

Summary
Immune checkpoint inhibitors (ICIs) have become a mainstay of cancer therapy, with over 80 FDA-approved indications. Used in a variety of settings and in combination with each other and with traditional chemotherapies, the hyperactive immune response induced by ICIs can often lead to immune-related adverse events in bystander normal tissues such as the kidneys, lungs, and the heart. In the kidneys, this immune-related adverse event manifests as acute interstitial nephritis (ICI-AIN). In the era of widespread ICI use, it becomes vital to understand the clinical manifestations of ICI-AIN and the importance of prompt diagnosis and management of these complications. In this review, we delve into the clinical phenotypes of ICI-AIN and how they differ from traditional drug-induced AIN. We also detail what is known about the mechanistic underpinnings of ICI-AIN and the important diagnostic and therapeutic implications behind harnessing those mechanisms to further our understanding of these events and to formulate effective treatment plans to manage ICI-AIN.
Keywords: acute interstitial nephritis, biomarkers, immune-mediated diseases, immunotherapies, Th1/Th2/Th17 cells
1 |. INTRODUCTION
Immune checkpoint inhibitor (ICI) therapy has revolutionized cancer care and its use is rapidly increasing. To date, 8 agents, targeting the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) signaling pathway or cytotoxic T lymphocyte antigen 4 (CTLA-4) pathway have been approved by the Food and Drug Administration (FDA) for over 20 cancer types and 80 indications.1 Figure 1 shows the dramatic increase in both FDA approvals (blue bars) and rise in prevalent ICI-treated patients within our healthcare network (solid line) between 2011 and 2022. ICIs are being increasingly used in the adjuvant setting; thus, not only will the number of patients receiving ICI therapy increase, but ICI-treated patients are also expected to live longer than before, and may be at risk for long-term complications of ICI treatment.2–6 Treatment-induced immune-mediated adverse events (irAEs) affect between 60% and 80% of patients who receive ICIs; in the kidney, irAEs manifest as acute tubulointerstitial nephritis (ICI-AIN) in the majority of cases.
FIGURE 1.
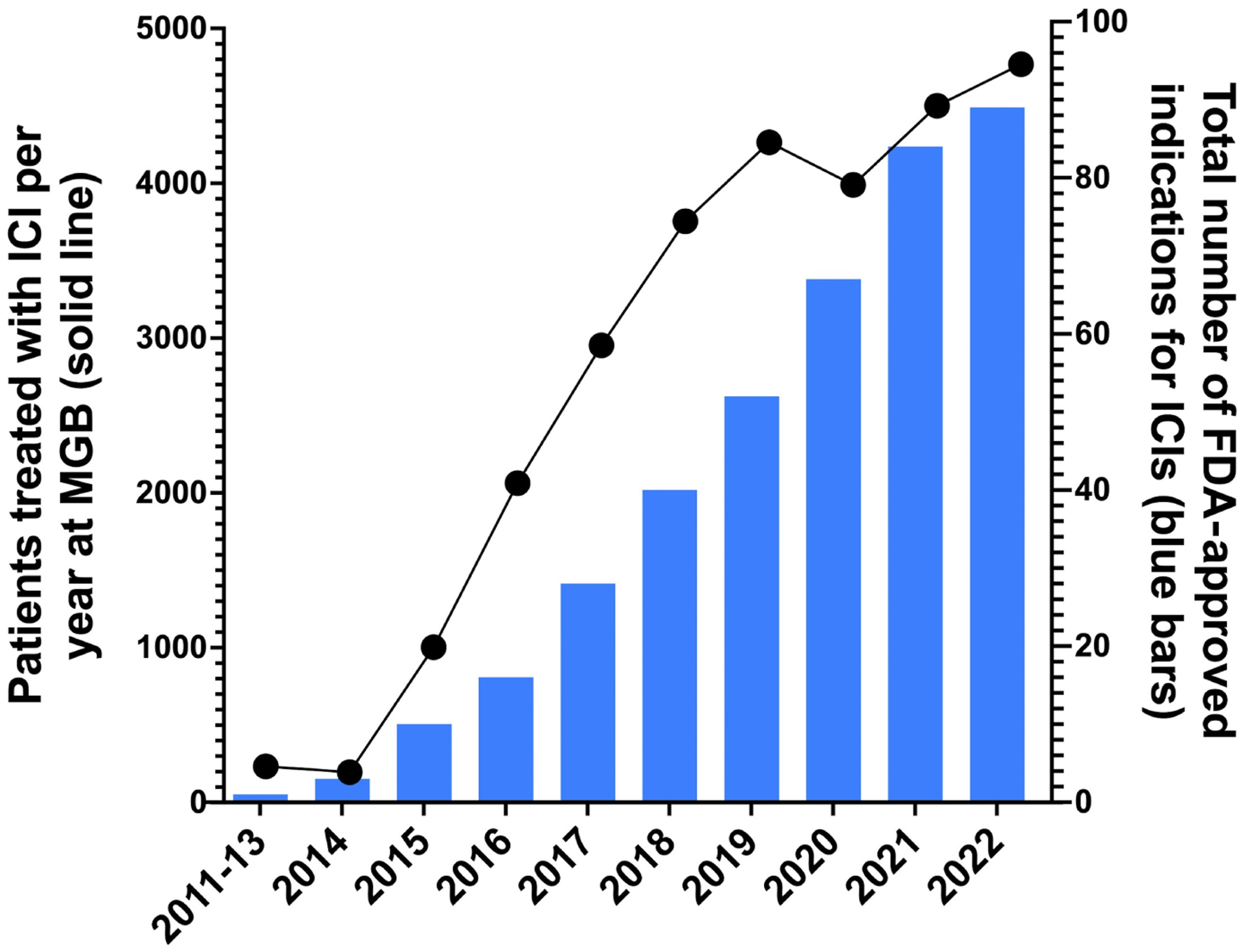
Trend in FDA indications and clinical use of immune checkpoint inhibitors at Mass General Brigham.
