

Abstract
Background
The application of high throughput technologies has enabled unravelling of unique differences between healthy mares and mares with endometritis at transcriptomic and proteomic levels. However, differences in the uterine microbiome are yet to be investigated.
Objectives
The present study was aimed at evaluating the differences in uterine microbiome between healthy mares and mares with endometritis.
Methods
Low‐volume lavage (LVL) samples were collected from the uterus of 30 mares classified into healthy (n = 15) and endometritis (n = 15) based on their reproductive history, intrauterine fluid accumulation, gross appearance of LVL samples, endometrial cytology and bacterial culture. The samples were subjected to 16S rRNA sequencing.
Results
Notable differences in the uterine microbiome were observed between healthy mares and mares with endometritis at various taxonomic levels. In healthy mares, the most abundant phylum, class, order and family were Firmicutes, Bacilli, Bacillales and Paenibacillaceae, respectively. In contrast, the most abundant corresponding taxonomic levels in mares with endometritis were Proteobacteria, Gammaproteobacteria, Enterobacterales and Enterobacteriaceae, respectively. At the genus level, Brevibacillus and Paenibacillus were more abundant in healthy mares, whereas Escherichia, Salmonella and Klebsiella were more abundant in mares with endometritis. In healthy mares, Brevibacillus brevis was the most abundant species, followed by Brevibacillus choshinensis and Paenibacillus sp JDR‐2. However, in mares with endometritis, Escherichia coli was the most abundant species, followed by Salmonella enterica and Klebsiella pneumoniae.
Conclusions
These results confirmed the previously reported presence of a uterine microbiome in healthy mares and helped unravel some alterations that occur in mares with endometritis. The findings can potentially help formulate new approaches to prevent or treat equine endometritis.
Keywords: bacteria, equine, inflammation, metagenomics, uterus
Metagenomic analysis of uterine fluid samples provided further support to refute the dogma that the healthy mare uterus is sterile. It also helped unravel novel differences in the uterine microbiome between healthy mares and mares with endometritis. The findings of this study can serve as a basis for future research aimed at developing new diagnostic, preventative and therapeutic approaches for equine endometritis.

1. INTRODUCTION
Endometritis, defined as inflammation of the endometrium, is one of the major problems in the equine industry. In a survey of equine practitioners, endometritis was ranked as the third most common medical problem in equine practice after colic and viral respiratory tract disease (Traub‐Dargatz et al., 1991). About 25%–60% of barren mares were reported to have endometritis in different studies (LeBlanc & Causey, 2009; Morris et al., 2020). Equine breeders suffer severe economic losses annually due to reduced pregnancy rates associated with endometritis (Riddle et al., 2007) combined with the diagnostic, therapeutic and rebreeding costs (LeBlanc, 2010). Endometritis is classified into acute or chronic, depending on the duration, and infectious or non‐infectious, depending on the aetiology (Hurtgen, 2006; LeBlanc, 2010). Bacteria are the major cause of infectious endometritis in mares and the most common isolates reported in various studies include Streptococcus equi subspecies zooepidemicus, Escherichia coli, Pseudomonas aeruginosa, Klebsiella species and Staphylococcus species (Albihn et al., 2003; Díaz‐Bertrana et al., 2021; Frontoso et al., 2008; LeBlanc et al., 2007). Despite several decades of research, mechanisms underlying equine endometritis are yet to be fully elucidated. The application of high throughput technologies in the recent years has advanced our understanding of the differences between uterine health and disease at genomic (Weber et al., 2021) and proteomic (Diel De Amorim et al., 2020) levels. Similar studies on the uterine microbiome are required to elucidate the etiological basis of endometritis and the variations in predisposition to this condition between different mares.
The dogma that the healthy uterus is sterile has been challenged by research studies conducted in various species, including cattle (Moore et al., 2017), horses (Heil et al., 2018; Holyoak et al., 2022) and dogs (Lyman et al., 2019). Several bovine studies have demonstrated that the uterine microbiome is dynamic with remarkable differences reported between a healthy state and disease conditions such as endometritis (Ballas et al., 2021; Bicalho et al., 2017; Pascottini et al., 2020). To our knowledge, differences in the uterine microbiome between healthy mares and mares with endometritis have not been investigated yet. Therefore, the objective of the present study was to evaluate the differences in uterine microbiome between healthy mares and mares with endometritis.
