

INTRODUCTION
Rare genetic diseases can be powerful tools for the discovery of specific biologic pathways relevant to human health and disease. COPA syndrome, a recently described autosomal dominant inborn error of immunity, has revealed the importance of intracellular protein trafficking to maintaining immune homeostasis. This review will explore the clinical presentation, genetics, molecular mechanisms, organ manifestations, and treatment approaches for COPA syndrome, ending with a diagnostic framework and remaining questions.
METHODS
Published articles and conference abstracts were identified through the literature search for “COPA syndrome.” No relevant results returned for “autoimmune interstitial lung joint and kidney disease” and “AILJK.” Three additional abstracts were identified through searching for “COPA syndrome” plus an acronym for pulmonology, immunology, rheumatology, and primary immunodeficiency-focused conferences. Two additional articles were identified through being referenced by other studies. Every effort was made to avoid duplicate reporting of individual patients. Scrutinizing author lists and patient-level information identified the following duplicate reports: 1. patients from original publication also reported upon in 2 early follow-up studies1–3; 2. kindred (1 affected, 1 unaffected) with the D243N mutation4,5; 3. Kindred (5 affected, 3 unaffected) with the R233H mutation5,6; 4. kindred (1 affected, 1 unaffected) with the R233H mutation5,7,8; 5. patients who underwent lung transplant from kindred (4 affected) with the V242G mutation9,10; 6: only novel patients from an institutional registry were reported (patients 3, 4, 5, 6, and 10 per T.P.V.).1,11–13 A P145S mutation is excluded as the referenced publication14,15 could not be found via search or via the website of the named journal. An abstract reporting 6 individuals with one of the 3 additional C terminal tail mutations16 was excluded due to insufficient patient information.
DETAILED DESCRIPTION
Clinical Presentation
COPA syndrome was first reported in 2015 after mutations in the coatomer subunit alpha (COPA) gene were identified in 5 kindreds in which multiple individuals had childhood onset interstitial lung disease, high titer autoantibodies, and inflammatory arthritis,1 almost half of whom also developed renal disease. Subsequently, additional reported patients generally share these features, with most showing childhood onset of symptoms, 64% up to age 5% and 89% up to age 12 for those with reported ages. Four individuals with symptom onset in their 50s have been reported, generally showing milder disease and identified through familial sequencing after mutations were identified in young probands.6,9,12 One individual, published as a case report, was identified through sequencing lung transplant recipients, with age available for ILD diagnosis but not arthritis onset.17
Presenting symptoms are typically pulmonary, ranging from tachypnea to respiratory failure, and/or musculoskeletal, ranging from arthralgias to deforming arthritis (Table 1). One patient presented with renal failure.6 Several infants and toddlers presented with nonspecific lethargy, failure to thrive, and/or anemia (from insidious alveolar hemorrhage), at times years prior to developing localizing symptoms.8 All patients have high levels of autoantibodies, including one or more of anti-nuclear antibodies (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), rheumatoid factor (RF), and anti-cyclic citrullinated peptide antibodies (CCP). Antibodies to extractable nuclear antigens (ENAs) and ANCA subtype results were more variable, both between patients and within an individual over time.
Table 1.
Features of patients by mutation
| Highest Level of Evidence | # Affected (% Male) | # Unaffected (% Male) | Cause of Death (Age) | Refs | Countries | Age (y) at Symptom Onset or Diagnosis | Presenting Symptoms | |
|---|---|---|---|---|---|---|---|---|
| Experimentally Validated | K230N | 6 (17%) | 1 (100%) | USA | 7 | Lethargy (n = 1) abdominal pain (n = 1) | 1,3 | |
| R233H | 29 (55%) | 6 (67%) | Lung disease (29) Lung disease (63) Suicide (unk) Unk (unk) |
USA France England |
0.83, 1, 2, 2, 2, 2, 2.5, 3, 3, 5, 5, 5, 7, 7, 8, 10, 11, 16, 26, 50a, 56a, 59b,c | Pulmonary (n = 15) Joint symptoms (n = 11) Anemia (n = 3) Renal failure (n = 1) Fever (n = 1) |
1,3,5,6,8,11–13,17,23,24 | |
| E241K | 9 (33%) | 3 (66%) | Unk (unk) | USA Iceland |
1.5, 2, 4, 11, 32c | Joint symptoms (n = 4) Pulmonary (n = 3) |
1,3,19 | |
| V242G | 4 (75%) | Lung disease (4) Lung disease(21) |
Japan | 0.17, 0.33, 0.58, 53a | Pulmonary (n = 3) Joint symptoms (n = 1) |
9,10 | ||
| D243G | 5 (0%) | 4 (100%) | Unk (unk) Unk (unk) | USA | 0.5 | Pulmonary (n = 1) | 1,3 | |
| D243N | 1 (0%) | 1 (0%) | England | 2.5 | Joint symptoms (n = 1) | 5,51 | ||
| Published Case Reports | H199R | 3 (100%) | 1 (50%) | China | 0.25 | Pulmonary (n = 2) Joint symptoms (n = 1) |
34 | |
| K238E | 1 (100%) | China | 7 | Pulmonary (n = 1) Joint symptoms (n = 1) |
34 | |||
| A239P | 3 (100%) | USA Canada China |
0.75, 2, 3 | Pulmonary (n = 3) FTT (n = 2) |
15,38,43 | |||
| W240L | 1 (0%) | Germany | 2 | Pulmonary (n = 1) | 40 | |||
| W240R | 1 (100%) | USA | 12 | Pulmonary (n = 1) FTT (n = 1) |
52 | |||
| W240S | 1 (0%) | Germany | 14 | Pulmonary (n = 1) Joint symptoms (n = 1) |
40 | |||
| E241A | 2 (100%) | USA | 0.5, 7 | Pulmonary (n = 2) | 39 | |||
| R281W | 5 (80%) | 2 (0%) | Lung disease (11) Lung disease (38) | India China Germany Italy |
1.5, 5, 7, 9, <12 | Joint symptoms (n = 3) Pulmonary (n = 2) Anemia (n = 1) | 14,20,36,40 | |
| Q285H | 1 (0%) | France | 6 | Joint symptoms (n = 1) | 35 | |||
| Mutations not Reported | 3 (33%) | USA Canada Brazil |
“childhood” | Pulmonary (n = 3) Joint symptoms (n = 1) | 18,41,53 | |||
Identified by family sequencing after mutation found in proband.