2 |. CLINICAL AND PATHOLOGIC FEATURES OF ICI-AIN
Acute kidney injury (AKI) is common in patients with cancer who are receiving immune checkpoint inhibitors (ICIs), affecting between 20 and 25 percent of patients within the first year; yet, ICI-AIN typically only affects 2%–3% of patients treated with anti-PD-1 or anti-PD-L1 monotherapy and 5% treated with anti-CTLA4 and anti-PD1/PDL1 combination therapy.7–11 The kidneys, while among the less commonly affected organ systems overall, are still the most common vital organ that could be affected by an irAE, with ICI-AIN being more frequent than myocarditis, pneumonitis, or hepatitis.12,13 When it occurs, ICI-AIN can be quite severe in a subset of patients. In a study of 429 patients with ICI-AIN, 49% of patients had Kidney Disease Improving Global Outcomes (KDIGO) stage 3 AKI (defined by a > 3-fold rise from baseline creatinine), and 8% developed dialysis-dependent AKI.14 Often, subclinical AKI, that is, AKI that does not meet the KDIGO threshold for Stage 1 AKI goes unrecognized, and there may also be delays in the recognition of AKI, since the serum creatinine may take several days to peak.
The diagnosis of traditional drug-induced AIN is aided by clinical signs and laboratory markers such as fever, rash, eosinophilia, arthralgia, etc.15 However, these signs are seldom present in ICI-AIN. Fever is not commonly reported in cases of ICI-AIN, and rash is often a sign of a concurrent dermatological irAE (~15% of cases).14 Leukocyturia is seen in around 56% of cases – this is in comparison to leukocyturia being found in 80% of those with drug-induced AIN.15 Proteinuria (≥0.3 g/g of creatinine) is seen in around 60% of patients, while peripheral eosinophilia is reported in <10% of patients.14 Concomitant irAEs provide an important clue to the diagnosis of ICI-AIN, with 57%–87% of patients with ICI-AIN experiencing a concomitant irAE episode affecting another organ, with skin, thyroid, colon and liver being the most commonly affected extra-renal organs.8,14
3 |. HISTOPATHOLOGIC FINDINGS
Kidney biopsy samples from patients on ICIs mostly reveal acute tubulointerstitial nephritis (>80%–90%).7,14 The interstitial infiltrate predominantly consisted of CD4+ T cells. In severe cases, granulomas can form in the interstitium. The inflammation is often described as diffuse or moderate to severe and is accompanied by tubulitis. Significant C3 or immunoglobulin staining is not seen on immunofluorescence; foot process effacement and electron-dense deposits are not typically present.16 In patients on PD-1 therapy, positive PD-L1 staining on tubular epithelial cells has been described as a differentiating factor for ICI-AIN as the staining has not been seen in ICI-treated patients with acute tubular necrosis.17 Apart from AIN, a variety of glomerular diseases have been described in the kidney biopsies of ICI-treated patients including pauci-immune glomerulonephritis, C3 glomerulopathy, amyloidosis, and podocytopathies; however, no glomerular diseases have been consistently associated with ICI.14,18
4 |. IS ICI-AIN DIFFERENT FROM DRUG-INDUCED AIN?
Prior to the widespread use of ICIs, drug-induced AIN was the most common cause of AIN. The most common medications that cause AIN include proton pump inhibitors (PPIs), nonsteroidal anti-inflammatory medications (NSAIDs), antibiotics (specifically penicillin, fluoroquinolones, and trimethoprim/sulfamethoxazole), and allopurinol. The mechanism of drug-induced AIN is considered to be a delayed hypersensitivity reaction; it was thought that there would be clinical and histological differences between both disease entities. Casals et al19 performed a study to differentiate between ICI-AIN and drug-induced AIN. They compared the histological findings of 11 patients with ICI-AIN to those with drug-induced AIN. Cortical involvement in ICI-AIN was much higher (50% vs. 25%), severe tubulitis was more common (64% vs. 25%), and staining for PD-1 and PD-L1 was positive in 10/11 ICI-AIN cases compared to just 1 in non-ICI cases.
It should be noted though that in many cases of ICI-AIN, many patients are concurrently receiving other AIN culprit drugs at the time of AKI. In a large single-center cohort study, PPIs were a significant risk factor for the occurrence of sustained AKI, that is, AKI lasting 72 h or more (HR 2.85, 95% CI 1.34–6.08) in patients receiving ICIs.8 The association extended to ICI-AIN and was confirmed by a large, multicenter study by Gupta et al. of 429 patients with ICI-AIN, where PPI use was significantly associated with the development of ICI-AIN (OR 2.4, 95% CI 1.8–3.2). Other drugs such as antibiotics and NSAIDs are also thought to contribute to the “second hit” that increases the risk of ICI-AIN.20 Guidelines now recommend cessation of other potentially AIN-causing medications (such as PPIs, NSAIDs, antibiotics, and allopurinol) in patients diagnosed with ICI-AIN.21
5 |. MANAGEMENT OF ICI-AIN
ICI-AIN poses a real threat of treatment interruption or termination, which is detrimental to the fight against cancer. Currently, guidelines suggest withholding ICI in patients with suspected ICI-AIN. When the first National Comprehensive Cancer Network (NCCN) guidelines for the treatment of irAEs were published in 2018, they recommended that for severe cases of ICI-AIN, patients should be treated with prednisone 1–2 mg/kg/day until the serum creatinine returns to <1.5-fold the upper limit of normal, and prednisone should subsequently be tapered slowly over the next 4–6 weeks.22 More recent guidelines published in 2021 from the Society of Immunotherapy for Cancer did not specify the duration of the prednisone treatment.21 However, nationally representative observational data suggest that in typical clinical practice, patients receive a 6–8 week duration of corticosteroids.23,24
ICI-AIN is typically responsive to corticosteroid treatment with 60%–80% of patients achieving partial or complete renal response with corticosteroids.14 Because of concerns that prolonged courses of corticosteroids may blunt the antitumor response, in 2018, we designed and implemented a novel clinical protocol for patients with ICI-AIN. In carefully selected patients without other concurrent organ-threatening irAEs, we tapered to prednisone 10 mg/day within 3 weeks instead of the conventional duration of 6–8 weeks. We compared 13 patients treated with this rapid corticosteroid taper to 14 historical control patients who received a conventional, NCCN guideline-based corticosteroid taper (6 weeks). To mitigate selection bias, the group assignment was based on the initially prescribed taper rather than the actual taper received. There were no differences between groups in rates of renal recovery or relapse of ICI-AIN.22 We corroborated this finding in a larger, international, multicenter cohort study of 165 patients with ICI-AKI to examine whether a shorter duration (4 weeks or less) of corticosteroids (N = 56) was associated with a higher risk of recurrence of ICI-AIN or death compared to a longer duration (4–12 weeks of corticosteroids) (N = 109). Renal recovery was similar between groups and there was no difference in the risk of recurrent ICI-AIN or death at 30 days following the taper.25 While these observational studies suggested equivalent outcomes among patients treated with shorter versus longer duration of glucocorticoids, a randomized clinical trial comparing treatment strategies that evaluate both kidney and cancer outcomes would benefit the field greatly.
