

Abstract
Purpose:
Individuals with urea cycle disorders (UCDs) may develop recurrent hyperammonemia, episodic encephalopathy, and neurological sequelae which can impact Health-related Quality of Life (HRQoL). To date, there have been no systematic studies of HRQoL in people with UCDs.
Methods:
We reviewed HRQoL and clinical data for 190 children and 203 adults enrolled in a multicenter UCD natural history study. Physical and psychosocial HRQoL in people with UCDs were compared to HRQoL in healthy people and people with phenylketonuria (PKU) and diabetes mellitus. We assessed relationships between HRQoL, UCD diagnosis, and disease severity. Finally, we calculated sample sizes required to detect changes in these HRQoL measures.
Results:
Individuals with UCDs demonstrated worse physical and psychosocial HRQoL than their healthy peers and peers with PKU and diabetes. In children, HRQoL scores did not differ by diagnosis or severity. In adults, individuals with decreased severity had worse psychosocial HRQoL. Finally, we show that a large number of individuals would be required in clinical trials to detect differences in HRQoL in UCDs.
Conclusion:
Individuals with UCDs have worse HRQoL compared to healthy individuals and those with PKU and diabetes. Future work should focus on the impact of liver transplantation and other clinical variables on HRQoL in UCDs.
INTRODUCTION
Urea cycle disorders (UCDs) are a group of inborn errors of metabolism (IEM) that have an estimated birth incidence of 1 in 35,000 in the United States.1 These disorders result from pathogenic variants in genes encoding one of six enzymes (i.e., N-acetylglutamate synthase [NAGS], carbamoyl phosphate synthetase 1 [CPS1], ornithine transcarbamylase [OTC], argininosuccinate synthetase 1 [ASS1], argininosuccinate lyase [ASL], arginase 1 [ARG1]) or two transporters (i.e., citrin or aspartate/glutamate carrier [CIT] and mitochondrial ornithine transporter 1 [ORNT1]) that are required for synthesis of urea.2 All UCDs are transmitted in autosomal recessive manner with the exception being OTC deficiency, the most common UCD, which is transmitted as an X-linked disorder. Disorders caused by the deficiency of urea cycle enzymes, i.e., NAGSD, CPS1D, OTCD, ASS1D, ASLD, and hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome caused by deficiency of ORNT1, are characterized by recurrent episodes of hyperammonemia that can cause significant neurological damage. The frequency and severity of the hyperammonemic episodes vary depending on the site of the block in ureagenesis and the degree of residual function of affected proteins.2 In addition to hyperammonemia-induced neurocognitive deficits, people with UCDs can also present with involvement of other organ systems. For example, individuals with OTCD may present with acute liver failure; and individuals with ASLD can present with chronic liver disease, cognitive impairment, and hypertension even in the absence of hyperammonemia.3–7 Individuals with ARG1 deficiency (ARG1D) typically present with neurological sequelae including progressive spasticity and movement disorders, though episodic hyperammonemia has been reported in a subset of individuals with this disorder.8 Citrin deficiency (CTLN2) is associated with a range of phenotypes including neonatal cholestasis, faltering growth, dyslipidemia, and episodic hyperammonemia with neuropsychiatric symptoms.9,10 The manifestations and sequelae of UCDs can confer significant morbidity to affected individuals and their families. Many individuals with UCDs develop intellectual and developmental disabilities (IDD). While the prevalence and degree of IDD vary depending on the type of UCD, disease severity, and access to care, a recent systematic review found that in some subtypes, IDD has been reported in up to two-thirds of affected individuals.12 In children, IDD, physical limitations, and behavioral abnormalities can affect learning and social functioning. In adolescents and adults, IDD and other chronic complications of UCDs can negatively impact the transition to independent living and integration into society. Furthermore, the mainstays of therapy for most UCDs, including protein-restricted diet, medical formula, amino acid supplementation, and nitrogen-scavenging medications, while effective in decreasing the frequency and severity of hyperammonemic episodes, can be burdensome and have a negative effect on the psychological health of parents of children with UCDs.13
Despite the recognition of the burden of UCDs, to date, only a few studies have systematically investigated the impact of UCDs on health-related quality of life (HRQoL). A study of 21 children with IEM in France that included 6 children with UCDs found that all participants reported lower general well-being than children with leukemia. In this sample, clinical factors that impacted HRQoL included presence of neurological disorders and types of feeding modalities.14 A study conducted in Japan found greater fatigue and worse scores on other aspects of HRQoL in 55 children and young adults with CTLN2 than healthy controls.15 European children with organic acidemias (n=100) and UCDs (n=52) were shown to experience higher rates of behavioral and emotional problems and worse physical HRQoL compared to controls. However, in this study, scores in children with UCDs were within 0.5 SD of the population norm.16 HRQoL in adults with UCDs has not yet been systematically analyzed.
This study reports HRQoL in a large cohort of children and adults with UCDs who were enrolled in an observational, longitudinal study conducted by the National Institutes of Health’s Rare Diseases Clinical Research Network’s (RDCRN) Urea Cycle Disorders Consortium (UCDC).17 HRQoL was assessed using two patient-reported outcome measures (PROMs): the Pediatric Quality of Life Inventory Generic Core Scales (PedsQL) for children, and the 36-item Short Form Survey version 2 (SF-36v2) for adults. This study represents the largest systematic investigation of HRQoL in children and adults with UCDs to date.
MATERIALS AND METHODS
Study Details
Data were collected from participants enrolled in the Longitudinal Study of Urea Cycle Disorders (NCT00237315) conducted by the UCDC.17 The UCDC consists of 13 clinical sites in the United States and three international clinical sites (one in Canada and two in Europe). Data were collected across all sites in accordance with a manual of operations and stored using online case report forms. Data management was performed by the Data Management and Coordinating Center of the RDCRN.
Individuals with diagnoses of NAGSD, CPS1D, ASS1D, ASLD, ARG1D, HHH syndrome, and CTLN2 based on previously published specific diagnostic criteria were enrolled in the study.18 Individuals with a diagnosis of an organic acidemia, lysinuric protein intolerance, mitochondrial disorder, congenital lactic acidemia, fatty acid oxidation defect, primary or secondary liver disease, those with rare and unrelated serious comorbidities (e.g., Down syndrome or a severe intraventricular hemorrhage in the newborn period), and those with extremely low birth weight (<1500g) were excluded from enrollment. Between 2006 and 2015, the study procedures were approved by the respective Institutional Review Boards (IRB) for each participating clinical site. From 2015 onward, the IRB at Children’s National Medical Center served as the IRB of record for all sites within the United States (US) with all participating US sites being under a reliance agreement with this central IRB. Informed consent was obtained from all participants or their parents or legal guardians.