Identified by the sequencing of lung transplant cohort.
COPA syndrome identified after additional organ involvement, age unknown for initial symptoms.
Disease manifestations can evolve in individual patients over time, with the detection of additional autoantibodies and/or involvement of additional organ systems. Organ-specific manifestations also change with time, especially for pulmonary disease in which interstitial lung disease involving cysts (on imaging) and follicular bronchitis, lymphocytic interstitial pneumonia, and/or interstitial fibrosis (on biopsy) are often later findings than alveolar hemorrhage. These findings can progress despite immune suppression, even while symptoms are controlled,3,18,19 and represent the main cause of death in COPA syndrome. Indeed, of the six patients with COPA syndrome with a reported cause of death, 5 passed away from lung disease (3 despite bilateral lung transplants),1,9–11,14,17,18,20 while one passed away from suicide.5 A total of eleven (14%) patients have passed away.
Genetics and Genotype-Phenotype Correlations
The first report of COPA syndrome identified four distinct disease-causing mutations with autosomal dominant inheritance and reduced penetrance. In the past 8 years, an additional 15 mutations have been described, often as case reports though 2 of these have been functionally validated experimentally and shown to statistically differ from unmutated COPA (see Table 1). The six validated mutations cluster tightly within exons 8 and 9 of the large COPA protein, all falling within the fifth and sixth WD repeats in the WD-40 domain that is involved in target protein recognition (Fig. 1). An additional mutation hotspot has been reported in exon 10 coding for part of the seventh WD repeat, with reported patients showing similar organ involvement.
Fig. 1.
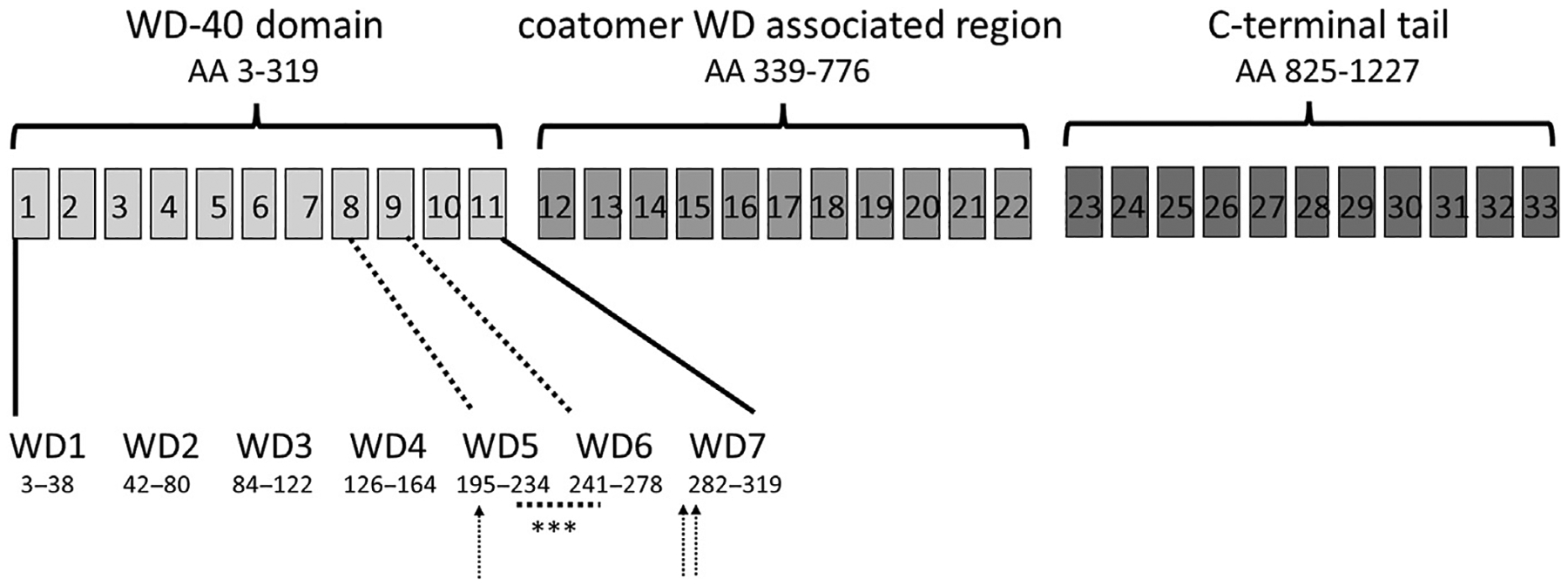
Structure of COPA locus, exons are marked in boxes shaded to correspond with protein motifs. Detail of WD regions containing COPA mutations highlighted. Validated mutations (K230 N, R233H, E241 K, V242 G, D243 G, D243 N) and variants from case report tightly cluster between amino acids 230 to 243, underlined and marked with asterisks. Arrows point to additional mutations from case reports describing patients with consistent phenotypic features (see Table 1 for full details). (Data from Quek et. al. in Cytogenetic and Genome Research, 1997.)