Although corticosteroid-refractory AIN is uncommon, successful treatment with other immunosuppressive agents such as infliximab or mycophenolate have been reported in patients who are refractory to corticosteroids, but these reports are rare due to the low frequency of corticosteroid-resistant ICI-AIN.26–28 A case series of 10 patients with corticosteroid-refractory or relapsed ICI-AKI demonstrated that 80% achieved renal recovery following treatment with IV infliximab, the majority of patients (6 of 8 responders) required only one dose.26
The occurrence of irAEs such as ICI-AIN may potentially be associated with improved tumor response.29,30 Hence, prompt recognition and treatment of a kidney irAE and minimizing corticosteroid dose and duration when possible is a crucial component of successful cancer care in these patients.
5.1 |. ICI rechallenge after an episode of ICI-AIN
Current guidelines recommend that after an episode of ICI-AIN, ICIs should be withheld until AKI recovery. However, after AKI recovery, ICI rechallenge can be considered in those whose kidney function recovers after an episode of ICI-AIN. It is extremely important to note that patients who were receiving a potential offending drug (PPI, NSAIDs, antibiotic) at the time they developed ICI-AIN should strictly avoid these agents during ICI rechallenge. Recently, a large, multicenter study shows that of the 121 patients with ICI-AIN who were rechallenged with an ICI, only 20 (16.5%) developed recurrent ICI-AIN.14 It should be also noted that patients who are rechallenged with ICI may be at risk of developing other irAEs, though that risk has been shown to be very low (<5%).31 Before rechallenging with ICI therapy, a detailed discussion about the risks and benefits should be discussed between the multidisciplinary team of oncologists and nephrologists with the patient, and patients should be closely monitored for AKI recurrence.
Figure 2 summarizes the clinical characteristics of ICI-mediated kidney disease.
FIGURE 2.
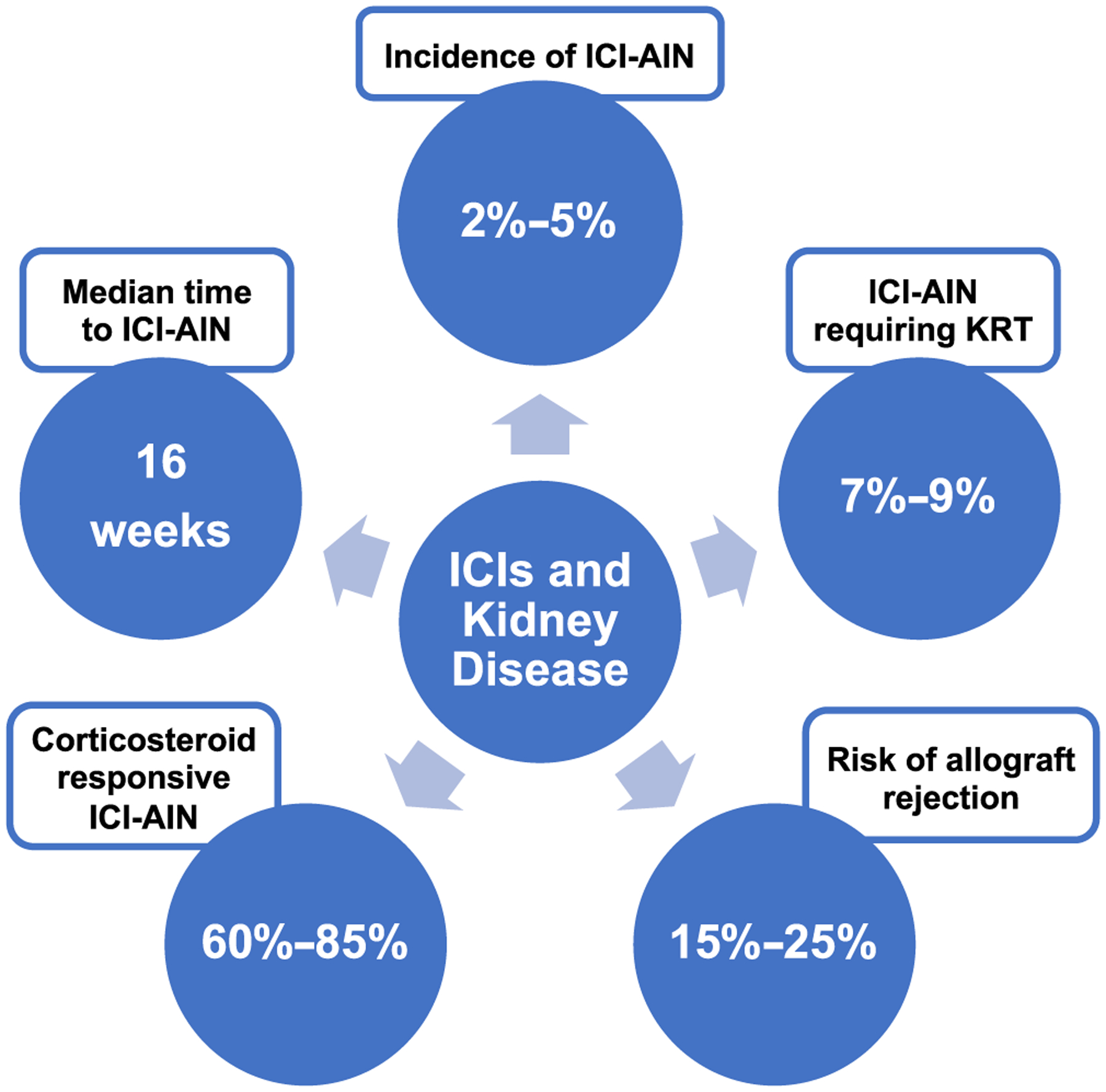
Statistics on immune checkpoint inhibitors and kidney disease.