Study Data
From the central database hosting all UCDC data, the following data were collected: age in years at enrollment, sex assigned at birth (male/female), UCD diagnosis (NAGSD, CPS1D, OTCD, ASS1D, ASLD, ARG1D, HHH, or CTLN2), PedsQL 4.0 generic core scale scores (both child-report and parent-report) for children 2-18 years, and SF-36v2 scale scores for adults. For this analysis, only data points that were collected from the baseline study visit, within one year of the enrollment visit, were extracted. As the disease course, treatment, and treatment-related burden are different for individuals who have undergone orthotopic liver transplantation (OLT) compared to those who have not, only responses to the PedsQL 4.0 and SF-36v2 from participants who had not undergone OLT prior to baseline enrollment visit were analyzed. Supplemental Figure 1 shows the inclusion and exclusion process.
The PedsQL 4.0 generic core scales (hereafter referred to as PedsQL) assess patient- or parent-reported HRQoL in the physical, emotional, social, and school domains.19 It is available for child-report in ages 5 -17 years, and for parent-report on behalf of the child ages ≥2 years. Scores range from 0-100 for each scale, with higher scores representing better HRQoL. The Physical Functioning scale is also used as the Physical Health Summary Score. Emotional, Social, and School Functioning scale scores are averaged to yield the Psychosocial Health Summary Score. The Total Scale Score results from averaging the responses across all answered items.
The SF-36v2 is a HRQoL measure designed for administration to adults. It consists of 36 items assessing eight health concepts, or scales, including 1) Limitations in physical activities due to health status; 2) Limitations in social activities due to physical or emotional problems; 3) Limitations in usual role activities due to physical health; 4) Bodily pain; 5) Mental health; 6) Limitations in usual role activities due to emotional problems; 7) Energy and fatigue; and 8) General health perceptions.20 Item responses are recoded linearly to 0-100 and z-scores for each scale are multiplied by factor scoring coefficients to yield the Physical Component Summary (PCS), which is an overall measure of physical HRQoL, and the Mental Component Summary (MCS), which is an overall measure of mental HRQoL. The PCS and MCS are represented as norm-based scores with a mean of 50 and standard deviation of 10 in the general US population.20,21
SF-36v2 and the PedsQL scores from individuals with UCD in our cohort were compared to scores from the literature in healthy children,19 general population adults,21 adults and children with phenylketonuria (PKU),22 children with type 1 and type 2 diabetes mellitus (T1 and 2 DM),23 and adults with type 1 diabetes (T1 DM).24 PKU and diabetes mellitus were chosen as comparison groups as these disorders also require a lifetime of dietary therapy and treatment. For healthy children, data from a 2001 study were used.19 For PKU, data from a 2015 study were used.22 For diabetes mellitus, data from two studies, conducted in 2003 and 2019, were used.23,24
Statistical Analyses
Comparisons between mean scores from PedsQL and SF-36v2 in individuals with UCDs, and published data from healthy individuals and individuals with PKU, type 1 diabetes mellitus (DM), and type 2 DM were performed using pairwise t-tests with a Bonferroni correction for the number of comparisons made. Comparisons among the various subtypes of UCDs were performed using an ANOVA with pairwise Bonferroni corrections; for this analysis, all subtypes of UCDs other than OTCD, ASS1D, and ASLD were grouped into one category, i.e., rare UCDs, as the numbers with individual subtypes were low. To evaluate the effect of clinical severity on HRQoL, we compared symptomatic vs asymptomatic individuals. Symptomatic was defined in the manual of operations as having: 1) at least one episode of hyperammonemia (NH3 ≥100 μmol/liter) associated with clinical symptoms; or 2) two of the following eight criteria: a) vomiting ≥ once monthly, b) protein intolerance, c) episodic lethargy, d) developmental or intellectual disability requiring special education or care, e) abnormal neurological examination (hypotonia, spasticity, hyperreflexia, or clonus), f) brain edema, g) ≥1 migraine headache per month, or h) episodic psychosis.18 Parent-report and child-report PedsQL Physical and Psychosocial Health Summary scores were compared using Pearson product-moment correlation and paired t-test. Comparisons of HRQoL in different child age groups was performed using a linear regression. From a clinical trial readiness standpoint, using the means and standard deviations from the Summary Scores, we estimated the sample sizes that would be required in a parallel group design to detect a mean difference of 5 and 10 in the PedsQL and SF-36v2 Summary scores. For this analysis, a two-sided, two-sample, equal-variance t-test was used to achieve a power of 0.8 to reject the null hypothesis of equal means with significance level (alpha) of 0.05. The sample size estimations were performed using PASS.25
RESULTS
HRQoL in UCDs
Table 1 provides the numbers and ages of participants on whom PedsQL responses were available in the UCDC. A total of 190 responses on PedsQL were from parents of children with UCDs aged 2 years and older, with a median age of 6 years. A total of 79 responses on PedsQL were from children with UCDs aged 5 years and older, with a median age of 9 years. Among parents and children, the majority of responses were from individuals with OTCD (Table 1). A total of 203 responses on SF-36v2 were from adults with UCDs; the median age of this subset was 33 years (range: 25-42 years). The majority of responses (n=166, 81%) were from individuals with OTCD (Table 1).
Table 1.
