

Abstract
INTRODUCTION
In addition to the accumulation of amyloid plaques and neurofibrillary tangles, the presence of excess neural activity is a pathological hallmark of Alzheimer's disease (AD) and a prognostic indicator for progression of AD pathology and clinical/cognitive worsening in mild cognitive impairment due to Alzheimer's disease (MCI due to AD). The HOPE4MCI clinical study tested the efficacy of a therapeutic with demonstrated ability to normalize heightened neural activity in the hippocampus in a randomized controlled trial of 78 weeks duration in patients with MCI due to AD.
METHODS
One hundred and sixty‐four participants were randomized to placebo (n = 83) or AGB101 (n = 81), an extended‐release formulation of low dose (220 mg) levetiracetam. The primary endpoint was the change in Clinical Dementia Rating Scale Sum of Boxes score (CDR‐SB) comparing follow up at 18 months to baseline. The goal of the primary efficacy analysis was to estimate the difference between the AGB101 and placebo arms in the mean change of the primary endpoint.
RESULTS
The mean change in CDR‐SB was estimated to be 1.12 (95% confidence interval [CI]: 0.66, 1.69) for the AGB101 arm and 1.22 (95% CI: 0.75, 1.78) for the placebo arm. The estimated difference between arms is ‐0.10 (95% CI: ‐0.85, 0.58), which was not statistically significant. In a prespecified analysis, the difference was ‐0.45 (95% CI: ‐1.43, 0.53) for ApoE‐4 noncarriers and ‐0.10 (95% CI: ‐0.92, 0.72) for apolipoprotein E (ApoE)‐4 carriers.
DISCUSSION
The possibility that ApoE‐4 carriers and noncarriers will respond differently to therapeutic intervention is consistent with recently reported findings from biologics and the present results show further testing of AGB101 in patients with MCI due to AD who are noncarriers of the ApoeE‐4 allele is warranted. Conclusions from the HOPE4MCI study are limited primarily due to the small sample size and results can only be regarded as a guide to future research.
Keywords: AGB101, Alzheimer's disease, clinical trial, hippocampus, levetiracetam, mild cognitive impairment
1. INTRODUCTION
It is well‐established that Alzheimer's disease (AD) pathology begins to accumulate in the brain many years prior to a diagnosis of dementia, providing a prolonged trajectory of disease‐related changes. 1 , 2 In amnestic mild cognitive impairment (aMCI), the early symptomatic phase of the disease, strong evidence indicates accumulation of AD pathology. 3 , 4 , 5 There is also strong evidence from preclinical models and human patients that, in addition to the accumulation of amyloid plaques and neurofibrillary tangles, neuronal circuits become hyperactive prior to the development of AD dementia, contributing to neuronal pathology and brain dysfunction. 6 These data direct attention to the potential for a novel therapeutic approach of targeting hyperactivity, especially in the MCI phase of AD when hippocampal hyperactivity is most pronounced and forecasts subsequent decline. 7 , 8
Basic research in support of hyperactivity as a target in early stages of AD includes direct chemogenetic reduction of neural activity in AD mouse models, demonstrating reduction of amyloid deposition, conferring beneficial effects on synaptic dysfunction and pathology 9 and demonstration that neural activity augments tau propagation and tau pathology in hippocampal circuits in vivo. 10 These data are consistent with evidence from a wide range of animal models and patients with aMCI that hyperactivity is a promising target for intervention to control amyloid and tau pathology. In that context, it is notable that both preclinical and clinical research has demonstrated that treatment with low, but not higher, doses of the atypical antiepileptic levetiracetam attenuates neural overactivity and has therapeutic effects on both cognition and AD pathophysiology, as summarized below and in a recent review. 11
In preclinical studies, low dose levetiracetam demonstrates efficacy on a range of molecular, synaptic, electrophysiological, functional, and behavioral endpoints across models (age‐related memory impairment, including the pathophysiology of amyloid and tau) and across species flies, 12 mice, 13 , 14 , 15 rats, 16 , 17 , 18 including in aMCI 19 , 20 and possibly in neurocognitive aging in humans. 21 Conversely, high doses of levetiracetam, such as those used in clinical practice to treat epilepsy, neither diminish hippocampal overactvity nor impact AD‐associated biomarkers. 13 , 20 Paralleling the preclinical data, low, but not high, dose levetiracetam normalized hippocampal activity and improved performance in a cross‐over study design in which a pattern separation task of episodic memory function in MCI was an outcome measure. 19 , 20
Levetiracetam is a molecule with high bioavailability, low metabolism, and high brain penetration 23 resulting in a very predictable relationship of plasma to brain concentration. AgeneBio, Inc. formulated AGB101 as a low dose extended‐release formulation (once‐a‐day medication) for therapeutic exposure in the current clinical trial. Pharmacokinetic studies show that this novel preparation of levetiracetam (AGB101) produces plasma concentrations of levetiracetam in a range shown in both preclinical models and in human patients to normalize hippocampal activity. 22 A preliminary report of a study using the once‐a‐day medication AGB101 at 220 mg found that the drug appeared to reduce increased hippocampal functional connectivity in cognitively normal elderly. 21 Currently, AGB101 is the first and only therapeutic to our knowledge being investigated to target hippocampal overactivity to slow progression and delay the onset of AD dementia.
