

Abstract
Environmental chemicals such as inorganic arsenic (iAs) significantly contribute to redox toxicity in the human body by enhancing oxidative stress. Imbalanced oxidative stress rapidly interferes with gut homeostasis and affects variety of cellular processes such as proliferation, apoptosis, and maintenance of intestinal barrier integrity. It has been shown that gut microbiota are essential to protect against iAs3+-induced toxicity. However, the effect of microbial metabolites on iAs3+ induced toxicity and loss of gut barrier integrity has not been investigated. The objectives of the study are to investigate impact of iAs on gut barrier function and determine benefits of gut microbial metabolite, urolithin A (UroA) against iAs3+-induced adversaries on gut epithelium. We have utilized both colon epithelial cells and in a human intestinal 3D organoid model system to investigate iAs3+ -induced cell toxicity, oxidative stress and gut barrier dysfunction in the presence or absence of UroA. Here, we report that treatment with UroA attenuated iAs3+-induced cell toxicity, apoptosis, and oxidative stress in colon epithelial cells. Moreover, our data suggest that UroA significantly reduces iAs3+-induced gut barrier permeability and inflammatory markers in both colon epithelial cells and in a human intestinal 3D organoid model system. Mechanistically, UroA protected against iAs3+-induced disruption of tight junctional proteins in intestinal epithelial cells through blockade of oxidative stress and markers of inflammation. Taken together, our studies for the first time suggest that microbial metabolites such as UroA can potentially be used to protect against environmental hazards by reducing intestinal oxidative stress and by enhancing gut barrier function.
Keywords: Microbiota, Microbial Metabolites, Urolithin A, Arsenic, Oxidative Stress, Gut barrier function
Introduction
The gastrointestinal (GI) tract harbors trillions of microbes (microbiota), which maintain a symbiotic relationship with the host in part by regulating essential metabolic processes. Microbes and their dietary metabolites strongly influence the homeostasis within the gut and are critical for the maintenance of an intact gut barrier [1]. The use of gut microbial metabolites has emerged as a novel treatment strategy to restore gut barrier homeostasis. Current evidence reveals that numerous environmental factors induce microbial dysbiosis in the human gut leading to loss of beneficial commensal microbiota and potentially their metabolites [2]. Altogether, such effects ultimately contribute to the etiology and severity of several diseases. Ongoing studies are defining the importance of microbiota in ameliorating adverse outcomes of exposure to environmental pollutants [3]. For example, GI microbiota can metabolize environmental chemicals such as metals (arsenic, bismuth etc.), polycyclic aromatic hydrocarbons (PAH), polychlorobiphenyls and pesticides [4, 5].
Inorganic arsenic induced toxicity causes major damage to human health following chronic or acute exposure. In the environment, trivalent (iAs3+) and pentavalent (iAs5+) forms of inorganic arsenic are abundant. Trivalent iAs3+ is more toxic to humans compared to iAs5+ [6, 7]. Chronic exposure to iAs3+ leads to an increase in both the onset and progression of several human diseases targeting multiple organs (liver, kidney, bladder, skin, intestines, and central nervous system), which is referred to as ‘arsenicosis’ [8–14]. Arsenic exposure is a major public health concern effecting over 225 million people globally [15, 16] and classified as a group I carcinogen by the International Agency for Research on Cancer (IARC). The susceptibility to arsenicosis varies among individuals despite similar level of arsenic exposure, which is attributed to host genetics, age, gender, and dietary habits [17, 18]. Interestingly, gastrointestinal symptoms and distress have been widely documented in human subjects exposed to arsenic chronically, acutely, or therapeutically. In a study on a chronically arsenic-exposed population from West Bengal, India, ~60% of all the study participants presented gastrointestinal symptoms [19]. In addition, gastrointestinal distress is a well-characterized effect of acute arsenic poisoning [20, 21]. A variety of clinical studies on subjects treated with As2O3 demonstrate that gastrointestinal symptoms are common side effects of such therapeutic use [22, 23].
Recent studies highlight the importance of gut microbiota in modulating As-induced toxicity [24, 25]. Studies have reported that populations exposed to iAs3+ exhibit gut microbial dysbiosis [26–28]. Importantly, arsenic exposure in mice following the depletion of microbiota or in germ free mice led to significant arsenic accumulation in tissues with concomitant reduction in its excretion [25]. The gut microbiome significantly influences arsenic metabolism and the outcome of arsenicosis [24, 29–32]. Though microbial dysbiosis is often reported in iAs3+ exposed individuals or in animal models [33–36], the functional dynamics of human gut microbiota in arsenic toxicity remain poorly explored.
Gut microbes modulate several facets of arsenic metabolism including the status of oxidation, methylation, thiolation, bioavailability and excretion [11]. Oxidative stress is a potent mechanism by which iAs3+ exposure induces its toxic effects, especially at high doses [37]. Such heightened oxidative stress causes cell damage, cell death and loss of cellular homeostasis [38]. Moreover, physiological imbalance of oxidative stress is also related to impairment of gut barrier function, which contributes to the pathogenesis of many GI-related diseases [39]. It has been demonstrated employing cell line models that acute exposure to both iAs3+ and iAs5+ leads to generation of oxidative stress along with a concomitant loss of tight junction protein ZO-1, resulting in alteration of membrane permeability [35, 40]. Therefore, inhibition of oxidative stress-induced loss of barrier integrity might be a crucial therapeutic target for arsenic induced toxicity.
Recent emerging studies suggest that microbial metabolites as promising therapeutics against the loss of gut barrier integrity [1, 41, 42]. However, it is not well understood whether gut microbial metabolites have protective effects against arsenic-induced oxidative stress and gut barrier dysfunction. Therefore, we undertook examination of one such beneficial microbial metabolite, urolithin A (UroA), which we previously demonstrated has gut barrier protective and anti-inflammatory activities [41, 43]. UroA (3,8-dihydroxybenzo[c]chromen-6-one) is derived from ellagic acid and ellagitannins (major poly phenolic components in berries and pomegranate) by gut commensal bacteria [44]. UroA exhibits a high rate of intestinal absorption [45] and exerts anti-inflammatory, anti-oxidative, and anti-ageing activities [43, 46–49]. The current study reports the efficacy of UroA against adverse effects of trivalent arsenic on human gut epithelia and potential mechanisms of action. These studies will pave a pathway for preventive and therapeutic applications to mitigate arsenic-elicited adverse health effects, which are currently unavailable.
Materials and Methods
2.1. Reagents and chemicals
General laboratory chemicals and reagent solutions were purchased from Sigma-Aldrich (St. Louis, MO) or VWR (Radnor, PA). ELISA kits for IL-8 and was procured from Bio-legend (San Diego, CA). Annexin V: FITC Apoptosis Detection Kit was purchased from BD biosciences (San Jose, CA). JC-1 Mitochondrial Membrane Potential Assay Kit was from Cayman chemicals (Ann Arbor, MI). LDH-Glo™ Cytotoxicity Assay Kit and GSH/GSSG-Glo™ Assay were procured from Promega (Madison, WI). All antibodies were purchased from Santacruz unless otherwise specified. UroA was custom synthesized as previously described [41, 43]. Sodium arsenite (NaAsO2, CAS 7784–46-5, > 99% pure) was obtained from Spectrum Chemical (New Brunswick, NJ). Sodium arsenite was used as a source of iAs3+ in all the experiments. Sodium arsenite was handled in accordance with the NIH Guidelines for the Use of Chemical Carcinogens. List of the antibodies and respective dilution for each method are provided in Table 1.
Table 1:
List of Antibodies:
| Antibody | Company | Catalogue number | Method* | Dilution |
|---|---|---|---|---|
| ZO-1 Antibody | ProteinTech | A21773-1-AP | WB IHC | 1:1000, 1:100 |
| claudin-4 Antibody (A-12) HRP | SCBT | sc-376643 HRP | WB | 1:500 |
| Occludin Rabbit Polyclonal antibody | ProteinTech | 3409-1-AP | WB, IHC | 1:1000, 1:100 |
| HRP-Conjugated Beta Actin Antibody | ProteinTech | RP-60008 | WB | 1:5000 |
| Goat anti-mouse IgG (H+L), HRP conjugate | ProteinTech | SA00001-1 | WB | 1:5000 |
| Goat anti-rabbit IgG (H+L), HRP conjugate | ProteinTech | SA00001-2 | WB | 1:5000 |
| IgG2a Cross-Adsorbed Goat anti-Mouse, Alexa Fluor® 594, Invitrogen™ | Invitrogen | A21135 | IHC | 1:200 |
| IgG (H+L) Cross-Adsorbed Goat anti-Rabbit, Alexa Fluor® 488, Invitrogen™ | Invitrogen | A11008 | IHC | 1:200 |
| Claudin-4 Antibody (A-12): sc-376643 | SCBT | sc-376643 | IHC | 1:50 |
WB: Western blot; IHC: Immunohistochemistry
2.2. Cell culture
The human colon epithelial carcinoma cell line T84 (ATCC # CCL-248™)) was maintained in DMEM: F-12 Medium (Cytiva # SH30261.01), supplemented with 10% fetal bovine serum, 1X penicillin-streptomycin solution (100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma Aldrich) in a humidified atmosphere (at 37 °C in a 5% CO2 incubator). The cells were seeded in 96-, 12- or 6-well plates at a density of 1 × 105 cells/mL using volumes of 100 μL, 1 mL, or 2 mL respectively, and cells were allowed to adhere overnight.
2.3. Monolayer Culture
Human colon epithelial cell monolayers were generated using T84 cells. The cells (2 × 104 cells/cm2) were seeded in 24-well Transwell® plates (Corning; USA) on 6.5 mm tissue diameter (surface area= 0.33 cm2) 0.4 μm pore sized polyester membrane filters. Culture medium was added to both apical and basal chambers and the medium was changed every other day up to 12 days for T84 cells. Each monolayer was assessed for the development of trans-epithelial electrical resistance (TEER) using a voltmeter (EVOM2, Epithelial Volt/Ohm meter, World Precision Instruments, Inc., Sarasota, Florida, USA) and STX2 electrode (World Precision Instruments, Inc., Sarasota, Florida, USA). T84 cell monolayers showing TEER more than 1000 Ω.cm2 were used for permeability studies.