Characteristics of children and adults with UCD for whom responses on PedsQL, and SF-36v2, respectively were analyzed.
| All children | OTCD (females) | OTCD (males) | ASLD | ASS1D | Rare UCDs1 | |
|---|---|---|---|---|---|---|
| PedsQL Parent Report | ||||||
| n | 190 | 74 | 32 | 35 | 32 | 17 |
| Age in years Median (IQR) | 5.8 (3.0-8.9) | 5.9 (3.3-9.3) | 6.5 (3.3-9.3) | 5.0 (2.6-8.4) | 6.1 (3.4-8.5) | 4.6 (3.3-7.1) |
| PedsQL Child Report | ||||||
| n | 79 | 41 | 12 | 12 | 11 | 3 |
| Age in years Median (IQR) | 8.6 (6.9-10.5) | 9.2 (7.2-11.0) | 9.0 (6.6-9.3) | 8.0 (7.4-10.1) | 7.8 (6.6-8.6) | 5.9 (5.8-6.5) |
| All adults | OTCD (females)1 | OTCD (males) | ASLD | ASS1 | Rare UCDs | |
| SF-36v2 | ||||||
| n | 203 | 141 | 25 | 13 | 11 | 13 |
| Age in years Median (IQR) | 32.9 (24.9-42.0) | 35.8 (28.5-42.8) | 30.0 (21.8-34.7) | 26.8 (23.1-38.5 | 23.9 (21.7-26.4) | 31.4 (20.2-51.2) |
Since the group sample sizes for individuals with NAGSD, CPS1D, ARG1D, HHH, and CTLN2 were too low to conduct statistical comparisons, they were grouped as “Rare UCDs”
To assess HRQoL in children with UCDs, we analyzed child-report and parent-report PedsQL Physical and Psychosocial Health Summary Scores and compared the scores with healthy children, children with PKU, and children with T1 and 2 DM. By child-report, the mean scores of children with UCDs were lower (worse) for physical health (79.8) compared to children with PKU (88.9) and T1 and 2 DM (86), and the psychosocial health scores in UCDs (72.7) were also lower (worse) than that of healthy children (82.4) and children with PKU (83.4) (Figure 1A). By parent report, the mean scores of children with UCDs were lower for physical health (76.4) compared to healthy children (89.3) and children with T1 and 2 DM (82) (Figure 1B). Regarding psychosocial health, the mean scores from child report were lower in children with UCDs in emotional functioning (74.1) compared to healthy children (80.7). Furthermore, the scores in children with UCDs in social and school functioning (76.4 and 67.8, respectively) were lower than scores in healthy children (87.4 and 78.6, respectively), and children with PKU (91.4 and 78.7, respectively) and T1 and 2 DM (85.8 and 74.2, respectively) (Figure 1C). Similarly, on parent report, the mean scores of children with UCDs were lower in emotional and social functioning (74.3 and 74.6, respectively) compared to healthy children (82.6 and 91.6, respectively), lower in social functioning children with T1 and 2 DM (81), and lower (67.8) than healthy children (85.4) in the school functioning domain (Figure 1D). To assess whether HRQoL differed by the type of UCD, we analyzed child-report and parent-report scores by UCD type. The mean composite scores of the Physical and Psychosocial Health Summary Scores did not differ among the UCD subtypes (p>0.0025, Bonferroni corrected p; Figures 1E and F; Supplemental table 1). Finally, to assess whether child and parental assessment of HRQoL were similar, we compared Physical and Psychosocial Health Summary scores for parent-report and child-report. This analysis showed that mean reported physical and psychosocial HRQoL did not differ between children with UCDs and their parents. Physical (r=0.51, p<0.0001) and Psychosocial (r=0.56, p<0.0001) Health Summary Scores between child-report and parent-report were significantly correlated.
Figure 1. Children with UCDs demonstrate lower physical and psychosocial health scores but these do not differ based type of UCD.
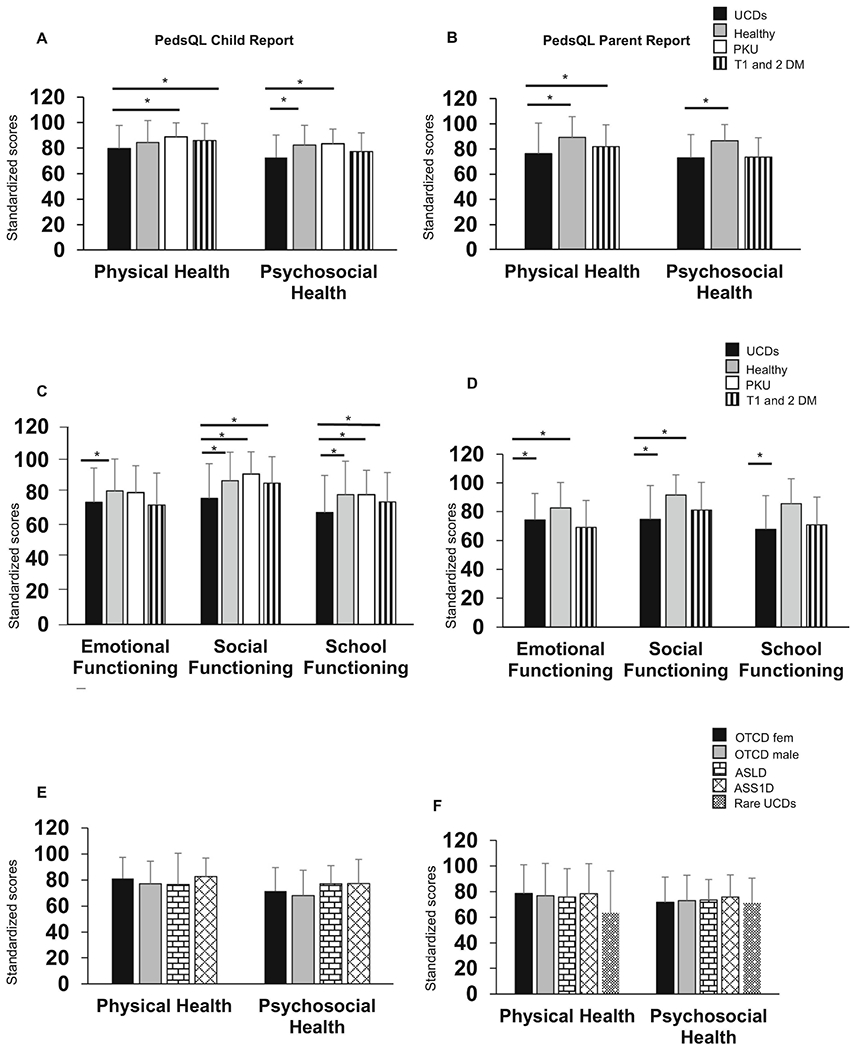
A) Physical Health and Psychosocial Health Summary child-report scores. B) Physical Health and Psychosocial Health Summary parent-report scores. C) Emotional, Social, and School functioning domains, child-report scores. D) Emotional, Social, and School functioning domain, parent-report scores. E) Physical Health and Psychosocial Health Summary child-report scores by UCD type. F) Physical Health and Psychosocial Health Summary parent-report scores by UCD type. “Rare UCDs” comprise NAGSD, CPS1D, ARG1D, CTLN2, and HHH syndrome. There were no child-report responses from children with rare UCD types. n for child report for UCDs = 79, Healthy Children = 401, PKU= 202, T1 and 2 DM = 300. n for parent report for UCDs = 190 Healthy Children = 717, T1 and 2 DM = 307. Bar graphs represent mean and standard deviation. *Bonferroni adjusted p<0.01 equivalent to an unadjusted p<0.05. The data for healthy children are derived from Varni et al. 2001. The data for children with PKU are derived from Bosch et al. 2015. The data for children with T1 and 2 DM are derived from Varni et al. 2003.