Here, we report the results of a randomized, double‐blind, placebo‐controlled, phase 2b trial of patients with MCI due to AD (NCT03486938). This trial, HOPE4MCI (Hippocampal Overactivity Prevention in the Elderly with MCI due to AD), was conducted to assess the efficacy of AGB101. While levetiracetam has been shown to improve specific aspects of episodic memory function even after 2 weeks of administration, we do not expect a short term effect of levetiracetam in improving all symptoms of AD. Our hypothesis tests whether diminishing hippocampal hyperactivity by AGB101 treatment will slow the overall progression of AD symptoms. The phase 2b program used a limited sample size to inform the design of a larger confirmatory phase 3 program targeting neural overactivity to slow progression in aMCI. As described in the statistical analysis plan (SAP) for the study, the change in Clinical Dementia Rating Scale Sum of Boxes score (CDR‐SB), comparing 18 months to baseline, was prespecified as the primary endpoint with additional secondary endpoints, including the Functional Activities Questionnaire (FAQ), and Mini–Mental State Examination (MMSE) for clinical/cognitive assessment. Considering previous data from trials for therapeutics in the MCI due to AD phase of disease, 24 , 25 the primary efficacy analysis in the SAP also prespecified a sensitivity analysis of the primary endpoint separately by ApoE4 carrier status.
2. METHODS
2.1. Study design
The HOPE4MCI study was an 18‐month, multinational, randomized, double‐blind, placebo‐controlled trial of AGB101 (a low dose extended release formulation of levetiracetam 220 mg) in patients with MCI due to AD. The study was conducted at 29 sites across the United States and Canada that screened and randomized participants for the study. The study was conducted in accordance with International Council for Harmonization guidelines and the ethical principles of the Declaration of Helsinki. For most sites, a central Institutional review board (IRB) approved the protocol. For some sites, a local IRB or independent ethics committee was utilized. All participants provided written informed consent. An independent data and safety monitoring board (DSMB) consisting of experts in AD and statistics reviewed unblinded safety data during the trial. The study was designed by AgeneBio in collaboration with academic co‐investigators at Johns Hopkins University and funded by the National Institutes of Health (R01AG048349, R56AG055416, and RO1AG061091). The prespecified statistical analysis plan and full reporting of adverse events for this study are provided in the Supplemental Materials.
2.2. Participants and eligibility
Patients were between 55 and 85 years old and met criteria for MCI due to AD based on the National Institute on Aging–Alzheimer's Association criteria. 26 Inclusion criteria included: (1) a Clinical Dementia Rating Scale (CDR) score of 0.5 with a memory box score of ≥ 0.5; (2) an MMSE score of 24–30; (3) a memory complaint reported by the participant or his/her study partner; (4) objective evidence of lower memory performance (z‐score greater than 1.4 SD below age‐matched controls) based on the delayed recall portion of the International Shopping List Test (ISLT); (5) essentially preserved activities of daily living, cognitive decline not primarily caused by vascular, traumatic, or medical problems (alternative causes of cognitive decline were ruled out); (6) a positive amyloid beta positron emission tomography (PET) scan and; (7) magnetic resonance imaging (MRI) scan results consistent with the diagnosis of amnestic MCI due to AD, with no clinically significant findings of non‐AD pathology that could account for the observed cognitive impairment. 27 Participants were excluded if they had evidence of vascular dementia as indicated by a clinical history, a score greater than 4 on the Hachinski scale, or evidence of cerebrovascular disease on T2 MRI readings by a neuroradiologist and the study physician. Exclusion criteria specifically related to use of levetiracetam included use a history of seizures or use of anticonvulsants, history of hypersensitivity or lack of tolerability to AGB101 (levetiracetam), severe renal impairment (creatinine clearance of < 30 mL/minute), or undergoing hemodialysis.
Following screening, eligible patients were randomly assigned to placebo or AGB101 in a 1:1 ratio based on a central randomization list. Participants were administered a single morning daily dose of placebo or AGB101 for 78 weeks completing a total of eight minor and major study visits (full schematic of trial design in supplemental materials). Given the low dose, AGB101 dosing was initiated without titration and discontinued without tapering. Participants were instructed to take AGB101 in the morning so that plasma concentrations during the day were in the range previously shown to diminish hippocampal overactivity. 19 , 20 , 21 Hippocampal overactivity has not been observed during sleep; therefore, the dosing regimen was likely sufficient to diminish all physiologically relevant overactivity. Participants were allowed to continue taking stable doses of standard of care symptomatic medications, such as acetylcholinesterase inhibitors and memantine, for the duration of the study.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. A significant literature supports the use of low dose levetiracetam as a novel therapeutic approach for targeting neuronal hyperactivity, especially in the mild cognitive impairment (MCI) phase of Alzheimer's disease (AD) when hippocampal hyperactivity is most pronounced and forecasts subsequent decline.
Interpretation: The HOPE4MCI clinical study tested the efficacy of AGB101, a novel therapeutic with demonstrated ability to normalize heightened neural activity in the hippocampus in a randomized controlled trial of 78 weeks duration in patients with MCI due to AD.
Future directions: Although the primary endpoint did not show a statistically significant benefit of treatment likely due to a small sample size, the present results support the view that further testing of AGB101 in patients with MCI due to AD who are noncarriers of ApoE‐4 is warranted.
2.3. Study endpoints
The primary endpoint was the change in CDR‐SB score from baseline to 78 weeks. The CDR is a semistructured interview including the patient and an informant assessing three domains of cognition (memory, orientation, judgment/reasoning) and three domains of function (community affairs, home/hobbies, and personal care) which are summed. The CDR‐SB shows progression, for example, longitudinal change, in the spectrum of MCI through to mild‐to‐moderate AD dementia. 28 , 29 , 30 , 31 Consistent with our inclusion criteria, patients with MCI who are amyloid positive by PET imaging show elevated hippocampal activity and progression on the CDR‐SB during the MCI phase of the disease. 7 Secondary clinical and cognitive endpoints included the change in score from baseline to 78 weeks on the FAQ 32 ; the MMSE 33 score. A repeated assessment on the Behavioral Pattern Separation Task‐Objects (BPS‐O) 34 was included as an instrument to assess memory performance dependent on hippocampal functioning using the Lure Discrimination Index (LDI) as the outcome measure for this task.