2.4. Primary human small intestinal epithelial cultures
EpiIntestinal 3D microtissues (SMI-100 and SMI-196) were obtained from MatTek Corporation (Ashland, MA, USA) and cultured according to the manufacturer’s instructions (24-well and 96-well format respectively) in a specially formulated culture medium (MatTek Corporation). Normal human 3D Small Intestinal Epithelium (SMI-196) were custom grown on 0.19 cm tissue diameter (surface area= 0.11 cm2) 0.4 μm pore sized Transwell™ inserts. For SMI-100, individual tissues were custom grown on 9 mm tissue diameter (surface area= 0.6 cm2) 0.4 μm pore sized Transwell™ inserts. All intestinal tissues for these studies were derived from a single healthy donor. Briefly, intestinal tissue cultured on Transwell™ inserts were received in agarose packaging and immediately transferred to a 96-well tissue culture plate containing 250 μl pre-warmed maintenance medium (SMI-100-MM). Following transfer to the 96-well plate, 20 μl SMI-100-MM was added to the apical surface and intestinal cultures, heretofore referred to as primary human small intestinal epithelial barriers, were incubated overnight in a 37 °C in a 5% CO2 incubator. For SMI-100, tissues were cultured with 5ml medium in HNG-TOP-12 plate supplied by manufacturer with 100 μL of media on apical side as per instructions. Each primary human small intestinal epithelial barrier was assessed for the development of TEER using an EVOM2 voltmeter. An STX100 electrode (World Precision Instruments, Inc., Sarasota, Florida, USA) was used for measurement of TEER in 96-well format and an STX2 electrode was used for 24 well format.
2.5. Cell viability
T84 cells were seeded at a density of 2×104 cells/well in 96 well plates (quadruplicate for each assay condition). 24 h post-seeding, the cells were treated with increased doses of iAs3+ (0–100 μM) in the presence of UroA at indicated doses. Treatments were carried out for 24, 48 and 72 h respectively, followed by removal of arsenic containing media and replacement with fresh media (180 μL) containing alamar blue reagent (20 μL/well). Cells were allowed to incubate for 4 h with the alamarBlue reagent (ThermoFisher Scientific, Rochester, NY) at 37 °C and readings were taken (OD at 570 nm) subsequently as per manufacturer’s instruction as described previously [50].
2.6. Determination of apoptosis by the annexin V/PI assay
The Annexin V-FITC Apoptosis Detection Kit with PI from BD Biosciences was used to identify cells undergoing apoptosis. The protocol was carried out as per instructions by the manufacturer. Then, a minimum of 30,000 cells per sample were acquired by flow cytometry (BD FACSCanto, BD biosciences, USA). Data was analyzed using the FlowJo v10 software.
2.7. Measurement of reactive oxygen species generation
Reactive oxygen species (ROS) generation was determined by utilizing an oxidation-dependent fluorescence dye with 6-carboxy-2′,7′-dichlorodihydroxy fluorescein diacetate (DCFDA). After 30 min incubation with DCFDA (2 μg/ml) in a 37°C 5% CO2 incubator, cells were washed twice with PBS. The ROS generation as green fluorescence was measured at excitation and emission wavelengths of 485 nm and 535 nm respectively by a Synergy HT Microplate Reader (Biotek, VT, USA), fluorescence imaging by Sapphire Biomolecular Imager (Azure Biosystems, USA), and flow cytometry (BD FACSCanto, BD biosciences, USA). Quantification of images was done using AzureSpot (Azure Biosystems, USA) and flow cytometry data was analyzed using the FlowJo v10 software.
2.8. Determination of GSH/GSSG
The ratio of reduced GSH and oxidized glutathione (GSSG) was quantified by using a GSH/GSSG-Glo™ Assay Kit (Promega, WI, USA). The samples were prepared according to the manufacturer’s manual and the change in luminescence recorded using a microplate reader (FLUOstar Omega, BMG Labtech).
2.9. Lactate Dehydrogenase (LDH) release assay
The release of LDH in the cell culture medium was determined with an LDH-Glo™ Cytotoxicity Assay Kit (Promega) as per manufactures instruction. The change in the luminescence was recorded using a microplate reader (FLUOstar Omega, BMG Labtech). LDH release (% control) was calculated from the equation:
2.10. Measurement of mitochondrial membrane potential (Δψ).
JC-1 mitochondrial membrane potential assay kits (Cayman Chemicals, MI, USA) were used to measure the mitochondrial membrane potential according to the manufacturer’s protocol. JC-1 activity related change in fluorescence was measured either in 96 well black plates using a Synergy HT Microplate Reader (Biotek, VT, USA) or fluorescence imaging using a Sapphire Biomolecular Imager (Azure Biosystems, USA). Fluorescence imaging was performed in 96 well clear bottom black plates and 8-well chambered slides (Nunc® Lab-Tek™ II Chamber Slide™ System). JC-1 aggregates (Red fluorescence) were measured at excitation and emission wavelengths of 540 nm and 570 nm respectively. JC-1 monomers (Green fluorescence) were measured at excitation and emission wavelengths of 485 nm and 535 nm respectively. Changes from red to green fluorescence indicate the decline in cell health as the monomeric form of JC-1 represents depolarization. The ratio of fluorescent intensity of J-aggregates and J-monomers (Red: Green) was used as an indicator of cell health. Quantification of image was done using AzureSpot (Azure Biosystems, USA).
2.11. In vitro permeability study
For in vitro cellular permeability studies, T84 cell monolayers or EpiIntestinal 3D microtissue were treated as required for each experiment. After treatment, the monolayer or microtissue was washed with PBS twice and 200 μL of FITC-Dextran (FD-4; Sigma Aldrich, USA) solution (1 mg/mL in medium) was added. After 2 h incubation in a 37 °C 5% CO2 incubator, a 100 μL sample from the basal chamber was analyzed for FD4 concentration. The change in fluorescence was determined along with FD4 standard dilutions using a Synergy HT Microplate Reader (Biotek, VT, USA).
2.12. Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was isolated from cells using Maxwell® 16 LEV simplyRNA tissue kits (Promega) and reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, CA, USA). The transcribed cDNA (after dilution) was mixed with 100 nM gene specific primers (Real time primers LLC) and 1X SYBR green reaction mix (PowerUp™ SYBR™ Green Master Mix; Applied Biosystems, CA, USA). Changes in gene expression of ZO-1, Occludin and Cldn4 were analyzed using CFX96TM Real-Time System (Bio Rad) and fold change in expression was calculated using the −ΔΔCT method using GAPDH/β-actin as a housekeeping gene control and normalized to the untreated control.
2.13. Immunohistochemistry
T84 cells (50,000 cells/well) were grown overnight on to 8-well chambered slides (Nunc® Lab-Tek™ II Chamber Slide™ System). The next day, the cells were induced with vehicle (0.01% DMSO) or iAs3+ (5 μM or 10 μM) with or without UroA (25 μM) for 24h and fixed with chilled methanol. The respective proteins were stained in the cells with primary anti-bodies (1:200 dilution) for ZO-1, Occludin, or Cldn4 followed by fluorescently labeled (Alexa flour 594 for Cldn4 and Alexa flour 488 for ZO-1 and Occludin) secondary antibodies (1:500 dilution; ThermoFisher Scientific). Stained cells were mounted with VECTASHIELD HardSet™ antifade mounting medium with DAPI (Vector Laboratories). The confocal images were captured using a Nikon A1R confocal microscope using ×60 magnification lenses with appropriate laser channels.
2.14. Western blot analysis
For immunoblotting, total protein lysates were collected and processed as described [41]. The immunoblots were probed with ZO-1, Occludin, Cldn-4 and GAPDH antibodies followed by respective secondary antibody conjugated with horseradish peroxidase (Proteintech, IL, USA). The protein bands were detected with chemiluminescent substrate and imaged using a Sapphire Biomolecular Imager (Azure Biosystems, USA). Densitometry analysis of bands were done using ImageJ software [51]. Anti-bodies for ZO-1, Occludin and GAPDH were purchased from Proteintech and Cldn4 from Santa Cruz Biotechnologies (USA).
2.15. Measurements of cytokines
For measurement of cytokine production via ELISA, after experimental time points and appropriate doses, the supernatant was collected form T84 monolayer or EpiIntestinal 3D microtissue. Cytokines were quantified from supernatant using human TNF-α and IL-8 specific ELISA kits (Biolegend) following manufacturer’s instruction.
2.16. Histopathology and immunohistochemistry for EpiIntestinal 3D microtissue.
The EpiIntestinal 3D microtissue were fixed in 10% buffered formaldehyde solution overnight followed by 70% alcohol. Fixed tissues were subjected to standard histopathological processing for paraffin embedding and 5μm paraffin sections were cut by Horus Scientific (MA, USA).
For ZO-1 staining, paraffin-embedded slides were processed as per standard methodology as described elsewhere [52]. ZO-1 was stained with primary anti-bodies (1:200 dilution) followed by Alexa flour 488 secondary antibody (1:500 dilution). Stained sections were mounted with VECTASHIELD HardSet™ antifade mounting medium with DAPI (Vector Laboratories). The confocal images were captured using a Nikon A1R confocal microscope using ×60 magnification lenses with appropriate laser channels.
2.17. Intracellular arsenic measurements:
Intra-cellular arsenic levels in T-84 cells treated with or without UroA in the presence of As3+ were determined from cell extracts. Briefly, T-84 cells were grown in 6-well plates up to 90–95% confluence and then treated with vehicle (0.05% DMSO) or iAs3+ (5 μM) or UroA (25 μM) or iAs3+ (5 μM) + UroA (25 μM) in quadruplicates for 24 h. The cells were washed twice with ice cold PBS and lysed using a solution of 10 mM Tris−HCl pH 7.4, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate (SDS), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM sodium vanadate and 1X protease inhibitor cocktail (Thermo Fisher Scientific). Lysates were sonicated and centrifuged at 4 °C for 15 min to remove insoluble debris. The protein was estimated using Pierce-BCA protein assay kit (ThermoFisher Scientific). Samples were diluted at 1 μg/μl in lysing solution and total of 50 μg of protein was used to determine arsenic levels. The protein extracts (50 μg in 50 μl) were mixed with 300 μl of 70% nitric acid (Fisher Scientific Cat#A509P500) and incubated in a shaking incubator at 65 °C for 3 h. Next, the samples were allowed to cool in the hood and then mixed with 100 μl of 30% H2O2 (Sigma Cat#95321) followed by incubation for an additional 3 h in a shaking incubator at 65 °C. The digested samples were allowed to cool down to room temperature and each sample was added to 4 ml of deionized (DI) water to bring the total volume up to 4.45 mL. Total arsenic was assayed using an Agilent 7800 ICP-MS instrument. The read counts were converted to concentrations by employing a standard curve (0–100 ppb), which was obtained by serial dilution of a commercially available standard stock solution (Cat # IV-STOCK-50, Inorganic Venture, Virginia USA). The limit of detection of arsenic for this instrument is 0.01 ppb.