In adults, the mean SF-36v2 PCS score in individuals with UCDs (48.6) was lower (worse) than in adults with PKU (55.9) but not different from healthy individuals (49.2) and those with T1 DM (50.2) (Figure 2A). The MCS mean norm-based score in adults with UCDs (46.3) was lower compared to healthy adults (53.77) and individuals with T1 DM (48.8) (Figure 2A). Adults with rare UCDs reported lower PCS (39.9) compared to those with ASS1D (55.4) and ASLD (53.6) (Figure 2B, Bonferroni corrected p<0.005). However, no statistically significant differences were found in the MCS based on UCD subtype.
Figure 2. HRQoL in adults with UCDs.
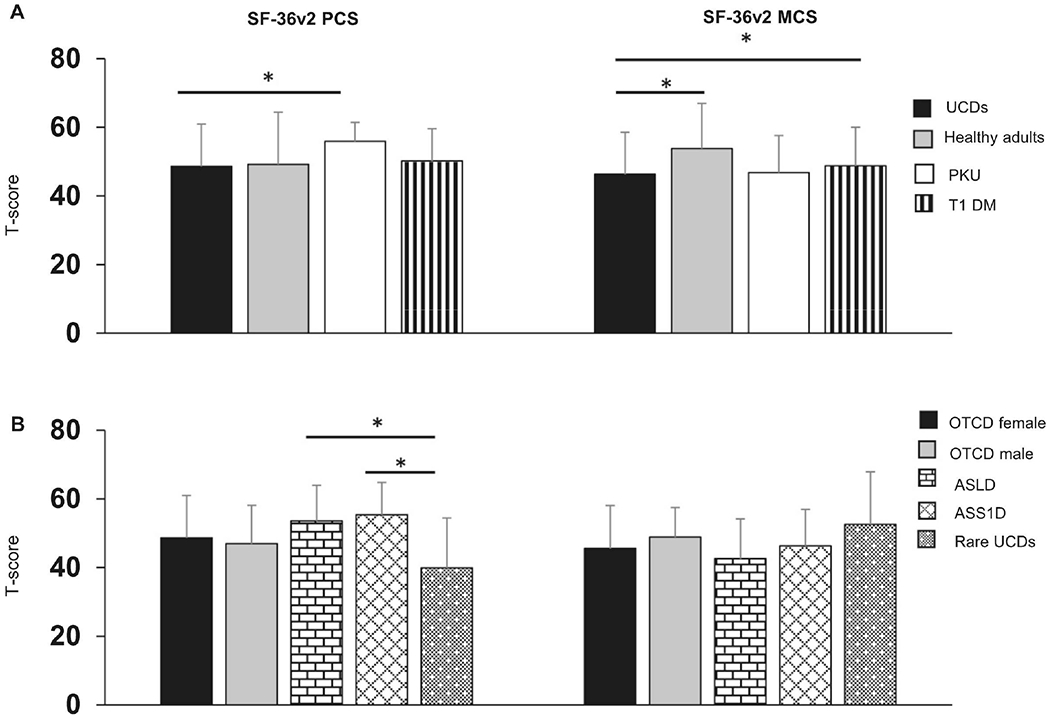
A) PCS and MCS in adults with UCDs compared to healthy adults and adults with PKU and T1 DM. B) PCS and MCS in adults with UCDs categorized by UCD type. n for UCDs = 203, healthy adults = 3828, PKU=104, and T1 DM =1373. Bar graphs represent mean and standard deviation. *denotes significant Bonferroni adjusted p-value. i.e., *p< 0.017. The data for healthy adults are derived from Maglinte et al 2012. The data for adults with PKU are derived from Bosch et al 2015. The data for adults with T1DM are derived from Svedbo Engstrom et al 2019.
Effect of age and UCD severity on HRQoL
As the natural history of UCDs varies as children age, we sought to determine whether age impacted HRQoL scores in children. On univariate linear regression, there was no statistically significant association between PedsQL physical HRQoL and age. However, Psychosocial Health Summary Score was negatively associated with age: every one-year increase in age was associated with −2.04 in score (p<0.001). A similar negative association was observed across all the domains within Psychosocial Health, including Emotional (Beta −1.098, p<0.01), Social (Beta −2.443, p<0.001), and School Functioning (Beta −2.529, p<0.001).
To assess whether individuals with more severe forms of UCDs had worse HRQoL, we categorized participants into symptomatic vs asymptomatic based on predefined criteria outlined in the manual of operations (see Materials and Methods for criteria). In children, by both child-report and parent-report, the mean summary scores for physical and psychosocial heath did not differ significantly between the symptomatic vs asymptomatic groups (Figure 3, Supplemental Table 2). Interestingly, adults with UCDs in the asymptomatic category reported lower (worse) mental HRQoL (45.7) than symptomatic adults (50.2; p<0.05) (Figure 3, Supplemental Table 3).
Figure 3. HRQoL in children and adults with UCDs in symptomatic vs asymptomatic categories.