One patient with a mutation in the C terminal tail, present in 172 individuals in gnomad21 and reported in association with a different phenotype, is not included in this review of COPA syndrome. Three additional C terminal mutations have also been reported, however available clinical information was not sufficient for evaluation.16 Indeed, there may be a spectrum of clinical phenotypes associated with an individual gene, as has been described for other immune dysregulation syndromes (ex: TREX1 mutations causing increased susceptibility to systemic lupus erythematosus (SLE), chilblain lupus, Aicardi-Goutières syndrome, or retinal vasculopathy with cerebral leukodystrophy22). However, “COPA syndrome” should typically be reserved for the clinical phenotype of lung disease in the presence or absence of inflammatory arthritis and/or renal disease. Future functional validation of COPA variants outside the WD-40 domain may provide support for the conclusion that additional human immune disease phenotypes are also the result of pathogenic mutations in COPA.
COPA mutations do not appear to show genotype-phenotype correlations for the common symptoms of COPA syndrome, though the strength of any conclusions is obviously limited by the small patient numbers. For example, 31 patients from 12 families (48% of all reported patients) have the R233H mutation.1,5,6,8,11–13,17,23,24 These individuals show variable disease expressivity, with clinical presentations ranging from severe recurrent alveolar hemorrhage7 to severe nephritis necessitating transplant in childhood.6 Disease manifestations differ within families as well, with relatives of the patient presenting with renal failure having pulmonary but not renal involvement.5,6 In contrast, none of 9 patients (12%) from 2 families with the E241K mutation are yet to be reported to have renal involvement, though it may subsequently develop.1,19 Several patients have additional findings, shared by at most 2 individuals, either first degree relatives or those bearing different COPA mutations, such that these findings may or may not be part of COPA syndrome.
Epidemiology
The true frequency of COPA syndrome is hard to determine given its recent discovery, as well as the absence of experimental validation for most of the mutations reported in the literature. Nevertheless, it appears to be ultra-rare, with a total of 77 patients reported (see Table 1). Patients sharing core COPA syndrome features do not show a gender preference among cases (38 males and 39 females). An additional 18 unaffected carriers have been reported, males twice as commonly as females, with a resultant 15% clinical non-penetrance rate that is almost certainly an underestimate as family members were not systematically tested in many reports. The cause of non-penetrance is currently unknown, with healthy mutation carriers variably reported as indistinguishable from other family members or with minor elevations in type I interferon (IFN) signature.1,5 Those with described racial and ethnic backgrounds are mostly Caucasian, with East Asian ancestry second most common. Cases have also been reported in individuals with South Asian, Native American, Hispanic, and African American ancestry (see Table 1).
Severe lung disease is a prominent feature of COPA syndrome, with the evaluation of lung transplant recipients potentially informative about disease frequency. A conference abstract reported sequencing results for 77 individuals transplanted for ILD, identifying one individual bearing the R233H COPA mutation,17 consistent with COPA syndrome being a rare clinical entity.
Mechanisms of Disease
The International Union of Immunologic Societies (IUIS) expert committee on Inborn Errors of Immunity has grouped COPA syndrome under the “auto-inflammatory, other” category since 2015, including in the most recent update in 2022.25 It is more accurate to describe it as a syndrome of dysregulated immunity, however, as both innate5,26,27 (autoinflammatory) and adaptive1,28 (autoimmune) immune aspects are aberrant.
COPA codes for the coatomer subunit alpha protein, a part of the coat protein complex I (COPI) that coats vesicles to mediate retrograde protein transport from the Golgi back to the endoplasmic reticulum (ER). How altered intracellular protein trafficking led to autoimmunity was unclear at the onset,1 however subsequent studies unveiled a critical role for failed retrieval of stimulator of interferon genes (STING) in pathogenesis.27 STING, a master regulator of innate immunity activated downstream of cytosolic DNA,26,27,29,30 requires ER to Golgi transport upon activation to initiate downstream signaling, including a potent type I interferon response.31 Termination of signaling requires transport out of the Golgi, either back to the ER, mediated by COPI, or to lysosomes for degradation.31 Of note, COPA does not bind STING directly, instead complexing with the intermediate cargo receptor protein surfeit 4 (SURF4).27,29
In the presence of pathogenic COPA mutations, activated STING accumulates in the Golgi, resulting in increased type I IFN signaling, altered T cell thymic selection, TH17 skewing, ER stress, and impaired autophagy.1,9,23,27,28 How these processes result in immune dysregulation and clinical symptoms remains poorly understood. Nevertheless, disease clearly depends on STING as knocking it out rescued mice from the embryonic lethality of a homozygous COPA E241K mutation.27 Data from humans further supports this link with STING as COPA syndrome shares many features with STING-associated vasculopathy with onset in infancy (SAVI), a Mendelian disorder driven by STING gain-of-function mutations.32 Interestingly, among monogenic type I interferonopathies ILD and alveolar hemorrhage are seen only in COPA syndrome and SAVI,33 suggesting interferon independent aspects of STING signaling may drive lung disease.
DETAILED DISEASE INVOLVEMENT BY ORGAN SYSTEM
Laboratory testing
Many patients with COPA syndrome have elevated inflammatory markers, with mild to moderately elevated erythrocyte sedimentation rate (ESR) seen in 95% of 22 reported patients, with results rarely above 90 mm/h. In contrast, C-reactive protein (CRP) was generally normal to mildly elevated, under 15 mg/L in 91% of 22 reported patients. Both ESR and CRP are reported to vary within an individual, with elevations somewhat tracking with disease activity, such as one patient with improvement but not the normalization of ESR a year after the resolution of severe arthritis.24
Levels of IFNα measured by ultra-sensitive digital ELISA were elevated in 100% of 6 patients, generally in the 150 to 1700 fg/mL range.5 Several patients had elevated interleukins (IL): IL-6 in 3 patients, IL-1β in one patient, and soluble IL2 receptor α in one patient. Additional results obtained from research-based testing included 100% of 12 patients having elevated interferon signatures,5,9,34–36 one of whom showed resolution with baricitinib treatment.9 An additional patient was reported to have elevated IFNα2.35
Autoantibodies
All but one patient with COPA syndrome had elevated ANA, ANCA, and/or RF/CCP. Autoantibody results were explicitly reported for 57 patients, 74% of whom were ANA positive, somewhat higher than the 67% to 70% reported previously.1,37 Titers, when reported, were almost universally 1:160 or higher, and often 1:640 or higher. Reported staining patterns were highly variable, including homogeneous, diffuse, and speckled appearances. ENAs were reported less frequently, though dsDNA, Smith, and RNP antibodies were each found in at least one patient. These can vary with time and disease activity, as demonstrated in an individual in which each of these antibodies was only identified once during serial testing over 14 months.38 In contrast, ANA positivity has been reported to be durable.