2.18. Statistical analyses
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, San Diego, USA) using ANOVA followed by Tukey’s Multiple Comparisons Test with p < 0.05 taken as significant. Details of the specific statistical tests are provided in the figure legends. Data are shown as mean ± SEM from triplicate determinations, unless otherwise stated in the figure legends.
All authors had access to the study data and had reviewed and approved the final manuscript.
Results
3.1. UroA protects against iAs3+ induced cytotoxicity.
The cytoprotective activities of UroA against iAs3+-induced cytotoxicity were examined in colon epithelial cells. Briefly, T84 cells (colon epithelial cells) were treated with iAs3+ (0.1–100 μM) in the presence or absence of UroA (25 μM) for 24 h (Figure 1A), 48 h (Figure 1B), or 72 h (Figure 1C) and cell viability were evaluated. As shown in Figure 1, treatment with iAs3+ induced significant cytotoxic effects at all three time points studied. However, co-treatment with UroA significantly protected the cells against iAs3+−induced toxicity at each time point. Unexpectedly, UroA significantly protected the colon epithelial cells against even the highest dose of iAs3+ (100 μM) and the longer exposure time (72 h). These results suggest that microbial metabolites such as UroA has beneficial activities against iAs3+ induced cytotoxicity. The experiments were performed using micromolar concentrations because UroA serum levels can reach up to micromolar levels without displaying toxicity in humans upon consumption of ellagic acid (EA)- or ellagitannins (ET)-rich diets [45, 53–56]. UroA levels in humans vary significantly among individuals based on dietary habits of consumption of EA/ET-containing diets such as pomegranate juice, walnuts and berries and the presence of UroA-producing bacteria. We and others have tested the effects of UroA at 1, 5, 10, 25 and 50 μM doses and reported anti-inflammatory activities, induction of tight junction proteins, and mitophagy in in vitro models [41, 48].
Figure 1:
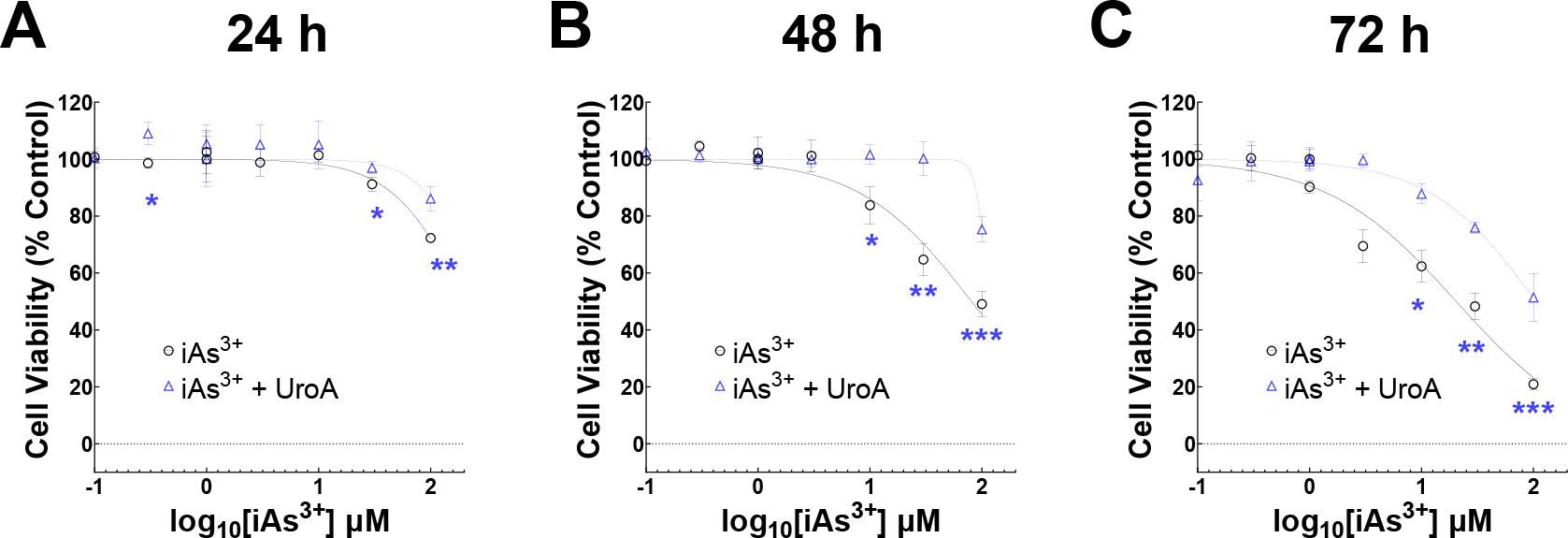
UroA protects against iAs3+ induced cytotoxicity in colon epithelial cells. T84 cells (2 × 104) per well were grown O/N in 96 well plate. Next day, cells were treated with iAs3+ (0.1, 0.3, 1, 3, 10, 30, 100 μM) in the presence of UroA (25 μM) or vehicle (DMSO, 0.05%) for (A) 24 h, (B) 48 h and (C) 72 h. Cell viability was determined using alamarBlue assay. Percent control cell viability against iAs3+ dose were plotted (log(inhibitor) vs. normalized response (variable slope) curve fit). **p < 0.01, ***p < 0.001, Two-way Repeated Measures ANOVA with Geisser-Greenhouse’s Correction followed by Tukey’s Multiple Comparisons Test between iAs3+ and iAs3++UroA. Each point in each curve represents the mean ± SD from three independent experiments.
3.2. UroA abrogates iAs3+ induced apoptosis of colon epithelial cells.
Exposure to iAs3+ effectively induces DNA damage and leads to apoptosis, which is potentially responsible for its cytotoxic effects and cell death [57, 58]. To investigate the effects of UroA on the regulation of iAs3+−induced apoptosis, we performed dose dependent apoptosis assays. The characteristic feature of apoptosis i.e., translocation of phosphatidylserine (PS) from inner to outer leaflet was evaluated by Annexin V/PI assay. The total apoptosis was determined as cumulative percentage of early apoptosis (Annexin V+/PI−) and late apoptosis (Annexin V+/PI+). As shown in Figure 2A and Supplementary Figure 1, UroA significantly protected colon epithelial cells against iAs3+-induced apoptosis. Moreover, disruption of mitochondrial function during iAs3+ medicated cellular apoptosis depends on the modulation of mitochondrial transmembrane potential (Δψ) [59]. The dissipation of the mitochondrial electrochemical potential gradient (Δψ) is an early event in apoptosis. Changes in mitochondrial inner-membrane electrochemical potential in living cells can be measured using the cationic, lipophilic dye, JC-1. In healthy cells, JC1 aggregates in the mitochondrial membrane matrix space and generates red fluorescence. In case of unhealthy (apoptotic) cells, JC1 released into the cytosol emits green fluorescence. Thus, the red to green ratio is a proxy for the level of apoptosis. As shown Figure 2B, treatment with iAs3+ decreased the red/green ration in a dose dependent manner, whereas UroA reversed the iAs3+-mediated changes. These results suggest that UroA ameliorates iAs3+- induced apoptosis in colon epithelial cells.
Figure 2: UroA protects colon epithelial cells against iAs3+ induced apoptosis and loss of mitochondrial membrane potential.
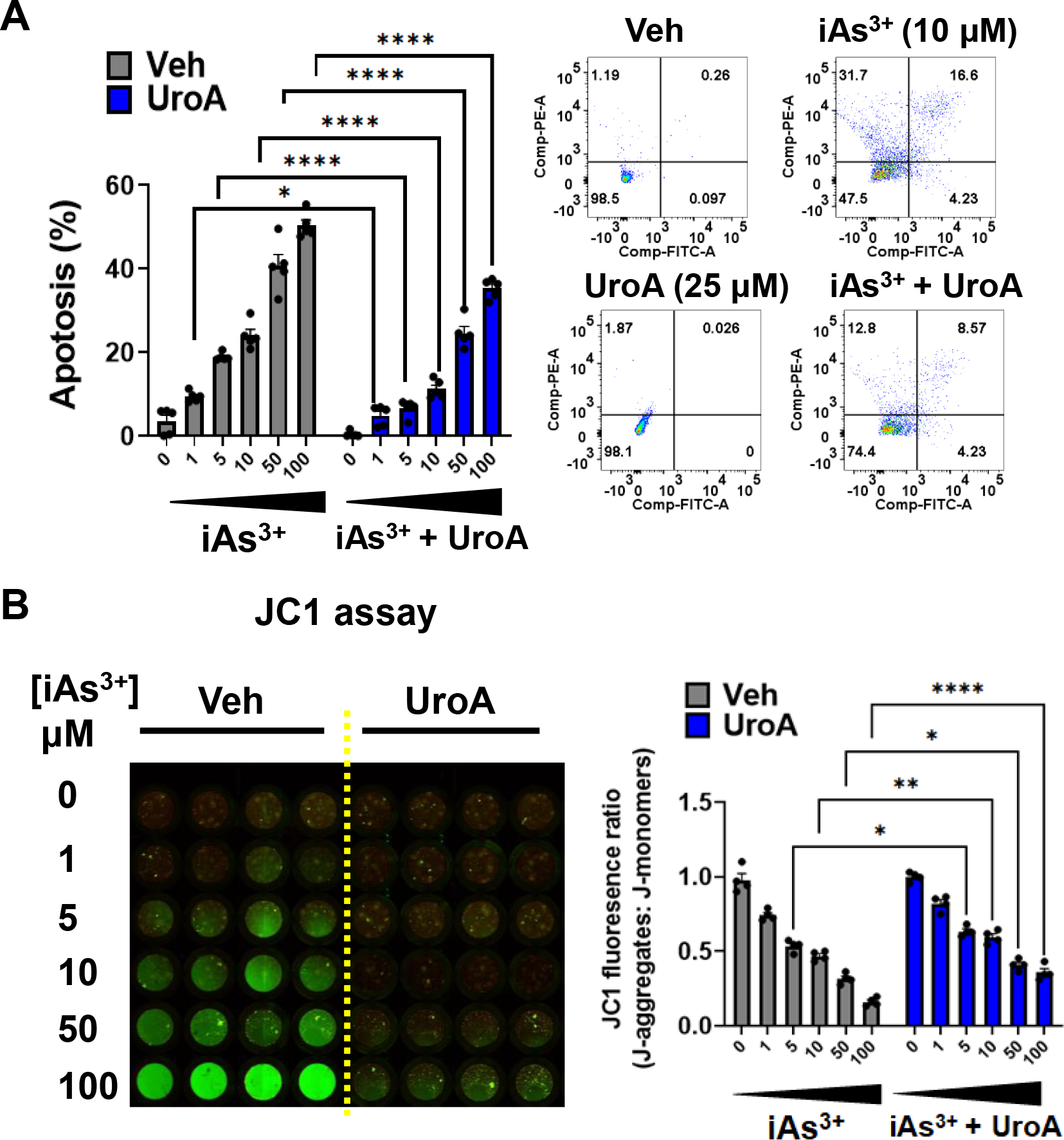
(A) T84 cells were treated with iAs+3 (0, 1, 5, 10, 50, 100 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) for 48 h. Bar graph and images representing apoptosis in T84 cells after the indicated treatment estimated by Annexin V/PI assay. (B) Representative fluorescence image of JC1 stained T84 cells representing the effect of iAs+3 in presence of vehicle or UroA on mitochondrial permeability (red to the green shift of fluorescence). Bar graphs showing the detection of JC-1 fluorescence as J aggregates (red) vs J monomers (green). Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using 2way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4–5)