2. MATERIALS AND METHODS
2.1. Animals
A total of 48 Thoroughbred mares (5–12‐year old, BCS 5–6) were examined during the breeding season to select 15 healthy mares and 15 mares with endometritis. The classification into healthy mares and mares with endometritis was based on breeding history and results of transrectal examination, low‐volume uterine lavage characteristics, endometrial cytology and bacterial culture. The examinations were conducted during oestrus, detected initially on the basis of behavioural signs and confirmed with transrectal ultrasonography. All examinations and sample collections were performed in the morning. Diagnosis of endometritis was based on a history of subfertility, excessive intrauterine fluid accumulation (>2 cm) noted on transrectal examinations, turbidity of low‐volume uterine lavage samples, inflammatory endometrial cytology (greater than 2 neutrophils per high power field or 1% neutrophil to epithelial cell ratio) and positive bacterial culture (Katila, 2016; LeBlanc & Causey, 2009).
2.2. Collection of low‐volume uterine lavage samples
After restraining each mare in stocks, the tail was wrapped, and the perineum was cleaned using soap and water. A low‐volume uterine lavage sample was collected transcervically from each mare using 100 mL sterile water and a sterile lavage assembly according to the procedure reported previously (Maloney et al., 2019). The samples were stored at −80°C until DNA extraction for metagenomic analysis. The samples from each group were pooled randomly using Microsoft Excel randomization function (pooled samples E1, E2, E3 and E4 from mares with endometritis and pooled samples H1, H2 and H3 from healthy mares; Table 1).
TABLE 1.
Clinical findings of healthy mares and mares with endometritis (n = 15 in each group).
| Mare ID (anonymised) | Metagenomic analysis sample pool | Intrauterine fluid accumulation (>2 cm) | Endometrial cytology (% neutrophils:epithelial cells) | Endometrial bacterial culture |
|---|---|---|---|---|
| Healthy mares | ||||
| a | H1 | No | 0.09 | Negative |
| b | H1 | No | 0.10 | Negative |
| e | H1 | No | 0.00 | Negative |
| f | H1 | No | 0.10 | Negative |
| h | H1 | No | 0.00 | Negative |
| c | H2 | No | 0.09 | Negative |
| d | H2 | No | 0.00 | Negative |
| j | H2 | No | 0.11 | Negative |
| l | H2 | No | 0.00 | Negative |
| o | H2 | No | 0.10 | Negative |
| g | H3 | No | 0.00 | Negative |
| i | H3 | No | 0.00 | Negative |
| k | H3 | No | 0.00 | Negative |
| m | H3 | No | 0.08 | Negative |
| n | H3 | No | 0.00 | Negative |
| Mares with endometritis | ||||
| B | E1 | Yes | 15.14 | Klebsiella spp. |
| D | E1 | Yes | 8.43 | Escherichia coli |
| E | E1 | Yes | 15.25 | Streptococcus spp. |
| F | E2 | Yes | 9.37 | Streptococcus spp. |
| G | E2 | Yes | 15.12 | Staphylococcus spp. |
| H | E2 | Yes | 10.72 | Escherichia coli |
| L | E2 | Yes | 11.85 | Streptococcus spp. |
| A | E3 | Yes | 9.14 | Klebsiella spp. |
| C | E3 | Yes | 13.84 | Escherichia coli |
| I | E3 | Yes | 14.22 | Escherichia coli |
| M | E3 | No | 18.79 | Staphylococcus spp. |
| J | E4 | Yes | 11.09 | Escherichia coli |
| K | E4 | No | 11.43 | Staphylococcus spp. |
| N | E4 | Yes | 8.77 | Escherichia coli |
| O | E4 | Yes | 17.17 | Staphylococcus spp. |
2.3. Metagenomic analysis
2.3.1. Library construction and sequencing
Total genomic DNA was extracted using the Qiagen DNeasy mini‐spin column (DNeasy Mini Kit, Qiagen) according to the manufacturer's instructions with molecular biology grade water as the negative control. The extracted DNA was quantified using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). For each PCR reaction, 20 ng of total DNA was used as a template. Amplification was performed with primers specific to V3–V4 regions of the bacterial 16S rRNA gene fragment (Forward 5′‐CCTAYGGGRBGCASCAG‐3′, Reverse 5′‐GGACTACNNGGGTATCTAAT‐3′). The amplification of 16S rRNA genes was conducted using the KAPA2G Robust HotStart ReadyMix PCR Kit (Kapa Biosystems) in a total volume of 25 µL. Amplification was performed with the following PCR conditions: initial denaturation at 95°C for 3 min, 5 cycles of 95°C for 15 s, 55°C for 15 s, 72°C for 30 s, 30 cycles of 95°C for 15 s, 62°C for 15 s and 72°C for 30 s, followed by a final extension at 72°C for 1 min using the grade PCR instrument BioRad T‐100. Amplified DNA was purified using AMPure XP (Beckman Coulter) and quantified by a NanoDrop 1000 (Thermo Fisher Scientific). Illumina TruSeq DNA PCR‐free Library Preparation Kits (Illumina) were used for library sequencing according to the manufacturer's protocol. Index codes were added to all the samples. The Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and the Agilent Bioanalyzer systems were used to assess library quality and the 250 bp paired‐end reads were obtained after sequencing on the Illumina HiSeq platform (Illumina).