ANCA or MPO/PR3 were positive in 61% of reported patients, including several patients with negative MPO and PR3 testing in whom ANCA testing was not performed. Most ANCA-positive individuals were MPO and PR3 negative (70%), potentially explaining the discrepancy from prior ANCA positivity in 64% to 71% of patients.1,37 MPO was identified in eleven individuals, generally at high titer, while PR3 was seen in six individuals, including at low titer and with variable positivity.38 MPO and PR3 can fluctuate with disease activity, while ANCA remains positive.38
RF and/or CCP were positive in 65% of reported patients, similar to the 43% to 61% previously reported.1,37 When reported, titers were generally markedly elevated to over 100 IU/mL for RF and over 50 U/mL for CCP, with multiple results above the limit of detection.
In conclusion, a high titer ANA is a feature of COPA syndrome, while extractable nuclear antigens are not common. When serially evaluated, ANA remained positive, while ENAs showed significant variability, with none predicting disease activity. ANCA is often positive, and can be seen in isolation or associated with MPO more commonly than PR3. RF and CCP are also common, again at high titer. A relatively unique feature of COPA syndrome is the presence of multiple autoantibodies regardless of phenotype, with many individuals reported to have high titer ANA, ANCA, and RF/CCP.
Pulmonary Disease
One of the more unique aspects of COPA syndrome is pulmonary involvement, especially among monogenic type I interferonopathies where it is only seen in COPA syndrome and SAVI. ILD, however, can be an extra-articular feature of rheumatoid arthritis (RA), while pulmonary hemorrhage can be seen in ANCA-associated vasculitis (AAV) and, less commonly, in SLE. Presentation with a combined pulmonary-renal syndrome, a hallmark of AAV, has also been reported.3,13 Pulmonary involvement is nearly universally reported, seen in 98% of 58 individuals in which clinical descriptions are available. The sole unaffected individual was aged 9 years at the time of report, with prior publications identifying a lag of up to 20 years between the development of arthritis and ILD.3,35
Alveolar hemorrhage (AH) is often the earliest pulmonary manifestation, occurring as early as infancy.9 AH recognition is often delayed as hemoptysis is infrequently seen in these children, who are often diagnosed with pulmonary infections in the setting of respiratory symptoms and patchy ground glass opacities (GGOs) on imaging.7,39 When it occurs, hemoptysis is a major clue to AH diagnosis.7 Interestingly AH was more common in earlier reports about COPA syndrome,1,3 likely reflecting phenotypic expansion, something that typically follows the identification of a novel monogenic disorder, into individuals with less early or dramatic pulmonary presentations. AH may be severe, and children have presented with respiratory failure.2 In mechanically ventilated individuals, bronchoalveolar lavage (BAL) has been demonstrated to be a sensitive tool for detecting occult AH.3 Capillaritis was seen on biopsy in the setting of AH in 2 related and 2 unrelated individuals, with 3 additional patients demonstrating alveolar hemorrhage without capillaritis, potentially due to resolution while awaiting biopsy.2,3,7,11
Cough, tachypnea, shortness of breath, wheezing, and/or exercise intolerance are also common in patients with COPA syndrome, associated either with occult AH or with findings of ILD on imaging. ILD develops over time in COPA syndrome,3 potentially from recurrent hemorrhage and inflammation and potentially from a yet unidentified direct injury to the lungs from COPA mutations. It is important to recognize that ILD may be clinically silent in young children, even when prominent on imaging.36 Imaging features relatively unique to COPA syndrome include cystic changes, while GGOs, septal thickening, centrilobular nodules, lymphadenopathy (LAD), and fibrosis can also be seen in other clinical entities.3,11 Crazy paving has also been reported, though it was seen in fewer than 20% of patients.11 Imaging findings often match nonspecific interstitial pneumonia (NSIP) or lymphocytic interstitial pneumonia (LIP) patterns.2,3,11 The usual interstitial pneumonia (UIP) pattern of fibrosis has only been reported, in a conference abstract, in one individual: an atypical case of a woman who developed lung disease diagnosed as RA-ILD in her 50s whose COPA mutation was identified through sequencing lung transplant recipients.17
Pulmonary function testing (PFT) results in COPA syndrome are variable, most often mixed obstruction and restriction, followed by restriction, and only occasionally isolated obstruction.3 Results of diffusion capacity of the lung for carbon monoxide (DLCO) are infrequently reported and almost always reduced (93% of 14 patients). For individuals old enough to perform them, serial PFTs can help screen for clinically silent progressive pulmonary impairment.