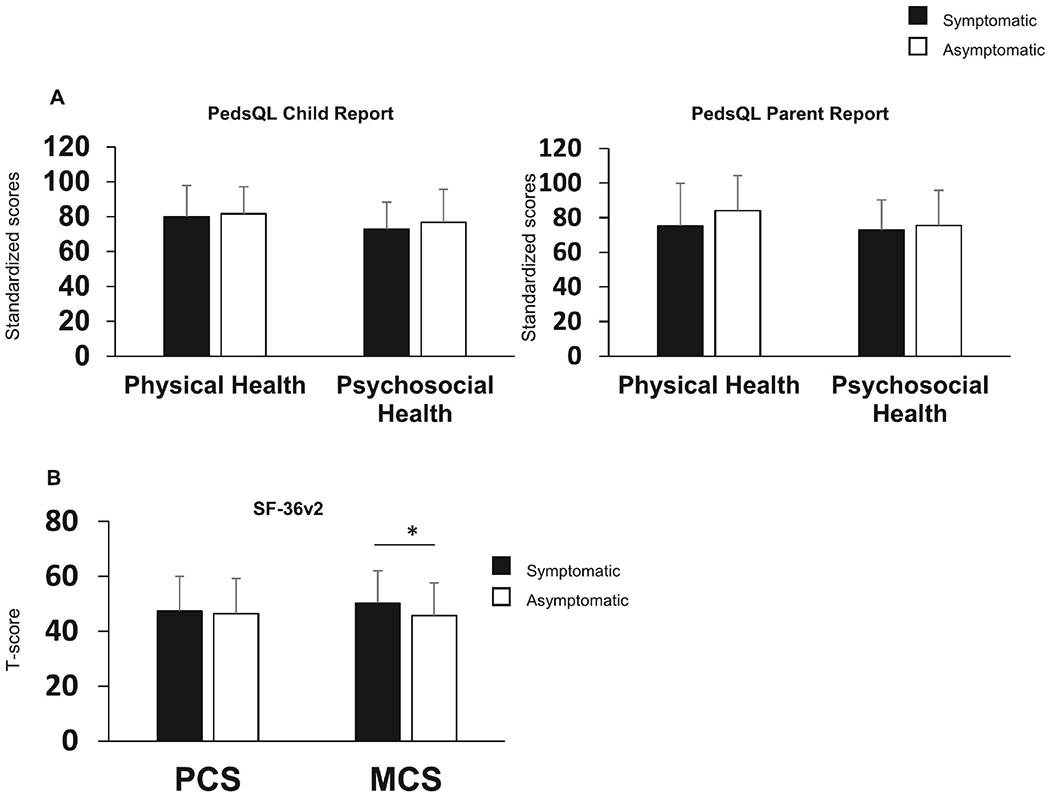
A) Child- and parent-report scores on PedsQL show no statistically significant differences between the symptomatic and asymptomatic children B) MCS on SF-36v2 is lower in asymptomatic as compared symptomatic adults with UCDs. n for child report – symptomatic = 59, asymptomatic =17; n for parent report symptomatic = 140, asymptomatic = 40; n for adults symptomatic = 98, asymptomatic = 97. Bar graphs represent mean and standard deviation. *denotes p<0.05
Another well-established marker of disease severity among UCDs is hyperammonemia. In children, while presence of hyperammonemia did not have an association with any HRQoL measures, the number of hyperammonemic events was significantly associated with parent-reported physical (p=0.008) and social HRQoL (p=0.048) on PedsQL. In adults, we found no significant association between HRQoL scores and presence of or number of hyperammonemic events.
Clinical Trial Readiness
To determine the applicability of these HRQoL measures in clinical trials, we determined the sample sizes that would be required to detect mean differences of 5 and 10 points on PedsQL and SF-36v2 composite scores (Table 2). These analyses show that only if interventions were to have a moderate effect size on these HRQoL measures, they could be used as endpoints in clinical trials with sample sizes that are feasible for enrollment.
Table 2:
Sample sizes required for detecting differences on HRQoL in UCDs based on data generated by this study.
| Total n required in a parallel group design to detect a mean difference of | ||||
|---|---|---|---|---|
| Mean score | SD | 5 | 10 | |
| PedsQL Child Report | ||||
| Physical Health Summary Score | 79.8 | 17.9 | 406 | 104 |
| Psychosocial Health Summary Score | 72.7 | 17.6 | 392 | 100 |
| PedsQL Parent Report | ||||
| Physical Health Summary Score | 76.4 | 24.2 | 736 | 186 |
| Psychosocial Health Summary Score | 73.0 | 18.5 | 432 | 110 |
| SF-36v2 | ||||
| Physical Component | 48.6 | 12.3 | 192 | 50 |
| Mental Component | 46.3 | 12.2 | 190 | 50 |
DISCUSSION
A few previous studies have measured HRQoL in children with various IEM, including UCDs, using the PedsQL. However, most of these studies have been limited by small sample sizes and limited distributions of ages and UCD subtypes; in fact, the largest study of HRQoL in children with UCDs was limited to one diagnosis, CTLN2, with 55 individuals aged 1-22 years.15,16,26 In contrast, our study represents data from a large group of children with UCDs who were enrolled at 16 sites in the U.S., Canada, and Europe and to date is the largest study to systematically assess HRQoL in this population. Furthermore, our study represents the first systematic study of HRQoL in adults with UCDs.
We found that average physical and psychosocial HRQoL in children with UCDs were worse than in healthy children. Psychosocial health was relatively worse than physical health. The differences in mean Physical Health and Psychosocial Health Summary Scores by parent report between children with UCDs and healthy children would translate into medium to large effect sizes, respectively (Cohen’s d, 0.625 and 0.85). We also found a negative association between age and parent reported psychosocial HRQoL. We hypothesized that this may be related to challenges with school, and thus we explored the effect of age on all three domains within psychosocial health, and found that in fact, increasing age was associated with worse social, emotional, and school functioning scores. On domains of psychosocial functioning, children with UCDs reported worse emotional, social, and school function than healthy children. However, when compared to children with PKU and T1 and 2 DM, children with UCDs reported worse scores on social and school functioning but not emotional functioning. This discrepancy may reflect the effect that a block in ureagenesis can have on neurocognitive functioning. The decompensations associated with hyperammonemia can cause long-term neurocognitive deficiencies that can affect social interaction and academic abilities. Yet, the similar emotional functioning scores may demonstrate that living with a chronic metabolic illness may have similar psychological effects in children independent of cognitive functioning. Previous studies have shown that dietary regimens predict lower HRQoL among children with IEM,26 suggesting that dietary restrictions may be one common contributing factor for decreased emotional functioning in children with UCDs, PKU, and DM. Future qualitative work investigating the impact of diet, medications, and other treatments upon HRQoL in children with UCDs could enrich our understanding of this phenomenon.