6 |. CHRONIC KIDNEY COMPLICATIONS OF ICI
A large retrospective cohort study that included more than 2500 ICI-treated patients that survived >1 year (median follow-up 688 days) demonstrated that long-term, sustained kidney function decline is common.2 In total, 13% developed either a new-onset decline in estimated GFR (eGFR) < 60 mL/min/1.73m2, 30% eGFR decline, or need for kidney replacement therapy sustained for at least 90 days. Among patients who survived at least 4 years, nearly 20% reached the composite kidney disease outcome. Recently, we demonstrated that ICI-AIN and chronic kidney disease risk is equivalent in patients with stage 3 or 4 melanoma receiving anti-PD-1 monotherapy, suggesting that patients with earlier-stage cancer that receive ICIs will also have to be monitored for long-term kidney impairment.32
7 |. MECHANISMS OF IMMUNE-MEDIATED INJURY
To understand the mechanisms of immune-mediated kidney injury in ICI-AIN, it is necessary to review the checkpoint pathways, which serve as the safeguards of the immune system against autoimmune injury. The immune system normally has mechanisms that keep immune responses within a tolerable physiologic range and prevent action against self-antigens. Central tolerance, whereby self-reactive T-cell clones are deleted in the thymus and peripheral tolerance by employment of regulatory T cells and T-cell anergy, are important mechanisms that protect against the development of autoimmunity. Tumor cells co-opt these immune suppressive mechanisms, engaging in negative T-cell costimulation, as an act of self-defense. Immune checkpoint blockade is a process that eliminates or diminishes negative costimulation signals that prevent T-cell activation. This allows the unshackled T cells to fight more efficiently against tumor cells.33
Presentation of an antigen by an antigen-presenting cell (APC) such as a macrophage or dendritic cell to a T-cell receptor (TCR) triggers downstream signaling pathways that then accentuate the immune response through cytokines and other molecules. CTLA-4 is a transmembrane receptor that is intrinsically linked to T-cell activation and its expression on the T cell surface peaks 2–3 days after activation.34 It dampens the immune response by competing with CD28 on T cells for the B7 ligands on APCs, inhibiting the costimulatory pathway and downstream signaling mediated by the PI3K-AKT pathway.35 This dampening response takes place at sites of T-cell priming where antigens are presented such as lymph nodes, tonsils, and the spleen, regulating T-cell activation. CTLA-4 also has additional extrinsic mechanisms to downplay the immune response, including its presence on Tregs, which decreases the availability of B7 ligands on APCs by endocytosis, and increasing T-cell motility, thereby decreasing interaction between APCs and effector T cells.36,37
CTLA-4 knockout mice develop a lethal lymphoproliferative disorder and die between 3 and 4 weeks of age, with lymphocytic infiltration seen in many organs.38 In humans, CTLA-4 haploinsufficiency is an inborn error of immune dysregulation that can cause variable phenotypes of autoimmune disorders, including lymphocytic infiltration of organs, such as pneumonitis and hematological autoimmune diseases such as immune thrombocytic purpura and hemolytic anemia.39,40 IrAEs caused by CTLA-4 blockade have some phenotypic similarities to CTLA-4 haploinsufficiency, reflecting the intensity or degree of negative costimulatory blockade caused by these agents. Patients with CTLA-4 haploinsufficiency also have B cell abnormalities and a high risk of infection, especially respiratory tract infections. However, an increased risk of infection has not been reported in patients receiving ICIs.
PD-1 is a transmembrane protein present in B cells and T cells that acts through its ligands PD-L1 and PD-L2 primarily to maintain peripheral tolerance and minimize autoimmunity in local tissues.41,42 This is because these ligands are predominantly expressed in nonlymphoid tissues. Both PD-L1 and PD-L2 are expressed in response to IFN-γ indicating downstream effects after activation of CD8 effector and CD4 helper T cells. The negative costimulatory signal occurs through recruitment of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP2), which then leads to the downregulation of proximal TCR signaling molecules and attenuation of the PI3K-AKT pathway.43 There is also evidence to suggest that PD-1 primarily acts to dephosphorylate CD28, thereby acting to downregulate T-cell activation in a similar manner to CTLA-4.44 Human deficiency phenotypes have not been found, except in one child with a homozygous PDCD1 mutation, who developed type 1 diabetes, juvenile arthritis, and hypothyroidism by age three, suffered severe infections including abdominal tuberculosis at age 10 and died at age 11 of pneumonitis.45 It is evident from mouse models that PD-1 signaling is vital for maintaining peripheral tolerance. In mice deficient in PD-1 (PDCD1 knockout mice), development of lupus-like autoimmune disease and autoimmune dilated cardiomyopathy have been described.46,47
It is important to note the differences between the two pathways. CTLA4 is predominantly intracellular until TCR is activated by APCs, after which it is transported to the surface of the activated T-cell. PD-1 is expressed on activated T cells and engages with its ligands (PD-L1 and PD-L2), which are predominantly located in peripheral tissues. CTLA-4 acts to downplay the overall T-cell response at its inception while PD-1 acts to decrease the peripheral effects of the T-cell response in tissues. Despite these differences, ICIs targeting either the CTLA4 or the PD-1/PD-L1 pathway can cause a wide and mostly overlapping spectrum of irAEs.13,48 In most cases, clinical features of irAEs overlap regardless of which class of ICI is used. Additionally, abatacept, an immunoglobulin fusion protein that serves as a CTLA4 agonist, has also been successfully used to treat severe irAEs induced by either CTLA4 or PD-1/PD-L1 inhibitors.49,50 This suggests a convergence of pathways of immune-related toxicity across classes of ICIs. This may be because the immune system dynamically recirculates and integrates peripherally received immune signals and these pathways converge to modulate the global state of peripheral T-cell activation and self-tolerance. However, when CTLA4 and PD1 or PDL1 are blocked at the same time (e.g., combination therapy of ipilimumab and nivolumab), they can lead to unchecked central and peripheral immune activity that can lead to irAEs that occur at a higher frequency and greater severity.