2.4. Amyloid PET acquisition
Positive historical amyloid PET scans utilizing research or commercial amyloid PET ligands (Neuroceq, Amyvid, Vizamyl) were acceptable for inclusion with documented evidence of amyloid positivity by a standard qualitative read. For participants without a historical scan, amyloid PET scans (predominantly Neuroceq) were conducted at site imaging facilities and were utilized for inclusion using recommended doses and procedures. Site neuroimaging facilities were trained on relevant study procedures by the central imaging laboratory (Clario, Inc.) and completed site qualification and assessment of scanner performance using phantom scans. Amyloid positivity was verified by Clario, Inc using a visual read of the PET scans by neuroradiologists trained in the assessment of amyloid according to the processes developed by the radiotracer vendors.
2.5. MRI data acquisition
Structural MRI scans were obtained for all participants enrolled during screening and at the week 52 and week 78 study visits. The MRI obtained during screening was used to assess any clinically significant findings of non‐AD pathology that could account for the observed cognitive impairment, as previously described, and served as the baseline scan for comparison with scans obtained at 52 and 78 weeks follow‐up. Site neuroimaging facilities were trained on relevant study procedures by the central imaging laboratory (Clario, Inc.) and completed site qualification and assessment of scanner performance for the duration of the study using American College of Radiology phantoms. All sites used 3T scanners (Philips, Siemens, or General Electric). Collected scans included a 3D T1 consisting of a sagittal magnetization‐prepared rapid gradient‐echo (Philips), sagittal 3D turbo field echo (Siemens), or a 3D fast spoiled gradient‐recalled sequence (General Electric) with an 1 × 1 mm2 in plane resolution, and 1.2 mm slice thickness. Two 3D T1 scans were collected during each session to increase the likelihood of obtaining good quality data. In addition, a FLAIR sequence with a 0.86 × 0.86 mm2 in plane resolution and 5 mm slice thickness and a T2* sequence with a 0.78 × 0.78 mm2 in plane resolution and 4 mm slice thickness were collected only during the screening visit. The FLAIR and T2* sequences were used to confirm study eligibility based on the exclusion criteria described previously.
2.6. Statistical analysis
The statistical analysis plan for the HOPE4MCI trial prespecified the change in CDR‐SB comparing 78 weeks to baseline as the primary efficacy endpoint. Analyses of the primary endpoint were done for the intention‐to‐treat (ITT) population consisting of all randomized participants (n = 164). The estimand (target of inference) is the average treatment effect defined as the difference between study arms of the (population) mean of the primary endpoint. A subgroup analysis looking separately at ApoE‐4 carriers versus ApoE‐4 noncariers was prespecified in the SAP as part of the primary efficacy analysis. No other subgroup analysis was planned or performed as part of this primary efficacy analysis. Specified secondary endpoints included FAQ, MMSE, and BPS‐O comparing change from baseline to 78 weeks between placebo and AGB 101 treatment.
To estimate the average treatment effect, we used a targeted minimum loss‐based estimator (TMLE). 35 This estimator is similar to commonly used methods including analysis of covariance (ANCOVA) in that it adjusts for chance imbalances in prespecified baseline variables between study arms to improve precision, and it has potential to better account for missing outcome data by a combination of regression modeling and inverse propensity score weighting. The prespecified baseline variables that were adjusted for by this estimator are: CDR‐SB score, APOE‐4 (carrier or noncarrier), Trail Making Test part A (number of errors), and Trail Making Test part B (number of errors). Also, study arm assignment and CDR‐SB change score at 26 and 52 weeks were used in the aforementioned regression and inverse propensity score models in order to account for participant dropout that may differ by study arm and by CDR‐SB progression during the study. The entire estimation procedure was prespecified in the statistical analysis plan, included in the Supplemental Materials.
The advantage of the TMLE estimator is that it relies on less stringent assumptions about missing data than are generally required by the unadjusted estimator (i.e., the difference in sample mean CDR‐SB change score comparing treatment vs. control), the ANCOVA estimator, and the mixed effects model for repeated measures (MMRM) estimator. The BCa bootstrap was used to construct confidence intervals. It was selected since it has theoretical and practical advantages compared to the basic and percentile bootstrap methods. 36
The study was originally planned to be adequately powered to serve as a phase 3 registration trial with a sample size of 830 participants (415 per treatment group). This was based on the calculation that 415 enrolled participants per treatment group is the minimum number required to detect a 30% relative reduction in the mean of the primary endpoint between the treatment and control arms based on an extrapolation of data obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. In protocol version 5 (amendment 4) the planned sample size was reduced to 160 (80 per treatment group) due to funding limitations and the need to obtain initial clinical efficacy data informing patient selection criteria for a confirmatory study.
3. RESULTS
3.1. Participants
A total of 739 subjects were screened for enrollment in the study according to the criteria described. Diagnosis of MCI due to AD 26 was supported by positive PET imaging for amyloid based on a qualitative read. The criterion for memory impairment based on 1.4 SD below the mean on delayed recall on the ISLT reduced screening failure for amyloid PET imaging to 10%. From January 14, 2019, to October 27, 2022, 164 participants were randomized across 29 sites in the United States and Canada. Of these participants 81 were randomized to AGB101 treatment and 83 were randomized to placebo. The ITT population included all 164 randomized participants and the safety population included 161 participants as three participants did not initiate treatment after randomization (see Consort Diagram in Figure 1).
FIGURE 1.
