

Abstract
Anthracyclines are effective chemotherapeutic agents, commonly used in the treatment of a variety of hematologic malignancies and solid tumors. However, their use is associated with a significant risk of cardiovascular toxicities and may result in cardiomyopathy and heart failure. Cardiomyocyte toxicity occurs via multiple molecular mechanisms, including topoisomerase II-mediated DNA double strand breaks and reactive oxygen species formation via effects on the mitochondrial electron transport chain, NADPH oxidases, and nitric oxide synthases. Excess reactive oxygen species may cause mitochondrial dysfunction, endoplasmic reticulum stress, calcium release, and DNA damage, which may result in cardiomyocyte dysfunction or cell death. These pathophysiologic mechanisms cause tissue-level manifestations, including characteristic histopathologic changes (myocyte vacuolization, myofibrillar loss, and cell death), atrophy, and fibrosis and organ-level manifestations including cardiac contractile dysfunction and vascular dysfunction. In addition, these mechanisms are relevant to current and emerging strategies to diagnose, prevent, and treat anthracycline-induced cardiomyopathy. This review details the established and emerging data regarding the molecular mechanisms of anthracycline-induced cardiovascular toxicity.
1. Background.
Anthracyclines are highly effective chemotherapeutic agents that remain a cornerstone of contemporary therapeutic regimens for a number of malignancies despite a remarkable number of recent advances in cancer therapeutics. Anthracyclines are used in up to 50-70% of adult lymphoma patients [1] and over half of childhood cancer patients [2], and remain the first-line therapeutic agent for soft tissue sarcomas [3]. Despite the increased and preferential use of taxane-based regimens, a significant proportion of breast cancer patients are still treated with anthracyclines [4].
Cardiovascular mortality is the leading non-cancer, health-related cause of death in childhood cancer survivors, and the prevalence of heart failure is not decreasing in this population despite improvements in cancer care and overall chronic disease burden [5,6]. Among long-term breast cancer survivors cardiovascular disease is the leading cause of death [7]. Cancer therapies, in particularly anthracyclines, contribute significantly to the higher incidence of cardiomyopathy and heart failure among cancer survivors. These effects are dose-dependent; there is a direct correlation between cumulative anthracycline dose and the incidence of cardiomyopathy [8]. In patients receiving 250 mg/m2 of doxorubicin the odds of developing cardiomyopathy are 25 times greater than in cancer patients with no anthracycline exposure [8,9]. According to the data from Cardinale et al., clinically detectable cardiotoxicity occurs in 9% of patients with a median time of onset of 3.5 months, and within the first year after chemotherapy in 98% of cases [10].
Anthracycline-induced cardiotoxicity significantly impairs patient’s quality of life and places a considerable financial burden on the healthcare system [11]. Congestive heart failure secondary to anthracyclines has a very poor outcome: a large population-based series reported median survival of 1 year [12]. Moreover, the development of cardiac dysfunction during cancer treatment may lead to anthracycline treatment delays and reductions, potentially impacting cancer outcomes [13]. Anthracycline-induced cardiotoxicities therefore have significant public health relevance.
Anthracyclines cause cardiovascular toxicities via multiple molecular mechanisms. An understanding of these mechanisms is critical to inform diagnostic and therapeutic strategies in patients treated with these chemotherapeutic agents. In this review, we describe the pathophysiologic mechanisms associated with the cardiovascular toxicities of anthracycline therapy, their tissue and organ-level effects, and the impact of these mechanisms on contemporary and emerging management strategies.
2. Molecular mechanisms of anthracycline toxicity.
2.1. Anthracycline effects on DNA topoisomerase.
One of the most important molecular mechanisms of cardiotoxicity is the effect of anthracycline therapy on topoisomerase IIβ (Top2β) [14]. The topological relationship between two DNA strands is critical for normal cellular function. Topoisomerases are DNA unwinding enzymes that are essential for cell replication and viability as they facilitate chromosome condensation, chromatid separation, and transient DNA strand breaks [15,16]. Two major topoisomerases are ubiquitously expressed: type I initiates cuts only in single-stranded DNA and type II enzyme can initiate double-stranded DNA breaks. In mammalian cells two topoisomerase II isoforms exist with distinct expression and localization of topoisomerase IIα and topoisomerase IIβ during the cell cycle [17,18]. Topoisomerase IIα is thought to be the main isoform expressed during mitotic processes because it is expressed at higher levels in proliferating cells [19], and in vitro Top2β was not required for mitotic events [20,21]. Top2β is predominantly expressed in quiescent cells (such as cardiac myocytes) and localized to the nuclear matrix and the nucleolus [22,23].
Anthracyclines target and inhibit Top2β, produce persistent DNA cleavable complexes, inducing DNA breaks, and thereby leading to cancer cell death [24,25]. During the past five decades several anti-neoplastic topoisomerase II inhibitors were generated with the hope of identifying anti-cancer agents with less overall toxicity [26]. In addition to resulting in anticancer activity, Top2β inhibition also causes cardiotoxicity, and may result in cardiomyocyte injury and cell death (Figure 1). Highlighting the importance of Top2β inhibition in doxorubicin cardiotoxicity, a cardiomyocyte-specific deletion of Top2β was shown to protect cardiomyocytes from doxorubicin-induced DNA double-strand breaks, reduced ROS generation, inhibited transcriptional changes responsible for defective mitochondrial biogenesis and oxidative stress, and protected from doxorubicin-induced cardiomyopathy [14]. Recent studies describe an important role of mitochondrial Top2β in mitochondrial biogenesis [27,28]. Anthracyclines bind to mitochondrial DNA [29], and cause acute reduction in mitochondrial DNA synthesis [30]. Mitochondrial DNA lesions have been documented in patients after anthracycline therapy [31]. Therefore, anthracyclines affect nuclear and mitochondrial DNA simultaneously. Since several different cell types exist in the heart, anthracycline-induced cleavage of double stranded-DNA may occur not only in cardiomyocytes, but also in endothelial cells, fibroblasts, and progenitor cells (Figure 2). In summary, Top2β-mediated DNA damage is an important and well-described mechanism of anthracycline cardiotoxicity.
Figure 1. Anthracycline effect on Topoisomerase 2β, and its inhibition by dexrazoxane.
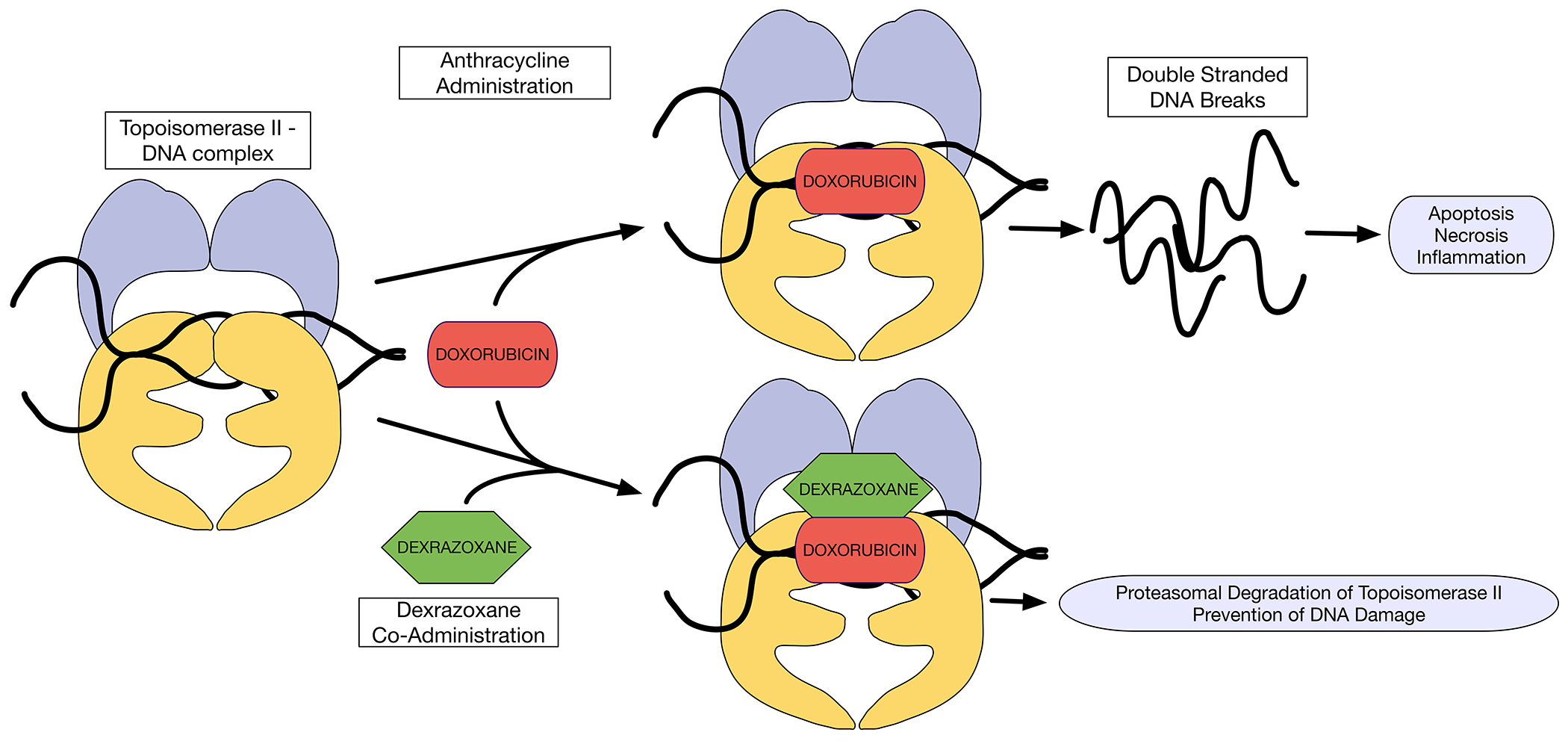
Doxorubicin intercalates DNA and inhibits Top2β. Doxorubicin forms cleavable complexes with DNA-bound Top2β, which may lead to DNA breaks and induce cell death. Dexrazoxane binds to DNA-bound Top2β, and triggers proteasomal degradation of Top2β, thereby reducing formation of doxorubicin-poisoned Top2β cleavage complexes.
DNA = deoxyribonucleic acid; Top2β = Topoisomerase 2β
Figure 2. Myocardial cells and sub-cellular compartments affected by anthracycline-induced topoisomerase II inhibition.

Transmission electron microscopic scan of murine cardiac tissue: A) Arrows indicate cardiac mitochondria (blue arrow, mitochondrial DNA), the cardiac nucleus (yellow arrow, nuclear DNA), and cardiac endothelial cells (red arrow, endothelial cell DNA, 2,500 x, scale bar = 5 μm). B + C) cardiomyocyte nucleus and cardiac capillary formed by a single endothelial cell, respectively (5,000x, scale bar = 2 μm), and D) cardiac interfibrillar mitochondria (10,000x, scale bar = 1 μm).
DNA = deoxyribonucleic acid
2.2. Anthracycline-induced generation of reactive oxygen species (ROS).
Increased oxidative stress is another important mechanism of anthracycline cardiotoxicity [32]. ROS are natural byproducts of cellular metabolism and under physiologic conditions a fine balance is maintained between ROS production and disposal. Oxidative stress occurs when ROS production overwhelms oxidative defense systems, or when antioxidant pathways are downregulated. Massive production of reactive oxygen and nitrogen species triggered by anthracyclines leads to oxidative stress [14,33,34], and may ultimately activate cell death signaling [35]. Anthracyclines affect ROS production via multiple cellular mechanisms, including through effects on the mitochondrial electron transport chain (NADH/Complex-1), nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), and dysfunctional nitric oxide synthases (NOS), as described here.
2.2.1. Effects on the mitochondrial electron transport chain.
The myocardium continuously consumes more energy per gram of tissue than any other organ. Cardiac metabolism is primarily aerobic with most of the energy supplied via an oxidative phosphorylation process in mitochondria. Electron carriers (complexes I-IV) embedded in the inner mitochondrial membrane export H+ out of the mitochondrial matrix, generating a H+ gradient across the membrane. ATP synthetase (also known as complex V) uses the energy released by H+ diffusion back into the mitochondrial matrix to synthesize ATP. To ensure high ATP production, the heart has the highest mitochondrial density among all the tissues [36,37]. The electron transport across the mitochondrial membrane is initiated in complex I (NADH dehydrogenase), when four protons (4H+) per molecule of oxidized NADH get translocated. In 1986 Davies and Doroshow showed that NADH dehydrogenase, associated with the mitochondrial complex I, is the site of anthracycline-induced free radical formation [38]. They demonstrated that in cardiomyocytes NADH dehydrogenase reduces the quinone moiety of anthracyclines to a semiquinone radical (Figure 3). In the presence of molecular oxygen, the semiquinone auto-oxidizes to generate the parent anthracycline and a superoxide anion [1]. This self-perpetuating redox cycling of anthracycline in complex I results in the accumulation of superoxide anions and reduced ATP production. Superoxide may be spontaneously converted to H2O2 by superoxide dismutase. Through this pathway, anthracycline-induced mitochondrial toxicity may result in cardiac dysfunction and cell death.
Figure 3. Molecular pathways affected by anthracyclines in cardiac myocytes.