8 |. DIAGNOSTIC CHALLENGE IN ICI-AIN AND NONINVASIVE TESTING INCLUDING BIOMARKERS OF ICI-AIN
While there are some clinical factors that may help identify ICI-AIN in a ICI-treated patient who develops AKI, such as leukocyturia on urinalysis, concurrent use of another AIN-causing medication, or concurrent irAE in another organ,14 all of these lack sensitivity and specificity, and the only confirmatory test is a kidney biopsy. Routine lab tests such as urinalysis and urine sediment exams are not sensitive and may be misleading.23,51,52 A kidney biopsy comes with a risk of bleeding complications, and may not be immediately feasible in a patient with cancer who is receiving antiplatelet or anticoagulant therapy.53,54
In recent years, several noninvasive biomarkers to detect ICI-AIN have been reported (Table 1). C-reactive protein (CRP) and urine retinol-binding protein (uRBP) by creatinine ratio have been evaluated as markers for detection of ICI-AIN.55 A study that included 37 patients with ICI-related AKI and 13 with non-ICI-related AKI found that the ratio of uRBP relative to urine creatinine was significantly higher (median [IQR] 1927 [1174, 46,522] vs. 233 [127, 989] μg/g Cr) in patients with ICI-related AKI compared to those with the non-ICI-related AKI. Serum CRP value was also higher in ICI-related AKI compared to non-ICI-related AKI (median [IQR] 55 [34, 90] vs. 3.5 [3, 8] mg/L). Wider use of these tests is required to confirm validity and their accessibility is currently limited; CRP is widely available and uRBP may be available as a send-out test at some hospitals.
TABLE 1.
Characteristics of novel diagnostic biomarkers for immune checkpoint inhibitor nephritis.
| Biomarker | Citation | Strengths | Limitations |
|---|---|---|---|
| Serum CRP | Isik et al | Clinically available | Single-center study, unlikely to distinguish between sepsis or other acute inflammatory syndromes |
| Urine RBP | Isik et al | Clinically available as a send-out test | Single-center study, nonspecific tubular injury marker, send-out test may delay results |
| Serum sIL-2R | Sise et al | Clinically available | Single-center study, unlikely to distinguish between sepsis or other acute inflammatory syndromes, send-out test may delay results |
| Flow cytometry (blood) | Sise et al | Clinically available, rapid turnaround | Single-center study, all flow cytometry panels studied are not available at every center |
| Urine TNF-alpha | Moledina et al | Rigorously studied in biopsy-proven AIN | Included few patients receiving ICIs, not clinically available |
| Urine IL-9 | Moledina et al | Rigorously studied in biopsy-proven AIN | Included few patients receiving ICIs, not clinically available |
| IFN-stimulated cytokines in urine (CXCL9, CXCL10) | Singh et al | Supported by mechanistic studies | Single-center study, not clinically available |
Abbreviations: CRP: C-reactive protein; CXCL: Chemokine Ligand; IFN: Interferon; RBP: Retinol Binding Protein; sIL-2R: Soluble Interleukin-2 Receptor; TNF: Tumor Necrosis Factor.
A recent study by Moledina and colleagues identified urine tumor necrosis factor-α and interleukin-9 as biomarkers that distinguished biopsy-proven AIN from those with other causes of AKI, with an area under the curve much higher than clinicians’ prebiopsy suspicion for AIN.56 While these are promising urine biomarkers of AIN, this study did not specifically focus on ICI-treated patients. Since ICI-treated patients have altered inflammatory milieu, including elevations in numerous cytokines and chemokines detected shortly after ICI initiation, previously discovered AIN biomarker cutoffs need to be revalidated in ICI users.
Novel diagnostic biomarkers under investigation may identify specific patterns of immune response or immunological patterns of molecular signature.
9 |. T-CELL LYMPHOID SIGNATURES
Singh et al57 studied gene expression patterns in tertiary lymphoid structures as a way to differentiate between ICI-AIN from other causes of AKI. Tertiary lymphoid structure are lymphoid aggregates in nonlymphoid tissue such as the kidney that forms as a sign of a robust antitumor response and the detection of these aggregates has been shown to be a predictive marker of ICI therapy response and is associated with improved clinical outcomes in multiple cancers, including melanoma, breast, pancreatic and lung cancer.58–62 They evaluated 36 patients on ICIs who underwent a kidney biopsy for AKI and the diagnosis of ICI-AIN was biopsy confirmed. They used the novel Nanostring nCounter PanCancer Immune Profiling Panel to evaluate gene expression profiles in the tissue samples and correlated that with blood and urine measurements of chemokines in paired samples. The PanCancer Immune Profiling Panel is a gene set specifically designed to comprehensively profile the innate and adaptive immune response, including markers for all major immune signaling pathways and immune cell types.
They found an upregulation of Th1 (T-helper cells) associated genes in ICI-AIN compared to other AKI. Since the differentiation of CD4+ T-helper cells into Th1 and Th2 is mediated by interferon pathways, they studied genes associated with the IFN-γ and found that those and the associated chemokines (CXCL9, CXCL10, CXCL11) were significantly upregulated in ICI-AIN too. The team then developed a customized 12-chemokine panel and correlated tissue immune signatures with that in the blood and urine. They found that the tertiary lymphoid structure signature in urine correlated much closer to tissue (R = 0.64) than blood (R = 0.35) and in particular, CXCL9 and CXCL10 correlated the closest and had the potential to be a novel biomarker for differentiating ICI-AIN from other AKI (area under the curve >0.75). This is important to know since chemokine secretion in the blood is not easily detectable, and hence, urine samples may provide the best chance of noninvasive detection of such immune signatures.
10 |. GENE EXPRESSION PROFILING
Adam et al63 performed a multicenter cohort study on gene expression profiling of kidney biopsies to differentiate between ICI-mediated transplant rejection and ICI-AIN. The authors used the NanoString nCounter gene expression system on formalin-fixed paraffin-embedded kidney biopsy samples and evaluated the molecular differences between ICI-AIN (native kidney, n = 10) and regular T-cell mediated rejection (TCMR) in a transplanted kidney, n = 15 from patients not on ICIs. 765 genes in total were analyzed, primarily based on the nCounter PanCancer Immune Profiling Panel. In this comparison, only 2 genes (IFI27, from the Interferon-α pathway, and FOS, from the TGF-β pathway) were expressed more in TCMR in comparison to ICI-AIN, with no genes being overexpressed in ICI-AIN compared to TCMR.