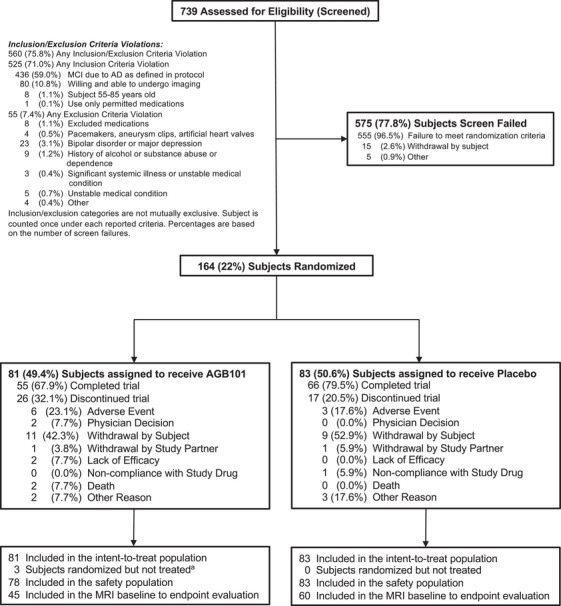
Screening, randomization, and follow‐up of participants in the Hope4MCI study. Participants who completed the week 78 visit were considered to have completed the trial. The intention‐to‐treat population included all participants randomized to treatment who completed a primary endpoint assessment at baseline and at least one dose of AGB101. Three participants were randomized to AGB101 but withdrew from the study before initiating treatment. Reasons for discontinuation included physician decision (n = 1), withdrawal by subject (n = 1), and other (n = 1)
The baseline characteristics including the prespecified variables to be compared at baseline were similar across study arms although the number (percent) of ApoE‐4 carriers was slightly higher with 54 (68%) in the AGB101 group compared to 45 (55%) in the placebo group (Table 1). Baseline characteristics also did not differ across study arms within the ApoE‐4 carrier and noncarrier subgroups (see Table S1).
TABLE 1.
Demographics and baseline characteristics
| Parameter | AGB101 (N = 81) | Placebo (N = 83) |
|---|---|---|
| Age at screening (years) | ||
| Mean (SD) | 72.2 (6.7) | 73.1 (7.0) |
| Gender, n (%) | ||
| Male | 49 (60.5) | 43 (51.8) |
| Female | 32 (39.5) | 40 (48.2) |
| Race, n (%) | ||
| White | 75 (92.6) | 81 (97.6) |
| Black or African American | 5 (6.2) | 2 (2.4) |
| American Indian or Alaska Native | 1 (1.2) | 0 (0) |
| ApoE carrier status, n (%) | ||
| Carrier | 54 (67.5) | 45 (54.9) |
| Non‐carrier | 26 (32.5) | 37 (45.1) |
| Missing | 1 (1.2) | 1 (1.2) |
| CDR sum of boxes | ||
| Mean (SD) | 2.72 (1.03) | 2.71 (1.06) |
| CDR global score | ||
| Mean (SD) | 0.51 (0.08) | 0.52 (0.11) |
| MMSE total score | ||
| Mean (SD) | 26.2 (2.1) | 26.0 (2.2) |
| FAQ total score | ||
| Mean (SD) | 7.6 (5.5) | 7.0 (5.2) |
| Missing | 5 (6.2%) | 2 (2.4%) |
| BPS‐O LDI | ||
| Mean (SD) | 0.22 (0.22) | 0.21 (0.24) |
| ISLT immediate recall | ||
| Mean (SD) | 14.2 (4.3) | 14.4 (3.7) |
| ISLT delayed recall | ||
| Mean (SD) | 2.3 (1.3) | 2.3 (1.4) |
Abbreviations: BPS‐O LDI, Behavioral Pattern Separation Task‐Objects Lure Discrimination Index; CDR, Clinical Dementia Rating Scale; CDR‐SB, Clinical Dementia Rating Scale Sum of Boxes score; FAQ, Functional Activities Questionnaire; ISLT, International Shopping List Test; MMSE, Mini‐Mental State Examination.
3.2. Primary endpoint
The mean change in CDR‐SB scores over 78 weeks is shown in Figure 2A and Table 2 for the ITT population, separately by study arm, computed by sample means and not adjusted for missing outcomes. When adjusting for missing outcomes using TMLE (the prespecified, primary efficacy analysis method), the estimated mean CDR‐SB change score at 78 weeks is 1.12 (95% confidence interval [CI]: 0.66, 1.69) for the AGB101 arm and 1.22 (95% CI: 0.75, 1.78) for the placebo arm. The corresponding difference between population means of the primary endpoint comparing AGB101 versus placebo, that is, the estimated average treatment effect, is −0.10 (95% CI: −0.85, 0.58). The point estimate, although not statistically significant, corresponds to an 8% relative reduction (the ratio of the −0.10 difference between study arms to the placebo arm mean change score 1.22) in the primary endpoint comparing AGB101 to placebo. The TMLE analysis and the unadjusted analysis gave qualitatively similar results.
FIGURE 2.
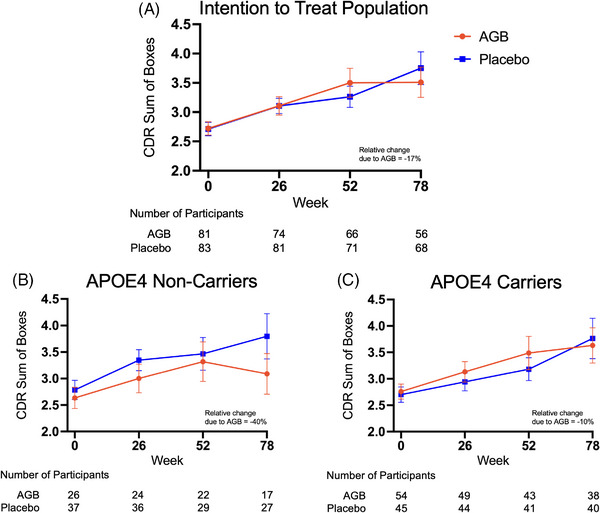
Primary endpoint. The score on the Clinical Dementia Rating‐Sum of Boxes (CDR‐SB) served as the primary endpoint in this study. CDR‐SB scores range from 0 to 18, with higher scores indicating greater impairment. Graphs show the progression of CDR‐SB scores over the 78‐week treatment period for both the placebo and AGB101 groups for the intention‐to‐treat population showing a relative improvement in CDR‐SB score due to AGB101 treatment of 17% (A), and by apolipoprotein E (ApoE)‐4 carrier status showing a relative improvement in CDR‐SB score due to AGB101 treatment of 40% for ApoE‐4 noncarriers (B) while ApoE‐4 carriers showed a relative improvement in the CDR‐SB score of 10% (C). Number of participants in each group contributing to each timepoint are provided below each graph. To facilitate comparison between the analyses all graphs show sample means ± SEM
TABLE 2.