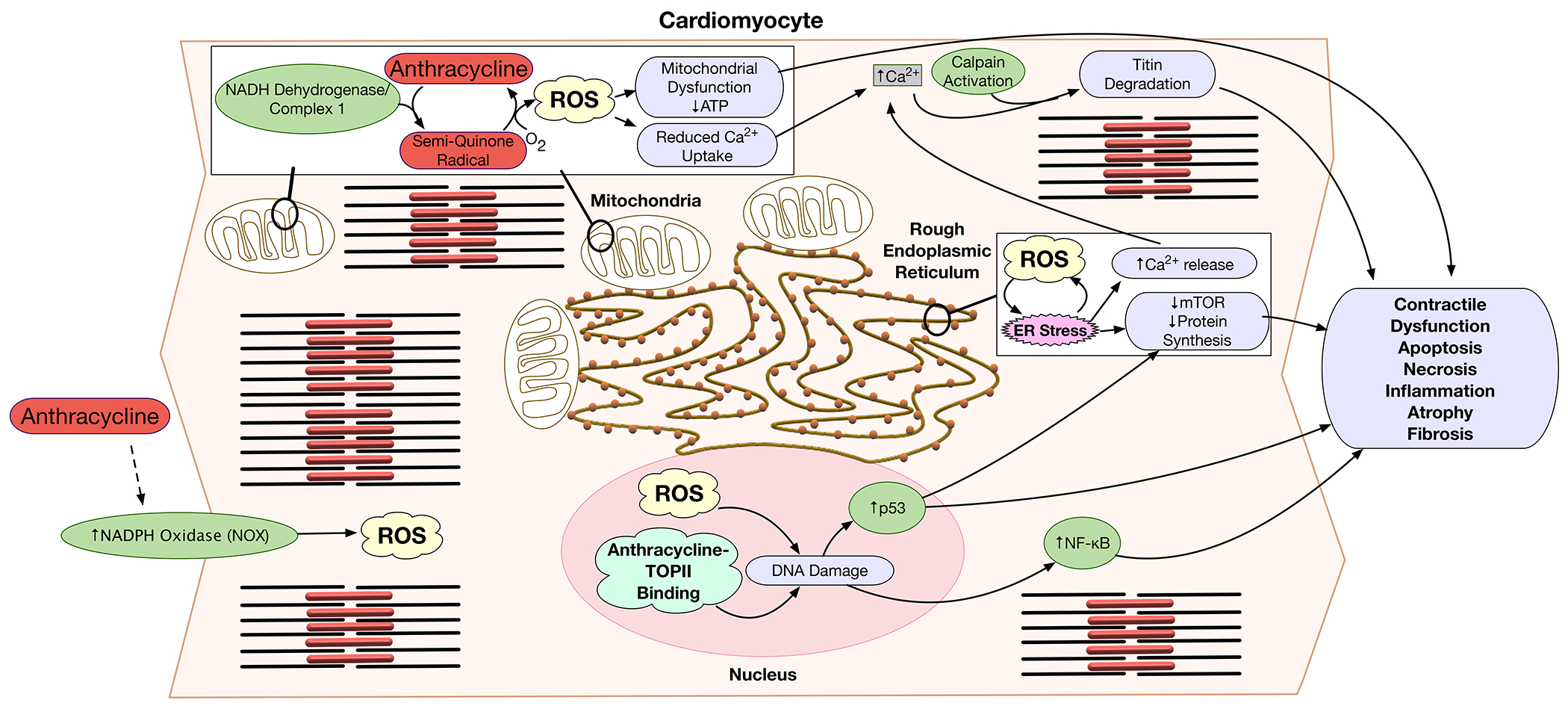
In myocytes anthracyclines induce ROS generation by mitochondria and NADPH oxidase (NOX), reduce mitochondrial respiration, induce ER-stress, alter Ca2+ signaling, and induce DNA damage. NADH dehydrogenase of complex I of the mitochondrial electron transport chain reduces the quinone moiety of anthracyclines to a semiquinone radical. In the presence of molecular oxygen, the semiquinone auto-oxidizes to generate the parent anthracycline and a superoxide anion. The effects of anthracyclines on the electron transport chain and increased ROS generation cause mitochondrial dysfunction, decreased ATP synthesis, and reduced Ca++ uptake. Increased ROS production induces ER-stress, further increasing cytoplasmic Ca++ concentrations due to ER Ca++ leak. ER stress also inhibits mTOR survival signaling and thereby reduces protein synthesis. ER-stress and mTOR inhibition trigger inflammation, and cell death signaling. In addition, anthracyclines induce activation of the NOX transmembrane enzyme complex, further increasing production of ROS. Anthracycline binding to TopII in the nucleus, and the direct effect of ROS may cause irreversible DNA damage. Anthracycline-induced DNA damage and oxidative stress lead to p53 tumor suppressor activation, that modulates mTOR pathway. ROS and DNA damage result in NF-kB activation. These molecular pathways lead to contractile dysfunction, cardiac atrophy, fibrosis, apoptosis, necrosis, and inflammation.
ATP = adenosine triphosphate; DNA = deoxyribonucleic acid; ER = endoplasmic reticulum; ROS = reactive oxygen species; TOPII = topoisomerase II
Catalase enzymes facilitate decomposition of H2O2 to water and oxygen to protect from oxidative insults. However, catalase activity levels in the heart are only 2-4% of activity levels in the liver and kidneys [39], and this low cardiac catalase activity may predispose the myocardium to doxorubicin-induced toxicity. Additional studies comparing the levels of other antioxidant enzymes including superoxide dismutase and glutathione peroxidase in cardiac and hepatic tissues have found that the heart has a much lower concentration of enzymatic defenses against free radicals [40]. Moreover, exposure to doxorubicin leads to a prolonged depression in cardiac glutathione peroxidase activity [40].
2.2.2. NADPH oxidase (NOX).
Besides mitochondria, key producers of cellular ROS are the NADPH oxidases (NOX). The NADPH oxidase is a multi-subunit transmembrane enzyme complex, and there are seven NOX family members (NOX 1-5, and dual oxidase 1-2) with different tissue distribution and activation mechanisms [41]. The primary NOX isoforms expressed in cardiac myocytes are the cell membrane-bound NOX2, and the organelle-bound NOX4. NOX2 is a mechano-sensor in cardiac tissue [42]. NOX-dependent ROS production regulates several key components of cardiac remodeling, including myocyte hypertrophy, contractile dysfunction, apoptosis, and fibrosis [43].
Anthracyclines induce NOX activation resulting in the increased production of ROS (Figure 3) [44]. Genetic disruption of NOX2 protected against the development of cardiac contractile dysfunction, atrophic cardiomyocyte degeneration, apoptosis, and interstitial fibrosis after chronic doxorubicin treatment [43]. NOX2-deficient mice reportedly exhibit resistance to DOX-induced cardiac atrophy as well [45].
Transient receptor potential (TRP) channels are multimodal cation channels regulated by several environmental factors, including mechanical stress, several of which have been implicated in the development of pathological hypertrophy caused by neurohormonal factors and mechanical stress [46]. TRPC3 interaction with NOX2 protects it from proteasome-dependent degradation [47]. NOX2 also stabilizes TRPC3 proteins to enhance mechanical stress-induced background Ca2+ entry into cardiomyocytes, leading to maladaptive remodeling. Shimauchi et al. demonstrated that increased levels of the TRPC3-NOX2 complex correlate well with the severity of anthracycline-induced cardiac atrophy and that inhibition of TRPC3-NOX2 functional coupling suppresses DOX-induced myocardial atrophy in mice. By contrast, voluntary exercise, which induces physiological cardiac hypertrophy, reduced levels of the TRPC3-NOX2 complex [46]. Ibudilast, an inhibitor of phosphodiesterase 4 (PDE 4), destabilizes the TRPC3-NOX2 complex, attenuates doxorubicin-induced NOX activation and ROS production, and was found to be a potent inhibitor of doxorubicin-induced cytotoxicity in mice [48].
2.2.3. Iron-dependent ROS production and ferroptosis.
ROS generation with doxorubicin exposure is further increased by free cellular iron [1]. Doxorubicin leads to accumulation of iron in the mitochondria by inhibiting ABCB8, a mitochondrial protein that facilitates iron export [49]. H2O2 may be converted to highly toxic hydroxyl radicals in the presence of heavy metals, such as iron. Additionally, doxorubicin can form complexes with iron, resulting in iron cycling between Fe(II) and Fe(III) forms, and substantial ROS production [50].
Mutations of the HFE gene result in the development of hereditary hemochromatosis, a condition that causes iron overload. HFE-deficient mice were found to be more susceptible to doxorubicin toxicity than wild-type mice potentially through exaggerated ROS response to anthracycline exposure in the setting of iron excess [51].
Ferroptosis is iron-dependent regulated cell death that occurs as a consequence of lethal lipid oxidation, and could be prevented by iron chelation [52]. Glutathione peroxidase 4 (GPx4) is an endogenous scavenger for lipid peroxides and is a key regulator of ferroptosis.
It was shown that doxorubicin downregulates GPx4 and induced excessive lipid peroxidation through doxorubicin-Fe(II) complex in mitochondria, leading to mitochondria-dependent ferroptosis [53]. GPx4 overexpression protected against anthracycline-induced myocardial dysfunction in mice. Additionally, inhibition of ferroptosis by ferrostatin-1 significantly reduced anthracycline-induced cardiomyopathy [54]. Furthermore, concomitant inhibition of ferroptosis and apoptosis with ferrostatin-1 and zVAD-FMK fully prevented anthracycline-induced cardiomyocyte death [53].
2.2.4. Nitric oxide synthase (NOS).
Nitric oxide synthase (NOS) expression in cardiomyocytes is induced following doxorubicin treatment, and leads to increased production of peroxynitrite (reactive nitrogen species; RNS) from nitric oxide (NO) and the superoxide anion [55]. RNS were shown to be a trigger of doxorubicin-induced cell death in cardiac myocytes [55]. In addition to its relevance to cardiomyocytes, endothelial nitric oxide synthase (eNOS) has particular importance in the pathophysiology of anthracycline-related endothelial toxicity. eNOS reduces doxorubicin to a semiquinone radical, which in turn leads to the generation of superoxide radicals and hydrogen peroxide, resulting in oxidative stress, and also leads to peroxynitrite formation and reduced nitric oxide levels, resulting in nitrosative stress (Figure 4.) [56–60]. These effects appear to be most important in the endothelial mitochondria, where post-anthracycline changes include an increase in mitochondrial ROS and oxidative stress metabolites with a lack of compensatory stimulation of superoxide dismutases 1 and 3 (endogenous antioxidants) in addition to decreased phosphorylated eNOS and NO levels [61,62]. The generation of ROS may be related to reduced circulating vascular endothelial growth factor (VEGF) levels [61,63,64]. Human cell culture and animal studies have found that treatment with L-arginine, mitochondrial-specific antioxidants, restoration of VEGF-A levels and VEGF-B gene therapy can reverse these effects and restore endothelial function [61,63–65].
Figure 4. Effects of anthracyclines on the vascular endothelium.
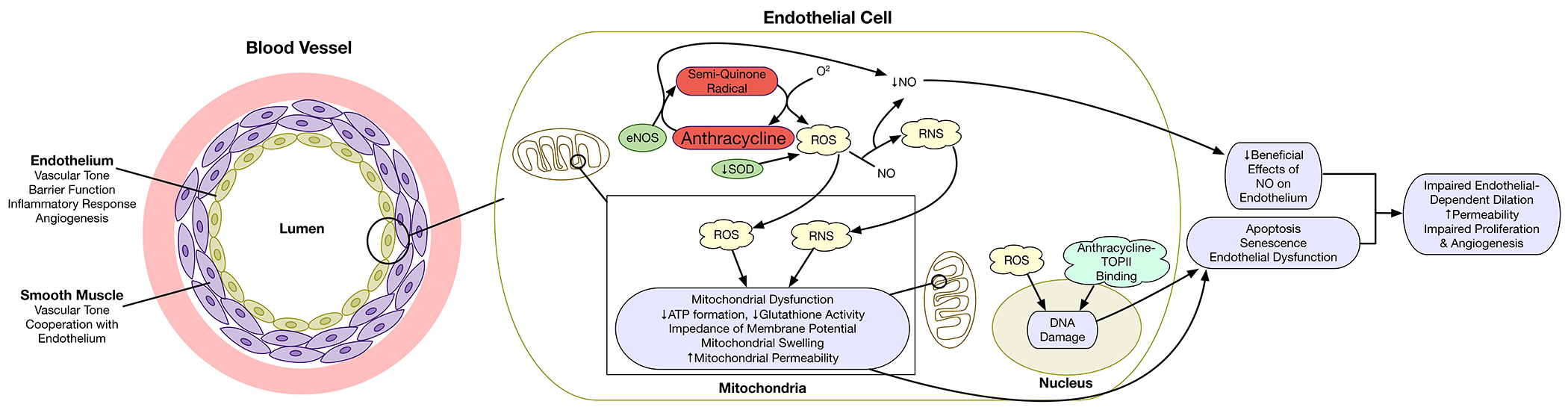
Within the vascular endothelium, anthracyclines result in the generation of reactive oxygen species (ROS) primarily via endothelial nitric oxide synthase (eNOS). Interactions of ROS with nitric oxide (NO) results in the formation of peroxynitrites (reactive nitrogen species; RNS) and result in reductions in NO. These reactive species result in mitochondrial dysfunction. DNA damage may result from ROS and topoisomerase II-mediated double stranded DNA breaks. Through these mechanisms, anthracyclines may cause endothelial apoptosis, senescence, and dysfunction resulting in impaired endothelial dependent vascular dilation, increased permeability, and impaired angiogenesis.
ATP = adenosine triphosphate; DNA = deoxyribonucleic acid; eNOS = endothelial nitric oxide synthase; RNS = reactive nitrogen species; ROS = reactive oxygen species; SOD = superoxide dismutase; TOPII = topoisomerase II
2.3. Anthracycline-induced cell signaling and myocardial cell death pathways.
2.3.1. Endoplasmic reticulum stress, apoptosis, and autophagy.
Increased ROS production leads to endoplasmic reticulum (ER) stress (Figure 3). Two kinds of ER-stress exist. Beneficial ER-stress inhibits mammalian target of rapamycin (mTOR) kinase signaling, that regulates cell growth, survival and metabolism. mTOR inhibition suppresses protein translation to improve protein folding and quality control. When these compensatory mechanisms are overwhelmed, detrimental ER-stress occurs and cell death signaling is triggered [66]. Because increased ROS production by stressed mitochondria and NOX activation induces ER-stress, and ER-stress ultimately can lead to increased ROS production a vicious cycle is initiated. ER stress signaling pathways intersect with oxidative stress signaling, and both pathways can trigger inflammatory processes [67,68].
It was confirmed experimentally that doxorubicin leads to ER stress in murine hearts. Doxorubicin disrupts expression of the ER chaperone, glucose-regulated protein 78 (GRP78, also called BiP), which plays a major role in adaptive responses to ER stress [69]. ER chaperones are involved in ensuring the quality control and post-translational modifications of newly synthesized proteins in addition to regulation of ER stress responses_[70]._Doxorubicin activates caspase-12, an ER membrane–resident apoptotic molecule, which can lead to cardiomyocyte apoptosis and myocardial dysfunction. Cardiac-specific overexpression of GRP78/BiP chaperone attenuates caspase-12 cleavage, and alleviates cardiac apoptosis and dysfunction induced by doxorubicin [69].