The authors then used a second cohort to study the differential gene expression between ICI-AIN and ICI-TCMR. The two genes that stood out in the previous cohort, IFI27 and FOS, were once again expressed much higher in ICI-TCMR when compared to ICI-AIN. The diagnostic performance of the genes as a two-gene set was high – accuracy of around 90% in the first comparison (ICI-AIN vs. regular TCMR), and 70% in the second comparison (ICI-AIN vs. ICI-TCMR). IFI27, which had the highest performance, was studied separately as well. However, while expression was high among regular TCMR and ICI-TCMR samples as noted above, the expression was no different in samples of ICI-AIN, other drug-induced AIN, or BK-virus nephropathy. The study also looked to differentiate between hypersensitivity and alloimmune gene expression patterns in instances of ICI-TCMR and ICI-AIN. Gene expression profiles overlapped significantly between the two conditions and were similar to drug-induced AIN, indicating that gene expression patterns in both ICI-TCMR and ICI-AIN more closely resembles a drug hypersensitivity reaction than an alloimmune disease.
11 |. BIOMARKERS OF CONGENITAL CTLA4 DEFICIENCY
Our group used a hypothesis-driven approach in the effort to find a novel biomarker for ICI-AIN. Using information previously uncovered in patients with congenital CTLA-4 deficiency, we profiled levels of cytokines and flow cytometry correlating with T-cell hyperactivation. Soluble interleukin-2 receptor alpha (sIL-2R/soluble CD25) is present at much higher levels in patients with CTLA-4 deficiency and is used as a marker of disease severity and treatment response in those patients.64
We prospectively identified clinically diagnosed and/or biopsy-proven cases of ICI-AIN (n = 24) and measured sIL-2R levels in the peripheral blood of those patients before initiation of corticosteroid therapy.65 We found that sIL-2R levels were much higher in patients with ICI-AIN when compared to two control groups that included patients with hemodynamic AKI (not receiving ICIs) or patients with cancer on ICIs without AKI or other irAEs. A sIL-2R cutoff point of 1.75-fold upper limit of normal offered maximal specificity (100%) and optimal sensitivity (81%) in differentiating ICI-nephritis from the ICI-treated controls and hemodynamic AKI controls in this study. We also developed a composite flow cytometry score that may help differentiate B-cell and T-cell signatures in patients with ICI-AIN when compared to AKI due to other causes. In ICI-AIN, there appears to be a decrease in total CD3+ T cells and decrease in naïve circulating CD8+ T cells, along with lower circulating absolute counts of memory CD27+ B cells and a relative expansion of plasmablasts. IL2RA gene expression, signifying T-cell hyperactivity, was also much higher in kidney biopsy samples of patients with IC-AIN in comparison with other AKI causes. Our findings suggest a consumptive state in the peripheral blood, with lower absolute T cells and an overactive state in the tissue, reflecting trafficking to the diseased organ. While sIL-2R was developed as a biomarker in congenital CTLA-4 deficiency, most of the patients in this cohort received PD-1 and PD-L1 inhibitors suggesting that serum markers and gene expression profiles are likely to be similar in patients on different checkpoint inhibitor classes. There are important limitations to using sIL-2R as a biomarker, as it may be elevated in patients with sepsis and also in those with lymphoma with high tumor burden.
12 |. USING IMAGING TECHNIQUES TO DIAGNOSE ICI-AIN
Imaging studies, such as positron emission tomography, that are routinely done to assess tumor response have also been proposed to help adjudicate ICI-AIN. A few reports have shown that uptake of18 F-fluorodeoxyglucose (FDG) on positron emission tomography (PET) scan can be increased in renal cortex in patients with ICI-associated AIN; however, this has not been rigorously studied.66–68 More advanced imaging techniques that are being developed to diagnose inflammation; however, none have been systematically studied in this setting.69 The fact that most tracers are renally excreted and accumulate in the collecting system affects the sensitivity and specificity of many imaging studies for identifying kidney inflammation.
13 |. PREDICTING irAES PRIOR TO DISEASE ONSET
The lack of tools to identify patients at highest risk of future irAEs or ICI-AIN severely limits our ability to predict and prevent these potentially devastating complications of ICI therapy. Recent work by Nunez et al. has sought to characterize the early immune response in patients receiving ICI therapy for melanoma and nonsquamous nonsmall cell lung cancer to identify biomarkers that predict irAE.70 Patients were grouped into those who responded to treatment and developed irAE (including one patient with ICI-AIN) and those who responded to treatment but did not develop irAE. Although the majority of types of peripheral blood mononuclear cells did not vary significantly in frequency between these groups for each cancer studied, a significant early increase in proliferating (Ki67+) T cells (including CD8+ T cells, CD4+ T cells, and Tregs) was observed in the group that went on to develop irAE, which could be identified at 1–2 weeks after start of ICI. Additionally, there was an early increase in serum expression of proteins associated with the IFN-γ signaling pathway (including CXCL9, CXCL10, CXCL11, FGF5, and IFN-y) in the group that went on to develop irAE.
Previous studies have demonstrated that development of irAE is associated with response to ICI therapy, suggesting that the immune response leading to irAE may overlap with antitumor activity. In line with these prior observations, development of irAE did correlate with patient survival in this study.70 However, Nunez et al. were able to resolve some key differences between treatment response-related markers and irAE-related markers that may signify the activation of different immune pathways in each of these conditions. In particular, they observed expansion of proliferating T cells and the increase in IFN-y-related proteins were both more strongly associated with irAE than with response to therapy. Of note, although these data provide novel insights into the immune cell populations and signaling pathways that predict the development of irAE, they do not demonstrate biological causality. Furthermore, although IFN-γ related proteins may constitute viable predictive biomarkers of irAE in the future, standardization to assess the expansion of proliferating T cells may prove to be challenging, given high variability of Ki67 staining from lab to lab.