CDR‐SB endpoint for ITT population
| AGB101 | Placebo | |||||
|---|---|---|---|---|---|---|
| Parameter |
Overall (N = 81) |
ApoE‐4 Non‐carriers (N = 26) |
ApoE‐4 Carriers (N = 54) |
Overall (N = 83) |
ApoE‐4 Non‐carriers (N = 37) |
ApoE‐4 Carriers (N = 45) |
| Change from BL to week 78 in CDR‐SB | ||||||
|
Mean (SD) Missing |
0.90 (1.50) 25 (31%) |
0.68 (1.36) 9 (35%) |
0.95 (1.55) 16 (30%) |
1.09 (2.02) 15 (18%) |
1.13 (1.96) 10 (27%) |
1.05 (2.11) 5 (11%) |
Abbreviations: Apo‐E, apolipoprotein E; BL, baseline; CDR‐SB, Clinical Dementia Rating Scale; ITT, intention‐to‐treat.
While the TMLE was used to analyze the ITT population, descriptive statistics (sample means) are provided when presenting results stratified by ApoE‐4 status. TMLE was not applied in the latter case due to the smaller sample sizes which could lead to regression model overfit and therefore unreliable results. Two participants had missing ApoE‐4 carrier status and were excluded from the analyses. As shown in Figures 2B and 2C, the point estimates are in the direction of an AGB101 benefit for both ApoE‐4 carriers and noncarriers but the estimated treatment effect for ApoE‐4 noncarriers is substantially larger than for carriers, though none of these results was statistically significant (see Figure S2 for individual subject data). As shown in Figure 3, the results were similar when considering the data separately for ApoE‐4 carrier and noncarrier participants who completed the study per protocol (Figure 3B and 3C).
FIGURE 3.
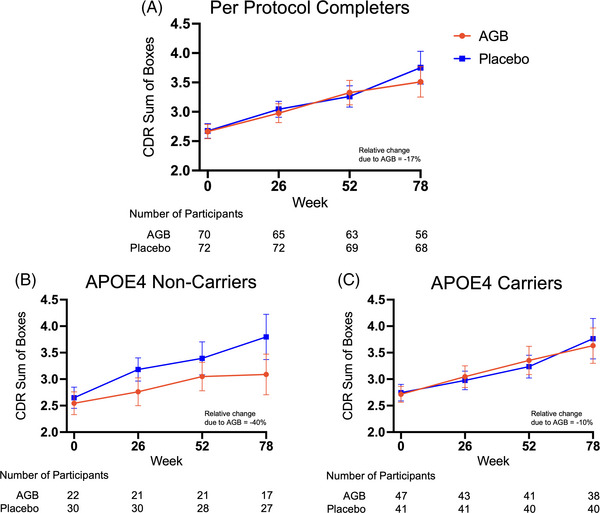
Primary endpoint for per protocol completers. Graphs show the progression of Clinical Dementia Rating‐Sum of Boxes (CDR‐SB) scores over the 78‐week treatment period for both the placebo and AGB101 groups for the per‐protocol completers (A), and for ApoE‐4 noncarrier completers (B), and apolipoprotein E (ApoE‐4) carrier completers (C). Number of participants in each group contributing to each timepoint are provided below each graph. Values represent sample means ± SEM
3.3. Secondary endpoints
Adjusted for missing outcomes using TMLE, the estimated mean FAQ score from baseline at 78 weeks is 3.82 (95% CI: 2.10, 5.17) for the AGB101 arm and 3.81 (95% CI: 2.38, 5.59) for the placebo arm. The corresponding estimated mean MMSE score at 78 weeks is −2.44 (95% CI: −3.29, −1.62) for the AGB101 arm and −1.97 (95% CI: −3.11, −1.18) for the placebo arm and the estimated mean BPS‐O score at 78 weeks is −0.003 (CI: −0.07, 0.06) for the AGB101 arm and 0.035 (CI: −0.02, 0.09) for the placebo arm (Figure 4 and Table S2). Mean change scores for the secondary endpoints are additionally provided for ApoE‐4 carriers and ApoE‐4 noncarriers separately (Figure 4 and Table S3). The same analyses using the per‐protocol population are provided in Table S4. For each of the primary and secondary endpoints, the corresponding ITT and per‐protocol analyses were qualitatively similar.
FIGURE 4.