CRTH2, a chemoattractant receptor-homologous molecule expressed on T helper type 2 cells, is highly expressed in the heart as well. CRTH2 activation facilitates ER stress-induced cardiomyocyte apoptosis via a caspase-12-dependent pathway. Doxorubicin significantly upregulates CRTH2 gene expression in cardiomyocytes. Deletion of CRTH2 markedly prevented left ventricular dysfunction and improved the survival rate in mice after doxorubicin treatment by suppressing cardiomyocyte apoptosis [71].
ER-stress activates autophagy, a cell defense mechanism for the removal of unfolded or damaged proteins [72]. Autophagosomes, vesicles surrounded by double membrane-structures, encapsulate defective cellular components. Ultimately the outer membrane of autophagosomes fuses with the lysosomal membrane so that all cargo together with the inner membrane of the autophagosome will get digested. There are conflicting reports on the role of autophagy in anthracycline-induced cardiotoxicity. Most, but not all, of the studies conclude that doxorubicin upregulates cardiac autophagy [73]. A number of studies indicated that anthracyclines inhibit autophagic flux, by inhibiting lysosomal biogenesis and function. The resulting accumulation of undegraded autolysosomes further stimulates ROS production [74,75]. Mitochondrial autophagy (mitophagy) degrades and recycles damaged mitochondria, and there is emerging evidence for defective mitophagy in doxorubicin-induced cardiotoxicity [74]. A recent study found that luteolin, a natural flavone found in vegetables, improved doxorubicin-induced cardiomyocyte contractile dysfunction in adult mouse cardiomyocytes, and attenuated doxorubicin-induced cardiotoxicity through promoting mitochondrial autophagy [76].
2.3.2. Effects on calcium handling.
In all cell types oxidative stress affects Ca2+- handing because mitochondria can uptake calcium and thereby buffer cytosolic Ca2+ concentrations [77]. Multiple studies have demonstrated that doxorubicin increases resting intracellular Ca2+ concentration (Figure 3) [78]. Excessive ROS production, combined with increased cytosolic calcium leads to opening of the mitochondrial permeability transition pore, and mitochondria lose their ability to develop and maintain electrochemical gradients across the inner membrane [79]. This could lead to the rupture of the mitochondrial outer membrane and release of proapoptotic factors from the mitochondrial intermembrane space, activating a mitochondrial-dependent apoptotic pathway [79].
Oxidative stress also activates calpains, a family of calcium-dependent cysteine proteases that play a role during myofibril remodeling. Since calcium-dependent calpain activation may trigger titin degradation, pathological remodeling of the sarcomere can be considered as a secondary effect of excessive ROS production. It was previously shown that the myofibrillar protein titin, which is the largest structural molecule in the cardiac sarcomere, was degraded after doxorubicin treatment and that pre-incubation with calpain inhibitors, but not caspase inhibitors, rescued titin degradation [80].
2.3.3. p53 signaling.
Anthracycline-induced DNA damage and oxidative stress lead to p53 tumor suppressor activation, which is a crucial protein for the induction of cell cycle arrest and apoptosis [81]. Numerous studies have shown that doxorubicin-induced cardiomyocyte apoptosis is associated with increased expression of the p53 protein [82,83]. Genetic deletion of p53 in mice protected animals against early anthracycline-induced cardiac dysfunction 2 weeks following exposure [83]. Zhu et al. found that acute anthracycline-induced cardiac dysfunction and cardiac mass loss occur via p53-dependent modulation of the mTOR pathway [82]. Anthracycline treatment significantly reduced level of activated mTOR in wild type mice. Transgenic animals with constitutively elevated mTOR activity in the heart were protected against anthracycline-induced cardiomyopathy. However, this effect was independent of the degree of cardiomyocyte apoptosis, as apoptotic signaling in transgenic mTOR animals was not significantly different from wild type animals [82].
Interestingly, although p53 inhibition offers cardioprotection early during anthracycline exposure, paradoxically, it predisposes to cardiotoxicity late after the exposure to the drug [84]. It was shown that p53 inhibition blocked activation of STAT3 (signal transducer and activator of transcription 3) transcription factor and led to late-stage cardiac dysfunction. STAT3 controls cell proliferation, growth and angiogenesis [85], but is also found in mitochondria, where it directly affects mitochondrial energetics [86]. Cardiac-specific overexpression of STAT3 in mice protected against doxorubicin-induced cardiomyopathy, and improved survival by preventing the reduction of contractile proteins [87]. Activation of STAT3 prevented doxorubicin-induced cardiomyopathy [88]. It was also shown that prostaglandin E2, a major COX-2 product, prevented doxorubicin-induced myocardial apoptosis and that this protection required the activation of STAT3 and ERK1/2 pathways [89].
Mice with homozygous knock-in of the p53 R172H (p53172H/H) mutation lack the prototypical tumor suppressor activities similarly to the p53 knock out state (p53 −/− ) [90]. However, p53 R172H retains its mitochondrial biogenesis capacity. Comparison of p53 R172H mutant with p53−/− mice revealed that p53 regulation of the mitochondrial genome following anthracycline exposure plays an important role in preventing cardiotoxicity [90]. Mutant p53 was protective against late-onset doxorubicin cardiotoxicity, however, this protective activity was associated with its regulation of the mitochondria and not with the loss of apoptosis or cell-cycle arrest activity. Both p53 R172H mutant and p53−/− null showed loss of cell cycle inhibitor p21 induction, indicating that they are similarly defective in cell cycle regulation and apoptosis. However, unlike p53−/− null cardiomyocytes, p53172H/H mutant cardiomyocytes show the increase in mtDNA transcripts in response to doxorubicin exposure [90].
2.3.4. NF-kB signaling.
ROS and RNS cause double strand breaks in DNA, thereby activating the nuclear enzyme PARP [55] . Overactivated PARP leads to depletion of intracellular ATP, slowing the rate of glycolysis and mitochondrial respiration, eventually resulting in cellular dysfunction and death, mostly by necrosis [55]. PARP-1 is also a known co-activator of a number of pro-inflammatory genes, including NF-kB.
The nuclear factor NF-kB superfamily of transcription factors regulates immune cell maturation, cell survival and inflammation. During acute ischemic conditions NF-kB has been shown to regulate cardiomyocyte survival through suppression of apoptotic cell death, but prolonged activation of NF-kB in the heart initiates inflammatory responses that lead to ER-stress induced cell death [91,92]. Increased inflammation further aggravates oxidative stress, resulting in cardiovascular dysfunction [55]. Doxorubicin administration increases NF-kB binding activities [93], and activation of NF-kB in cardiomyocytes and endothelial cells may lead to apoptosis [94].
3. Tissue and organ-level manifestations of anthracycline toxicity.
3.1. Tissue-level manifestations.
3.1.1. Histopathologic hallmarks of anthracycline cardiotoxicity.
Histopathologic studies have demonstrated characteristic abnormalities resulting from anthracycline exposure [95,96]. Progressive sarcoplasmic reticular swelling and vacuolization may occur, resulting in distension and ballooning appearance of severely affected cells. The size and number of vacuoles may vary considerably within affected cells. Interstitial edema and perivascular fibrosis may accompany these findings. Myofibrils are displaced by vacuoles and degraded, leading to a reduced number of myofibrils in moderately to severely affected cells. In more severely affected cells, mitochondrial degradation occurs, and mitochondrial abnormalities may be observed such as prominent cristae, increased electron density of outer membranes, myelin figures, and mitochondrial dense bodies. Severely affected cells undergoing necrosis may have nuclear changes as well. Myocytes are not uniformly affected such that cells adjacent to affected cells may appear normal. In human autopsy studies of patients who had a long-term anthracycline cardiomyopathy, cell death may lead to a depleted number of cardiomyocytes, loss of normal myocardial fiber architecture, and interstitial and replacement fibrosis with a lack of hypertrophy of remaining cells may be observed [97,98]. In addition to these classically described findings, there has been a recent focus on anthracycline-induced development of cardiomyocyte atrophy and the development of cardiac fibrosis; we highlight recent research in these areas below.
3.1.2. Atrophy.
Reduction in left ventricular (LV) mass is observed both early after anthracycline therapy and during survivorship [99,100]. In addition to cell death, this finding may be related to cardiomyocyte atrophy. CMR imaging of breast cancer patients revealed that a reduction in LV mass index after anthracycline therapy was associated with an ≈30% decrease in intracellular lifetime of water (τic) [101], a noninvasive way for estimating the size of cardiomyocytes. τic measures the time it takes a water molecule to diffuse to the cell membrane, and in cardiomyocytes it is primarily affected by the cell diameter [102]. This finding suggests that LV mass reduction with anthracycline therapy is at least partly related to myocyte atrophy.
In a murine model of anthracycline-induced cardiomyopathy Willis et al. showed that there was a 44% decrease in cardiomyocyte cross-sectional area, suggesting that cell atrophy was one of the causes of myocardial mass loss [103]. Expression of MuRF1, a striated muscle-specific ubiquitin ligase, and its upstream regulator, BNIP3 were increased in the myocardium of doxorubicin-treated animals. As typically seen in atrophy, expression of myosin heavy chain (MHC) switched from the α to the β isoform. MuRF1 has been previously described as a mediator of cardiac atrophy in other mouse models [104]. Global MuRF1-deficient mice were resistant to doxorubicin-induced cardiomyopathy, they had preserved myocardial function and heart weight after anthracycline exposure.
Similarly, Xia et al. observed significant up-regulation of MuRF1 protein in myocardium of doxorubicin-treated mice, and proposed that the forkhead box family of transcription factors (FOXO) mediate myocardial atrophy through transcriptional activation of MuRF1 [105]. Administration of selective FOXO1 inhibitor AS1842856 to mice treated with doxorubicin prevented anthracycline-induced decline in ejection fraction and LV mass. Additionally, anthracycline-induced reduction in cardiomyocyte cross-sectional area was attenuated by AS1842856. Treatment with FOXO1 inhibitor suppressed doxorubicin-induced MuRF1 expression, suggesting that FOXO1-mediated transcription of MuRF1 is a potential mechanism of anthracycline-induced cardiac atrophy. Further studies are needed to confirm involvement of these molecular mechanisms of anthracycline toxicity in humans.
3.1.3. Fibrosis.
Cardiac interstitial fibrosis has been reported in both animal and human studies of doxorubicin cardiotoxicity and may be related to oxidative stress. ROS generation is a driving force in transforming-growth-factor beta (TGF-ß) stimulation, that leads to activation of activin-linked kinase (ALK). ALK phosphorylates Smad2 and 3 signal transducers, which then bind Smad4 and induce a translocation to the nucleus where it activates transcriptional regulation [106]. TGF-ß upregulation was described with physiological tissue repair as well as with pathological cardiac fibrosis [107,108]. NOX4 was shown to activate TGF-ß, thereby inducing the differentiation from fibroblasts to myofibroblasts [109]. Doxorubicin administration increased pathological remodeling through NOX2-dependent mechanisms [43]. Doxorubicin upregulated the expression of TGF-ß/Smad3, induced myofibroblast differentiation, and induced collagen-1 deposition [110].
Sirtuins belong to the family of nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylases that promote DNA repair, and induce signaling that is pro-survival, stress-adaptive, and anti-aging. Sirt1 is highly expressed in cardiomyocytes, shuttles between the cytoplasm and the nucleus, and its activation by resveratrol was shown to reduce anthracycline-induced fibrosis, and protected from doxorubicin-induced cardiac dysfunction [110]. Sirt3, is localized to mitochondria, and adenovirus-mediated overexpression of Sirt3 restored mitochondrial respiratory chain defects thereby protecting from anthracycline-induced cell death [111]. Pillai et al. report that Sirt3 overexpression protected mitochondria from doxorubicin-induced DNA damage [112]. Because Sirt1 and Sirt3 are highly expressed in cardiac myocytes, Sirt1 and Sirt3 activation during anthracycline therapy may mitigate anthracycline-induced cardiotoxic effects [113]. Signaling involved in the regulation of pro-survival and anti-aging genes may be altered by anthracyclines, including Sirt [32].
Histopathologic animal studies have demonstrated the progression of fibrosis after anthracycline therapy and its correlation with CMR-derived T1 values and derived-extracellular volume (ECV) measures [114]. A daunorubicin-induced rabbit heart failure model showed ultrastructural myocardial changes due to reduced mitochondrial size and number as well as increase caveolae (membrane lipid rafts) formation [115]. Fibrosis was unchanged after 3 weeks of daunorubicin administration but after daunorubicin-induced heart failure at 9 weeks post treatment, increased fibrosis, activated beta-1 integrin, and elevated membrane repair protein MG53 were found. A number of studies in anthracycline-exposed human cancer populations have demonstrated increased CMR-based measures of ECV, and that these parameters may have prognostic relevance [116–119]. However, it is possible that the increase in ECV fraction in human cancer survivors may be related to cardiomyocyte loss or atrophy rather than the progression of fibrosis [101,119,120]. As stated above, histopathologic findings in cancer survivors with significant heart failure have suggested that interstitial and replacement fibrosis may be an important pathologic finding in patients exposed to anthracyclines, however it is unclear whether cardiac fibrosis is a late finding that manifests primarily after the development of end-stage heart failure [97,121,122].
3.2. Organ-level manifestations.
3.2.1. Contractile Dysfunction.
Systolic cardiac dysfunction and heart failure is the most clinically important manifestation of anthracycline cardiotoxicity. Both acute and chronic systolic dysfunction may occur after anthracycline therapy [123–125]. Acutely, contractile dysfunction may result from multiple causes including mitochondrial dysfunction, altered calcium handling, and titin degradation. In the medium to long-term, the reduced number of contractile units due to cardiomyocyte and myofibrillar loss contribute to contractile dysfunction. In addition, myocardial wall thinning resulting in increased systolic wall stress, and secondary stressors such as hypertension may further exacerbate systolic dysfunction [125,126]. Systolic dysfunction and heart failure with reduced ejection fraction may manifest during and early after anthracycline chemotherapy, and/or in long-term survivorship [125–129]. Rates of systolic dysfunction have been reported to be 9-17% during and shortly after therapy [10,130]. There may be some degree of improvement seen after cessation of anthracycline therapy, though systolic function may not recover to baseline levels [128]. In long-term survivors of childhood cancer, progressive incidence of heart failure over time leads to an average of ~9% of childhood cancer survivors developing heart failure by the age of 50 years [131]. Due to the dose dependence of anthracycline cardiotoxicity, heart failure rates are higher with higher dose exposure [132]. The development of heart failure after anthracycline therapy portends a poor prognosis [12,133].