14 |. CONCLUSION
There is an important, unmet need for noninvasive diagnosis of ICI-AIN, and recent research using gene expression profiling and immunophenotyping will improve our understanding of disease mechanisms and may nominate promising biomarker candidates. Rigorous studies with prospective sample collection of consecutive patients with AKI and are needed to validate potential biomarkers of ICI-AIN. We are hopeful that liquid biopsy will supplant the need for percutaneous kidney biopsy, which is challenging in patients with risk factors for bleeding complications. As we better understand the immunological profiles of these toxicities better, it will better enable us to personalize treatment strategies and avoid downstream effects of overimmunosuppression patients with ICI-AIN.
CONFLICT OF INTEREST STATEMENT
The authors report no conflicts of interest. MES has received research funding from Angion, Gilead, Merck, Abbvie, EMD Serono, Otsuka and has served as a scientific advisory board member for Mallinckrodt, Travere, Vera, and Novartis and DMC member for Alpine Immunosciences. HS and KM report no funding sources or disclosures.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable as no new data are generated.
REFERENCES
- 1.Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. 2021;23:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chute DF, Zhao S, Strohbehn IA, et al. Incidence and predictors of CKD and estimated GFR decline in patients receiving immune checkpoint inhibitors. Am J Kidney Dis. 2022;79:134–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson DB, Nebhan CA, Moslehi JJ, Balko JM. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat Rev Clin Oncol. 2022;19:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nigro O, Pinotti G, de Galitiis F, et al. Late immune-related adverse events in long-term responders to PD-1/PD-L1 checkpoint inhibitors: a multicentre study. Eur J Cancer. 2020;134:19–28. [DOI] [PubMed] [Google Scholar]
- 5.Schulz TU, Zierold S, Sachse MM, et al. Persistent immune-related adverse events after cessation of checkpoint inhibitor therapy: prevalence and impact on patients’ health-related quality of life. Eur J Cancer. 2022;176:88–99. [DOI] [PubMed] [Google Scholar]
- 6.Lutgens E, Seijkens TTP. Cancer patients receiving immune checkpoint inhibitor therapy are at an increased risk for atherosclerotic cardiovascular disease. J Immunother Cancer. 2020;8:e000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortazar FB, Marrone KA, Troxell ML, et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016;90:638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seethapathy H, Zhao S, Chute DF, et al. The incidence, causes, and risk factors of acute kidney injury in patients receiving immune checkpoint inhibitors. Clin J Am Soc Nephrol. 2019;14:1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espi M, Teuma C, Novel-Catin E, et al. Renal adverse effects of immune checkpoints inhibitors in clinical practice: ImmuNoTox study. Eur J Cancer. 2021;147:29–39. [DOI] [PubMed] [Google Scholar]
- 10.Meraz-Muñoz A, Amir E, Ng P, et al. Acute kidney injury associated with immune checkpoint inhibitor therapy: incidence, risk factors and outcomes. J Immunother Cancer. 2020;8:e000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelrahim M, Mamlouk O, Lin H, et al. Incidence, predictors, and survival impact of acute kidney injury in patients with melanoma treated with immune checkpoint inhibitors: a 10-year single-institution analysis. Onco Targets Ther. 2021;10:1927313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos-Casals M, Brahmer JR, Callahan MK, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour-and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. 2017;28:2377–2385. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Short SAP, Sise ME, et al. Acute kidney injury in patients treated with immune checkpoint inhibitors. J Immunother Cancer. 2021;9:e003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Praga M, González E. Acute interstitial nephritis. Kidney Int. 2010;77:956–961. [DOI] [PubMed] [Google Scholar]
- 16.Hultin S, Nahar K, Menzies AM, et al. Histological diagnosis of immune checkpoint inhibitor induced acute renal injury in patients with metastatic melanoma: a retrospective case series report. BMC Nephrol. 2020;21:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassol C, Satoskar A, Lozanski G, et al. Anti-PD-1 immunotherapy may induce interstitial nephritis with increased tubular epithelial expression of PD-L1. Kidney Int Rep. 2019;4:1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitchlu A, Jhaveri KD, Wadhwani S, et al. A systematic review of immune checkpoint inhibitor–associated glomerular disease. Kidney Int Rep. 2021;6:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casals J, Acosta Y, Caballero G, et al. Differentiating acute interstitial nephritis from immune checkpoint inhibitors from other causes. Kidney Int Rep. 2022;8:672–675. doi: 10.1016/j.ekir.2022.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seethapathy H, Zhao S, Strohbehn IA, et al. Incidence and clinical features of immune-related acute kidney injury in patients receiving programmed cell death Ligand-1 inhibitors. Kidney Int Rep. 2020;5:1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahmer JR, Abu-Sbeih H, Ascierto PA, et al. Society for Immunotherapy of cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer. 2021;9:e002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J Clin Oncol. 2018;36:1714–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perazella MA, Shirali AC. Immune checkpoint inhibitor nephrotoxicity: what do we know and what should we do? Kidney Int. 2020;97:62–74. [DOI] [PubMed] [Google Scholar]
- 24.Perazella MA, Sprangers B. Checkpoint inhibitor therapy-associated acute kidney injury: time to move on to evidence-based recommendations. Clin Kidney J. 2021;14:1301–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta S, Garcia-Carro C, Prosek JM, et al. Shorter versus longer corticosteroid duration and recurrent immune checkpoint inhibitor-associated AKI. J Immunother Cancer. 2022;10:e005646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JS, Mamlouk O, Selamet U, et al. Infliximab for the treatment of patients with checkpoint inhibitor-associated acute tubular interstitial nephritis. Onco Targets Ther. 2021;10:1877415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mamlouk O, Selamet U, Machado S, et al. Nephrotoxicity of immune checkpoint inhibitors beyond tubulointerstitial nephritis: single-center experience. J Immunother Cancer. 2019;7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jessel S, Austin M, Kluger HM. Mycophenolate as primary treatment for immune checkpoint inhibitor induced acute kidney injury in a patient with concurrent immunotherapy-associated diabetes: a case report. Clin Oncol Case Rep. 2021;4:156. [PMC free article] [PubMed] [Google Scholar]