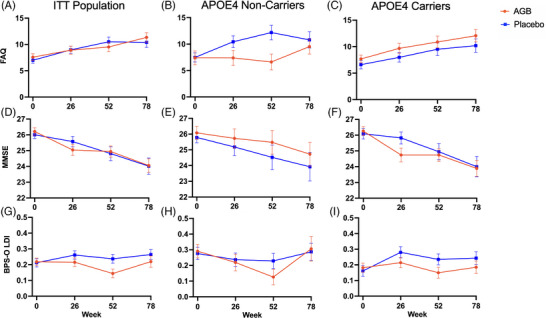
Secondary endpoints. Scores on the Functional Activities Questionnaire (FAQ; A, B, and C), Mini‐Mental Status Exam (MMSE; D, E, and F), and Lure Discrimination Index (LDI) from the Behavioral Pattern Separation‐Objects (BPS‐O) Task served as secondary endpoints in this study. For FAQ higher scores indicate greater impairment, while higher scores on the MMSE, and BPS‐O indicate better performance. Graphs show progression of scores over the 78‐week treatment period for both the placebo and AGB101 groups for the intention‐to‐treat population (A, D, and G), and by apolipoprotein E (ApoE)‐4 carrier status showing longitudinal change in secondary endpoints for ApoE‐4 noncarriers (B, E, and H) and ApoE‐4 carriers (C, F, and I). Values represent sample means ± SEM
3.4. Safety
The safety profile of AGB101 was generally consistent with the known safety profile from previous clinical studies and AGB101 was generally well‐tolerated in adults with MCI due to AD. Notably, much higher dosing in elderly patients with CNS disorders is well‐tolerated. 37 The overall incidence of treatment‐emergent adverse events (TEAEs) was similar in the two groups with 67.9% in AGB101 and 71.1% in placebo (Table 3). The most common adverse events (affecting > 5% of the participants) in the AGB101 group were urinary tract infection (6.4% with AGB101 and 3.6% with placebo), fall (7.7% with AGB101 and 9.6% with placebo), and anxiety (5.1% with AGB101 and 2.4% with placebo). TEAEs leading to discontinuation of the trial agent occurred in 7.7% of the participants in the AGB101 group and 3.6% of those in the placebo group (Table 3). All reported TEAEs are provided in the Tables S5–S7.
TABLE 3.
Overall summary of adverse events‐safety analysis set
| AE category |
AGB101 (N = 78) n (%) / Events |
Placebo (N = 83) n (%) / Events |
Overall (N = 161) n (%) / Events |
|---|---|---|---|
| TEAEs | 53 (67.9) / 148 | 59 (71.1) / 232 | 112 (69.6) / 380 |
| Treatment‐related TEAEs | 9 (11.5) / 15 | 3 (3.6) / 5 | 12 (7.5) / 20 |
| Serious TEAEs | 13 (16.7) / 16 | 10 (12.0) / 12 | 23 (14.3) / 28 |
| Treatment‐related serious TEAEs | 0 | 0 | 0 |
| TEAEs leading to drug withdrawal (with ‘adverse event’ as primary reason for early treatment termination) | 6 (7.7) / 8 | 3 (3.6) / 3 | 9 (5.6) / 11 |
| TEAEs leading to death | 2 (2.6) / 2 | 0 | 2 (1.2) / 2 |
Abbreviations: AE, adverse event; TEAE, treatment‐emergent adverse event.
4. DISCUSSION
The HOPE4MCI double‐blind placebo‐controlled trial was designed to examine the hypothesis that diminishing hippocampal overactivity in patients with MCI due to AD, using a low dose of extended release levetiracetam (AGB101), would slow the progression of disease over a 78‐week observation period. The study was motivated by extensive preclinical data indicating that hippocampal overactivity is a driver of neurodegeneration and the spread of tau pathology and clinical data showing that patients with MCI clinical symptoms and amyloid deposits in brain have levels of hippocampal overactivity that exceed those associated with normal aging. 38
Analysis of the ITT population of 164 participants did not find a statistically significant effect of AGB101 on progression determined as change from baseline to 18 months for the primary efficacy measure (CDR‐SB), nor secondary clinical measures (FAQ, MMSE, and BPS‐O). Similarly, the descriptive analyses stratified by ApoE‐4 carrier status were not statistically significant, though the point estimates of the primary endpoint for APOE4 noncarriers correspond to a 40% reduction in 18 month CDR‐SB change score comparing AGB101 to placebo. The same pattern was also observed on the secondary endpoints of FAQ, MMSE, and BPS‐O. Among ApoE‐4 carriers, there was essentially no difference on any measure between AGB101 treated patients and those on placebo.
The primary analysis of all participants does not provide sufficient evidence to reject the null hypthosis that that AGB101 is equivalent to placebo. Nevertheless, the point estimates are in the direction of a positive AGB101 benefit. The treatment effect point estimate in the direction of a positive AGB101 benefit among ApoE‐4 noncarriers is intriguing and worth further investigation. It is notable that across aging and AD models in laboratory animals that do not naturally carry the ApoE‐4 allele, low doses of levetiracetam have consistently demonstrated beneficial neurocognitive and behavioral effects. 11 It is worth noting that currently insufficient data are available to know whether hippocampal overactivity occurs earlier or later in those who are ApoE‐4 carriers relative to noncarriers nor is it clear that the response to levetiracetam will show the same dose‐response characteristics by ApoE‐4 genotype. Future studies are needed to examine the pharmacodynamic response to AGB101 treatment as a function of both ApoE‐4 genotype and level of hippocampal overactivity.
With respect to ApoE‐4, autopsy studies show lower levels of synaptic proteins such as synaptophysin in brains from older persons without cognitive impairment carrying an ApoE‐4 allele compared with those not carrying ApoE‐4. 39 Synaptic proteins are also lower in mild cases of AD dementia who are ApoE‐4 positive, 40 but the difference in synaptic protein levels is not found in brains from cases of advanced AD dementia. 39 It is well established that levetiracetam exerts its clinical effect by binding to the SV2A receptor, a membrane protein present on synaptic vesicles and a regulator of neurotransmitter release, 41 and the preclinical profile of levetiracetam is quite different from other antiepileptic drugs. 23 SV2A is reduced in the hippocampus of AD brains and is co‐localized with amyloid precursor protein (APP), 42 suggesting that SV2A is involved in amyloid processing. SV2A is likely to be involved in processing of the tau protein as well. 42 Human PET imaging studies show an inverse association of Aβ deposits with SV2A binding but only in patients at the MCI stage of AD and not in patients with more advanced disease. 43 Ongoing clinical studies with SV2A PET ligands are examining the possibility of a direct relationship of ApoE genotype with SV2A density. 44 It seems likely that SV2A is one of the downstream mechanisms mediating the effect of ApoE‐4. Thus, it is not unexpected that the effect of AGB101 should be different in noncarriers than carriers of the ApoE‐4 allele.