3.2.2. Vascular Dysfunction.
In addition to cardiomyocytes, anthracycline chemotherapy has direct toxic effects on the vasculature (Figure 4). The vascular wall contains both an intimal layer of endothelial cells and medial smooth muscle cells. Endothelial cells serve multiple functions including regulation of vascular tone, the passage of fluid and solutes, angiogenesis, and inflammatory responses, and smooth muscle cells cooperate with the endothelium to regulate vascular tone and blood pressure [134–136]. Although anthracyclines may cause vascular smooth muscle cell toxicity and autophagy, particularly at high doses and with repetitive and long-term treatment, the vascular endothelium is more sensitive to their effects [61,137,138]. The direct endotheliotoxic effects of anthracyclines result in reduced responsiveness to endothelium-dependent vasodilators such as acetylcholine and are distinct from the indirect vascular dysfunction caused by doxorubicin-induced nephrosis, which may cause impairment in bradykinin-mediated coronary artery dilation and enhanced vasoconstrictor response of the aorta to angiotensin II [139,140].
Similar to their effects on cardiomyocytes, anthracyclines cause vascular endothelial toxicity through multiple mechanisms including topoisomerase II-related DNA damage, and ROS and RNS generation, with particular importance of eNOS and associated effects on endothelial mitochondria [141]. The oxidative and nitrosative stresses within the mitochondria cause reduced ATP formation and glutathione activity, mitochondrial dysfunction, impedance of the mitochondrial membrane potential, swelling, increased permeability, and release of cytochrome C into the cytosol, potentially resulting in caspase activation-mediated endothelial cell apoptosis, which is independent of Fas/FasL [58,62,142]. Cells that remain viable after anthracycline exposure show evidence of senescence, reduced proliferative ability, reduced metabolic activity, and impaired function [143]. These effects are associated with endothelial dysfunction, including reduced endothelial-dependent vascular dilation, increased permeability, impaired wound closure, reduced migratory capacity, altered angiogenic potential and reduced vascular density [57,59,142,143].
Vascular disease is relevant to the clinical course of cancer survivors treated with anthracycline therapy. Several studies have shown that aortic stiffness increases shortly after anthracycline exposure, and there are both short- and longer-term deficiencies in brachial artery flow mediated dilation [59,144–149]. Moreover, ventricular-vascular interactions may contribute to the development of heart failure. Altered ventricular-arterial coupling has been demonstrated early after doxorubicin therapy in breast cancer patients and is predictive of subsequent cardiac dysfunction [128,150]. Arterial hypertension is highly prevalent among long-term childhood cancer survivors and is an important risk factor for anthracycline-related heart failure [126,151]. However, an association between anthracycline exposure and the development of hypertension in cancer survivors has not been established [151,152].
4. Clinical implications.
In this final section we aim to link the described molecular mechanisms of anthracycline-induced cardiotoxicity to currently available clinical data on risk assessment, prevention and treatment of toxicity (Table 1). Here we also seek to describe how underlying genetic polymorphisms affect these molecular mechanisms, and lead to alteration of individual susceptibility to anthracyclines.
Table 1.
Cardioprotective strategies and their proposed mechanisms.
| Cardioprotective strategies | Proposed mechanisms |
|---|---|
| Dexrazoxane | • Inhibits formation of anthracydine-DNA-Top2β cleavable complexes and prevents DNA damage |
| Angiotensin-converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ARB) | • Afterload reduction • Improved endothelial function • Antioxidant properties • Improvement of intracellular Ca++ handling |
| Beta blockers | • Normalization of β1 adrenergic receptor expression and sensitivity • Improvement of intracellular Ca++ handling • Antioxidant properties (carvedilol) |
| HMG-CoA reductase inhibitors (statins) | • Rho GTPase Rac1 inhibition • Antioxidant properties • Anti-inflammatory effect |
| Exercise | • Antioxidant properties • Improvement of intracellular Ca++ handling • Inhibition of apoptotic signaling • Increased NO production • Improvement in mobilization and homing of cardiac progenitor cells |
4.1. Topoisomerase 2β.
Top2β expression in peripheral blood was proposed as a biomarker of susceptibility to anthracycline-induced cardiotoxicity. Top2β levels in 21 patients who had evidence of cardiomyopathy with low doses of doxorubicin (< 250 mg/m2) were significantly higher than among 15 patients who had preserved cardiac function despite exposure to high doses of doxorubicin (450 mg/m2) [153]. This finding suggested that patients with higher level of Top2β expression might be more susceptible to anthracycline cardiotoxicity.
Retinoic acid receptors (RARG) bind to DNA regulatory sequences and regulate gene expression in response to their agonist all-trans retinoic acid (ATRA) [154]. RARG has been shown to bind to the Top2β promoter, and Top2β expression in cell culture was significantly decreased when human RARG was added, and this effect was further exacerbated with the addition of ATRA [155]. A genome-wide association study among children treated with anthracyclines revealed that a RARGS427L variant of retinoic acid receptor gamma (RARG) had a strong association with anthracycline-induced cardiomyopathy [155]. RARGS427L variant did not repress Top2β expression as effectively as wild type RARG, therefore, leading to higher levels of Top2β in cardiac cells, predisposing them to anthracycline-induced cardiotoxicity.
Dexrazoxane is a Top2β inhibitor that is approved for use for the prevention of anthracycline-induced cardiomyopathy in certain specific cancer populations. Initial interest in studying dexrazoxane and related substances was fueled by their in vitro antineoplastic effects [156]. But subsequent series of preclinical studies revealed that dexrazoxane has the potential to prevent anthracycline-related cardiotoxicity [157]. Its cardioprotective effect was later confirmed in a number of clinical trials [158,159]. Initially, the protective mechanism of dexrazoxane had been attributed to iron chelation. However, that hypothesis remains controversial, as other iron chelators (deferasirox) do not offer protection against doxorubicin-induced cardiotoxicity [160]. It is more widely accepted that dexrazoxane prevents doxorubicin-induced DNA damage via two potential mechanisms (Figure 1) [160]. Dexrazoxane forms a complex with free Top2β, stabilizing its closed-clamp conformation, therefore, preventing Top2β binding to chromosomal DNA. As a result, doxorubicin is unable to bring Top2β into the DNA cleavage complexes. Additionally, dexrazoxane can bind to DNA-bound Top2β and trigger its proteasomal degradation [160]. This process results in fewer doxorubicin-Top2β DNA cleavage complexes. Meta-analysis of 8 placebo-controlled trials of 1358 adult patients showed that dexrazoxane significantly reduced the risk of anthracycline-induced cardiotoxicity [161]. Most importantly, dexrazoxane use was associated with significantly reduced risk of clinical heart failure, with pooled OR of 0.12 (95% CrI 0.06 – 0.23) [161]. It is important to mention that a majority of clinical trials showing a benefit of dexrazoxane in adult patients focused on subjects receiving very high doses of anthracycline (over 400 mg/m2 of doxorubicin). A meta-analysis of dexrazoxane randomized controlled trials in pediatric populations demonstrated a benefit in preventing subclinical cardiotoxicity, represented by asymptomatic LV function declines and adverse remodeling [162]. Although an initial signal suggesting that increased secondary malignancies may occur in pediatric patients receiving dexrazoxane, subsequent studies and a meta-analysis did not find this association [162]. Currently, the FDA restricts dexrazoxane use to metastatic breast cancer patients, who have received more than 300 mg/m2 of doxorubicin, and require further therapy with doxorubicin [163]. Though the FDA has not approved its use in children, it has designated dexrazoxane as an orphan drug for the prevention of cardiomyopathy in children treated with anthracyclines, allowing its clinical use in pediatric patients within the United States [164]. The European Medicines Agency has also lifted its dexrazoxane contraindication to children under the age of 18 years who are intended to receive at least 300 mg/m2 of doxorubicin equivalents [165].
4.2. ROS production and mitochondrial dysfunction.
Wang et al. identified a common SNP, rs2232228, in the hyaluronan synthase 3 (HAS3) gene that modifies the risk of anthracycline-induced cardiomyopathy [166]. HAS3 gene encodes hyaluronan, which plays an active role in tissue remodeling, and reduces ROS-induced cardiac injury. Among individuals with rs2232228 GG genotype, cardiomyopathy was infrequent and not dose-dependent. However, in individuals exposed to high-dose (> 250 mg/m2) anthracyclines, the rs2232228 AA genotype was associated with 8.9 times higher risk of cardiomyopathy compared with the GG genotype. Relative HAS3 mRNA levels measured in healthy hearts tended to be lower among individuals with AA compared with GA genotype. The authors hypothesized that higher risk of cardiomyopathy associated with AA genotype could be due to inadequate protection of the myocardium from ROS-mediated injury and/or inadequate remodeling following high dose anthracycline exposure [166].
Iron-dependent ROS production.
The hypothesis of iron-dependent ROS production as a mechanism of anthracycline-related cardiotoxicity has been challenged by a number of studies that did not prove effectiveness of iron chelators in preventing anthracycline-induced cardiomyopathy [167]. However, there is an association between mutations predisposing to iron-overload, and susceptibility to doxorubicin damage supports this mechanism of toxicity [168]. The most common gene affected in humans with hereditary hemochromatosis is HFE: most affected individuals carry at least one copy of C282Y gene mutation. Genetic testing of pediatric patients treated with doxorubicin for acute lymphoblastic leukemia revealed that C282Y HFE carriers had higher incidence of myocardial injury during doxorubicin therapy, detected by troponin elevation [168]. Additionally, children with C282Y and/or H63D HFE variants had significantly lower LV function, mass, and wall thickness 2 years after diagnosis [168].
NOX-dependent ROS production.
Genotyping of adult non-Hodgkin lymphoma patients revealed that anthracycline-induced heart failure was associated with a variant of the NOX subunit NCF4, which is responsible for downregulation of the enzyme [45]· This association between NOX variants and risk of cardiotoxicity was replicated in a number of other studies [169,170], suggesting that increased NOX activity could predispose to anthracycline-induce cardiomyopathy through increased ROS production. Cascales et al. correlated histopathological findings with NOX polymorphisms, and revealed that certain variants protected against focal myocardial necrosis, whereas others were strongly associated with cardiac fibrosis [171].
Mitochondrial dysfunction.
Enough evidence indicates that the effects on mitochondrial function and biogenesis are one of the key manifestations of doxorubicin toxicity [79]. Cardiac mitochondria are affected by anthracycline therapy via multiple mechanisms, including irreversible mtDNA breaks, anthracycline interaction with NADH, and increased ROS production. Ghrelin is a signaling peptide that prevented impairment of mitochondrial bioenergetics and reduced mitochondrial pathways of apoptosis in cultured cardiac myocytes and mice following doxorubicin exposure [172,173]. The mitochondria-specific antioxidant, MitoQ, was shown to be protective against vascular dysfunction in mice [61]. However, the translational value of these findings remains unclear, as it is recommended that cancer patients avoid antioxidant medications and supplements during cancer treatments due to their potential to interfere with cancer therapies. A recent study found that antioxidant supplementation among participants was associated with worse breast cancer outcomes [174]. These findings limit the potential for antioxidants to be beneficial in the prevention of anthracycline-induced vascular toxicity.
The role of mitochondrial dysfunction in the pathogenesis of anthracycline-induced cardiomyopathy is underlined by the finding of an association between a missense variant of the electron transfer flavoprotein beta subunit (ETFB) gene and the risk of cardiomyopathy in anthracycline treated patients [175]. ETFB is involved in mitochondrial β-oxidation and ATP production [175]. Deciphering how to protect solely cardiac mitochondria, but not mitochondria residing in cancer cells, would enable us to design novel preventive therapies to reduce anthracycline-cardiotoxicity.
4.3. Neurohormonal antagonists.
Angiotensin-converting enzyme inhibitors (ACEI)/angiotensin receptor blockers (ARBs) and ß-blockers are the first line therapies in the treatment of anthracycline-related cardiac dysfunction and heart failure, based on broader heart failure treatment guidelines, not specific to cardio-oncology populations [176]. Afterload reduction is the primary target of ACEI/ARBs, though pre-clinical studies also showed that ACEIs have an antioxidant effect [177,178], and protect against doxorubicin-induced abnormal calcium handling [179]. Blockade of adrenergic overstimulation and heart rate reduction are the primary targets of ß-blockers, however they also have antioxidant properties and may reverse maladaptive effects on Ca2+ signaling [180]. Pre-clinical studies have demonstrated that cardiomyocyte ß1AR expression and responsiveness decrease due to doxorubicin-indued oxidative stress, and these effects may be prevented by carvedilol [181].
One large observational study found that most patients who received early ACEI and beta-blocker therapy for anthracycline-induced cardiac dysfunction had successful recovery of LVEF [182]. However, these results have not been widely replicated. In survivorship, anthracycline-related heart failure is associated with a poor prognosis despite standard HF therapy [12,133]. ACEI/ARBs and carvedilol have all demonstrated some benefit in primary prevention trials [182–186]. However, these agents are not widely used for the primary prevention of cardiotoxicity during therapy with anthracyclines due to their relatively modest benefit in these trials, which is outweighed by potential side effects.
4.4. HMG-CoA reductase inhibitors (statins).
Retrospective studies indicate that statin use is associated with reduced risk of anthracycline-induced heart failure [187,188]. A small prospective trial showed a modest effect on EF preservation in atorvastatin-treated cohort, when compared to a placebo group [189]. Statins exhibit pleiotropic anti-inflammatory and anti-oxidative effects linked to indirect inhibition of small Ras homologous (Rho) GTPases. Rho GTPase Rac1 has been shown to be a major factor in the regulation of the NOX, and Top2 [190]. Therefore, Rac1 inhibition by statins is a proposed mechanism that could explain their cardioprotective effect with anthracycline use, however further studies are needed to confirm this hypothesis. PREVENT (Preventing Anthracycline Cardiovascular Toxicity With Statins) is a randomized controlled clinical trial that is currently under way, and it aims to evaluate atorvastatin effect on LVEF preservation in 250 anthracycline-treated breast cancer patients [191].