Interestingly, while adults with UCDs reported worse mental HRQoL compared to the general population, their physical HRQoL was not different. This result may reflect the preponderance of females with OTCD in our adult population, a group that, while at risk for hyperammonemia, can be asymptomatic or mildly symptomatic from a physical perspective.27 On the other hand, the worse mental HRQoL among adults with UCDs may reflect the known neuropsychiatric phenotypes which can be observed even in mildly symptomatic females with OTCD.28–31
No previous studies have compared HRQoL among individuals with different UCD diagnoses. In our study, we did not find any differences between the UCD subtypes among children. This could represent a limitation of the analyses with smaller sample sizes within each UCD category. However, the mean scores within each individual UCD type were worse than in healthy children alluding to the fact that UCDs may affect HRQoL, especially psychosocial health, irrespective of the subtype. In adults, we found statistically significantly worse SF-36v2 PCS in individuals with rare UCDs compared to ASS1D and ASLD. These results must be interpreted with caution as the numbers of individuals within each of these categories was less than 15. However, this may reflect the fact that individuals with argininemia, who experience spasticity and significant mobility challenges, were included in the rare UCDs group.
A single-center longitudinal study has demonstrated that children with more severe IEM as rated by clinicians had worse HRQoL than those with milder IEM.32 Notably, at one-year follow-up after care in a multidisciplinary center, the HRQoL of children with severe IEM had improved significantly, whereas there was no change in HRQoL of children with milder IEM, implying that those with more severe disease may stand to benefit more from comprehensive care. In our sample, we compared HRQoL scores based on severity of UCD. It can be argued that there are many ways to categorize the severity of UCDs: by number of hyperammonemic episodes, the severity of intellectual disability, neurocognitive and neurological abnormalities, degree of behavioral abnormalities, the presence or absence of seizures, and severity of liver disease, among others. However, no such definition is bound to be all-encompassing. Within the UCDC, a predefined set of criteria are used to categorize severity of UCDs into symptomatic vs asymptomatic. This classification not only uses hyperammonemia as a feature of severity but also accounts for the consequences of such decompensation. Furthermore, individuals who have neuropsychological deficits and symptoms even in the absence of hyperammonemia are also classified as being symptomatic. In children, counterintuitive to expectation, the HRQoL measures assessed did not differ between symptomatic and asymptomatic individuals. However, this categorization did not consider the number and frequency of hyperammonemic episodes. Future research focused on HRQoL after hyperammonemic episodes and the trajectory of HRQoL after such episodes will be important for the field. Intriguingly, asymptomatic adults reported worse mental HRQoL than their symptomatic peers. This may indicate that increased disease burden and severity may not negatively impact HRQoL in individuals with UCD, or it may indicate that the clinical variables we used to stratify disease severity are incomplete. Future work should examine the impact of other factors that may reflect disease severity, such as age of symptom onset, use of nitrogen-scavenging medications, frequency of hyperammonemic episodes, and need for OLT.
OLT has become an accepted standard treatment option for patients with certain IEM with severe disease burden. Data on the impact of OLT on HRQoL in individuals with IEM are unclear. While several case reports and case series document improved clinical status and suggest that liberalized dietary restrictions and decreased need for metabolic medications after OLT improve HRQoL,33–36 these findings have not always been quantified in a systematic manner. A study comparing PedsQL scores in children with methylmalonic acidemia who had and had not undergone OLT demonstrated no significant differences between the two groups.37 In our study, we excluded participants who had undergone OLT due to the potential for sampling bias with regards to differences in disease severity and comorbidities of OLT. Future studies should investigate the effects of OLT on HRQoL in patients with UCDs. Longitudinal, paired comparisons of HRQoL before and after OLT would reduce possible confounding by individual patients’ clinical variability.
Especially when studying children living with diseases that may cause IDD, questions may arise regarding the validity of self-reported responses.38 Therefore, researchers may prefer to rely on proxy reports when utilizing PROMs in these populations. However, prior research has shown that parents of children with genetic diseases tend to rate their children’s HRQoL lower than the affected children rate their own HRQoL.39 Our work found that parents and children do not differ significantly in their perception of the physical and psychosocial HRQoL in children with UCDs. We further demonstrated that parent-report and child-report HRQoL were correlated, which indicates that it might be reasonable to rely on only parent-report on children’s HRQoL, when such responses cannot be collected from children.
Finally, we show that with the variance seen in PedsQL and SF-36v2 scores in individuals with UCDs, only effect sizes that are moderate (Cohen’s d 0.5 - 0.8) can be detected using sample sizes that would be feasible for enrollment. Whereas these HRQoL measures are not likely to be primary outcome measures in pivotal interventional trials, they can be used as secondary measures that can provide a patient experience outcome to support a more robust clinical event or biomarker-driven primary endpoint.
The strengths of our study are the following: 1) This represents the largest cohort of children and adults with UCDs; 2) The HRQoL and clinical data were collected in a systematic manner across the sites; 3) Data management was conducted by the NIH RDCRN’s Data Management Coordinating Center that has policies for data queries and quality check; 4) Data were collected in geographically diverse regions and thus may likely represent the UCD population at large; 5) The large sample size allowed for robust analyses. The limitations of the study are the following: 1) We only analyzed the data from the enrollment visit and thus we cannot comment on the trajectory of HRQoL measures in each individual over time; 2) The categorization into symptomatic and asymptomatic groups does not take into account, the number of hyperammonemic episodes (or annualized number of episodes). Thus, this analysis cannot discern the effect of hyperammonemia, especially recurrent hyperammonemia, on HRQoL; 3) Participants reported their generic HRQoL. While PedsQL has been widely used in the IEM population, and SF-36v2 has been widely used in many populations (though less so in intoxication-type IEM), these generic measures do not include items inquiring about the impact of dietary restrictions, enteral feeding, symptoms specific to IEM, and burden of disease management (e.g., blood draws, medications, follow-up visits). Future work would benefit from use of a QoL measure that is more specific to IEM, such as the recently developed MetabQoL.40
Despite these limitations, this work adds valuable information to the literature regarding HRQoL in children and adults with UCDs. Our findings provide objective support to what has long been appreciated by clinicians that individuals (and families) with UCDs require comprehensive mental healthcare and psychological support in addition to careful metabolic management.
In summary, our study illustrates the importance of natural history studies in uncovering the opportunities and limitations with the use of generic HRQoL measures in clinical trials in UCDs. We believe that the insights from this study will be important for the development and evaluation of UCD-specific HRQoL measures.