Conclusions from the HOPE4MCI study are limited primarily because of the small sample size. Since the difference between study arms in the primary endpoint was not statistically significant, any results can only be regarded as potential guides to future research. The possibility that ApoE‐4 carriers and noncarriers respond differently to therapeutic intervention is consistent with the recent findings with the amyloid plaque clearing antibody lecanemab as secondary analyses of the phase 3 trial of lecanemab showed a smaller estimated therapeutic effect in ApoE‐4 carriers relative to noncarriers. 25 While lecanemab and donanemab 45 , 46 ‐ both have been shown to slow disease progression in early AD, neither stops disease progession or reverses symptoms, indicating that other mechanisms must be tested to halt or reverse progression. The present results support the view that further testing of AGB101 in patients with MCI due to AD who are noncarriers of ApoE‐4 is warranted. If one assumes that the observed nominal effect and variability in the ApoE‐4 carriers is the true effect, detecting an effect of AGB101 to slow disease progression by 30% with 80% power in a phase 3 clinical trial, would require approximately 1000 participants with 500 participants assigned to the active drug and placebo conditions each.
CONFLICT OF INTEREST STATEMENT
M.G. is the founder of AgeneBio. M.G. and A.B. are inventors on Johns Hopkins University intellectual property with patents pending and licensed to AgeneBio. M.G. consults for the company and owns company stock, which is subject to certain restrictions under University policy. M.G. and A.B's role in the current study was in compliance with the conflict of interest policies of the Johns Hopkins School of Medicine. R.M., S.R.L., M.R., R.L.B., M.S.A., S.C., and S.Z. have nothing to disclose. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants in this study provided written informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank all the patients, their families, and caregivers who participated in the HOPE4MCI trial as well as the site staff, raters, and site investigators, members of the data and safety monitoring board, and vendor partners including World Wide Clinical Trials, Clinical Inc, Cogstate, Eurofins, Suvoda, Clario, and Alcemi. The authors would also like to thank Ken Payie at KGP‐Biotech for the production and manufacture of the extended‐release medication, Kevin Arauz at World Wide Clinical Trials for project management, Carrie L. Speck at Johns Hopkins University for project coordination and Sarita Foster at AgeneBio for administrative support. This work was supported by NIH grants R01AG061091 to RM and SRL, R56AG055416 to RM and SRL, R01AG048349 to MG and MSA, and the Phyllis F. Albstein Fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Mohs R, Bakker A, Rosenzweig‐Lipson S, et al. The HOPE4MCI study: A randomized double‐blind assessment of AGB101 for the treatment of MCI due to AD. Alzheimer's Dement. 2024;10::e12446. 10.1002/trc2.12446
REFERENCES
- 1. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183‐194. doi: 10.1111/j.1365-2796.2004.01388.x [DOI] [PubMed] [Google Scholar]
- 2. Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS‐ADRDA criteria. Lancet Neurol. 2007;6(8):734‐746. doi: 10.1016/S1474-4422(07)70178-3 [DOI] [PubMed] [Google Scholar]
- 3. Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64(5):834‐841. doi: 10.1212/01.WNL.0000152982.47274.9E [DOI] [PubMed] [Google Scholar]
- 4. Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63(1):38‐46. doi: 10.1001/archneur.63.1.38 [DOI] [PubMed] [Google Scholar]
- 5. Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63(5):665‐672. doi: 10.1001/archneur.63.5.665 [DOI] [PubMed] [Google Scholar]
- 6. Busche MA, Konnerth A. Neuronal hyperactivity—A key defect in Alzheimer's disease? BioEssays. 2015;37(6):624‐632. doi: 10.1002/bies.201500004 [DOI] [PubMed] [Google Scholar]
- 7. Huijbers W, Mormino EC, Schultz AP, et al. Amyloid‐β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain. 2015;138(Pt 4):1023‐1035. doi: 10.1093/brain/awv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry. 2008;79(6):630‐635. doi: 10.1136/jnnp.2007.124149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan P, Grutzendler J. Attenuation of β‐amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J Neurosci. 2016;36(2):632‐641. doi: 10.1523/JNEUROSCI.2531-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu JW, Hussaini SA, Bastille IM, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19(8):1085‐1092. doi: 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haberman RP, Branch A, Gallagher M. Targeting neural hyperactivity as a treatment to stem progression of late‐onset Alzheimer's disease. Neurotherapeutics. 2017;14(3):662‐676. doi: 10.1007/s13311-017-0541-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tabuchi M, Lone SR, Liu S, et al. Sleep interacts with aβ to modulate intrinsic neuronal excitability. Curr Biol : CB. 2015;25(6):702‐712. doi: 10.1016/j.cub.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanchez PE, Zhu L, Verret L, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Nat Acad Sci USA. 2012;109(42):E2895—E2903. doi: 10.1073/pnas.1121081109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi JQ, Wang BR, Tian YY, et al. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci Ther. 2013;19(11):871‐881. doi: 10.1111/cns.12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suberbielle E, Sanchez PE, Kravitz AV, et al. Physiologic brain activity causes DNA double‐strand breaks in neurons, with exacerbation by amyloid‐β. Nat Neurosci. 2013;16(5):613‐621. doi: 10.1038/nn.3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology. 2010;35(4):1016‐1025. doi: 10.1038/npp.2009.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gallagher M, Koh MT. Episodic memory on the path to Alzheimer's disease. Curr Opin Neurobiol. 2011;21(6):929‐934. doi: 10.1016/j.conb.2011.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haberman RP, Koh MT, Gallagher M. Heightened cortical excitability in aged rodents with memory impairment. Neurobiol Aging. 2017;54:144‐151. doi: 10.1016/j.neurobiolaging.2016.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bakker A, Krauss GL, Albert MS, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74(3):467‐474. doi: 10.1016/j.neuron.2012.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage. Clinical. 2015;7:688‐698. doi: 10.1016/j.nicl.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li SJ, Bakker A, Ward BD, et al. Lilypadd trial: targeting hippocampal hyperconnectivity in cognitively normal older adults at risk for alzheimer's disease with AGB101. J Prev Alzheimers Dis. 2022;9(1):S87. [Poster presentation] [Google Scholar]