4.5. Cell transport and hepatic metabolism of anthracyclines.
Besides the genes controlling Top2β activity, and redox pathways, there are also a number of genetic variants that affect cell transport and hepatic metabolism of anthracyclines that have been associated with anthracycline-induced cardiotoxicity. UDP-glucuronosyltransferase family 1A, isoform 6 (UGT1A6) is involved in hepatic anthracycline glucuronidation pathway [192,193]. UGT1A6 variants (rs17863783, V209V) with reduced enzymatic activity lead to accumulation of toxic anthracycline metabolites. They were found to be associated with an increased risk of anthracycline-induced cardiomyopathy in pediatric patient cohorts.
A number of studies identified an association between anthracycline-induced cardiotoxicity and polymorphisms in ABCC genes [194–196]. Human ABCC genes encode membrane-bound glycoprotein that is almost ubiquitously expressed in different cell types, and primarily involved in energy-dependent transport of cytotoxic agents out of the cell [197]. Genetic variants of the solute carrier (SLC) transporter SLC28A3 (rs7853758, rs885004) were also associated with anthracycline-induced cardiotoxicity in three independent pediatric cohorts [192,198]. Canadian Pharmacogenomics Network for Drug Safety (CPNDS) recommendations suggest routine screening for RARG rs2229774, SLC28A3 rs7853758 and UGT1A6*4 rs17863783 variants in pediatric patients requiring anthracycline-based chemotherapy [199].
4.6. Exercise training.
Anthracycline chemotherapy is associated with decreased functional capacity measured as the peak oxygen uptake during exercise (peak VO2)[200]. Exercise training has been proposed as an effective and easily accessible cardioprotective strategy among patients undergoing anthracycline chemotherapy [201]. Meta-analysis of 48 randomized controlled trials of exercise training among cancer patients showed improvement in peak VO2 with exercise therapy [202]. Costello et all. found that there is an improvement in CMR-based global longitudinal strain among patients undergoing exercise training while receiving anthracycline chemotherapy [203]. Exercise training has been shown to affect many of the pathways implicated in anthracycline-induced cardiomyopathy (for review see [200]). Exercise blunts ROS production from cardiac mitochondria in murine models, and also significantly increases expression of antioxidant enzymes such as glutathione peroxidase 1, catalase, and manganese superoxide dismutase in cardiac tissue [204]. Exercise reduced levels of the TRPC3-NOX2 complex, that was shown to correlate with the severity of anthracycline-induced cardiac atrophy [46]. Physical exercise in rodents reduces doxorubicin-induced p53 expression and prevents cardiomyocyte apoptosis [205], improves intracellular Ca2+ handling, and stimulates mobilization and homing of cardiac progenitor cells [200].
The growing body of evidence supporting the beneficial effects of exercise in preventing anthracycline-induced cardiotoxicity makes it an attractive potential modality for primary prevention. A recent statement from American Heart Association proposed the development of a comprehensive cardio-oncology rehabilitation model that will use the multimodality approach of cardiac rehabilitation for prevention of cardiovascular events in patients at high risk for chemotherapy-related cardiotoxicity [201].
5. Future directions.
As detailed in this review, there are a number of active areas of investigation to further elucidate the mechanisms of anthracycline cardiovascular toxicity and their relevance to clinical management. We seek to highlight a few interesting areas of future research.
Dosing, time-course, and validation in human studies.
A majority of the described studies investigating mechanisms of anthracycline cardiotoxicity utilized cell culture and small animal models. However, doses of doxorubicin used in these studies are frequently above the typical doses utilized in clinical practice.
Additionally, studies in human induced pluripotent stem cell derived cardiomyocytes indicate that mechanisms of toxicity are dose dependent, as different molecular pathways are involved with different anthracycline doses [206]. In addition, these studies have generally focused on acute effects of doxorubicin therapy, with findings that are not necessarily applicable to the cardiovascular health of long-term human survivors of cancer. This discrepancy highlights that basic science studies should attempt to examine dosing that is comparable to clinical use and should seek to utilize animal models of chronic effects of anthracycline therapy.
Computational modeling.
Multiple specific effects of anthracyclines at the biochemical, cellular, and tissue levels play roles in the development of cardiovascular toxicity. However, there is a limited understanding of the complex interplay between different mechanisms, and their relative significance. Computational modeling of anthracycline-induced toxicity could offer a unique in silico platform for exploring the complex interactions between multiple pathogenetic mechanisms [207].
Primary prevention studies.
Robust clinical studies in primary prevention seeking to alter the pharmacokinetics and cardiotoxic specificity of anthracyclines such as liposomal anthracycline formulations, dexrazoxane, and exercise training are needed. A notable limitation in prior primary prevention studies with neurohormonal antagonist therapy is that relatively low risk patients were enrolled. The incidence of cardiomyopathy was low even in the placebo groups, making it challenging to demonstrate any cardioprotective effect in the treatment groups. In future primary prevention trials, targeted enrollment of individuals at higher risk for anthracycline-induced cardiomyopathy should be considered, focusing on individuals with pre-existing cardiovascular risk factors and cardiac disease. It is also unknown whether inherited forms of cardiomyopathies (dilated, hypertrophic cardiomyopathy) predispose to anthracycline-induced heart failure.
Risk prediction and secondary prevention.
Secondary prevention has been limited by late diagnosis of disease, at a time at which prognosis has been suboptimal. Improved risk prediction strategies are needed, which may include multi-marker analyses of clinical factors, imaging, and biomarkers to better predict an individual’s propensity to develop heart failure to guide therapy. In addition, a growing body of evidence indicates that genetic polymorphisms may determine individual susceptibility to anthracycline cardiotoxicities. However, studies of genetic predisposition have generally been derived from relatively small patient cohorts, often restricted to a certain geographic region. Therefore, additional studies among larger and more diverse patient populations are needed before these findings are generalized and applied in clinical practice. The ability to predict an individual’s risk for anthracycline-induced cardiotoxicity is one of the ultimate goals in clinical practice; and incorporating genetic factors into the models for risk assessment could improve the accuracy of prediction [193].
6. Conclusion.
Anthracycline-related cardiovascular toxicities are an important public health concern relevant to a substantial number of cancer patients both during therapy and in survivorship. Anthracyclines cause cardiovascular toxicities through multiple molecular mechanisms, with important clinical implications. Through further basic and translational research a more comprehensive understanding of the molecular mechanisms of the cardiotoxic effects of anthracyclines is necessary to inform the development of innovative strategies for the prevention and treatment of anthracycline-induced cardiomyopathy.
Funding Sources:
Dr. Narayan was supported by Padres Pedal the Cause/RADY #PTC2020 and UC San Diego Moore Cancer Center, Specialized Cancer Control Support Grant NIH/NCI P30CA023100. Dr. E. Zemljic-Harpf was funded by a VMRF Pilot Project Award, and UC San Diego ANES Research Seed Award.
Conflict of interest:
Dr. Zemljic-Harpf receives funding from the Merck Investigator Studies Program and Scientific Engagements.
References.
- 1.McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM and Yellon DM (2017) Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther 31, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mulrooney DA, Hyun G, Ness KK, Ehrhardt MJ, Yasui Y, Duprez D, Howell RM, Leisenring WM, Constine LS, Tonorezos E, et al. (2020) Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: report from the Childhood Cancer Survivor Study cohort. The BMJ 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf.
- 4.Giordano SH, Lin Y-L, Kuo YF, Hortobagyi GN and Goodwin JS (2012) Decline in the Use of Anthracyclines for Breast Cancer. J. Clin. Oncol 30, 2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armstrong GT, Liu Q, Yasui Y, Neglia JP, Leisenring W, Robison LL and Mertens AC (2009) Late Mortality Among 5-Year Survivors of Childhood Cancer: A Summary From the Childhood Cancer Survivor Study. J. Clin. Oncol 27, 2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bates JE, Howell RM, Liu Q, Yasui Y, Mulrooney DA, Dhakal S, Smith SA, Leisenring WM, Indelicato DJ, Gibson TM, et al. (2019) Therapy-Related Cardiac Risk in Childhood Cancer Survivors: An Analysis of the Childhood Cancer Survivor Study. J. Clin. Oncol., American Society of Clinical Oncology 37, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patnaik JL, Byers T, DiGuiseppi C, Dabelea D. and Denberg TD (2011) Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: a retrospective cohort study. Breast Cancer Res. BCR 13, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armenian SH, Sun C-L, Shannon T, Mills G, Francisco L, Venkataraman K, Wong FL, Forman SJ and Bhatia S. (2011) Incidence and predictors of congestive heart failure after autologous hematopoietic cell transplantation. Blood 118, 6023–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco JG, Sun C-L, Landier W, Chen L, Esparza-Duran D, Leisenring W, Mays A, Friedman DL, Ginsberg JP, Hudson MM, et al. (2012) Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes--a report from the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 30, 1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, Civelli M, Lamantia G, Colombo N, Curigliano G, et al. (2015) Early Detection of Anthracycline Cardiotoxicity and Improvement With Heart Failure Therapy. Circulation CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 11.Cox CL, Rai SN, Rosenthal D, Phipps S. and Hudson MM (2008) Subclinical late cardiac toxicity in childhood cancer survivors: impact on self-reported health. Cancer 112, 1835–1844. [DOI] [PubMed] [Google Scholar]
- 12.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL and Kasper EK (2000) Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med 342, 1077–1084. [DOI] [PubMed] [Google Scholar]
- 13.Getz KD, Sung L, Ky B, Gerbing RB, Leger KJ, Leahy AB, Sack L, Woods WG, Alonzo T, Gamis A, et al. (2019) Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 37, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF and Yeh ETH (2012) Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med 18, 1639–1642. [DOI] [PubMed] [Google Scholar]
- 15.Depew RE, Liu L-F and Wang JC (1978) Interaction between DNA and Escherichia coli protein omega. Formation of a complex between single-stranded DNA and omega protein. J. Biol. Chem 253, 511–518. [PubMed] [Google Scholar]
- 16.Gellert M, Mizuuchi K, O’Dea MH and Nash HA (1976) DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci 73, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adachi N, Miyaike M, Kato S, Kanamaru R, Koyama H. and Kikuchi A. (1997) Cellular distribution of mammalian DNA topoisomerase II is determined by its catalytically dispensable C-terminal domain. Nucleic Acids Res. 25, 3135–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake FH, Zimmerman JP, McCabe FL, Bartus HF, Per SR, Sullivan DM, Ross WE, Mattern MR, Johnson RK and Crooke ST (1987) Purification of topoisomerase II from amsacrine-resistant P388 leukemia cells. Evidence for two forms of the enzyme. J. Biol. Chem 262, 16739–16747. [PubMed] [Google Scholar]
- 19.Capranico G, Tinelli S, Austin CA, Fisher ML and Zunino F. (1992) Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta BBA-Gene Struct. Expr 1132, 43–48. [DOI] [PubMed] [Google Scholar]
- 20.Taagepera S, Rao PN, Drake FH and Gorbsky GJ (1993) DNA topoisomerase II alpha is the major chromosome protein recognized by the mitotic phosphoprotein antibody MPM-2. Proc. Natl. Acad. Sci 90, 8407–8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakaguchi A. and Kikuchi A. (2004) Functional compatibility between isoform α and β of type II DNA topoisomerase. J. Cell Sci 117, 1047–1054. [DOI] [PubMed] [Google Scholar]
- 22.Zini N, Santi S, Ognibene A, Bavelloni A, Neri LM, Valmori A, Mariani E, Negri C, Astaldi-Ricotti GC and Maraldi NM (1994) Discrete localization of different DNA topoisomerases in HeLa and K562 cell nuclei and subnuclear fractions. Exp. Cell Res 210, 336–348. [DOI] [PubMed] [Google Scholar]
- 23.Kimura K, Saijo M, Ui M. and Enomoto T. (1994) Growth state-and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J. Biol. Chem 269, 1173–1176. [PubMed] [Google Scholar]
- 24.Zunino F. and Capranico G. (1990) DNA topoisomerase II as the primary target of anti-tumor anthracyclines. Anticancer. Drug Des 5, 307–317. [PubMed] [Google Scholar]
- 25.Bodley A, Liu LF, Israel M, Seshadri R, Koseki Y, Giuliani FC, Kirschenbaum S, Silber R. and Potmesil M. (1989) DNA topoisomerase II-mediated interaction of doxorubicin and daunorubicin congeners with DNA. Cancer Res. 49, 5969–5978. [PubMed] [Google Scholar]
- 26.Marinello J, Delcuratolo M. and Capranico G. (2018) Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci 19, 3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Low RL, Orton S. and Friedman DB (2003) A truncated form of DNA topoisomerase IIβ associates with the mtDNA genome in mammalian mitochondria. Eur. J. Biochem 270, 4173–4186. [DOI] [PubMed] [Google Scholar]
- 28.Goffart S, Hangas A. and Pohjoismäki JLO (2019) Twist and Turn-Topoisomerase Functions in Mitochondrial DNA Maintenance. Int. J. Mol. Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashley N. and Poulton J. (2009) Mitochondrial DNA is a direct target of anti-cancer anthracycline drugs. Biochem. Biophys. Res. Commun 378, 450–455. [DOI] [PubMed] [Google Scholar]
- 30.Hixon SC, Ellis CN and Daugherty JP (1981) Heart mitochondrial DNA synthesis: Preferential inhibition by adriamycin. J. Mol. Cell. Cardiol 13, 855–860. [DOI] [PubMed] [Google Scholar]
- 31.Lebrecht D. and Walker UA (2007) Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc. Toxicol 7, 108–113. [DOI] [PubMed] [Google Scholar]