3.3. UroA impairs iAs3+ induced ROS generation and protects against oxidative stress.
Numerous studies suggested that enhancement of oxidative stress leads to an increase in the generation of reactive oxygen species (ROS) and a decrease in mitochondrial transmembrane potential, ultimately triggering apoptosis [60, 61]. It is also well documented that induction of ROS generation through the mitochondrial transport chain contributes to arsenic triggered cellular toxicity [38, 60, 61]. Previously, we and others showed that UroA can act as an anti-oxidative and anti-inflammatory agent [43, 62]. To examine the effects of UroA on iAs3+ induced ROS production, we measured ROS production and related levels of oxidative stress. iAs3+-induced ROS in dose dependent manner in T84 cells, but co-treatment with UroA (25 μM) significantly reduced iAs3+-induced ROS production (Figure 3A and Supplementary Figure 2).
Figure 3: UroA impairs iAs3+ induced ROS generation and protects against oxidative stress.
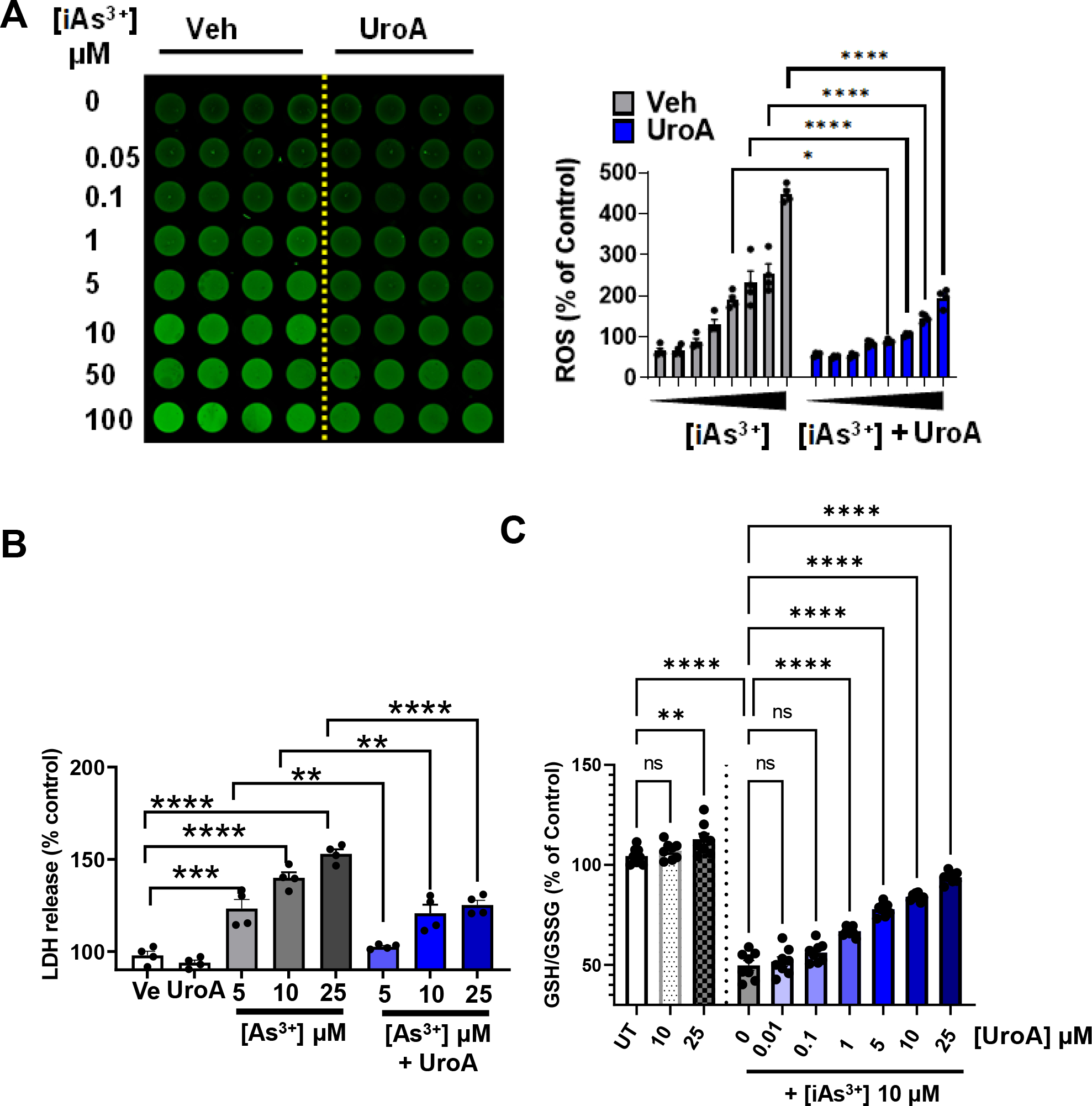
(A) T84 cells were treated with iAs+3 (0, 0.05, 0.1, 1, 5, 10, 50, 100 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) for 12 h. Representative fluorescence image and bar graph are showing ROS generation as green fluorescence from DCFDA stained T84 cells. (B) LDH release after 24 h incubation of T84 cells with iAs+3 (5, 10, 20 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) (C). T-84 cells were treated with UroA (10, 25 μM). T84 cells were treated with iAs+3 (10 μM) and different concentration of UroA (0, 0.01, 0.1, 1, 5, 10, 25 μM) for 24 h. The levels of GSH and GSSG were measured and the GSH/GSSG ratio was calculated. Untreated (UT) cells were used as control (100%). Results are representative of three independent experiments. Statistics performed using 2way ANOVA in GraphPad Prism software. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bar, mean ± SEM (n=4–8)
We also measured the levels of iAs3+-induced lactate dehydrogenase (LDH) release in the presence or absence of UroA. Previous studies showed that oxidative stress in general and iAS3+ specifically triggers elevated LDH activity, suggesting that LDH levels positively correlate with cell damage [63, 64]. As shown in Figure 3B, treatment with iAs3+ led to increased LDH activity in T84 cells, whereas co-treatment with UroA significantly reduced iAs3+-induced LDH levels. Similarly, we sought to measure the levels of reduced glutathione (GSH) in iAs3+-treated cells, which is an important scavenger of ROS. In healthy cells, GSH and oxidized glutathione (GSSG) both exist, with a high GSH to GSSG ratio. The GSH/GSSG ratio is indicative of the extent of oxidative stress. We found that iAs3+ treatment reduced the GSH/GSSG ratio in a dose dependent manner (Supplementary Figure 3), whereas UroA co-treatment significantly blocked the iAs3+-induced decrease in the GSH/GSSG ratio (Figure 3C), suggesting that GSH may play an important role in UroA-mediated rescue from cell death. Treatment with UroA alone at 10 μM, caused modest but significant increase in the GSH/GSSG ratio suggesting it has a beneficial physiological function. Therefore, we suggest that UroA-mediated anti-oxidative properties may be responsible for its protective activities against iAs3+-induced oxidative stress in colon epithelial cells.
3.4. UroA ameliorates iAs3+ induced intestinal permeability and disruption of tight junctional proteins.
Previously, we showed that UroA induces the expression of intestinal tight junction proteins and protects the intestinal epithelium from inflammation-induced increases in gut permeability [41]. Since increased ROS levels are known to enhance intestinal permeability [39], we determined whether exposure of iAs3+ leads to changes in the permeability of T84 monolayers grown on transwell membranes [65]. We utilized a transwell FITC-dextran permeability assay to monitor barrier function (Figure 4A). We found that iAs3+ exposure led to a dose dependent increase in permeability. However, co-treatment with UroA significantly mitigated iAs3+-induced permeability (Figure 4B and Supplementary Figure 4) as well as enhancing transepithelial electrical resistance (TEER) (Figure 4C). Further investigation of LDH release revealed that UroA also protected against iAs3+-induced oxidative stress in the T84 monolayers (Figure 4D).
Figure 4: UroA mitigates iAs3+ mediated oxidative stress, inflammation and impairment of gut barrier permeability.