- 29.Shimozaki K, Sukawa Y, Beppu N, et al. Multiple immune-related adverse events and anti-tumor efficacy: real-world data on various solid tumors. Preprint at 10.21203/rs.2.21095/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankar B, Zhang J, Naqash AR, et al. Multisystem immune-related adverse events associated with immune checkpoint inhibitors for treatment of non–small cell lung cancer. JAMA Oncol. 2020;6:1952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolladille C, Ederhy S, Sassier M, et al. Immune checkpoint inhibitor Rechallenge after immune-related adverse events in patients with cancer. JAMA Oncol. 2020;6:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, Strohbehn IA, Zhao S, et al. Effect of cancer stage on adverse kidney outcomes in patients receiving immune checkpoint inhibitors for melanoma. Kidney Int Rep. 2022;7:2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.cd-18-0367 [DOI] [PubMed] [Google Scholar]
- 34.Brunner MC, Chambers CA, Chan FKM, Hanke J, Winoto A, Allison JP. CTLA-4-mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–5820. [PubMed] [Google Scholar]
- 35.Pagès F, Ragueneau M, Rottapel R, et al. Binding of phosphatidyl-inositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369:327–329. [DOI] [PubMed] [Google Scholar]
- 36.Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972–1975. [DOI] [PubMed] [Google Scholar]
- 38.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. [DOI] [PubMed] [Google Scholar]
- 39.Schwab C, Gabrysch A, Olbrich P, Patiño V. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4–insufficient subjects. J Allergy Clin Immunol. 2018;142(6):1932–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schubert D, Bode C, Kenefeck R, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20:1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. [DOI] [PubMed] [Google Scholar]
- 43.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogishi M, Yang R, Aytekin C, et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat Med. 2021;27:1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. [DOI] [PubMed] [Google Scholar]
- 47.Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. [DOI] [PubMed] [Google Scholar]
- 48.Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563–580. [DOI] [PubMed] [Google Scholar]
- 49.Salem J-E, Allenbach Y, Vozy A, et al. Abatacept for severe immune checkpoint inhibitor–associated myocarditis. N Engl J Med. 2019;380:2377–2379. [DOI] [PubMed] [Google Scholar]
- 50.Liu X, Wu W, Fang L, Liu Y, Chen W. TNF-α inhibitors and other biologic agents for the treatment of immune checkpoint inhibitor-induced myocarditis. Front Immunol. 2022;13:922782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutgarts V, Glezerman IG. Kidney biopsy should Be performed to document the cause of immune checkpoint inhibitor-associated acute kidney injury: CON. Kidney360. 2020;1:162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sise ME, Seethapathy H, Reynolds KL. Diagnosis and Management of Immune Checkpoint Inhibitor-Associated Renal Toxicity: illustrative case and review. Oncologist. 2019;24:735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corapi KM, Chen JLT, Balk EM, Gordon CE. Bleeding complications of native kidney biopsy: a systematic review and meta-analysis. Am J Kidney Dis. 2012;60:62–73. [DOI] [PubMed] [Google Scholar]
- 54.Palsson R, Short SAP, Kibbelaar ZA, et al. Bleeding complications after percutaneous native kidney biopsy: results from the Boston kidney biopsy cohort. Kidney Int Rep. 2020;5:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isik B, Alexander MP, Manohar S, et al. Biomarkers, clinical features, and Rechallenge for immune checkpoint inhibitor renal immune-related adverse events. Kidney Int Rep. 2021;6:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moledina DG, Wilson FP, Pober JS, et al. Urine TNF-α and IL-9 for clinical diagnosis of acute interstitial nephritis. JCI Insight. 2019;4:e127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh S, Long JP, Tchakarov A, Dong Y, Yee C, Lin JS. Tertiary lymphoid structure signatures are associated with immune checkpoint inhibitor related acute interstitial nephritis. JCI Insight. 2022;e165108. doi: 10.1172/jci.insight.165108. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 58.Trüb M, Zippelius A. Tertiary lymphoid structures as a predictive biomarker of response to cancer immunotherapies. Front Immunol. 2021;12:674565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. [DOI] [PubMed] [Google Scholar]
- 60.Zhang N-N, Qu FJ, Liu H, et al. Prognostic impact of tertiary lymphoid structures in breast cancer prognosis: a systematic review and meta-analysis. Cancer Cell Int. 2021;21:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. 2015;112:1782–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang J, Ramis-Cabrer D, Curull V, et al. B cells and tertiary lymphoid structures influence survival in lung cancer patients with Resectable tumors. Cancer. 2020;12:2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adam BA, Murakami N, Reid G, et al. Gene expression profiling in kidney transplants with immune checkpoint inhibitor–associated adverse events. Clin J Am Soc Nephrol. 2021;16:1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lo B, Zhang K, Lu W, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349:436–440. doi: 10.1126/science.aaa1663 [DOI] [PubMed] [Google Scholar]
- 65.Sise ME, Wang Q, Seethapathy H, et al. Soluble and cell-based markers of immune checkpoint inhibitor-associated nephritis. J Immunother Cancer. 2023;11:e006222. doi: 10.1136/jitc-2022-006222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qualls D, Seethapathy H, Bates H, et al. Positron emission tomography as an adjuvant diagnostic test in the evaluation of checkpoint inhibitor-associated acute interstitial nephritis. J Immunother Cancer. 2019;7:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Awiwi MO, Abudayyeh A, Abdel-Wahab N, et al. Imaging features of immune checkpoint inhibitor-related nephritis with clinical correlation: a retrospective series of biopsy-proven cases. Eur Radiol. 2023;33:2227–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heybeli C, Nathan MA, Herrmann SM. Renal injury in the setting of immune checkpoint inhibitor: report of a case of hypothyroidism and the role of positron emission tomography. J Onco-Nephrol. 2020;4:112–116. [Google Scholar]
- 69.Ferreira CA, Heidari P, Ataeinia B, et al. Non-invasive detection of immunotherapy-induced adverse events. Clin Cancer Res. 2021;27:5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nuñez NG, Berner F, Friebel E, et al. Immune signatures predict development of autoimmune toxicity in patients with cancer treated with immune checkpoint inhibitors. Med. 2023;4:113–129.e7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable as no new data are generated.