- 22. Rosenzweig‐Lipson S, Mulcahy S, Payie P, et al. Pharmacokenetic profile of a novel low‐dose extended release formulation of AGB101 (levetiracetam). 2015. CTAD P2‐50
- 23. Klitgaard H. Levetiracetam: the preclinical profile of a new class of antiepileptic drugs? Epilepsia. 2001;42 Suppl 4:13‐18. [PubMed] [Google Scholar]
- 24. Egan MF, Kost J, Voss T, et al. Randomized trial of verubecestat for prodromal Alzheimer's disease. N Engl J Med. 2019;380(15):1408‐1420. doi: 10.1056/NEJMoa1812840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388(1):9‐21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 26. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology. 2008;70(19 Pt 2):1740‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daly E, Zaitchik D, Copeland M, Schmahmann J, Gunther J, Albert M. Predicting conversion to Alzheimer disease using standardized clinical information. Arch Neurol. 2000;57(5):675‐680. doi: 10.1001/archneur.57.5.675 [DOI] [PubMed] [Google Scholar]
- 29. Coley N, Andrieu S, Jaros M, Weiner M, Cedarbaum J, Vellas B. Suitability of the clinical dementia rating‐sum of boxes as a single primary endpoint for Alzheimer's disease trials. Alzheimers Dement. 2011;7(6):602‐610.e2. doi: 10.1016/j.jalz.2011.01.005 [DOI] [PubMed] [Google Scholar]
- 30. Cedarbaum JM, Jaros M, Hernandez C, et al. Rationale for use of the clinical dementia rating sum of boxes as a primary outcome measure for Alzheimer's disease clinical trials. Alzheimers Dement. 2013;9(1 Suppl):S45‐S55. doi: 10.1016/j.jalz.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 31. Williams MM, Storandt M, Roe CM, Morris JC. Progression of Alzheimer's disease as measured by clinical dementia rating sum of boxes scores. Alzheimers Dement. 2013;9(1 Suppl):S39‐S44. doi: 10.1016/j.jalz.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pfeffer RI, Kurosaki TT, Harrah CH, Jr , Chance, JM , Filos S. Measurement of functional activities in older adults in the community. J Gerontol. 1982;37(3):323‐329. doi: 10.1093/geronj/37.3.323 [DOI] [PubMed] [Google Scholar]
- 33. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189‐198. doi: 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- 34. Stark SM, Yassa MA, Lacy JW, Stark CE. A task to assess behavioral pattern separation (BPS) in humans: data from healthy aging and mild cognitive impairment. Neuropsychologia. 2013;51(12):2442‐2449. doi: 10.1016/j.neuropsychologia.2012.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van der Laan MJ, Gruber S. Targeted minimum loss based estimation of causal effects of multiple time point interventions. Int J Biostat. 2012;8(1). doi: 10.1515/1557-4679.1370 [DOI] [PubMed] [Google Scholar]
- 36. Efron B, Tibshirani R. Statistical data analysis in the computer age. Science. 1991;253(5018):390‐395. doi: 10.1126/science.253.5018.390 [DOI] [PubMed] [Google Scholar]
- 37. Love S, Siew LK, Dawbarn D, Wilcock GK, Ben‐Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27(6):797‐803. doi: 10.1016/j.neurobiolaging.2005.04.008 [DOI] [PubMed] [Google Scholar]
- 38. Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR. Selective loss of synaptic proteins in Alzheimer's disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49(7):631‐639. doi: 10.1016/j.neuint.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 39. deToledo‐Morrell L, Stoub TR, Wang C. Hippocampal atrophy and disconnection in incipient and mild Alzheimer's disease. Prog Brain Res. 2007;163:741‐753. doi: 10.1016/S0079-6123(07)63040-4 [DOI] [PubMed] [Google Scholar]
- 40. Harrison TM, Maass A, Adams JN, Du R, Baker SL, Jagust WJ. Tau deposition is associated with functional isolation of the hippocampus in aging. Nat Commun. 2019;10(1):4900. doi: 10.1038/s41467-019-12921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vossel K, Ranasinghe KG, Beagle AJ, et al. Effect of levetiracetam on cognition in patients with Alzheimer disease with and without epileptiform activity: a randomized clinical trial. JAMA Neurol. 2021;78(11):1345‐1354. doi: 10.1001/jamaneurol.2021.3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Nat Acad Sci USA. 2004;101(26):9861‐9866. doi: 10.1073/pnas.0308208101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kong Y, Huang L, Li W, et al. The synaptic vesicle protein 2A interacts with key pathogenic factors in Alzheimer's disease: implications for treatment. Front Cell Dev Biol. 2021;9:609908. doi: 10.3389/fcell.2021.609908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Dell RS, Mecca AP, Chen MK, et al. Association of Aβ deposition and regional synaptic density in early Alzheimer's disease: a PET imaging study with [11C]UCB‐J. Alzheimers Res Ther. 2021;13(1):11. doi: 10.1186/s13195-020-00742-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Snellman A, Ekblad LL, Koivumäki M, et al. ASIC‐E4: interplay of beta‐amyloid, synaptic density and neuroinflammation in cognitively normal volunteers with three levels of genetic risk for late‐onset Alzheimer's disease—study protocol and baseline characteristics. Front Neurol. 2022;13:826423. doi: 10.3389/fneur.2022.826423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691‐1704. doi: 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information