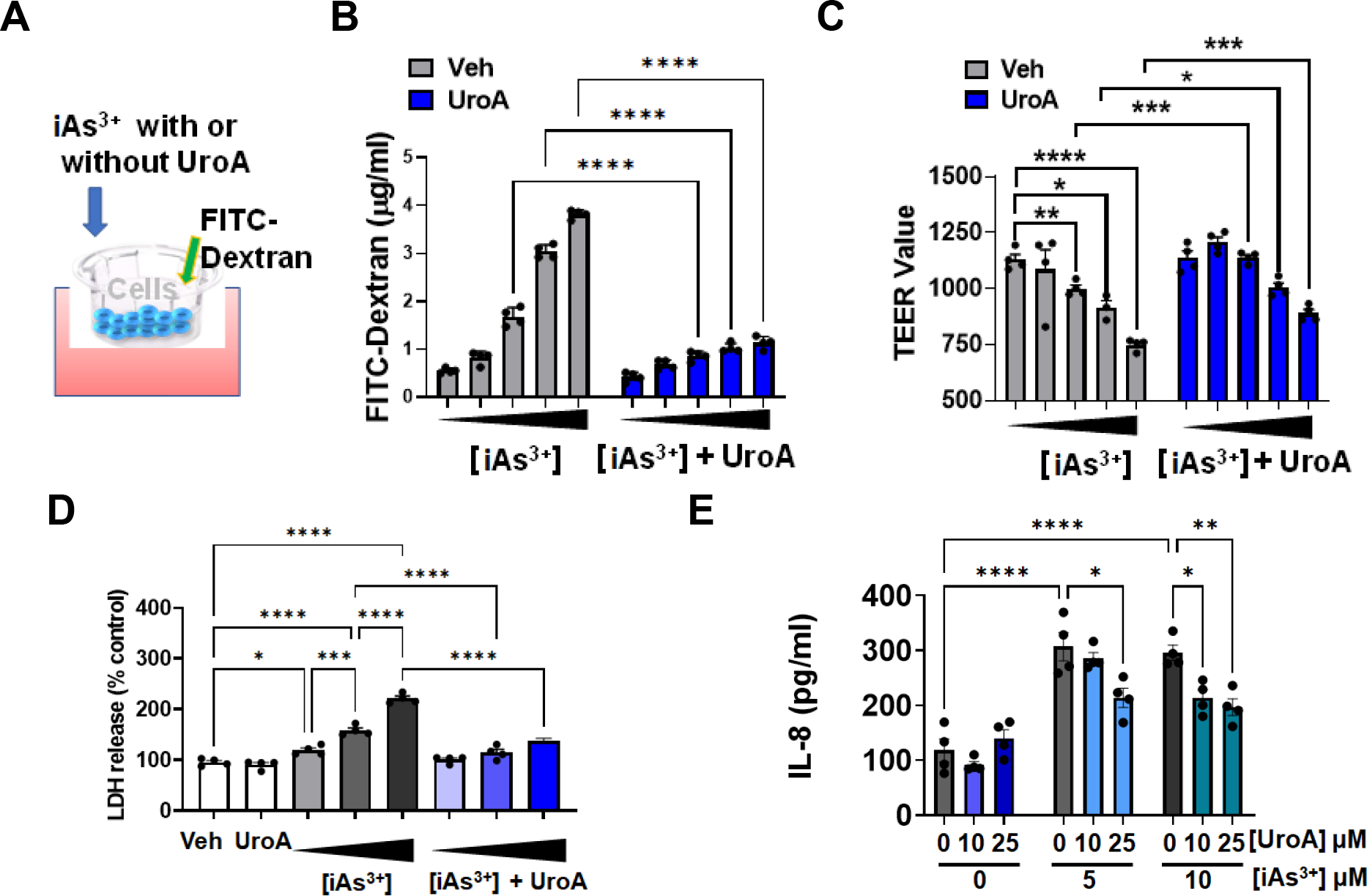
(A) Schematic representation of in vitro permeability study with T84 monolayers (B) and (C) Monolayer T84 cells on transmembrane were treated with iAs+3 (0, 1, 5, 10, 20 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) for 24 h. FITC-dextran was added to these cells (top of the membrane) and incubated for 2 h at 37°C and FITC-dextran levels in the bottom chamber well was measured. TEER values were also measured. (B) LDH release after 24 h incubation of T84 monolayer cells with iAs+3 (5, 10, 20 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) (C) T84 monolayer cells were incubated with iAs+3 (5, 10 μM) without or with UroA (0, 10, 25 μM) for 24 h. IL-8 levels in supernatants were measured. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using 2way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4).
ROS-triggered signaling pathways have been shown to be involved in the progression of inflammatory disorders and that inflammation in turn contributes to gut barrier dysfunction [66]. Measuring the levels of inflammatory cytokines such as IL-8 release by T84 monolayer revealed that UroA downregulated iAs3+-triggered inflammation (Figure 4E). To elucidate potential underlying molecular mechanisms of barrier integrity maintenance, we examined expression of the tight junction (TJ) proteins zona occludin 1 (ZO-1), claudin 4 (Cldn4) and occludin (Ocln) in T84 monolayers treated with iAS3+. As shown Figure 5A and 5B, UroA blocked iAs3+-induced downregulation of TJ proteins both at the protein and mRNA levels. Moreover, immunofluorescent confocal imaging confirmed that UroA treatment resulted in enhanced levels of intercellular TJ proteins in iAs3+-treated T84 monolayers (Figure 5C). Collectively, the results described in this section demonstrate that treatment with UroA significantly rescues T84 colonic monolayer cells from iAs3+-induced oxidative stress, enhanced gut barrier permeability, TJ protein disruption, and increased levels of inflammation.
Figure 5: UroA protects gut barrier from iAs3+ mediated permeability through regulation of tight junction proteins.
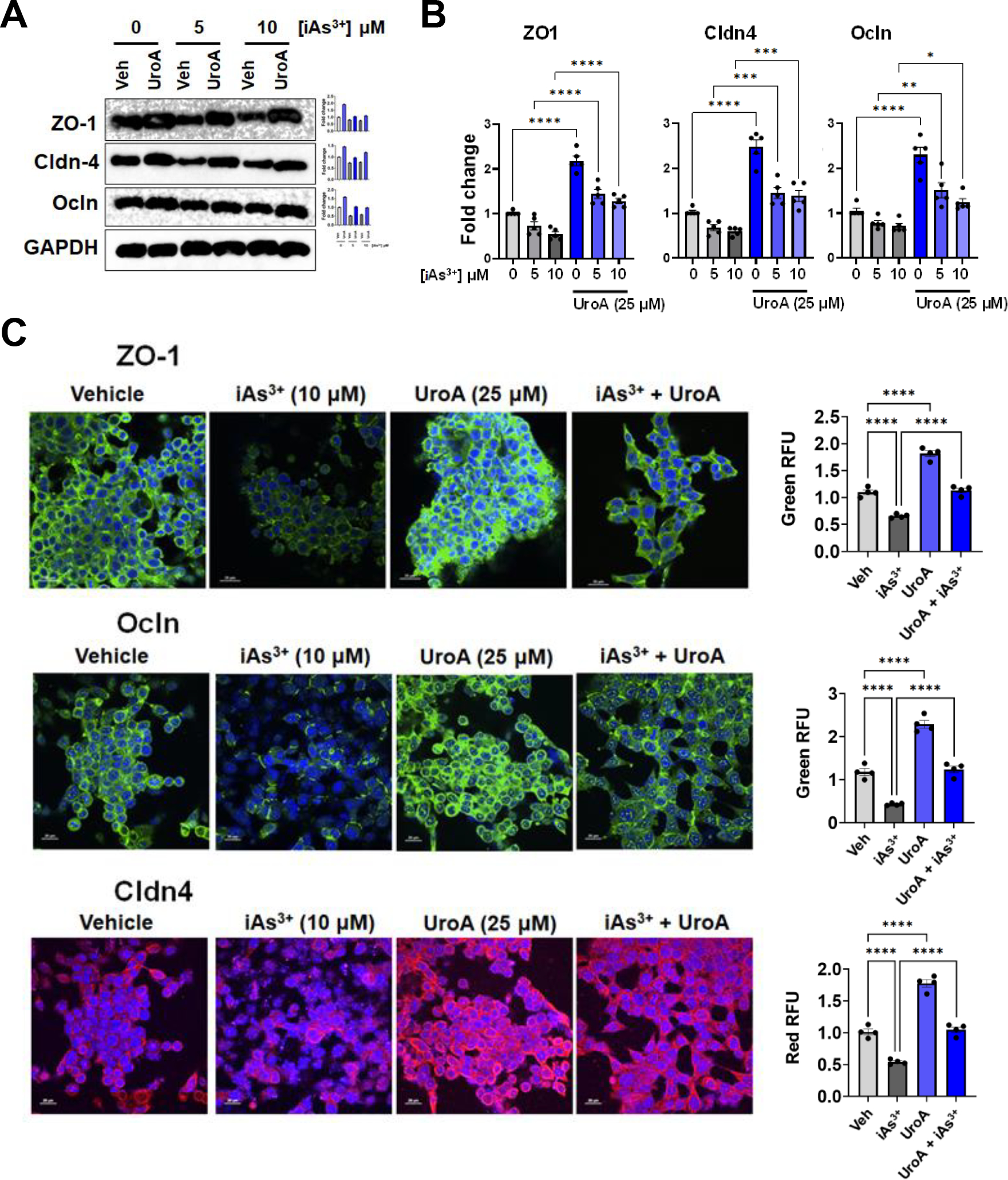
T84 cells were treated with iAs+3 (0, 5, 10 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) for 24 h. (A) Protein expression of Zona occludens 1 (ZO1), claudin 4 (Cldn4) and occludin (Ocln) in T84 cells were determined by immunoblots. (B) The fold changes in mRNA levels of ZO1, Cldn4, Ocln, and ZO1 in T84 cells were determined by RT PCR method. (C) T84 cells were grown on chambered slides and treated with iAs+3 (10 μM) in presence of vehicle (DMSO-0.01%) or UroA (25 μM) for 24 h. The cells were stained with rabbit anti ZO-1, rabbit anti Ocln and mouse anti-Cldn4, followed by secondary antibody tagged with anti-rabbit Alexa 488 for ZO-1, Ocln and anti-mouse Alexa-594 for cldn-4. Nucleus was stained using DAPI. The confocal images were captured. Scale bars for T84 cells indicate 20 μm respectively. The fluorescence intensity (n = 15–20 cell membrane regions) was measured. Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using one-way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=3–5).