Immune suppression is associated with improvement in GGOs, nodules, and LAD, but not in cystic changes or fibrosis.3,11 This has enormous clinical implications as lung disease, the major cause of death in COPA syndrome, can have subclinical progression.3,18,19 This progression has been documented in the context of multiple combinations of immune suppressive medications, including in patients receiving a janus kinase (JAK) inhibitor.37
Lung pathology, whether by surgical or transbronchial approaches, can show features of alveolar hemorrhage, as discussed above, with additional common patterns of follicular bronchiolitis (lymphoid follicles associated with bronchioles and the peri-bronchiolar interstitium) and airspace enlargement with cystic changes.1,3,12,40 Fibrosis patterns were in non-usual interstitial pneumonitis pattern (histologic NSIP), other than the above-mentioned adult-onset individual that was reported in a conference abstract to have UIP on imaging and histology.17 Histologic and radiographic patterns for individual patients are generally concordant.3 Three individuals had additional neuroendocrine cell abnormalities on biopsy, including neuroendocrine hyperplasia and a carcinoid tumor in a 56-year-old man12 and diffuse neuroendocrine cell hyperplasia (DIPNECH) in 2 individuals.2
Several patients with COPA syndrome have progressed to bilateral lung transplant (BLTx). Nine patients have been reported to undergo transplant, many with a limited duration of follow up. Patient outcomes vary from a standard post-transplant course2,41 to death, including 3 patients who passed away within the first post-transplant year9,18 and a fourth who passed away later in her post-transplant course.17 Two of these early deaths were from related individuals in Japan whose detailed peri-transplant courses are published.9,10 While there is a wide range of survival between lung transplant centers, 1- year survival in the United States is roughly 85%,42 higher than the 67% seen in these patients with COPA syndrome. In general, sicker patients have poorer transplant outcomes, and 2 post-transplant deaths in this COPA syndrome cohort were in patients bridged to transplant on long-term mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO),9,18 one of whom further required increased immune suppression due to sensitization and presence of a positive antibody crossmatch.18 The third early death was in a young boy with severe mycobacterial infection pre transplant.9 Therefore, it is not clear that outcomes for patients transplanted for COPA syndrome are inferior to those transplanted for other indications. While obviously limited by sample size, outcomes in COPA syndrome are better than in SAVI, where only one of the 5 BLTx recipients survived the first post-transplant year.10
The question of whether COPA syndrome recurs post-transplant is important. One of the Japanese transplant patients died from overwhelming infection, without recurrence of ILD, and the other had diffuse alveolar damage, associated with pulmonary hemorrhage, and mild bronchiolitis obliterans (BO), overall consistent with chronic lung allograft dysfunction (CLAD). Follicular bronchitis and cystic changes consistent with COPA syndrome were not seen.9,10 A third patient had CLAD on autopsy, after several documented infections and after treating cellular and antibody-mediated rejection.18 Detailed information was not reported for the fourth patient.
Overall, there is no definitive proof of COPA syndrome recurrence post-transplant, though immune dysregulation from COPA syndrome could have contributed to accelerated CLAD and/or heightened susceptibility or severity of post-transplant infections. More information about post-transplant clinical courses is needed, including the evaluation of interferon signatures and potentially trials of JAK inhibitors (see therapy section later in discussion). Future research is also needed to determine the extent to which ILD in COPA syndrome is lung intrinsic, driven by hematopoietic cells, or both, and whether combined lung and bone marrow transplants should be considered.
Arthritis
Joint symptoms are very common in COPA syndrome, and have been reported to appear before, after, or in conjunction with pulmonary disease. Of 44 patients for whom clinical data were available, 82% had joint symptoms, ranging from arthralgias to symmetric polyarthritis to deforming arthritis, both erosive and non-erosive. Joint involvement in 3 additional patients may have resulted from steroid exposure, including avascular necrosis in 2 individuals1,12 and a spinal compression fracture in a third.14 If these individuals are included, 89% of patients had musculoskeletal involvement.
Affected joints in patients with arthritis are variable, including proximal interphalangeal joints, metacarpophalangeal joints, wrists, ankles, knees, hips, and spine. Non-erosive arthritis was almost always seen, 87% of 15 patients for which the type was specified, but can be severely deforming.20 Fortunately, immune suppression of various sorts can be effective at controlling symptoms and debility from joint disease. Erosive disease which was only reported in 2 patients,11,39 including one in whom it evolved from non-erosive arthritis while he was lost to follow up and off therapy for 8 years.39
Renal Disease
Renal disease as the predominant presenting symptom of COPA syndrome is atypical but has been reported, with COPA syndrome suspected due to a known familial mutation. That patient did have pulmonary involvement, albeit asymptomatic, with alveolar hemorrhage seen on BAL.5,6 Published details on renal disease are sparser than for other main COPA syndrome manifestations, often limited to presence or absence and to biopsy results. Nephritis can, nevertheless, be severe, with the above individual and a second patient requiring renal transplantation.1,6 It is largely unclear from the literature whether kidney biopsies were performed to further evaluate abnormal laboratory testing or to obtain a tissue diagnosis in the setting of elevated autoantibodies. Within these limitations, 40% of 43 patients have renal involvement, representing 22% of the full patient cohort. Given underreporting on renal phenotypes, a prevalence rate of 40% is likely more accurate based on prior cohort studies reporting rates of 42% (9 of 21) and 47% (9 of 19).2,37
All renal biopsies reported show an immune-mediated glomerulopathy, though various histopathological patterns have been seen. Three patients showed crescentic glomerulonephritis with negative immune fluorescence, while 2 showed crescentic glomerulonephritis with positive immunofluorescence (one low positive mesangial IgM, IgG, and C3, and one with “full house” IgM, IgG, IgA, C3, and C1q).1,6,11,13 A sixth patient showed focal mesangial hypercellularity with C3 and C4 staining, a seventh showed IgA nephropathy, and an eight individual (with asymptomatic proteinuria biopsied as a potential kidney donor) had membranous glomerulonephritis.1,2,6 One individual with normal renal function underwent a partial nephrectomy for a nodule identified on imaging, finding clear cell carcinoma in the background of normal kidney tissue,12 a finding that may or may not relate to COPA syndrome.
Other Findings
Various additional clinical findings have been reported in 1 or 2 individuals with COPA syndrome. Neurologic involvement includes paroxysmal exertional dyskinesia,1 neuromyelitis optica,12,15 and disruptive behavior disorder.40 Gastrointestinal involvement includes severe gastroesophageal reflux necessitating surgery,24,41 hepatic cysts with cytolytic hepatitis,5 and persistent transaminitis.43 Cutaneous manifestations include leukocytoclastic vasculitis,1 chilblain lupus,15 polymorphic rash,19 eczema,34 and vitiligo.5 Immune and infectious manifestations include macrophage activation syndrome,5 thyroiditis,1 and bacterial and viral infections.9,12 Cardiac hypertrophy5 has also been reported. The extent to which these represent manifestations of COPA syndrome is unclear.