2.3.2. Metagenome assembly, mapping and taxonomical assignment
MEGAHIT (Version 1.1.3), an assembler that can assemble large and complex metagenomics data, was used to assemble the processed reads. Minimum multiplicity for filtering (k_min+1)‐mers was set as 2 and minimum length of contigs to output was 200. The assembled reads were used for mapping using Mega BLAST against target database (nt_2018‐01‐22). Kraken2 (Version 2.1.1) was applied for taxonomical classification using the Kraken2 database pluspf2021‐05.
2.3.3. Statistical analysis
Data was analysed using QIIME (Quantitative Insights into Microbial Ecology). Alpha diversity indices (Simpson, Shannon and Chao1) were calculated to assess the variability of species within a sample, whereas beta diversity was calculated using Bray–Curtis dissimilarity to assess the differences in composition between samples. Principal component analysis (PCA) plot was used to visualize the variance between the samples.
3. RESULTS
Metagenomic analysis revealed the existence of diverse microbial communities in uterine samples from all mares included in the study (Tables 2 and 3). As shown in the PCA plot, there was a difference in the microbiome composition between healthy mares and mares with endometritis (Figure 1). The relative abundance of the various taxonomic levels in the pooled samples from the two groups of mares is shown in Figure 2 (panels a–g). The most abundant kingdom across all samples in both groups was bacteria (Figure 2a). In healthy mares, Firmicutes was consistently the major phylum followed by Proteobacteria. There was an exactly opposite trend in mares with endometritis where the major phylum was Proteobacteria followed by Firmicutes (Figure 2b). Bacilli were found to be the most abundant class followed by Gammaproteobacteria across all samples from healthy mares, whereas Gammaproteobacteria were the most abundant class followed by Bacilli across all samples from mares with endometritis. Other relatively less abundant classes in both groups included Clostridia, Betaproteobacteria and Alphaproteobacteria (Figure 2c). The most abundant order in healthy mares was Bacillales followed by Lactobacillales and relatively less abundant Clostridiales and Enterobacterales, whereas in mares with endometritis, the most abundant order was Enterobacterales followed by Xanthomonadales and Pseudomonadales (Figure 2d). Paenibacillaceae was the most abundant family across all samples from healthy mares followed by Streptococcaceae and Clostridiaceae. On the other hand, in mares with endometritis, the most abundant family was Enterobacteriaceae followed by Xanthomonadaceae and Moraxellaceae (Figure 2e).
TABLE 2.
Alpha diversity indices of the microbiome in uterine samples from healthy mares (H1–H3) and mares with endometritis (E1–E4).
| Sample | Simpson (1‐D) | Shannon (H) | Chao1 |
|---|---|---|---|
| H1 | 0.87 | 2.51 | 1827 |
| H2 | 0.95 | 3.95 | 2388 |
| H3 | 0.95 | 3.85 | 2113 |
| E1 | 0.87 | 2.38 | 1886 |
| E2 | 0.89 | 2.65 | 2083 |
| E3 | 0.88 | 2.49 | 2042 |
| E4 | 0.88 | 2.58 | 1868 |
TABLE 3.