- 32.Cappetta D, De Angelis A, Sapio L, Prezioso L, Illiano M, Quaini F, Rossi F, Berrino L, Naviglio S. and Urbanek K. (2017) Oxidative stress and cellular response to doxorubicin: a common factor in the complex milieu of anthracycline cardiotoxicity. Oxid. Med. Cell. Longev 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Songbo M, Lang H, Xinyong C, Bin X, Ping Z. and Liang S. (2019) Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol Lett 307, 41–48. [DOI] [PubMed] [Google Scholar]
- 34.Lenneman CG and Sawyer DB (2016) Cardio-Oncology: An Update on Cardiotoxicity of Cancer-Related Treatment. Circ Res 118, 1008–20. [DOI] [PubMed] [Google Scholar]
- 35.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M. and Voipio-Pulkki L-M (2000) Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 60, 1789–1792. [PubMed] [Google Scholar]
- 36.Page E. and McCallister LP (1973) Quantitative electron microscopic description of heart muscle cells: application to normal, hypertrophied and thyroxin-stimulated hearts. Am. J. Cardiol 31, 172–181. [DOI] [PubMed] [Google Scholar]
- 37.Weiss RG, Gerstenblith G. and Bottomley PA (2005) ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci 102, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies KJ and Doroshow JH (1986) Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem, American Society for Biochemistry and Molecular Biology 261, 3060–3067. [PubMed] [Google Scholar]
- 39.Thayer WS (1977) Adriamycin stimulated superoxide formation in submitochondrial particles. Chem. Biol. Interact 19, 265–278. [DOI] [PubMed] [Google Scholar]
- 40.Doroshow JH, Locker GY and Myers CE (1980) Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J. Clin. Invest 65, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panday A, Sahoo MK, Osorio D. and Batra S. (2015) NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol 12, 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prosser BL, Ward CW and Lederer WJ (2011) X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ and Grieve DJ (2010) Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 70, 9287–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghigo A, Li M. and Hirsch E. (2016) New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim. Biophys. Acta BBA-Mol. Cell Res 1863, 1916–1925. [DOI] [PubMed] [Google Scholar]
- 45.Leszek Wojnowski, Bettina Kulle, Markus Schirmer, Gregor Schlüter, Albrecht Schmidt, Albert Rosenberger, Stefan Vonhof, Heike Bickeböller, Toliat Mohammad Reza Suk Eun-Kyung, et al. (2005) NAD(P)H Oxidase and Multidrug Resistance Protein Genetic Polymorphisms Are Associated With Doxorubicin-Induced Cardiotoxicity. Circulation, American Heart Association 112, 3754–3762. [DOI] [PubMed] [Google Scholar]
- 46.Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, Hoka S, Ide T, Koitabashi N, Uchida K, et al. TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitajima N, Numaga-Tomita T, Watanabe M, Kuroda T, Nishimura A, Miyano K, Yasuda S, Kuwahara K, Sato Y, Ide T, et al. (2016) TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep, Nature Publishing Group 6, 37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishiyama K, Numaga-Tomita T, Fujimoto Y, Tanaka T, Toyama C, Nishimura A, Yamashita T, Matsunaga N, Koyanagi S, Azuma Y-T, et al. (2019) Ibudilast attenuates doxorubicin-induced cytotoxicity by suppressing formation of TRPC3 channel and NADPH oxidase 2 protein complexes. Br. J. Pharmacol 176, 3723–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Prasad SVN, Mutharasan RK, Naik TJ and Ardehali H. (2014) Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest, American Society for Clinical Investigation 124, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu X, Persson HL and Richardson DR (2005) Molecular Pharmacology of the Interaction of Anthracyclines with Iron. Mol. Pharmacol, American Society for Pharmacology and Experimental Therapeutics 68, 261–271. [DOI] [PubMed] [Google Scholar]
- 51.Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, Bertrand R, Andrews NC and Santos MM (2003) Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood, American Society of Hematology 102, 2574–2580. [DOI] [PubMed] [Google Scholar]
- 52.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. (2017) Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A 116, 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mukhopadhyay P, Rajesh M, Bátkai S, Kashiwaya Y, Haskó G, Liaudet L, Szabó C. and Pacher P. (2009) Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol 296, H1466–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vásquez-Vivar J, Martasek P, Hogg N, Masters BSS, Pritchard KA and Kalyanaraman B. (1997) Endothelial Nitric Oxide Synthase-Dependent Superoxide Generation from Adriamycin †. Biochemistry 36, 11293–11297. [DOI] [PubMed] [Google Scholar]
- 57.Kalivendi SV, Kotamraju S, Zhao H, Joseph J. and Kalyanaraman B. (2001) Doxorubicin-induced Apoptosis Is Associated with Increased Transcription of Endothelial Nitric-oxide Synthase: EFFECT OF ANTIAPOPTOTIC ANTIOXIDANTS AND CALCIUM. J. Biol. Chem 276, 47266–47276. [DOI] [PubMed] [Google Scholar]
- 58.Wu S. (2002) Adriamycin-induced Cardiomyocyte and Endothelial Cell Apoptosis: In Vitro and In Vivo Studies. J. Mol. Cell. Cardiol 34, 1595–1607. [DOI] [PubMed] [Google Scholar]
- 59.Duquaine D, Hirsch GA, Chakrabarti A, Han Z, Kehrer C, Brook R, Joseph J, Schott A, Kalyanaraman B, Vasquez-Vivar J, et al. (2003) Rapid-onset endothelial dysfunction with adriamycin: evidence for a dysfunctional nitric oxide synthase. Vasc. Med 8, 101–107. [DOI] [PubMed] [Google Scholar]
- 60.Finkelman BS, Putt M, Wang T, Wang L, Narayan H, Domchek S, DeMichele A, Fox K, Matro J, Shah P, et al. (2017) Arginine-Nitric Oxide Metabolites and Cardiac Dysfunction in Patients With Breast Cancer. J. Am. Coll. Cardiol 70, 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clayton ZS, Brunt VE, Hutton DA, VanDongen NS, D’Alessandro A, Reisz JA, Ziemba BP and Seals DR (2020) Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC CardioOncology 2, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, Yin D, Huang Q. and He M. (2020) Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front. Pharmacol 10, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Räsänen M, Degerman J, Nissinen TA, Miinalainen I, Kerkelä R, Siltanen A, Backman JT, Mervaala E, Hulmi JJ, Kivelä R, et al. (2016) VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc. Natl. Acad. Sci. U. S. A 113, 13144–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin Z, Zhao Y, Li H, Yan M, Zhou L, Chen C. and Wang DW (2016) miR-320a mediates doxorubicin-induced cardiotoxicity by targeting VEGF signal pathway. Aging 8, 192–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X, Gu J, Zhang Y, Feng S, Huang X, Jiang Y, Xia Y, Liu Y. and Yang X. (2019) l-arginine alleviates doxorubicin-induced endothelium-dependent dysfunction by promoting nitric oxide generation and inhibiting apoptosis. Toxicology 423, 105–111. [DOI] [PubMed] [Google Scholar]
- 66.Glembotski CC (2007) Endoplasmic reticulum stress in the heart. Circ Res 101, 975–84. [DOI] [PubMed] [Google Scholar]
- 67.Zhang K. (2010) Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int. J. Clin. Exp. Med 3, 33. [PMC free article] [PubMed] [Google Scholar]
- 68.Pahl HL and Baeuerle PA (1996) Activation of NF-κB by ER stress requires both Ca2+ and reactive oxygen intermediates as messengers. FEBS Lett 392, 129–136. [DOI] [PubMed] [Google Scholar]
- 69.Fu Hai Ying Sanada Shoji, Takashi Matsuzaki, Yulin Liao, Keiji Okuda, Masaki Yamato, Shota Tsuchida, Ryo Araki, Yoshihiro Asano, Hiroshi Asanuma, et al. (2016) Chemical Endoplasmic Reticulum Chaperone Alleviates Doxorubicin-Induced Cardiac Dysfunction. Circ. Res, American Heart Association 118, 798–809. [DOI] [PubMed] [Google Scholar]
- 70.Halperin L, Jung J. and Michalak M. (2014) The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 66, 318–326. [DOI] [PubMed] [Google Scholar]
- 71.Zuo S, Kong D, Wang C, Liu J, Wang Y, Wan Q, Yan S, Zhang J, Tang J, Zhang Q, et al. (2018) CRTH2 promotes endoplasmic reticulum stress-induced cardiomyocyte apoptosis through m-calpain. EMBO Mol. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li J, Ni M, Lee B, Barron E, Hinton DR and Lee AS (2008) The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 15, 1460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dirks-Naylor AJ (2013) The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 93, 913–916. [PubMed] [Google Scholar]
- 74.Koleini N and Kardami E (2017) Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 8, 46663–46680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al. (2016) Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 133, 1668–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu H, Yu W, Sun S, Li C, Zhang Y and Ren J (2020) Luteolin Attenuates Doxorubicin-Induced Cardiotoxicity Through Promoting Mitochondrial Autophagy. Front. Physiol. 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rizzuto R, De Stefani D, Raffaello A and Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol, Nature Publishing Group 13, 566–578. [DOI] [PubMed] [Google Scholar]
- 78.Ikeda S, Matsushima S, Okabe K, Ikeda M, Ishikita A, Tadokoro T, Enzan N, Yamamoto T, Sada M, Deguchi H, et al. (2019) Blockade of L-type Ca 2+ channel attenuates doxorubicin-induced cardiomyopathy via suppression of CaMKII-NF-κB pathway. Sci. Rep, Nature Publishing Group 9, 9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallace Kendall B, Sardão Vilma A, and Oliveira Paulo J (2020) Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res, American Heart Association 126, 926–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R and Sawyer DB (2004) Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J. Biol. Chem 279, 8290–8299. [DOI] [PubMed] [Google Scholar]
- 81.L’Ecuyer T, Sanjeev S, Thomas R, Novak R, Das L, Campbell W and Heide RV (2006) DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am. J. Physiol.-Heart Circ. Physiol, American Physiological Society 291, H1273–H1280. [DOI] [PubMed] [Google Scholar]
- 82.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W and Field LJ (2009) Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shizukuda Y, Matoba S, Mian OY, Nguyen T and Hwang PM (2005) Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol. Cell. Biochem 273, 25–32. [DOI] [PubMed] [Google Scholar]
- 84.Zhu W, Zhang W, Shou W and Field LJ (2014) P53 inhibition exacerbates late-stage anthracycline cardiotoxicity. Cardiovasc. Res 103, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klampfer L (2006) Signal transducers and activators of transcription (STATs): Novel targets of chemopreventive and chemotherapeutic drugs. Curr. Cancer Drug Targets 6, 107–121. [DOI] [PubMed] [Google Scholar]
- 86.Wegrzyn J, Potla R, Chwae Y-J, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M and Gornicka A (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, Okabe M, Kishimoto T and Yamauchi-Takihara K (2000) Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci 97, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu J, Guo W, Lin SZ, Wang ZJ, Kan JT, Chen SY and Zhu YZ (2016) Gp130-mediated STAT3 activation by S-propargyl-cysteine, an endogenous hydrogen sulfide initiator, prevents doxorubicin-induced cardiotoxicity. Cell Death Dis. 7, e2339–e2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frias MA, Somers S, Gerber-Wicht C, Opie LH, Lecour S and Lang U (2008) The PGE2-Stat3 interaction in doxorubicin-induced myocardial apoptosis. Cardiovasc. Res 80, 69–77. [DOI] [PubMed] [Google Scholar]
- 90.Li J, Wang P-Y, Long NA, Zhuang J, Springer DA, Zou J, Lin Y, Bleck CKE, Park J-H, Kang J-G, et al. (2019) p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc. Natl. Acad. Sci. U. S. A 116, 19626–19634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B and Prabhu SD (2011) Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc. Res 89, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gordon JW, Shaw JA and Kirshenbaum LA (2011) Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ. Res 108, 1122–1132. [DOI] [PubMed] [Google Scholar]
- 93.Li D, Li J, An Y, Yang Y and Zhang SQ (2013) Doxorubicin-induced apoptosis in H9c2 cardiomyocytes by NF-κB dependent PUMA upregulation. Eur Rev Med Pharmacol Sci 17, 2323–2329. [PubMed] [Google Scholar]
- 94.Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J and Kalyanaraman B (2002) Activation of nuclear factor-κB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem. J 367, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olson HM, Young DM, Prieur DJ, LeRoy AF and Reagan RL (1974) Electrolyte and morphologic alterations of myocardium in adriamycin-treated rabbits. Am. J. Pathol 77, 439–454. [PMC free article] [PubMed] [Google Scholar]
- 96.Bristow MR, Mason JW, Billingham ME and Daniels JR (1978) Doxorubicin cardiomyopathy: evaluation by phonocardiography, endomyocardial biopsy, and cardiac catheterization. Ann. Intern. Med 88, 168–175. [DOI] [PubMed] [Google Scholar]
- 97.Bernaba BN, Chan JB, Lai CK and Fishbein MC (2010) Pathology of late-onset anthracycline cardiomyopathy. Cardiovasc. Pathol 19, 308–311. [DOI] [PubMed] [Google Scholar]
- 98.Lefrak EA, Pitha J, Rosenheim S and Gottlieb JA (1973) A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 32, 302–314. [DOI] [PubMed] [Google Scholar]
- 99.Jordan JH, Castellino SM, Meléndez GC, Klepin HD, Ellis LR, Lamar Z, Vasu S, Kitzman DW, Ntim WO, Brubaker PH, et al. (2018) Left Ventricular Mass Change After Anthracycline Chemotherapy. Circ. Heart Fail 11, e004560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Neilan TG, Coelho-Filho OR, Pena-Herrera D, Shah RV, Jerosch-Herold M, Francis SA, Moslehi J and Kwong RY (2012) Left Ventricular Mass in Patients with a Cardiomyopathy after Treatment with Anthracyclines. Am. J. Cardiol 110, 1679–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.de Souza TF, Silva TQAC, Costa FO, Shah R, Neilan TG, Velloso L, Nadruz W, Brenelli F, Sposito AC, Matos-Souza JR, et al. (2018) Anthracycline Therapy Is Associated With Cardiomyocyte Atrophy and Preclinical Manifestations of Heart Disease. JACC Cardiovasc. Imaging 11, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coelho-Filho OR, Shah RV, Mitchell R, Neilan TG, Moreno H, Simonson B, Kwong R, Rosenzweig A, Das S and Jerosch-Herold M (2013) Quantification of Cardiomyocyte Hypertrophy by Cardiac Magnetic Resonance: Implications on Early Cardiac Remodeling. Circulation 128, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Willis MS, Parry TL, Brown DI, Mota RI, Huang W, Beak JY, Sola M, Zhou C, Sean T, Hicks ST, C. Caughey MC, D’agostino RB Jr, Jordan JH, Hundley WG, Jensen BC. (2019) Doxorubicin exposure causes subacute cardiac atrophy dependent upon the striated muscle-specific ubiquitin ligase Muscle Ring Finger-1. Circ Heart Fail. 12:e005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Willis MS, Rojas M, Li L, Selzman CH, Tang R-H, Stansfield WE, Rodriguez JE, Glass DJ and Patterson C (2009) Muscle ring finger 1 mediates cardiac atrophy in vivo. Am. J. Physiol. - Heart Circ. Physiol 296, H997–H1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xia P, Chen J, Liu Y, Fletcher M, Jensen BC and Cheng Z (2020) Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J. Biol. Chem 295, 4265–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leask A (2015) Getting to the heart of the matter: new insights into cardiac fibrosis. Circ. Res 116, 1269–1276. [DOI] [PubMed] [Google Scholar]