THERAPIES
There is no specific therapeutic at this time for COPA syndrome, and manifestations are treated with immune suppression as extrapolated from the care of other immune disorders with overlapping features. Life-threatening alveolar hemorrhage is generally controlled with pulse dose IV steroids that are subsequently converted to an oral maintenance regimen, with doses in many patients significantly weaned.3 Some patients have also received cyclophosphamide and/or intravenous immunoglobulin (IVIG). Various steroid-sparing agents have been reported, often as part of a 2- to 3-drug regimen using standard dosing, with variable success for any given agent or combination even among related individuals. No single regimen has emerged to be superior among standard immunosuppressives including corticosteroids, azathioprine, mycophenolate mofetil/mycophenolic acid, methotrexate, hydroxychloroquine, cyclophosphamide, cyclosporine, azithromycin, sirolimus, and IVIG. Many patients also received combination therapy with biologics, including tumor necrosis factor alpha inhibitors, IL-1 blockers, rituximab, and abatacept, again with varying success reported. It should be noted that many patients were able to achieve symptom remission with combination therapies, but without preventing ILD progression.
Jakinibs are a relatively new drug class of small molecule inhibitors of JAKs, kinases that activate intracellular signaling in response to surface receptor engagement by cytokines. Type I interferon signaling, clearly elevated in the blood of patients with COPA syndrome, is modulated by baricitinib, ruxolitinib, and upadacitinib, all of which have been used in patients with COPA syndrome.
Baricitinib was reported to require higher dosing in pediatric patients with other interferonopathies (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature and SAVI).44 Following this approach, Krtuzke and colleagues increased baricitinib dosing from 4 to 6 mg daily in a 10-year-old girl with refractory arthritis, resulting in rapid symptom resolution while the stabilization of lung disease occurred more gradually.24 This patient had previously failed multiple agents in combination therapy that included corticosteroids, methotrexate (continued alongside baricitinib), golimumab, etanercept, adalimumab, abatacept, tocilizumab, rituximab, leflunomide, and azithromycin. Success with baricitinib has been subsequently reported for 2 more patients, one of whom transitioned to upadacitinib for unspecified reasons.9,36
Ruxolitinib was described in an 11-year-old girl with recurrent steroid-refractive severe alveolar hemorrhage failing the addition of hydroxychloroquine, azathioprine, mycophenolate mofetil, and cyclophosphamide. She was transitioned to ruxolitinib 0.25 mg/kg twice daily, with steroids weaned within 3 months and exercise intolerance resolving. She was able to avoid hospitalization, in stark contrast to prior severe exacerbations requiring ECMO, though she required brief steroid courses for 2 mild exacerbations. Careful examination showed ongoing alveolar hemorrhage, with PFTs initially reported to be stable8 but with subsequent progression of fibrosis necessitating change to baricitinib and addition of IL-1 inhibition.37
In summary, patients may respond to standard immune suppression regimens, especially in the setting of life-threatening alveolar hemorrhage where other regimens have not been trialed. It is important to evaluate for occult progression of lung disease even when patients achieve disease control, with the consideration of escalation or change in therapy if identified. Jakinibs hold promise for COPA syndrome, at least in the short-term outcomes currently reported, though more clinical experience is needed before these can be broadly recommended. More robust results for jakinibs have been reported in SAVI, with most patients improving lung function though some patients did not respond or worsened.45–47 In COPA syndrome it remains to be seen whether these can durably control disease manifestations, with treatment periods longer than 1 year needed to truly evaluate ILD.
STING inhibitors, which are in late phase clinical development, may be a more promising drug class when available, targeting not only interferon-dependent but also interferon-independent functions of STING. Another potential treatment approach worth exploring is the adjunct use of antifibrotics, whether nintedanib, which is FDA approved to slow the progression of scleroderma ILD48 and, more broadly, of progressive pulmonary fibrosis,49 or pirfenidone, which has been shown to slow ILD progression in rheumatoid arthritis ILD.50
DISCUSSION AND RECOMMENDATIONS FOR TESTING
COPA syndrome is a relatively new genetic disease of immune dysregulation with common features of high autoantibody titers, pulmonary disease, and arthritis. Diagnosis is confirmed via sequencing of COPA. In Table 2, we outline a framework for when to consider testing an individual with lung disease for COPA syndrome. Familial testing of at-risk healthy individuals should be considered once a mutation is identified as there is a non-penetrance rate of at least 15%. Additional research is needed to inform the causes of disease non-penetrance. The hallmark of treatment is immune suppression, with active evaluation for progressive lung disease needed even if symptoms are otherwise controlled. Further understanding of COPA syndrome would benefit from patient registries to help clarify rates of disease prevalence and penetrance, to enrich the understanding of disease manifestations, and to guide treatments.
Table 2.
When to consider genetic testing for COPA syndrome in a patient with lung disease
| Pulmonary Features | Non-Pulmonary Features |
|---|---|
|
|
| Consider testing: | |
| At least 1 pulmonary and 2 non-pulmonary features -or- recurrent alveolar hemorrhage in infancy | |
| Strongly consider testing: | |
| At least 1 pulmonary and 3 non-pulmonary features | |
Many of the reported COPA mutations have not undergone experimental validation, and it remains to be determined whether all mutations cause disease through identical disease mechanisms. Future work is needed to characterize each reported mutation, including future novel variants obtained from sequencing additional individuals with lung disease.