Supplementary Material
Acknowledgements
We are indebted to individuals with UCDs and their families for their participation in the longitudinal study. All Urea Cycle Disorders Consortium (UCDC) sites contributed to the Longitudinal Study dataset used in this publication.
Study coordinators include: Kim Bardillon, Kia Bryan, Liora Caspi, Kiaira, Coles, Christine Deng, Sara Elsbecker, Debbie Fu, Jinsung Jeon, Elijah Kravets, Ursula Kuhn, Saima Ali, Genya Kisin, Roland Posset, Thu Quan, Kara Simpson, Julia Sloan, Hayden Vreugdenhil, and Ashley Wilson, and Melissa Zerofsky. Study neuropsychologists include: Fabienne Dietrich Alber, Christopher Boys, David Breiger, Benjamin Goodlett, Elizabeth Kerr, Casey Krueger, Eva Mamak, Ami Norris-Brilliant, David Schwartz, Yuri Shishido, Arianna K. Stefanatos, Rachel Tangen, Magdalena Walter, and Greta Wilkening.
We would also like to acknowledge the contributions of former longitudinal study PIs: Mark L. Batshaw, Peter Burgard, Stephen Cederbaum, Annette Feigenbaum, Douglas S. Kerr, Brendan Lee, Uta Lichter-Konecki, Margretta R. Seashore, Marshall L. Summar, Mendel Tuchman, Susan Waisbren, James Weisfeld-Adams, and Marc Yudkoff.
The Urea Cycle Disorders Consortium (UCDC; U54HD061221) is part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through a collaboration between the Office of Rare Diseases Research (ORDR), the National Center for Advancing Translational Science (NCATS) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The Urea Cycle Disorders Consortium has also been supported by the O’Malley Foundation, the Rotenberg Family Fund, the Dietmar Hopp Foundation, the Kettering Fund and the National Urea Cycle Disorders Foundation. Research reported in this publication was also supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number P50HD103555 for use of the BCM IDDRC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. CNM was partly supported by the Medical Genetics Research Fellowship Program (T32 GM07526-42). LCB is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK126786 and by a Burroughs Wellcome Career Award for Medical Scientists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1-2):179–180. doi: 10.1016/J.YMGME.2013.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsumoto S, Häberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K. Urea cycle disorders-update. J Hum Genet. 2019;64(9):833–847. doi: 10.1038/S10038-019-0614-4 [DOI] [PubMed] [Google Scholar]
- 3.Baruteau J, Diez-Fernandez C, Lerner S, et al. Argininosuccinic aciduria: Recent pathophysiological insights and therapeutic prospects. J Inherit Metab Dis. 2019;42(6):1147–1161. doi: 10.1002/JIMD.12047 [DOI] [PubMed] [Google Scholar]
- 4.Kho J, Tian X, Wong WT, et al. Argininosuccinate Lyase Deficiency Causes an Endothelial-Dependent Form of Hypertension. Am J Hum Genet. 2018;103(2):276–287. doi: 10.1016/J.AJHG.2018.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrage LC, Madan S, Li X, et al. Chronic liver disease and impaired hepatic glycogen metabolism in argininosuccinate lyase deficiency. JCI Insight. 2020;5(4). doi: 10.1172/JCI.INSIGHT.132342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallagher RC, Lam C, Wong D, Cederbaum S, Sokol RJ. Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J Pediatr. 2014;164(4). doi: 10.1016/J.JPEDS.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laemmle A, Gallagher RC, Keogh A, et al. Frequency and Pathophysiology of Acute Liver Failure in Ornithine Transcarbamylase Deficiency (OTCD). PLoS One. 2016;11(4). doi: 10.1371/JOURNAL.PONE.0153358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain-Ghai S, Nagamani SCS, Blaser S, Siriwardena K, Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol Genet Metab. 2011;104(1-2):107–111. doi: 10.1016/J.YMGME.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohura T, Kobayashi K, Tazawa Y, et al. Neonatal presentation of adult-onset type II citrullinemia. Hum Genet. 2001;108(2):87–90. doi: 10.1007/S004390000448 [DOI] [PubMed] [Google Scholar]
- 10.Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 2002;47(7):333–341. doi: 10.1007/S100380200046 [DOI] [PubMed] [Google Scholar]
- 11.Mew NA, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea Cycle Disorders Overview. GeneReviews®. Published online June 22, 2017. Accessed May 27, 2022. https://www.ncbi.nlm.nih.gov/books/NBK1217/ [Google Scholar]
- 12.Waisbren SE, Stefanatos AK, Kok TMY, Ozturk-Hismi B. Neuropsychological attributes of urea cycle disorders: A systematic review of the literature. J Inherit Metab Dis. 2019;42(6):1176–1191. doi: 10.1002/JIMD.12146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeowell G, Burns DS, Fatoye F. The burden of pharmacological treatment on health-related quality of life in people with a urea cycle disorder: a qualitative study. J Patient Rep Outcomes. 2021;5(1):1–13. doi: 10.1186/S41687-021-00387-X/TABLES/3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabre A, Baumstarck K, Cano A, et al. Assessment of quality of life of the children and parents affected by inborn errors of metabolism with restricted diet: preliminary results of a cross-sectional study. Health QualLife Outcomes. 2013;11(1). doi: 10.1186/1477-7525-11-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okano Y, Kobayashi K, Ihara K, et al. Fatigue and quality of life in citrin deficiency during adaptation and compensation stage. Mol Genet Metab. 2013;109(1):9–13. doi: 10.1016/J.YMGME.2013.01.020 [DOI] [PubMed] [Google Scholar]
- 16.Jamiolkowski D, Kölker S, Glahn EM, et al. Behavioural and emotional problems, intellectual impairment and health-related quality of life in patients with organic acidurias and urea cycle disorders. J Inherit Metab Dis. 2016;39(2):231–241. doi: 10.1007/s10545-015-9887-8 [DOI] [PubMed] [Google Scholar]