- 107.Leask A and Abraham DJ (2004) TGF-β signaling and the fibrotic response. FASEB J. 18, 816–827. [DOI] [PubMed] [Google Scholar]
- 108.Kane CJ, Hebda PA, Mansbridge JN and Hanawalt PC (1991) Direct evidence for spatial and temporal regulation of transforming growth factor β1 expression during cutaneous wound healing. J. Cell. Physiol 148, 157–173. [DOI] [PubMed] [Google Scholar]
- 109.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S and Sorescu D (2005) NAD (P) H oxidase 4 mediates transforming growth factor-β1–induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res 97, 900–907. [DOI] [PubMed] [Google Scholar]
- 110.Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, Berrino L, Rossi F, De Angelis A and Urbanek K (2016) SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int. J. Cardiol 205, 99–110. [DOI] [PubMed] [Google Scholar]
- 111.Du Q, Zhu B, Zhai Q and Yu B (2017) Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am. J. Transl. Res 9, 3360. [PMC free article] [PubMed] [Google Scholar]
- 112.Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S and Gupta MP (2016) Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol.-Heart Circ. Physiol 310, H962–H972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dolinsky VW (2017) The role of sirtuins in mitochondrial function and doxorubicin-induced cardiac dysfunction. Biol. Chem 398, 955–974. [DOI] [PubMed] [Google Scholar]
- 114.Hong YJ, Park HS, Park JK, Han K, Park CH, Kim TK, Yoo SJ, Lee JY, Kim PK, Hur J, et al. (2017) Early Detection and Serial Monitoring of Anthracycline-Induced Cardiotoxicity Using T1-mapping Cardiac Magnetic Resonance Imaging: An Animal Study. Sci. Rep 7, 2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ichikawa Y, Zemljic-Harpf AE, Zhang Z, McKirnan MD, Manso AM, Ross RS, Hammond HK, Patel HH and Roth DM (2017) Modulation of caveolins, integrins and plasma membrane repair proteins in anthracycline-induced heart failure in rabbits. PLoS One 12, e0177660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jordan JH, D’Agostino RB Jr, Hamilton CA, Vasu S, Hall ME, Kitzman DW, Thohan V, Lawrence JA, Ellis LR and Lash TL (2014) Longitudinal assessment of concurrent changes in left ventricular ejection fraction and left ventricular myocardial tissue characteristics after administration of cardiotoxic chemotherapies using T1-weighted and T2-weighted cardiovascular magnetic resonance. Circ. Cardiovasc. Imaging 7, 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jordan JH, Vasu S, Morgan TM, D’Agostino RB, Meléndez GC, Hamilton CA, Arai AE, Liu S, Liu C-Y, Lima JAC, et al. (2016) Anthracycline-Associated T1 Mapping Characteristics Are Elevated Independent of the Presence of Cardiovascular Comorbidities in Cancer Survivors. Circ. Cardiovasc. Imaging 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tham EB, Haykowsky MJ, Chow K, Spavor M, Kaneko S, Khoo NS, Pagano JJ, Mackie AS and Thompson RB (2013) Diffuse myocardial fibrosis by T1-mapping in children with subclinical anthracycline cardiotoxicity: relationship to exercise capacity, cumulative dose and remodeling. J. Cardiovasc. Magn. Reson 15, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Muehlberg F, Funk S, Zange L, von Knobelsdorff-Brenkenhoff F, Blaszczyk E, Schulz A, Ghani S, Reichardt A, Reichardt P and Schulz-Menger J (2018) Native myocardial T1 time can predict development of subsequent anthracycline-induced cardiomyopathy: Native T1 time can predict anthracycline-induced cardiomyopathy. ESC Heart Fail. 5, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hundley WG and Jordan JH (2018) When Left Ventricular Extracellular Volume Fraction Changes After Anthracyclines. JACC Cardiovasc. Imaging 11, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE and Sanders SP (1991) Late Cardiac Effects of Doxorubicin Therapy for Acute Lymphoblastic Leukemia in Childhood. N. Engl. J. Med 324, 808–815. [DOI] [PubMed] [Google Scholar]
- 122.Berry GJ and Jorden M (2005) Pathology of radiation and anthracycline cardiotoxicity. Pediatr. Blood Cancer 44, 630–637. [DOI] [PubMed] [Google Scholar]
- 123.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M and Voipio-Pulkki L-M Acute Doxorubicin Cardiotoxicity Involves Cardiomyocyte Apoptosis 5. [PubMed] [Google Scholar]
- 124.Ganame J, Claus P, Eyskens B, Uyttebroeck A, Renard M, D’hooge J, Gewillig M, Bijnens B, Sutherland GR and Mertens L (2007) Acute Cardiac Functional and Morphological Changes After Anthracycline Infusions in Children. Am. J. Cardiol 99, 974–977. [DOI] [PubMed] [Google Scholar]
- 125.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD and Colan SD (2004) Chronic Progressive Cardiac Dysfunction Years After Doxorubicin Therapy for Childhood Acute Lymphoblastic Leukemia. J. Clin. Oncol 23, 2629–2636. [DOI] [PubMed] [Google Scholar]
- 126.Armstrong GT, Oeffinger KC, Chen Y, Kawashima T, Yasui Y, Leisenring W, Stovall M, Chow EJ, Sklar CA, Mulrooney DA, et al. (2013) Modifiable Risk Factors and Major Cardiac Events Among Adult Survivors of Childhood Cancer. J. Clin. Oncol 31, 3673–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, Civelli M, Lamantia G, Colombo N, Curigliano G, et al. (2015) Early Detection of Anthracycline Cardiotoxicity and Improvement With Heart Failure Therapy. Circulation CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 128.Narayan HK, Finkelman B, French B, Plappert T, Hyman D, Smith AM, Margulies KB and Ky B (2017) Detailed Echocardiographic Phenotyping in Breast Cancer Patients: Associations With Ejection Fraction Decline, Recovery, and Heart Failure Symptoms Over 3 Years of Follow-Up. Circulation 135, 1397–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Getz KD, Sung L, Ky B, Gerbing RB, Leger KJ, Leahy AB, Sack L, Woods WG, Alonzo T, Gamis A, et al. (2019) Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children’s Oncology Group. J. Clin. Oncol 37, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla J-P, Sami A, Glaspy J, Juhos E, Wardley A, Fornander T, et al. (2013) Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 14, 72–80. [DOI] [PubMed] [Google Scholar]
- 131.Chow EJ, Chen Y, Kremer LC, Breslow NE, Hudson MM, Armstrong GT, Border WL, Feijen EAM, Green DM, Meacham LR, et al. (2015) Individual Prediction of Heart Failure Among Childhood Cancer Survivors. J. Clin. Oncol 33, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bates JE, Howell RM, Liu Q, Yasui Y, Mulrooney DA, Dhakal S, Smith SA, Leisenring WM, Indelicato DJ, Gibson TM, et al. (2019) Therapy-Related Cardiac Risk in Childhood Cancer Survivors: An Analysis of the Childhood Cancer Survivor Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 37, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lipshultz SE, Lipsitz SR, Sallan SE, Simbre VC, Shaikh SL, Mone SM, Gelber RD and Colan SD (2002) Long-Term Enalapril Therapy for Left Ventricular Dysfunction in Doxorubicin-Treated Survivors of Childhood Cancer. J. Clin. Oncol, American Society of Clinical Oncology 20, 4517–4522. [DOI] [PubMed] [Google Scholar]
- 134.Pries AR and Kuebler WM (2006) Normal Endothelium. In The Vascular Endothelium I (Moncada S, and Higgs A, eds.), pp 1–40, Springer Berlin Heidelberg, Berlin, Heidelberg. [Google Scholar]
- 135.Soultati A (2012) Endothelial vascular toxicity from chemotherapeutic agents: Preclinical evidence and clinical implications. Cancer Treat. Rev 11. [DOI] [PubMed] [Google Scholar]
- 136.Lilly B (2014) We Have Contact: Endothelial Cell-Smooth Muscle Cell Interactions 29, 8. [DOI] [PubMed] [Google Scholar]
- 137.Murata T, Yamawaki H, Yoshimoto R, Hori M, Sato K, Ozaki H and Karaki H (2001) Chronic effect of doxorubicin on vascular endothelium assessed by organ culture study. Life Sci. 69, 2685–2695. [DOI] [PubMed] [Google Scholar]
- 138.Murata T, Yamawaki H, Hori M, Sato K, Ozaki H and Karaki H (2001) Chronic vascular toxicity of doxorubicin in an organ-cultured artery. Br. J. Pharmacol 132, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ito M, Oguri M, Naruse A, Ito H, Suzuki Y, Satake N and Shibata S (1991) Impaired endothelium-dependent relaxation in isolated thoracic aorta of rats with daunomycin-induced nephrosis. J. Pharmacol. Exp. Ther 258, 388–395. [PubMed] [Google Scholar]
- 140.Ulu N, Buikema H, Gilst W. H. v. and Navis G (2008) Vascular dysfunction in adriamycin nephrosis: different effects of adriamycin exposure and nephrosis. Nephrol. Dial. Transplant 23, 1854–1860. [DOI] [PubMed] [Google Scholar]
- 141.Damrot J, Nübel T, Epe B, Roos WP, Kaina B and Fritz G (2006) Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br. J. Pharmacol 149, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wolf MB and Baynes JW (2006) The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim. Biophys. Acta BBA - Gen. Subj 1760, 267–271. [DOI] [PubMed] [Google Scholar]
- 143.Eakin AJ, Mc Erlain T, Burke A, Eaton A, Tipping N, Allocca G and Branco CM (2020) Circulating Levels of Epirubicin Cause Endothelial Senescence While Compromising Metabolic Activity and Vascular Function. Front. Cell Dev. Biol 8, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jordan JH, Castellino SM, Meléndez GC, Klepin HD, Ellis LR, Lamar Z, Vasu S, Kitzman DW, Ntim WO, Brubaker PH, et al. (2018) Left Ventricular Mass Change After Anthracycline Chemotherapy. Circ. Heart Fail 11, e004560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chaosuwannakit N, D’Agostino R, Hamilton CA, Lane KS, Ntim WO, Lawrence J, Melin SA, Ellis LR, Torti FM, Little WC, et al. (2010) Aortic Stiffness Increases Upon Receipt of Anthracycline Chemotherapy. J. Clin. Oncol 28, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grover S, Lou PW, Bradbrook C, Cheong K, Kotasek D, Leong DP, Koczwara B and Selvanayagam JB (2015) Early and late changes in markers of aortic stiffness with breast cancer therapy: Chemotherapy-related aortic remodelling. Intern. Med. J 45, 140–147. [DOI] [PubMed] [Google Scholar]
- 147.Drafts BC, Twomley KM, D’Agostino R, Lawrence J, Avis N, Ellis LR, Thohan V, Jordan J, Melin SA, Torti FM, et al. (2013) Low to Moderate Dose Anthracycline-Based Chemotherapy Is Associated With Early Noninvasive Imaging Evidence of Subclinical Cardiovascular Disease. JACC Cardiovasc. Imaging 6, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nagy L, Szabó F, Iványi J, Németh L, ScD GLK, Palatka J, Tarján J and Tóth K (2001) A method for detection of doxorubicin-induced cardiotoxicity: Flow-mediated vasodilation of the brachial artery 6, 6. [PMC free article] [PubMed] [Google Scholar]
- 149.Okur A, Karadeniz C, Özhan Oktar S, Pınarlı FG, Aral A and Oğuz A (2016) Assessment of brachial artery reactivity, carotid intima-media thickness, and adhesion molecules in pediatric solid tumor patients treated with anthracyclines. Pediatr. Hematol. Oncol 33, 178–185. [DOI] [PubMed] [Google Scholar]
- 150.Narayan HK, French B, Khan AM, Plappert T, Hyman D, Bajulaiye A, Domchek S, DeMichele A, Clark A, Matro J, et al. (2016) Noninvasive Measures of Ventricular-Arterial Coupling and Circumferential Strain Predict Cancer Therapeutics-Related Cardiac Dysfunction. JACC Cardiovasc. Imaging 9, 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gibson TM, Li Z, Green DM, Armstrong GT, Mulrooney DA, Srivastava D, Bhakta N, Ness KK, Hudson MM and Robison LL (2017) Blood Pressure Status in Adult Survivors of Childhood Cancer: A Report from the St. Jude Lifetime Cohort Study. Cancer Epidemiol. Biomarkers Prev 26, 1705–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koelwyn GJ, Lewis NC, Ellard SL, Jones LW, Gelinas JC, Rolf JD, Melzer B, Thomas SM, Douglas PS, Khouri MG, et al. (2016) Ventricular-Arterial Coupling in Breast Cancer Patients After Treatment With Anthracycline-Containing Adjuvant Chemotherapy. The Oncologist 21, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pimprapa Vejpongsa, Massey Mona R, Acholonu Sandra A, Zhang Sui, and Yeh Edward T. (2013) Abstract 11619: Topoisomerase 2ß Expression in Peripheral Blood Predicts Susceptibility to Anthracycline-Induced Cardiomyopathy. Circulation, American Heart Association 128, A11619–A11619. [Google Scholar]
- 154.Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 10, 940–954. [PubMed] [Google Scholar]
- 155.The Canadian Pharmacogenomics Network for Drug Safety Consortium, Aminkeng F, Bhavsar AP, Visscher H, Rassekh SR, Li Y, Lee JW, Brunham LR, Caron HN, van Dalen EC, et al. (2015) A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat. Genet 47, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Langer SW (2014) Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag. Res 6, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Herman E, Ardalan B, Bier C, Waravdekar V and Krop S (1979) Reduction of daunorubicin lethality and myocardial cellular alterations by pretreatment with ICRF-187 in Syrian golden hamsters. Cancer Treat. Rep 63, 89–92. [PubMed] [Google Scholar]
- 158.Lopez M, Vici P, Di Lauro K, Conti F, Paoletti G, Ferraironi A, Sciuto R, Giannarelli D and Maini CL (1998) Randomized prospective clinical trial of high-dose epirubicin and dexrazoxane in patients with advanced breast cancer and soft tissue sarcomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 16, 86–92. [DOI] [PubMed] [Google Scholar]
- 159.Marty M, Espié M, Llombart A, Monnier A, Rapoport BL, Stahalova V, and Dexrazoxane Study Group. (2006) Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 17, 614–622. [DOI] [PubMed] [Google Scholar]
- 160.Lyu YL, Kerrigan JE, Lin C-P, Azarova AM, Tsai Y-C, Ban Y and Liu LF (2007) Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 67, 8839–8846. [DOI] [PubMed] [Google Scholar]
- 161.Abdel-Qadir H, Ong G, Fazelzad R, Amir E, Lee DS, Thavendiranathan P and Tomlinson G (2017) Interventions for preventing cardiomyopathy due to anthracyclines: a Bayesian network meta-analysis. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 28, 628–633. [DOI] [PubMed] [Google Scholar]
- 162.Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L and Nathan PC (2016) Cardioprotection and Second Malignant Neoplasms Associated With Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst 108. [DOI] [PubMed] [Google Scholar]
- 163.ZINECARD® (dexrazoxane) for injection package insert. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020212s017lbl.pdf13.