Beta diversity of the microbiome in uterine samples from healthy mares (H1–H3) and mares with endometritis (E1–E4).
| Sample | H1 | H2 | H3 | E1 | E2 | E3 | E4 |
|---|---|---|---|---|---|---|---|
| H1 | 0 | 0.38 | 0.44 | 0.28 | 0.30 | 0.31 | 0.33 |
| H2 | 0.38 | 0 | 0.31 | 0.39 | 0.39 | 0.40 | 0.41 |
| H3 | 0.44 | 0.31 | 0 | 0.45 | 0.44 | 0.44 | 0.44 |
| E1 | 0.28 | 0.39 | 0.45 | 0 | 0.32 | 0.33 | 0.30 |
| E2 | 0.30 | 0.39 | 0.44 | 0.32 | 0 | 0.26 | 0.33 |
| E3 | 0.31 | 0.40 | 0.44 | 0.33 | 0.26 | 0 | 0.33 |
| E4 | 0.33 | 0.41 | 0.44 | 0.30 | 0.33 | 0.33 | 0 |
FIGURE 1.
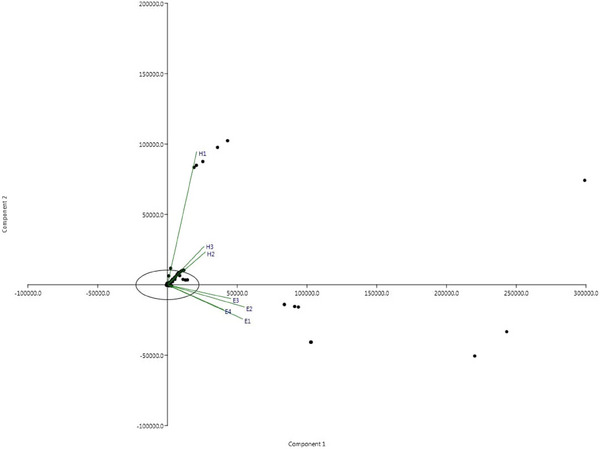
Principal component analysis (PCA) plot of the uterine proteome from healthy mares and mares with endometritis.
FIGURE 2.
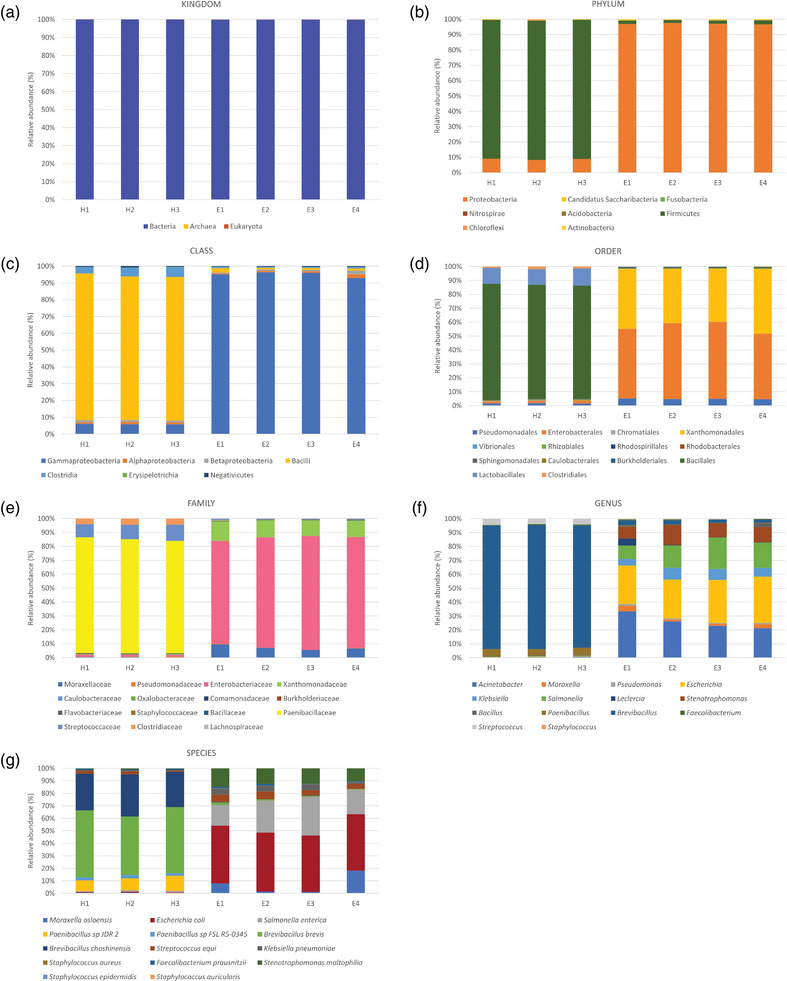
(Panels a–g) Taxonomical classification of the uterine proteome of pooled samples from healthy mares (H1–H3) and mares with endometritis (E1–E4).
