy. Nature 585, 357–362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.pandas-dev/pandas: Pandas 1.4.1. Zenodo; 2022. [Google Scholar]
- 51.Matplotlib: A 2D Graphics Environment | IEEE Journals & Magazine | IEEE Xplore. [Google Scholar]
- 52.Waskom M Seaborn: Statistical Data Visualization. JOSS 2021, 6 (60), 3021. [Google Scholar]
- 53.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang Z, Liu X & Peltz G GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Patients ID and clinical data
Supplementary Table 2: Number of high-quality cells for each patient
Supplementary Table 3: Differentially expressed genes defined by median log2 fold change of ≥ 1 or ≤−1 between SDp and D patients by pairwise comparisons.
Supplementary Table 4: Total number of viral RNA harboring cells
Supplementary Table 5: Candidate cell-cell interactions in SDp vs. D patients
Supplementary Table 6: Cytokine concentration in patients serum
Supplementary Table 7: DEGs between SDp and D patients are comparably detected via KS test and pairwise comparison.
Supplementary Table 8: Additional cell-cell interactions - source table
Data Availability Statement
Sequencing data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO accession number GSE220969. Fcs file used for CyTOF data reanalysis are available at FlowRepositiry: https://flowrepository.org/id/FR-FCM-Z5MQ, IDFR-FCM-Z5MQ. Source data are provided with this manuscript.