SUMMARY
COPA syndrome is a recently described autosomal dominant inborn error of immunity characterized by high titer autoantibodies and interstitial lung disease, with many individuals also having arthritis and nephritis. Onset is usually in early childhood, with unique disease features including alveolar hemorrhage, which can be insidious, pulmonary cyst formation, and progressive pulmonary fibrosis in nonspecific interstitial pneumonia or lymphocytic interstitial pneumonia patterns. This review explores the clinical presentation, genetics, molecular mechanisms, organ manifestations, and treatment approaches for COPA syndrome, and presents a diagnostic framework of suggested indications for patient testing.
CLINICS CARE POINTS.
COPA syndrome is a rare autosomal dominant disorder characterized by high autoantibody levels and interstitial lung disease in the presence or absence of inflammatory arthritis and renal disease.
Prominent pulmonary manifestations of COPA syndrome include cough, tachypnea/exercise intolerance, alveolar hemorrhage (which can be insidious), respiratory failure, and interstitial lung disease characterized by cysts, centrilobular nodules, and/or pulmonary fibrosis.
Arthritis in COPA syndrome is indistinguishable from rheumatoid arthritis or juvenile arthritis.
Immune suppression is critical for patients with COPA syndrome.
JAK inhibitors may be an option to slow the progression of ILD, the most common cause of death for patients with COPA syndrome and typically the most refractory aspect of the disease.
ACKNOWLEDGMENTS
A.K. Shum is supported by NIH, United States grants R21AI160107 and R01AI168299, and the ATS/chILD Foundation, referring to the American Thoracic Society and to the Children’s Interstitial Lung Disease Foundation N. Simchoni is supported by NIH T32AR007304.
DISCLOSURE
T.P. Vogel has consulted for Novartis, Pfizer, Moderna, and SOBI and receives research support from AstraZeneca.
REFERENCES
- 1.Genomics BHC for M, Watkin LB, Jessen B, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 2015;47(6):654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vece TJ, Watkin LB, Nicholas SK, et al. Copa Syndrome: a Novel Autosomal Dominant Immune Dysregulatory Disease. J Clin Immunol 2016;36(4):377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsui JL, Estrada OA, Deng Z, et al. Analysis of pulmonary features and treatment approaches in the COPA syndrome. Erj Open Res 2018;4(2):00017–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan M, McDougall C, Walsh J, et al. G426 A Case report: Copa mutation – a new condition to consider with polyarthritis and interstitial lung disease. Arch Dis Child 2017;102(Suppl 1):A167. [Google Scholar]
- 5.Lepelley A, Martin-Niclós MJ, Bihan ML, et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J Exp Med 2020;217(11):e20200600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalifi SBE, Viel S, Lahoche A, et al. COPA Syndrome as a Cause of Lupus Nephritis. Kidney Int Reports 2019;4(8):1187–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathan N, Legendre M, Amselem S, et al. COPA syndrome restricted to life-threatening alveolar hemorrhages: clinical, pathological, molecular and biological characterization. Rare Ild Dpld 2018;PA2236. 10.1183/13993003.congress-2018.pa2236. [DOI] [Google Scholar]
- 8.Frémond ML, Legendre M, Fayon M, et al. Use of ruxolitinib in COPA syndrome manifesting as life-threatening alveolar haemorrhage. Thorax 2020;75(1):92. [DOI] [PubMed] [Google Scholar]
- 9.Kato T, Yamamoto M, Honda Y, et al. Augmentation of Stimulator of Interferon Genes–Induced Type I Interferon Production in COPA Syndrome. Arthritis Rheumatol 2021;73(11):2105–15. [DOI] [PubMed] [Google Scholar]
- 10.Matsubayashi T, Yamamoto M, Takayama S, et al. Allograft dysfunction after lung transplantation for COPA syndrome: A case report and literature review. Mod Rheumatology Case Reports 2022;6(2):rxac004. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen HN, Salman R, Vogel TP, et al. Imaging findings of COPA Syndrome. Pediatr Radiol 2023;1–10. 10.1007/s00247-023-05600-1. [DOI] [PubMed] [Google Scholar]
- 12.Taveira-DaSilva AM, Markello TC, Kleiner DE, et al. Expanding the phenotype of COPA syndrome: a kindred with typical and atypical features. J Med Genet 2019; 56(11):778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cabrera-Pérez JS, Branch J, Reyes A, et al. A Zebra at the Rodeo: Dyspnea, Hematuria, and a Family History of Arthritis. Arthrit Care Res 2022;74(2):165–70. [DOI] [PubMed] [Google Scholar]
- 14.Zeng J, Hao J, Zhou W, et al. A Novel Mutation c.841C>T in COPA Syndrome of an 11-Year-Old Boy: A Case Report and Short Literature Review. Frontiers Pediatrics 2021;9:773112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Tang Y, Zhang L, et al. Case report: COPA syndrome with interstitial lung disease, skin involvement, and neuromyelitis spectrum disorder. Frontiers Pediatrics 2023;11:1118097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delafontaine S, Bigley T, Iannuzzo A, et al. C-Terminal Domain COPA Mutations in Six Children From Three Unrelated Families With Autosomal Dominant COPA Syndrome. Gothenburg, Sweden: on October 2022;12:2022. [Google Scholar]
- 17.Beshay S, Smith J, Osuna I, et al. Copa syndrome-associated mutations in lung transplant recipients for interstitial lung disease. Chest 2021;160(4):A1265–6. [Google Scholar]
- 18.Riddell P, Moshkelgosha S, Levy L, et al. IL-6 receptor blockade for allograft dysfunction after lung transplantation in a patient with COPA syndrome. Clin Transl Immunol 2021;10(2):e1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensson BO, Hansdottir S, Arnadottir GA, et al. COPA syndrome in an Icelandic family caused by a recurrent missense mutation in COPA. BMC Med Genet 2017; 18(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banday AZ, Kaur A, Jindal AK, et al. Splice-site mutation in COPA gene and familial arthritis – a new frontier. Rheumatology 2020;60(1):e7–9. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Francioli LC, Goodrich JK, et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022;2022: 485034. [Google Scholar]