At the genus level, Brevibacillus and Paenibacillus were more abundant across all samples from healthy mares, whereas Escherichia, Salmonella and Klebsiella were more abundant in mares with endometritis (Figure 2f). In healthy mares, Brevibacillus brevis was the most abundant species across all samples, followed by Brevibacillus choshinensis and Paenibacillus sp JDR‐2. However, in mares with endometritis, E. coli was the most abundant species, followed by Salmonella enterica and Klebsiella pneumoniae (Figure 2g).
As shown in Table 1, the traditional bacterial culture approach detected E. coli, Staphylococcus spp., Streptococcus spp. and Klebsiella spp. in 6, 4, 3 and 2 mares with endometritis, respectively. No bacterial growth was detected in any of the 15 healthy mares.
4. DISCUSSION
The detection of a diverse uterine microbiome in healthy mares in the present study, together with the findings of previous studies (Heil et al., 2018; Holyoak et al., 2022), strongly refutes the dogma of a sterile uterus in healthy mares. The composition of the uterine microbiome in the healthy mares in this study was mostly similar to that reported in a previous study in horses (Holyoak et al., 2022) and studies in other domestic animal species such as cows (Clemmons et al., 2017; Moore et al., 2017) and dogs (Lyman et al., 2019). Similar to the findings of Holyoak et al. (2022), Proteobacteria and Firmicutes constituted a vast majority of the uterine microbiome. However, there were some differences at the genus and species levels, which could be attributed to different geographical locations. The traditional bacterial culture approach used in the present study was able to detect only a few bacterial species in mares with endometritis and unable to detect any microbial growth in healthy mares. Importantly, the species detected using the traditional bacterial culture were also detected through the metagenomic approach with E. coli being the predominantly detected pathogen in each method. However, the metagenomic analysis also identified other species that were not detected on traditional bacterial culture. These findings underscore the importance of metagenomic approaches in unravelling the diverse resident microbiomes in healthy and diseased organ systems that could not be detected using traditional microbial culture methods.
The results showed notable differences in the uterine microbiome at various taxonomic levels between the two groups of mares. Interestingly, the major components of the uterine microbiome detected in healthy mares have been shown to be previously associated with defence mechanisms against pathogenic bacteria and fungi. Several studies have reported that Brevibacillus and Paenibacillus species produce a variety of peptides such as gramicidin and tostadin with documented broad‐spectrum antimicrobial activity (Jianmei et al., 2015; Song et al., 2012; Wu et al., 2011; Yang & Yousef, 2018). These findings suggest that the resident microbiome might have an important role in the uterine defence mechanism of mares and, consequently, their potential fertility.
Endometritis continues to be one of the major causes of infertility in mares and the findings of this study could potentially serve as a basis for novel diagnostic and therapeutic approaches to address this important problem. As suggested recently by Morrell and Rocha (2022), the interactions among various microbes rather than the presence of a single microbial species may play an important role in maintaining uterine health. The diverse uterine microbiome detected in the present study makes a strong case for potential inclusion of metagenomic profiling in the diagnostic work up of infertile mares. The findings also have potential implications for treating mares diagnosed with endometritis. Traditionally, mares with endometritis are treated with intrauterine antimicrobials in conjunction with approaches to improve uterine clearance such as uterine lavage and systemic oxytocin treatment (LeBlanc, 2010; Morris et al., 2020). Based on the findings of this study, re‐establishment of the resident microbiome using probiotics could be suggested as a potential adjunct to the traditional therapeutic approaches used for equine endometritis.
The pooling of uterine lavage samples from individual mares within each group in this study was necessitated by the high cost of metagenomic analysis for individual samples. Although an apparent limitation, the use of pooled samples has also been reported in previous microbiome research (Santos et al., 2011; Ray et al., 2019). The use of an Excel randomization function in the present study was intended to reduce any bias while assigning individual samples to the various pools within each group. A limitation of the 16S rRNA sequencing approach used in the present study is its inability to detect less abundant taxa in comparison to the relatively advanced and more sensitive metagenomic next generation sequencing (NGS) approach (Durazzi et al., 2021; Lamoureux et al., 2022). Future studies involving metagenomic NGS of individual samples are required to further our understanding of the uterine microbiome in healthy mares and mares with endometritis. It might also be worthwhile to investigate the effect of severity of endometritis on the uterine microbiome in mares. Another possible limitation of the study is the slight difference in number of mares within the various sample pools. However, as the comparisons between healthy mares and mares with endometritis were based on per cent relative abundances of the taxa rather than the absolute values, the difference in sample pool sizes is unlikely to have confounded the results of this study.