- 164.(2020, December 16) Orphan Drug Designations and Approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=441314.
- 165.(2018, September 17) Cardioxane; European Medicines Agency: https://www.ema.europa.eu/en/medicines/human/referrals/cardioxane. Eur. Med. Agency, Text. [Google Scholar]
- 166.Wang X, Liu W, Sun C-L, Armenian SH, Hakonarson H, Hageman L, Ding Y, Landier W, Blanco JG, Chen L, et al. (2014) Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: a report from the children’s oncology group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 32, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rao VA, Zhang J, Klein SR, Espandiari P, Knapton A, Dickey JS, Herman E and Shacter EB (2011) The iron chelator Dp44mT inhibits the proliferation of cancer cells but fails to protect from doxorubicin-induced cardiotoxicity in spontaneously hypertensive rats. Cancer Chemother. Pharmacol 68, 1125–1134. [DOI] [PubMed] [Google Scholar]
- 168.Lipshultz SE, Lipsitz SR, Kutok JL, Miller TL, Colan SD, Neuberg DS, Stevenson KE, Fleming MD, Sallan SE, Franco VI, et al. (2013) Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer 119, 3555–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rossi D, Rasi S, Franceschetti S, Capello D, Castelli A, De Paoli L, Ramponi A, Chiappella A, Pogliani EM, Vitolo U, et al. (2009) Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia, Nature Publishing Group 23, 1118–1126. [DOI] [PubMed] [Google Scholar]
- 170.Reichwagen A, Ziepert M, Kreuz M, Gödtel-Armbrust U, Rixecker T, Poeschel V, Reza Toliat M, Nğrnberg P, Tzvetkov M, Deng S, et al. (2015) Association of NADPH oxidase polymorphisms with anthracycline-induced cardiotoxicity in the RICOVER-60 trial of patients with aggressive CD20+ B-cell lymphoma. Pharmacogenomics, Future Medicine 16, 361–372. [DOI] [PubMed] [Google Scholar]
- 171.Cascales A, Pastor-Quirante F, Sánchez-Vega B, Luengo-Gil G, Corral J, Ortuño-Pacheco G, Vicente V and Peña FA de la. (2013) Association of Anthracycline-Related Cardiac Histological Lesions With NADPH Oxidase Functional Polymorphisms. The Oncologist 18, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang X, Wang X-L, Chen H-L, Wu D, Chen J-X, Wang X-X, Li R-L, He J-H, Mo L, Cen X, et al. (2014) Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem. Pharmacol 88, 334–350. [DOI] [PubMed] [Google Scholar]
- 173.Xu Z, Lin S, Wu W, Tan H, Wang Z, Cheng C, Lu L and Zhang X (2008) Ghrelin prevents doxorubicin-induced cardiotoxicity through TNF-alpha/NF-κB pathways and mitochondrial protective mechanisms. Toxicology 247, 133–138. [DOI] [PubMed] [Google Scholar]
- 174.Ambrosone CB, Zirpoli GR, Hutson AD, McCann WE, McCann SE, Barlow WE, Kelly KM, Cannioto R, Sucheston-Campbell LE, Hershman DL, et al. (2020) Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients With Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J. Clin. Oncol 38, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ruiz-Pinto S, Pita G, Martín M, Alonso-Gordoa T, Barnes DR, Alonso MR, Herraez B, García-Miguel P, Alonso J, Pérez-Martínez A, et al. (2018) Exome array analysis identifies ETFB as a novel susceptibility gene for anthracycline-induced cardiotoxicity in cancer patients. Breast Cancer Res. Treat 167, 249–256. [DOI] [PubMed] [Google Scholar]
- 176.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. (2013) 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol 62, e147–e239. [DOI] [PubMed] [Google Scholar]
- 177.Cernecka H, Doka G, Srankova J, Pivackova L, Malikova E, Galkova K, Kyselovic J, Krenek P and Klimas J (2016) Ramipril restores PPARβ/δ and PPARγ expressions and reduces cardiac NADPH oxidase but fails to restore cardiac function and accompanied myosin heavy chain ratio shift in severe anthracycline-induced cardiomyopathy in rat. Eur. J. Pharmacol. 791, 244–253. [DOI] [PubMed] [Google Scholar]
- 178.El-Aziz MAA, Othman AI, Amer M and El-Missiry MA (2001) Potential protective role of angiotensin-converting enzyme inhibitors captopril and enalapril against adriamycin-induced acute cardiac and hepatic toxicity in rats. J. Appl. Toxicol 21, 469–473. [DOI] [PubMed] [Google Scholar]
- 179.Maeda A, Honda M, Kuramochi T, Tanaka K and Takabatake T (1997) An angiotensin-converting enzyme inhibitor protects against doxorubicin-induced impairment of calcium handling in neonatal rat cardiac myocytes. Clin. Exp. Pharmacol. Physiol 24, 720–726. [DOI] [PubMed] [Google Scholar]
- 180.Feuerstein GZ and Ruffolo RR (1995) Carvedilol, a novel multiple action antihypertensive agent with antioxidant activity and the potential for myocardial and vascular protection. Eur. Heart J, Oxford Academic 16, 38–42. [DOI] [PubMed] [Google Scholar]
- 181.Misun Park and Steinberg Susan F. (2018) Carvedilol Prevents Redox Inactivation of Cardiomyocyte β1-Adrenergic Receptors. JACC Basic Transl. Sci, American College of Cardiology Foundation 3, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Daniela Cardinale, Alessandro Colombo, Sandri Maria T., Lamantia Giuseppina, Colombo Nicola, Maurizio Civelli, Martinelli Giovanni, Veglia Fabrizio, Fiorentini Cesare, and Cipolla Carlo M. (2006) Prevention of High-Dose Chemotherapy-Induced Cardiotoxicity in High-Risk Patients by Angiotensin-Converting Enzyme Inhibition. Circulation, American Heart Association 114, 2474–2481. [DOI] [PubMed] [Google Scholar]
- 183.Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, Gravdehaug B, von Knobelsdorff-Brenkenhoff F, Bratland Å, Storås TH, et al. (2016) Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur. Heart J 37, 1671–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bosch X, Rovira M, Sitges M, Domènech A, Ortiz-Pérez JT, de Caralt TM, Morales-Ruiz M, Perea RJ, Monzó M and Esteve J (2013) Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (prevention of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive Chemotherapy for the treatment of Malignant hEmopathies). J. Am. Coll. Cardiol 61, 2355–2362. [DOI] [PubMed] [Google Scholar]
- 185.Nabati M, Janbabai G, Baghyari S, Esmaili K and Yazdani J (2017) Cardioprotective Effects of Carvedilol in Inhibiting Doxorubicin-induced Cardiotoxicity. J. Cardiovasc. Pharmacol 69, 279–285. [DOI] [PubMed] [Google Scholar]
- 186.Kalay N, Basar E, Ozdogru I, Er O, Cetinkaya Y, Dogan A, Inanc T, Oguzhan A, Eryol NK, Topsakal R, et al. (2006) Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J. Am. Coll. Cardiol 48, 2258–2262. [DOI] [PubMed] [Google Scholar]
- 187.Seicean S, Seicean A, Plana JC, Budd GT and Marwick TH (2012) Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J. Am. Coll. Cardiol 60, 2384–2390. [DOI] [PubMed] [Google Scholar]
- 188.David Bobrowski, Limei Zhou, Peter Austin, Arguelles Oscar Calvillo Amir Eitan, Douglas Lee, Paaladinesh Thavendiranathan, and Husam Abdel-Qadir. (2020) Statins are associated with lower risk of heart failure after anthracycline and trastuzumab chemotherapy for early stage breast cancer. J. Am. Coll. Cardiol, American College of Cardiology Foundation 75, 7–7. [Google Scholar]
- 189.Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, Meriç M and Ağaç MT (2011) Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J. Am. Coll. Cardiol 58, 988–989. [DOI] [PubMed] [Google Scholar]
- 190.Henninger C and Fritz G (2017) Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell Death Dis. 8, e2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Preventing Anthracycline Cardiovascular Toxicity With Statins (PREVENT) - Full Text View - ClinicalTrials.gov.
- 192.Visscher H, Ross CJD, Rassekh SR, Barhdadi A, Dubé M-P, Al-Saloos H, Sandor GS, Caron HN, van Dalen EC, Kremer LC, et al. (2011) Pharmacogenomic Prediction of Anthracycline-Induced Cardiotoxicity in Children. J. Clin. Oncol, American Society of Clinical Oncology 30, 1422–1428. [DOI] [PubMed] [Google Scholar]
- 193.Visscher H, Ross CJD, Rassekh SR, Sandor GSS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Rogers PC, Rieder MJ, et al. (2013) Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr. Blood Cancer 60, 1375–1381. [DOI] [PubMed] [Google Scholar]
- 194.Vulsteke C, Pfeil AM, Maggen C, Schwenkglenks M, Pettengell R, Szucs TD, Lambrechts D, Dieudonné A-S, Hatse S, Neven P, et al. (2015) Clinical and genetic risk factors for epirubicin-induced cardiac toxicity in early breast cancer patients. Breast Cancer Res. Treat 152, 67–76. [DOI] [PubMed] [Google Scholar]
- 195.Hertz DL, Caram MV, Kidwell KM, Thibert JN, Gersch C, Seewald NJ, Smerage J, Rubenfire M, Henry NL, Cooney KA, et al. (2016) Evidence for association of SNPs in ABCB1 and CBR3, but not RAC2, NCF4, SLC28A3 or TOP2B, with chronic cardiotoxicity in a cohort of breast cancer patients treated with anthracyclines. Pharmacogenomics, Future Medicine 17, 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Armenian SH, Ding Y, Mills G, Sun C, Venkataraman K, Wong FL, Neuhausen SL, Senitzer D, Wang S, Forman SJ, et al. (2013) Genetic Susceptibility to Anthracycline-Related Congestive Heart Failure in Survivors of Haematopoietic Cell Transplantation. Br. J. Haematol 163, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dean M, Rzhetsky A and Allikmets R (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166. [DOI] [PubMed] [Google Scholar]
- 198.Visscher H, Rassekh SR, Sandor GS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Rogers PC, Rieder MJ, Carleton BC, et al. (2015) Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 16, 1065–1076. [DOI] [PubMed] [Google Scholar]
- 199.Aminkeng F, Ross CJD, Rassekh SR, Hwang S, Rieder MJ, Bhavsar AP, Smith A, Sanatani S, Gelmon KA, Bernstein D, et al. (2016) Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharmacol 82, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Scott JM, Khakoo A, Mackey JR, Haykowsky MJ, Douglas PS and Jones LW (2011) Modulation of Anthracycline-induced Cardiotoxicity by Aerobic Exercise in Breast Cancer: Current Evidence and Underlying Mechanisms. Circulation 124, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gilchrist Susan C, Barac, Ades Philip A., Alfano Catherine M., Franklin Barry A., Jones Lee W., La Gerche Andre, Ligibel Jennifer A., Lopez Gabriel, Madan Kushal, et al. (2019) Cardio-Oncology Rehabilitation to Manage Cardiovascular Outcomes in Cancer Patients and Survivors: A Scientific Statement From the American Heart Association. Circulation, American Heart Association 139, e997–e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Scott JM, Zabor EC, Schwitzer E, Koelwyn GJ, Adams SC, Nilsen TS, Moskowitz CS, Matsoukas K, Iyengar NM, Dang CT, et al. (2018) Efficacy of Exercise Therapy on Cardiorespiratory Fitness in Patients With Cancer: A Systematic Review and Meta-Analysis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 36, 2297–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Costello Benedict T, Roberts Timothy J, Howden Erin J, Bigaran Ashley, Foulkes Steven J., Beaudry Rhys I., Janssens Kristel, Haykowsky Mark J., Antill Yoland, Nightingale Sophie, et al. (2019) Exercise Attenuates Cardiotoxicity of Anthracycline Chemotherapy Measured by Global Longitudinal Strain. JACC CardioOncology, American College of Cardiology Foundation 1, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kavazis AN, Smuder AJ, Min K, Türner N and Powers SK (2010) Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am. J. Physiol. - Heart Circ. Physiol 299, H1515–H1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Werner C, Hanhoun M, Widmann T, Kazakov A, Semenov A, Pöss J, Bauersachs J, Thum T, Pfreundschuh M, Müller P, et al. (2008) Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J. Am. Coll. Cardiol 52, 470–482. [DOI] [PubMed] [Google Scholar]
- 206.Burridge PW, Li YF, Matsa E, Wu H, Ong S-G, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, et al. (2016) Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med, Nature Publishing Group 22, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lewalle A, Land S and Niederer S (2017) Development of a Patient-Based Computational Modeling Framework for Analyzing the Mechanisms of Doxorubicin Cardiotoxicity. FASEB J. 31, lb713–lb713. [Google Scholar]