- 22.Rice GI, Rodero MP, Crow YJ. Human Disease Phenotypes Associated With Mutations in TREX1. J Clin Immunol 2015;35(3):235–43. [DOI] [PubMed] [Google Scholar]
- 23.Volpi S, Tsui J, Mariani M, et al. Type I interferon pathway activation in COPA syndrome. Clin Immunol 2018;187:33–6. [DOI] [PubMed] [Google Scholar]
- 24.Germany D of GP Centre for Paediatric Rheumatology, Clinic Sankt Augustin, Sankt Augustin, Krutzke S, Rietschel C, Germany D for PR Clementine Kinderhospital, Horneff G, Germany D of P and A medicine University Hospital of Cologne, Cologne. Baricitinib in therapy of COPA syndrome in a 15-year-old girl. European J Rheumatology 2019;7(1):78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bousfiha A, Moundir A, Tangye SG, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol 2022;42(7): 1508–20. [DOI] [PubMed] [Google Scholar]
- 26.Rivara S, Ablasser A. COPA silences STING. J Exp Med 2020;217(11): e20201517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng Z, Chong Z, Law CS, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med 2020;217(11): e20201045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng Z, Law CS, Ho FO, et al. A Defect in Thymic Tolerance Causes T Cell–Mediated Autoimmunity in a Murine Model of COPA Syndrome. J Immunol 2020;204(9):2360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukai K, Ogawa E, Uematsu R, et al. Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat Commun 2021;12(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steiner A, Hrovat-Schaale K, Prigione I, et al. Deficiency in coatomer complex I causes aberrant activation of STING signalling. Nat Commun 2022;13(1):2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeltema D, Abbott K, Yan N. STING trafficking as a new dimension of immune signaling. J Exp Med 2023;220(3):e20220990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frémond ML, Crow YJ. STING-Mediated Lung Inflammation and Beyond. J Clin Immunol 2021;41(3):501–14. [DOI] [PubMed] [Google Scholar]
- 33.Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 2022;22(8):471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan Y, Liu H, Tang X, et al. Effective sirolimus treatment of 2 COPA syndrome patients. J Allergy Clin Immunol Pract 2021;9(2):999–1001.e1. [DOI] [PubMed] [Google Scholar]
- 35.Bader-Meunier B, Bustaffa M, Iskounen T, et al. Rheumatoid factor positive polyarticular juvenile idiopathic arthritis associated with a novel COPA mutation. Rheumatology 2020;60(5):e171–3. [DOI] [PubMed] [Google Scholar]
- 36.Basile P, Gortani G, Taddio A, et al. A toddler with an unusually severe polyarticular arthritis and a lung involvement: a case report. BMC Pediatr 2022;22(1):639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frémond ML, Nathan N. COPA syndrome, 5 years after: Where are we? Joint Bone Spine 2021;88(2):105070. [DOI] [PubMed] [Google Scholar]
- 38.Psarianos P, Kwan JYY, Dell S, et al. COPA Syndrome (Ala239Pro) Presenting with Isolated Follicular Bronchiolitis in Early Childhood: Case Report. J Clin Immunol 2021;41(7):1660–3. [DOI] [PubMed] [Google Scholar]
- 39.Patwardhan A, Spencer CH. An unprecedented COPA gene mutation in two patients in the same family: comparative clinical analysis of newly reported patients with other known COPA gene mutations. Pediatric Rheumatology Online J 2019; 17(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prenzel F, Harfst J, Schwerk N, et al. Lymphocytic interstitial pneumonia and follicular bronchiolitis in children: A registry-based case series. Pediatr Pulmonol 2020;55(4):909–17. [DOI] [PubMed] [Google Scholar]
- 41.Mallea JM, Kornafeld A, Khoor A, et al. Lung Transplantation in a Patient with COPA Syndrome. Case Reports Transplant 2020;2020:3624795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valapour M, Lehr CJ, Schladt DP, et al. OPTN/SRTR 2021 Annual Data Report: Lung. Am J Transplant 2023;23(2):S379–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thaivalappil SS, Garrod AS, Borowitz SM, et al. Persistent Unexplained Transaminitis in COPA Syndrome. J Clin Immunol 2021;41(1):205–8. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Brooks KM, Tang CC, et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin Pharmacol Ther 2018; 104(2):364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez GAM, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128(7): 3041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frémond ML, Hadchouel A, Berteloot L, et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract 2021;9(2):803–18.e11. [DOI] [PubMed] [Google Scholar]
- 47.Gómez-Arias PJ, Gómez-García F, Hernández-Parada J, et al. Efficacy and Safety of Janus Kinase Inhibitors in Type I Interferon-Mediated Monogenic Auto-inflammatory Disorders: A Scoping Review. Dermatology Ther 2021;11(3): 733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Distler O, Highland KB, Gahlemann M, et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. New Engl J Med 2019;380(26):2518–28. [DOI] [PubMed] [Google Scholar]
- 49.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. New Engl J Med 2019;381(18):1718–27. [DOI] [PubMed] [Google Scholar]
- 50.Solomon JJ, Danoff SK, Woodhead FA, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir Medicine 2023;11(1):87–96. [DOI] [PubMed] [Google Scholar]
- 51.Brennan M, McDougall C, Walsh J, et al. COPA syndrome - a new condition to consider when features of polyarthritis and interstitial lung disease are present. Rheumatology 2017;56(suppl_6). 10.1093/rheumatology/kex356.059. [DOI] [Google Scholar]
- 52.Noorelahi R, Perez G, Otero HJ. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr Radiol 2018;48(2):279–82. [DOI] [PubMed] [Google Scholar]
- 53.Oliveira FR, Sciortino ADS, Nascimento Y, et al. Copa Syndrome in a 20-Year-Old Man: A Case Report. D23 Not Usual Suspects Case Reports Rare Lung Dis 2022;A5128. 10.1164/ajrccm-conference.2022.205.1_meetingabstracts.a5128. [DOI] [Google Scholar]