In conclusion, metagenomic analysis of uterine fluid samples confirmed the previously reported presence of a uterine microbiome in healthy mares and helped unravel some alterations that occur in mares with endometritis. The findings of this study can serve as a basis for future research aimed at developing new diagnostic, preventative and therapeutic approaches for equine endometritis. Future studies using better sequencing techniques such as metagenomic NGS are required to get more detailed information of the uterine microbiome in mares.
AUTHOR CONTRIBUTIONS
Conceptualization: Aeknath Virendra, Sarita U. Gulavane and Firdous A. Khan. Data curation: Aeknath Virendra. Formal analysis: Firdous A. Khan. Funding acquisition: Sarita U. Gulavane and Shailesh D. Ingole. Investigation: Aeknath Virendra, Zulfikar A. Ahmed, Ravi Reddy and Varsha D. Thorat. Methodology: Aeknath Virendra and Firdous A. Khan. Project administration: Sarita U. Gulavane and Firdous A. Khan. Resources: Sarita U. Gulavane, Zulfikar A. Ahmed and Shailesh D. Ingole. Software: Firdous A. Khan. Supervision: Sarita U. Gulavane, Zulfikar A. Ahmed, Varsha D. Thorat, Ravindra J. Chaudhari, Sandeep M. Gaikwad, Raju R. Shelar, Shailesh D. Ingole and Firdous A. Khan. Visualization: Aeknath Virendra, Afroza Khanam and Firdous A. Khan. Writing – original draft: Aeknath Virendra and Afroza Khanam. Writing – review and editing: Afroza Khanam and Firdous A. Khan.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
FUNDING INFORMATION
None.
ETHICS STATEMENT
This study was conducted at the Department of Animal Reproduction, Gynecology and Obstetrics, Mumbai Veterinary College located in Maharashtra, India. All animal handling and clinical procedures used in this study were approved by the Institutional Ethics Committee for Veterinary Clinical Research (BVC/IEC‐VCR/2021/dated 15/06/2021).
ACKNOWLEDGEMENTS
The authors are thankful to the equine farm owners for their participation and the Mumbai Veterinary College for providing research facilities and financial assistance to conduct the study.
Virendra, A. , Gulavane, S. U. , Ahmed, Z. A. , Reddy, R. , Chaudhari, R. J. , Gaikwad, S. M. , Shelar, R. R. , Ingole, S. D. , Thorat, V. D. , Khanam, A. , & Khan, F. A. (2024). Metagenomic analysis unravels novel taxonomic differences in the uterine microbiome between healthy mares and mares with endometritis. Veterinary Medicine and Science, 10, e1369. 10.1002/vms3.1369
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Albihn, A. , Båverud, V. , & Magnusson, U. (2003). Uterine microbiology and antimicrobial susceptibility in isolated bacteria from mares with fertility problems. Acta Veterinaria Scandinavica, 44(3–4), 121–129. 10.1186/1751-0147-44-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas, P. , Reinländer, U. , Schlegl, R. , Ehling‐Schulz, M. , Drillich, M. , & Wagener, K. (2021). Characterization of intrauterine cultivable aerobic microbiota at the time of insemination in dairy cows with and without mild endometritis. Theriogenology, 159, 28–34. 10.1016/j.theriogenology.2020.10.018 [DOI] [PubMed] [Google Scholar]
- Bicalho, M. L. S. , Lima, S. , Higgins, C. H. , Machado, V. S. , Lima, F. S. , & Bicalho, R. C. (2017). Genetic and functional analysis of the bovine uterine microbiota. Part II: Purulent vaginal discharge versus healthy cows. Journal of Dairy Science, 100(5), 3863–3874. 10.3168/jds.2016-12061 [DOI] [PubMed] [Google Scholar]