- 17.Seminara J, Tuchman M, Krivitzky L, et al. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol Genet Metab. 2010;100 Suppl 1(Suppl 1). doi: 10.1016/J.YMGME.2010.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuchman M, Lee B, Lichter-Konecki U, et al. Cross-Sectional Multi-Center Study of Patients with Urea Cycle Disorders in the United States. Mol Genet Metab. 2008;94(4):397. doi: 10.1016/J.YMGME.2008.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varni JW, Seid M, Kurtin PS. PedsQL™ 4.0: Reliability and Validity of the Pediatric Quality of Life Inventory™ Version 4.0 Generic Core Scales in Healthy and Patient Populations. Med Care. 2001;39(8):800–812. doi: 10.1097/00005650-200108000-00006 [DOI] [PubMed] [Google Scholar]
- 20.Ware JE, Sherbourne CD. The MOS 36-item short-form health survey (Sf-36): I. conceptual framework and item selection. Med Care. 1992;30(6):473–483. doi: 10.1097/00005650-199206000-00002 [DOI] [PubMed] [Google Scholar]
- 21.Maglinte GA, Hays RD, Kaplan RM. US general population norms for telephone administration of the SF-36v2. J Clin Epidemiol. 2012;65(5):497–502. doi: 10.1016/j.jclinepi.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosch AM, Burlina A, Cunningham A, et al. Assessment of the impact of phenylketonuria and its treatment on quality of life of patients and parents from seven European countries. Orphanet J Rare Dis. 2015;10(1):80. doi: 10.1186/s13023-015-0294-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varni JW, Burwinkle TM, Jacobs JR, Gottschalk M, Kaufman F, Jones KL. The PedsQL in type 1 and type 2 diabetes: reliability and validity of the Pediatric Quality of Life Inventory Generic Core Scales and type 1 Diabetes Module. Diabetes Care. 2003;26(3):631–637. doi: 10.2337/DIACARE.26.3.631 [DOI] [PubMed] [Google Scholar]
- 24.Svedbo Engstrom M, Leksell J, Johansson UB, et al. Health-related quality of life and glycaemic control among adults with type 1 and type 2 diabetes - a nationwide cross-sectional study. Health Qual Life Outcomes. 2019;17(1). doi: 10.1186/S12955-019-1212-Z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hintz J PASS. Published online 2011. [Google Scholar]
- 26.Bösch F, Landolt MA, Baumgartner MR, et al. Health-related quality of life in paediatric patients with intoxication type inborn errors of metabolism: analysis of an international dataset. J Inherit Metab Dis Published online August 12, 2020:jimd.12301. doi: 10.1002/jimd.12301 [DOI] [PubMed] [Google Scholar]
- 27.Batshaw ML, Msall M, Beaudet AL, Trojak J. Risk of serious illness in heterozygotes for ornithine transcarbamylase deficiency. J Pediatr. 1986;108(2):236–241. doi: 10.1016/S0022-3476(86)80989-1 [DOI] [PubMed] [Google Scholar]
- 28.Sprouse C, King J, Helman G, et al. Investigating neurological deficits in carriers and affected patients with ornithine transcarbamylase deficiency. Mol Genet Metab. 2014; 113(1-2): 136–141. doi: 10.1016/J.YMGME.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niwinski P, Remberk B, Rybakowski F, Rokicki D. Psychiatric Symptoms as the First or Solitary Manifestation of Somatic Illnesses: Hyperammonaemia Type II. Neuropsychobiology. 2021;80(3):271–275. doi: 10.1159/000508679 [DOI] [PubMed] [Google Scholar]
- 30.Muzammil SM, Chrusciel D, Katyal R. Undiagnosed Late-onset Ornithine transcarbamylase (OTC) Deficiency Presenting With Psychiatric Symptoms (P4.6-067). Neurology. 2019;92(15 Supplement). [Google Scholar]
- 31.Kim SH, Lee JS, Lim BC, et al. A female carrier of ornithine carbamoyltransferase deficiency masquerading as attention deficit-hyperactivity disorder. Brain Dev. 2014;36(8):734–737. doi: 10.1016/J.BRAINDEV.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 32.Dimitrova N, Glaus J, Urben S, Wüthrich V, Morisod Harari M, Ballhausen D. The impact of disease severity on the psychological well-being of youth affected by an inborn error of metabolism and their families: A one-year longitudinal study. Mol Genet Metab Rep. 2021. ;29:100795. doi: 10.1016/J.YMGMR.2021.100795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foschi FG, Morelli MC, Savini S, et al. Urea cycle disorders: A case report of a successful treatment with liver transplant and a literature review. World J Gastroenterol. 2015;21(13):4063–4068. doi: 10.3748/wjg.v21.il3.4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasahara M, Sakamoto S, Shigeta T, et al. Living-donor liver transplantation for carbamoyl phosphate synthetase 1 deficiency. Pediatr Transplant. 2010; 14(8): 1036–1040. doi: 10.111l/j.1399-3046.2010.01402.x [DOI] [PubMed] [Google Scholar]
- 35.Ban K, Sugiyama N, Sugiyama K, et al. A pediatric patient with classical citrullinemia who underwent living-related partial liver transplantation. Transplantation. 2001;71(10): 1495–1497. doi: 10.1097/00007890-200105270-00026 [DOI] [PubMed] [Google Scholar]
- 36.Kawagishi N, Satoh K, Enomoto Y, et al. Improved quality of life and unchanged magnetic resonance brain imaging after living donor liver transplantation for late-onset ornithine transcarbamylase deficiency: Report of a case. Surg Today. 2005;35(12): 1087–1091. doi: 10.1007/s00595-005-3071-y [DOI] [PubMed] [Google Scholar]
- 37.Splinter K, Niemi AK, Cox R, et al. Impaired Health-Related Quality of Life in Children and Families Affected by Methylmalonic Acidemia. J Genet Couns. 2016;25(5):936–944. doi: 10.1007/s10897-015-9921-x [DOI] [PubMed] [Google Scholar]
- 38.Greenberg KK, Schwartz AE, Kramer JM. Adoption of patient-reported outcome measures with youth with intellectual/developmental disabilities: Contextual influences and practice patterns. Child Care Health Dev. 2021;47(4):501–508. doi: 10.1m/CCH.12862 [DOI] [PubMed] [Google Scholar]
- 39.Cohen JS, Biesecker BB. Quality of life in rare genetic conditions: a systematic review of the literature. Am J Med Genet A. 2010;152A(5):1136–1156. doi: 10.1002/ajmg.a.33380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeltner NA, Baumgartner MR, Bondarenko A, et al. Development and psychometric evaluation of the metabQoL 1.0: A quality of life questionnaire for paediatric patients with intoxication-type inborn errors of metabolism. In: JIMD Reports. Vol 37. Springer; 2017:27–35. doi: 10.1007/8904201711 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.