3.5. UroA mitigates the barrier damage induced by arsenic exposure in a human 3D small intestinal tissue model.
MatTek Life Sciences, Inc. has developed a unique technology to grow 3-D human intestine tissues in transwell membranes that can be used to examine gut barrier activities and inflammation (https://www.mattek.com/products/epiintestinal/) [67]. This 3D intestinal model incorporates enterocytes, Paneth cells, M cells, tuft cells and intestinal stem cells into a highly differentiated, polarized epithelium as well as an underlying lamina propria containing normal human intestinal fibroblasts. This “EpiIntestinal™” model recapitulates many aspects of normal intestinal function including gut barrier, metabolism, inflammatory and toxicity responses [68]. EpiIntestinal™ organoid 3D tissue cultures were grown on transwell plates for 21 days and then supplied to our laboratory by MatTek Life Science. These 3D tissues with TEER values at ~1000–1200 per well (24 well or 96 well) were exposed to iAs3+ in the presence or absence of UroA in a dose dependent manner. At 24 or 48 h post exposure, we evaluated permeability using FITC-dextran. As shown in Figure 6A and 6C, iAs3+ exposure increased permeability of the EpiIntestinal tissue in a dose dependent manner both at 24 and 48 h. However, similar to the results described above with T84 monolayer cells, co-treatment with UroA significantly reduced iAs3+-induced intestinal permeability in a dose dependent manner at both 24 and 48 h (Figure 6B and 6D). UroA treatment alone either at 10 or 25 μM concentrations did not alter FITC-dextran permeability suggesting that UroA treatment did not cause any additional stress leading to barrier dysfunction. In agreement with these permeability data, exposure of the EpiIntestinal tissue to iAs3+ disrupted the levels of ZO1, whereas treatment with UroA increased the expression of ZO1 (Figure 7). Moreover, LDH levels were also significantly upregulated in iAs3+ treated EpiIntestinal tissues, whereas UroA treatment resulted in a decrease in LDH levels (Figure 8A). UroA also effectively reduced the levels of iAs3+-induced inflammatory cytokines such as IL-8 and TNF-α in the EpiIntestinal tissue (Figure 8B and 8C). These results are consistent with the T84 monolayer data described above, showing that UroA significantly reduces iAs3+-induced gut barrier dysfunction, enhances TJ protein expression, decreases oxidative stress, and reduces inflammatory markers.
Figure 6: UroA ameliorates iAs3+ mediated gut barrier permeability in human 3D small intestinal tissue model.
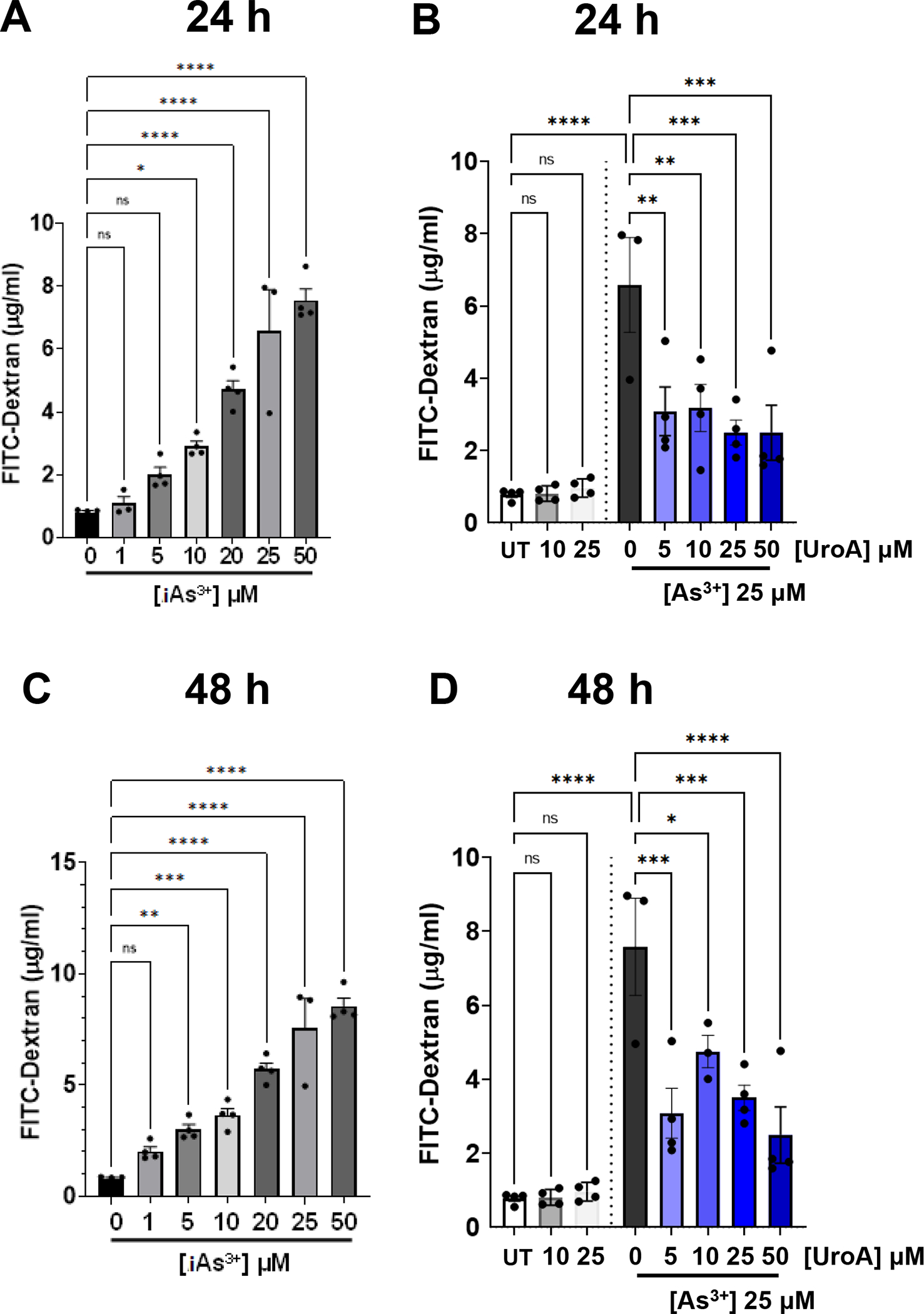
(A) and (C) iAs+3 (0, 1, 5, 10, 20, 25, 50 μM) (B) and (D) iAs+3 (25 μM) with UroA (0, 5, 10, 25, 50 μM) were added to the apical compartment of EpiIntestinal 3D microtissues grown on Transwell inserts and incubated at 37 °C for 24 h and 48 h, respectively. FITC-dextran was added to these cells (apical compartment) and incubated for 2 h at 37°C and FITC-dextran levels in the bottom chamber well (basal compartment) was measured. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using one-way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4).
Figure 7: UroA protects from iAs3+ induced TJ protein disruption in primary human 3D tissue organoids.
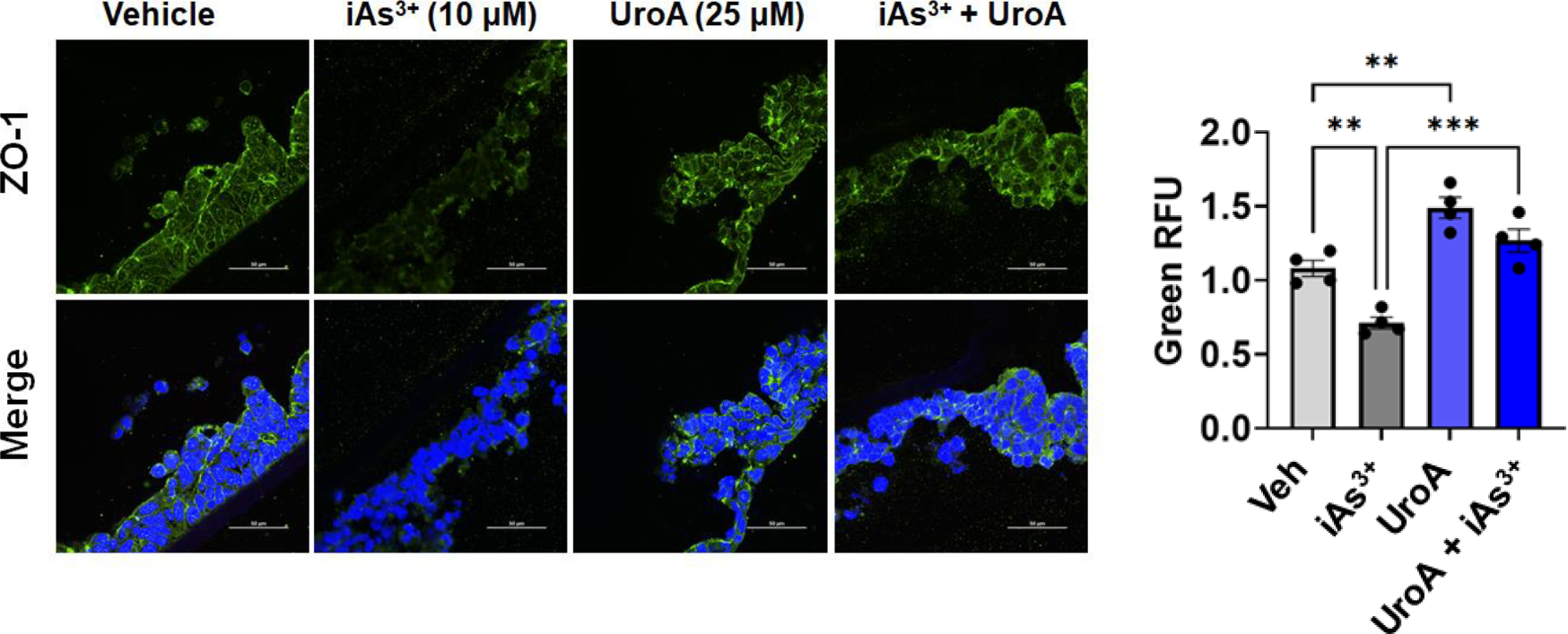
Microphotographs EpiIntestinal3D microtissues treated with iAs+3 (10 μM) in presence or absence of UroA (25μM) were stained with rabbit anti ZO-1 followed by secondary antibody tagged with anti-rabbit Alexa 488. Nucleus was stained using DAPI. The confocal images were captured. Scale bars indicate 50 μm. The fluorescence intensity (n = 15–20 cell membrane regions) was measured. Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using one-way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4).