- Clemmons, B. A. , Reese, S. T. , Dantas, F. G. , Franco, G. A. , Smith, T. P. L. , Adeyosoye, O. I. , Pohler, K. G. , & Myer, P. R. (2017). Vaginal and uterine bacterial communities in postpartum lactating cows. Frontiers in Microbiology, 8, 1047. 10.3389/fmicb.2017.01047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz‐Bertrana, M. L. , Deleuze, S. , Pitti Rios, L. , Yeste, M. , Morales Fariña, I. , & Rivera Del Alamo, M. M. (2021). Microbial prevalence and antimicrobial sensitivity in equine endometritis in field conditions. Animals, 11(5), 1476. 10.3390/ani11051476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diel de Amorim, M. , Khan, F. A. , Chenier, T. S. , Scholtz, E. L. , & Hayes, M. A. (2020). Analysis of the uterine flush fluid proteome of healthy mares and mares with endometritis or fibrotic endometrial degeneration. Reproduction, Fertility, and Development, 32(6), 572–581. 10.1071/RD19085 [DOI] [PubMed] [Google Scholar]
- Durazzi, F. , Sala, C. , Castellani, G. , Manfreda, G. , Remondini, D. , & De Cesare, A. (2021). Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Scientific Reports, 11(1), 3030. 10.1038/s41598-021-82726-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontoso, R. , De Carlo, E. , Pasolini, M. P. , van der Meulen, K. , Pagnini, U. , Iovane, G. , & De Martino, L. (2008). Retrospective study of bacterial isolates and their antimicrobial susceptibilities in equine uteri during fertility problems. Research in Veterinary Science, 84(1), 1–6. 10.1016/j.rvsc.2007.02.008 [DOI] [PubMed] [Google Scholar]
- Heil, B. A. , Thompson, S. K. , Kearns, T. A. , Davolli, G. M. , King, G. , & Sones, J. L. (2018). Metagenetic characterization of the resident equine uterine microbiome using multiple techniques. Journal of Equine Veterinary Science, 66, 111. 10.1016/j.jevs.2018.05.156 [DOI] [Google Scholar]
- Holyoak, G. R. , Premathilake, H. U. , Lyman, C. C. , Sones, J. L. , Gunn, A. , Wieneke, X. , & DeSilva, U. (2022). The healthy equine uterus harbors a distinct core microbiome plus a rich and diverse microbiome that varies with geographical location. Scientific Reports, 12(1), 14790. 10.1038/s41598-022-18971-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtgen, J. P. (2006). Pathogenesis and treatment of endometritis in the mare: A review. Theriogenology, 66(3), 560–566. 10.1016/j.theriogenology.2006.04.006 [DOI] [PubMed] [Google Scholar]
- Jianmei, C. , Bo, L. , Zheng, C. , Huai, S. , Guohong, L. , & Cibin, G. (2015). Identification of ethylparaben as the antimicrobial substance produced by Brevibacillus brevis FJAT‐0809‐GLX. Microbiological Research, 172, 48–56. 10.1016/j.micres.2014.11.007 [DOI] [PubMed] [Google Scholar]
- Katila, T. (2016). Evaluation of diagnostic methods in equine endometritis. Reproductive Biology, 16, 189–196. [DOI] [PubMed] [Google Scholar]
- Lamoureux, C. , Surgers, L. , Fihman, V. , Gricourt, G. , Demontant, V. , Trawinski, E. , N'Debi, M. , Gomart, C. , Royer, G. , Launay, N. , Le Glaunec, J. M. , Wemmert, C. , La Martire, G. , Rossi, G. , Lepeule, R. , Pawlotsky, J. M. , Rodriguez, C. , & Woerther, P. L. (2022). Prospective comparison between shotgun metagenomics and sanger sequencing of the 16S rRNA gene for the etiological diagnosis of infections. Frontiers in Microbiology, 13, 761873. 10.3389/fmicb.2022.761873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc, M. M. , & Causey, R. C. (2009). Clinical and subclinical endometritis in the mare: Both threats to fertility. Reproduction in Domestic Animals = Zuchthygiene, 44, 10–22. [DOI] [PubMed] [Google Scholar]
- LeBlanc, M. M. , Magsig, J. , & Stromberg, A. J. (2007). Use of a low‐volume uterine flush for diagnosing endometritis in chronically infertile mares. Theriogenology, 68(3), 403–412. 10.1016/j.theriogenology.2007.04.038 [DOI] [PubMed] [Google Scholar]
- LeBlanc, M. M. (2010). Advances in the diagnosis and treatment of chronic infectious and post‐mating‐induced endometritis in the mare. Reproduction in Domestic Animals = Zuchthygiene, 45(Suppl 2), 21–27. 10.1111/j.1439-0531.2010.01634.x [DOI] [PubMed] [Google Scholar]
- Lyman, C. C. , Holyoak, G. R. , Meinkoth, K. , Wieneke, X. , Chillemi, K. A. , & DeSilva, U. (2019). Canine endometrial and vaginal microbiomes reveal distinct and complex ecosystems. PLoS ONE, 14(1), e0210157. 10.1371/journal.pone.0210157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney, S. E. , Khan, F. A. , Chenier, T. S. , Diel de Amorim, M. , Anthony Hayes, M. , & Scholtz, E. L. (2019). A comparison of the uterine proteome of mares in oestrus and dioestrus. Reproduction in Domestic Animals = Zuchthygiene, 54(3), 473–479. 10.1111/rda.13375 [DOI] [PubMed] [Google Scholar]
- Moore, S. G. , Ericsson, A. C. , Poock, S. E. , Melendez, P. , & Lucy, M. C. (2017). Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. Journal of Dairy Science, 100(6), 4953–4960. 10.3168/jds.2017-12592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell, J. M. , & Rocha, A. (2022). A novel approach to minimising acute equine endometritis that may help to prevent the development of the chronic state. Frontiers in Veterinary Science, 8, 799619. 10.3389/fvets.2021.799619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, L. H. A. , McCue, P. M. , & Aurich, C. (2020). Equine endometritis: A review of challenges and new approaches. Reproduction (Cambridge, England), 160(5), R95–R110. 10.1530/REP-19-0478 [DOI] [PubMed] [Google Scholar]
- Pascottini, O. B. , Van Schyndel, S. J. , Spricigo, J. F. W. , Rousseau, J. , Weese, J. S. , & LeBlanc, S. J. (2020). Dynamics of uterine microbiota in postpartum dairy cows with clinical or subclinical endometritis. Scientific Reports, 10(1), 12353. 10.1038/s41598-020-69317-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, K. J. , Cotter, S. Y. , Arzika, A. M. , Kim, J. , Boubacar, N. , Zhou, Z. , Zhong, L. , Porco, T. C. , Keenan, J. D. , Lietman, T. M. , & Doan, T. (2019). High‐throughput sequencing of pooled samples to determine community‐level microbiome diversity. Annals of Epidemiology, 39, 63–68. 10.1016/j.annepidem.2019.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle, W. T. , LeBlanc, M. M. , & Stromberg, A. J. (2007). Relationships between uterine culture, cytology and pregnancy rates in a thoroughbred practice. Theriogenology, 68(3), 395–402. 10.1016/j.theriogenology.2007.05.050 [DOI] [PubMed] [Google Scholar]
- Santos, T. M. , Gilbert, R. O. , & Bicalho, R. C. (2011). Metagenomic analysis of the uterine bacterial microbiota in healthy and metritic postpartum dairy cows. Journal of Dairy Science, 94(1), 291–302. 10.3168/jds.2010-3668 [DOI] [PubMed] [Google Scholar]
- Song, Z. , Liu, Q. , Guo, H. , Ju, R. , Zhao, Y. , Li, J. , & Liu, X. (2012). Tostadin, a novel antibacterial peptide from an antagonistic microorganism Brevibacillus brevis XDH. Bioresource Technology, 111, 504–506. 10.1016/j.biortech.2012.02.051 [DOI] [PubMed] [Google Scholar]
- Traub‐Dargatz, J. L. , Salman, M. D. , & Voss, J. L. (1991). Medical problems of adult horses, as ranked by equine practitioners. Journal of the American Veterinary Medical Association, 198(10), 1745–1747. [PubMed] [Google Scholar]
- Weber, K. S. , Wagener, K. , Blanco, M. , Bauersachs, S. , & Bollwein, H. (2021). A comparative analysis of the intrauterine transcriptome in fertile and subfertile mares using cytobrush sampling. BMC Genomics [Electronic Resource], 22(1), 377. 10.1186/s12864-021-07701-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. C. , Qian, C. D. , Fang, H. H. , Wen, Y. P. , Zhou, J. Y. , Zhan, Z. J. , Ding, R. , Li, O. , & Gao, H. (2011). Paenimacrolidin, a novel macrolide antibiotic from Paenibacillus sp. F6‐B70 active against methicillin‐resistant Staphylococcus aureus . Microbial Biotechnology, 4(4), 491–502. 10.1111/j.1751-7915.2010.00201.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , & Yousef, A. E. (2018). Antimicrobial peptides produced by Brevibacillus spp.: Structure, classification and bioactivity: A mini review. World Journal of Microbiology and Biotechnology, 34(4), 57. 10.1007/s11274-018-2437-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.