Figure 8: UroA reduces iAs3+ induced oxidative stress and inflammatory cytokines in human intestinal organoids.
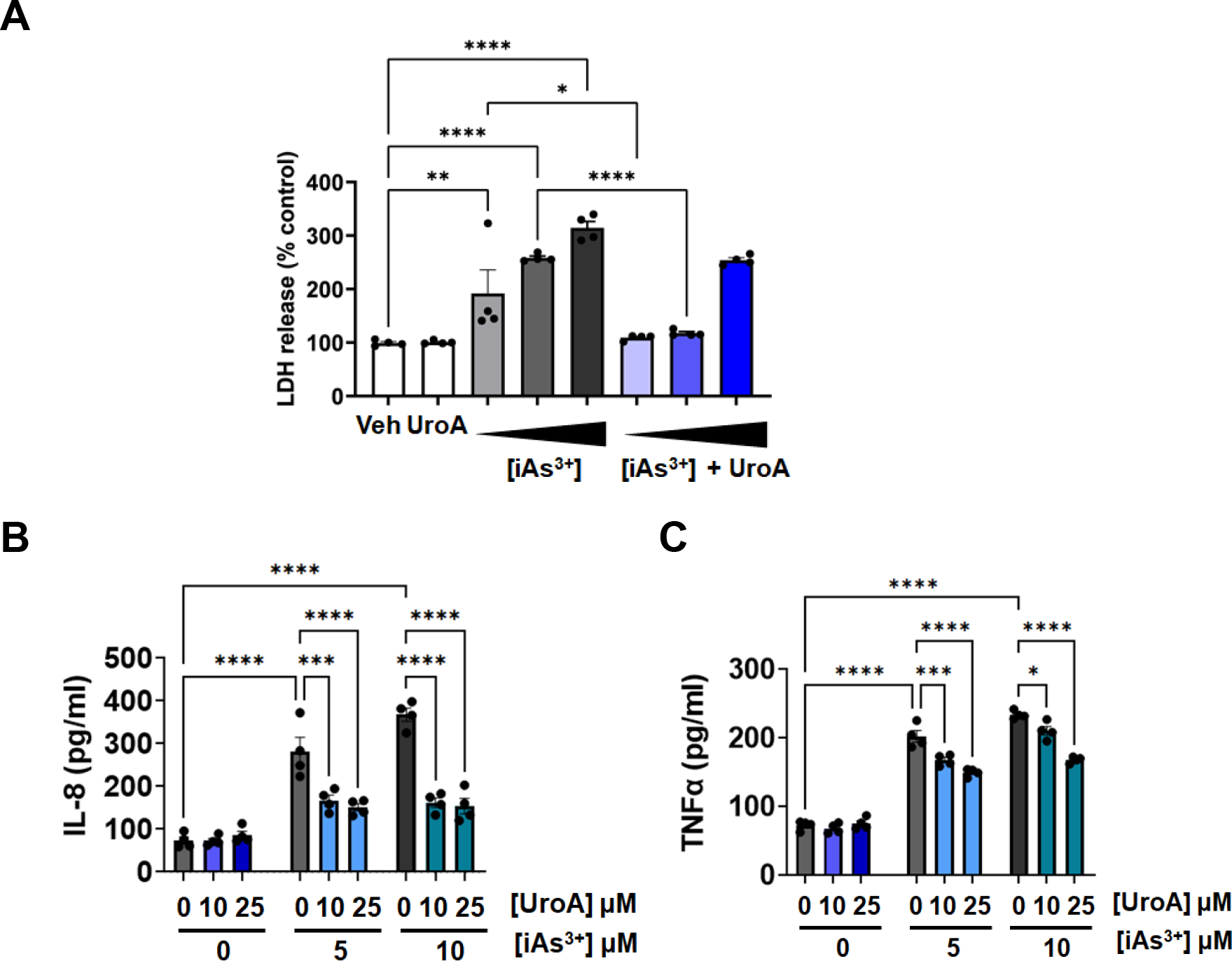
EpiIntestinal3D microtissues were treated with iAs+3 (5,10,20 μM) in presence or absence of UroA (25μM) for 24h. (A) LDH release (B) IL-8 levels and (C) TNFα levels in supernatants were measured. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using 2way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4).
3.6. UroA protects gut barrier against iAs3+ chronic exposure.
Finally, we asked whether UroA provides long-term protection against chronic exposure to low doses of iAs3+. For this purpose, we exposed EpiIntestinal™ 3D organoid tissues to iAs3+ (1 or 5 μM) for 7 and 14 days in the presence or absence of UroA (10 μM). As shown in Figure 9, exposure to iAs3+ induced increased permeability over time in a dose dependent manner. At day 14 there was a significant loss of barrier for both iAS3+ doses. However, UroA treatment reduced the levels of iAs3+-induced permeability (Figure 9A). Further, to confirm the health of cells, we measured LDH levels in pooled corresponding supernatants that were collected during experimental period. As shown in Figure 9B, treatment with UroA alone was similar to vehicle. suggesting that UroA is not toxic. As expected, we observed that treatment with iAs3+ at 1 μM or 5 μM led to an increase in LDH levels over the control treatment, suggesting that iAs3+ led to cell damage/cytotoxicity of the intestinal organoids. Treating with UroA significantly attenuated iAs3+-induced cell damage/cytotoxicity suggesting UroA-mediated protective activities against iAs3+ toxcity (Figure 9B). Importantly, UroA treatment also reduced iAs3+-induced inflammatory cytokines levels in the supernatants (Figure 9C and 9D), suggesting its long-term protective activity against chronic exposure of iAs3+.
Figure 9: Chronic iAs3+ exposure induces barrier disruption and inflammation in human intestinal organoids and UroA provides protection against it.
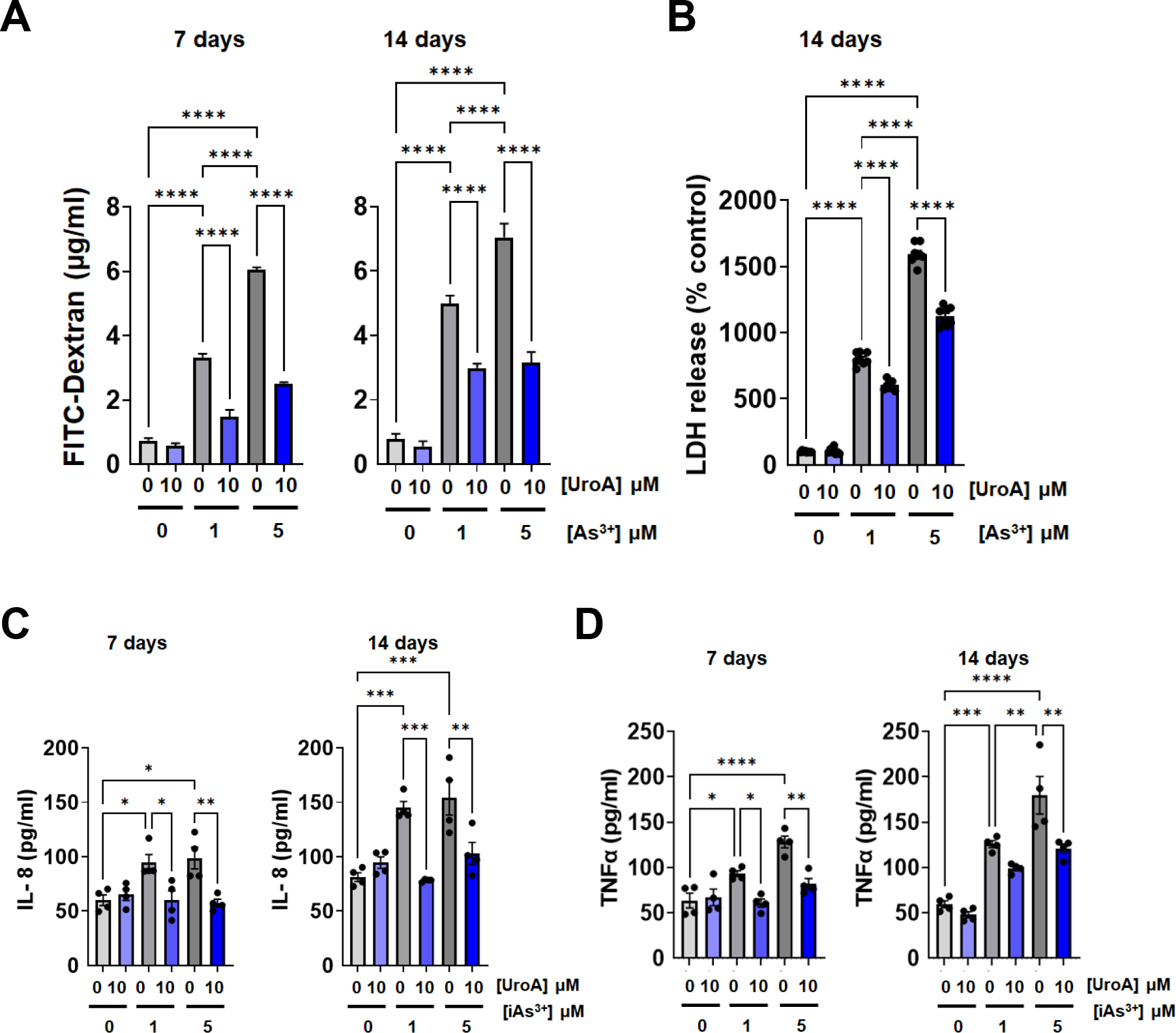
For chronic exposure of iAs+3 in human intestinal organoids, iAs+3 (0, 1, 5 μM) with or without UroA (10 μM) were added to the apical compartment of EpiIntestinal 3D microtissues grown on Transwell inserts and incubated at 37 °C for 14 days. Fresh treatments were added every alternate days and supernatants were stored. (A) On 7th and 14th day, FITC-dextran was added to these cells (apical compartment) and incubated for 2 h at 37°C and FITC-dextran levels in the bottom chamber well (basal compartment) was measured. (B) EpiIntestinal 3D microtissues were treated with iAs3+ (1, 5 μM) in presence or absence of UroA (10μM) for 14 days. The supernatants were collected every 3 days for 14 days and pooled. LDH release in these cumulative supernatants at day 14 was measured. (C) IL-8 levels and (D) TNFα levels in supernatants were measured. *p < 0.05, **p < 0.01, ***p < 0.001, Statistics performed using one-way ANOVA in GraphPad Prism software. Error bar, mean ± SEM (n=4).
Discussion
Epidemiological and clinical reports suggest that arsenic exposure, whether chronic, acute or therapeutic leads to gastrointestinal disorders in humans [14, 19–21, 23, 69, 70]. While a handful of studies have attempted to understand the underlying mechanisms by which arsenic induces gut barrier dysfunction [35, 36, 40, 71], a detailed understanding of the mechanisms of action of arsenic toxicity in the gut are poorly understood. Furthermore, no therapeutic intervention strategy currently exists to reverse arsenic-induced adverse health outcomes. Reducing source of exposure is the only available mitigation strategy [72]. However, removal of arsenic is often difficult to achieve and sustain especially from widespread implementation and economic feasibility standpoints, especially in the developing countries [72–76]. This necessitates the study and development of putative therapeutic interventions to combat the effects of arsenic exposure and toxicity. In this vein, the current study focused on examining the effect of the gut microbial metabolite UroA on iAs3+-induced adverse activities on gut barrier function.
UroA is a microbial metabolite derived from ellagitannins/ellagic acid rich diets (e.g., pomegranate, berries, walnuts etc.). It was reported that UroA can only be detected in ~ 30–40% of the population upon consumption of EA containing diets [54, 77–84]. Importantly, UroA can reach up to micromolar levels both in serum and urine without exerting any toxicity. Therefore, we tested the effects of UroA at micromolar concentrations, which appears to be a physiologically relevant dose in certain individuals. Phase I clinical study reported that human oral consumption of UroA, either as a single dose or multiple doses (500 mg and 1000 mg daily) over a 4-week time period by healthy or sedentary elders was not toxic and showed favorable safety profiles [85]. Recent human studies highlighted that direct supplementation of UroA overcomes limitations of dietary exposure (e.g., EA or ET containing diets) and gut microbiome variability in healthy adults to achieve consistent levels of UroA [86]. Importantly, they demonstrated that UroA producers display more bacterial diversity compared to non-producers [86]. Our group has recently elucidated the molecular mechanism by which UroA enhances gut barrier function. We showed that UroA treatment upregulated gut epithelial tight junction proteins in an aryl hydrocarbon receptor (AhR)- nuclear factor erythroid 2–related factor 2 (Nrf2) dependent manner and attenuated chemical induced colitis [41]. Many studies have shown that exposure to iAs3+ induces cell death and apoptosis and enhances oxidative stress in cellular systems [38, 87, 88]. As shown in Figure 1, iAs3+ induced cell death (T-84 cells) in a dose dependent manner, whereas co-treatment with UroA protected against iAs3+ induced cell death. UroA showed protective activities against iAs3+ even at relatively high doses at longer time period. Arsenic is known for induction of ROS in various cell types, which leads to increased oxidative stress and mitochondrial dysfunction [38, 88–90]. It has been suggested that iAs3+-induced apoptotic cell death is mediated through mechanisms involving the production of ROS. In addition, several studies suggest that redox-active enzymes such as LDH get elevated in response to oxidative stress induced damage in mitochondrial respiration [64]. Under oxidative stress anaerobic glycolytic enzymes like LDH reversibly convert pyruvate to lactate for ATP production.
Interestingly, UroA effectively countered iAs3+-mediated modulation of mitochondrial transmembrane potential, ROS and LDH production in gut epithelial cells (Fig. 2–3). It is possible that targeting iAs3+-induced adverse events by supplementing with the non-toxic microbial metabolite UroA may provide protective activities. Moreover, our data demonstrate that exposure to iAs3+ significantly decreased GSH/GSSG in a dose-dependent manner indicating that iAs3+ induces oxidative stress in colon epithelial cells. Glutathione (GSH), a tripeptide antioxidant is a vital mediator for cellular defense against oxidative stress. Previously, we have demonstrated that UroA upregulates and activates Nrf2 and its dependent pathways in colon epithelial cells and macrophages leading to induction of heme oxydenase-1 (HO1), Glutamate-Cysteine Ligase Modifier Subunit (GCLM) and NAD(P)H Quinone Dehydrogenase 1 (NQO1) [41]. Activation of Nrf2 leads to production of anti-oxidative mediators and protects the host cells from oxidative stress. In agreement with these observations, UroA significantly reduced iAs3+-induced oxidative stress and rescued loss of GSH/GSSG levels in a dose-dependent manner in colon epithelial cells (Figure 3C). It is possible that UroA-dependent activation of Nrf2 is responsible for anti-oxidative properties and cytoprotective activities against iAs3+-induced oxidative stress. This hypothesis requires further investigation by knocking down AhR/Nrf2 in colon epithelial cells and evaluating iAs3+ and UroA mediated activities.
Gut epithelial cells form a single cell layer gut barrier that separates the host from external challenges in the gut. A comprehensive body of literature demonstrates that exposure to iAs3+ at chronic, sub-chronic, acute or therapeutic levels induce several GI-related symptoms of dyspepsia, gastroenteritis and chronic diarrhea [19–21, 23]. Our study employed iAs3+ dosage of 0.1–100 μM for most of the experiments. To put this dose into perspective, subjects treated with As2O3 for APL have a plasma arsenic level of 5 μM [91], while those exposed chronically through drinking water have a mean blood arsenic level of ~100 nM [92–94]. We acknowledge that iAs concentrations above 5 μM are supratherapeutic and may not be physiologically or toxicologically relevant. Our experiments were designed (with doses below and above 5 μM of iAs) to demonstrate the efficacy of UroA as a mitigation strategy. Our data show unequivocally that UroA treatment is capable of mitigating variety of toxic cellular effects of subtherapeutic to supratherapeutic iAs exposure to a considerable extent. In general, iAs-induced toxic effects in humans are seen upon chronic exposure to low doses for extended time periods lasting as long as several years. It would be challenging to recapitulate these chronic low dose exposures using cellular models. Therefore, we tested doses as low as 1 μM for 14 days using intestinal organoids (Figure 9), where we observed that UroA attenuated iAs-induced cellular damage. We postulate that iAs3+-induced ROS could potentially have significant impact on gut barrier function. Previous studies showed that enhanced oxidative stress can induce gut barrier permeability leading to gut barrier dysfunction in various models [39, 95]. Our results demonstrate that exposure to iAs3+ increased permeability and decreased TEER in T84 monolayered cells in a dose-dependent manner. UroA treatment protected against iAs3+-induced permeability and loss of TEER (Figure 4). Epithelial tight junction (TJ) proteins such as ZO1, Clnd4 and Ocln play an important role in maintenance of gut barrier function [96, 97] (Figure 5). TJ proteins form a barrier to entry of external insults such as allergens, toxins and pathogens across the epithelium into the interstitial tissues including GI tract. It is possible that iAs3+-induced disruption of TJ proteins could lead to increased gut permeability and inflammation [1, 98, 99]. Mounting evidence suggests that disruption of TJ proteins and increased paracellular permeability play a critical role in pathogenesis of numerous GI-related disorders including IBD, alcoholic liver disease, infectious enterocolitis, necrotizing enterocolitis etc. [100–103]. We postulated that iAs3+-induced permeability is potentially a consequence of down regulation of TJ proteins or disruption of TJ barriers. As predicted, iAs3+ treatment significantly downregulated TJ proteins both at protein and mRNA levels. Further, we confirmed the disruption TJ proteins by immunofluorescence staining and confocal imaging. Treatment with UroA rescued the downregulation and disruption of TJ proteins mediated by iAs3+ exposure. These results suggest that UroA provides gut barrier protection against iAs3+-induced barrier dysfunction in colon epithelial cells. Further, we determined whether UroA-mediated mitigation of iAs3+-induced damage is due to altered ability of iAs3+ to enter the cells. To address this question, we determined intra-cellular arsenic levels in T-84 cells treated with or without UroA (25 μM) in the presence of As3+ (5 μM) for 24 h as described in Methods. As expected arsenic was not detected in lysates of control cells treated with Vehicle or UroA alone (Supplementary Figure 5). We found a significant increase in arsenic levels in cell lysates treated with iAs3+ or iAs3+ + UroA suggesting that UroA treatment did not affect arsenic entry into cells. In fact, we unexpectedly observed that intracellular arsenic levels were modestly increased in cells co-treated with UroA. We postulate that increased intracellular iAs3 in iAs3+ + UroA samples could be attributed to UroA-mediated down regulation of drug transporters, which was observed in colon cancer cell lines in an independent study (unpublished results from our laboratory). Previously, it was shown that arsenic efflux transporters are responsible for efflux of iAs3+ from variety of cells. These efflux systems include members of the multidrug resistance ATP binding cassette (ABC) transporters protein family and the bacterial exchangers Acr3 and ArsB [104, 105]. UroA-mediated regulation of these proteins and the correlation with increased intracellular arsenic level require further investigation. We postulate that despite a slight increase in arsenic levels, UroA treatment mitigates arsenic-induced cell damage/toxicity potentially through its anti-oxidative and some unknown mechanisms.
To recapitulate human intestinal tissue physiology, we established the MatTek’s 3D EpiIntestinal™ tissue model of the human small intestine and tested the effects of iAs3+ exposure on gut barrier function. The major advantages of this model system over monolayer 2D cell cultures are that this system incorporates enterocytes, Paneth cell, M cells, tuft cells and intestinal stem cells into a highly differentiated, polarized epithelium. This system recapitulates many aspects of normal intestinal function including gut barrier, metabolism, inflammatory and toxicity responses, similar to native human intestinal tissue [68]. Acute exposure of EpiIntestinal™ tissues grown on Transwell chambers to iAs3+ led to an increase in permeability in a dose-dependent manner (1 to 50 μM). Importantly, UroA treatment significantly reduced iAs3+-induced permeability in a dose-dependent manner (5 to 50 μM) suggesting potential translational applications. Moreover, we also tested effects of iAs3+ chronic exposure for 14 days in this system on gut permeability and inflammation (Figure 9). Again, as expected, lower doses of iAs3+ (1 and 5 μM) for longer time (14 days) induced excessive permeability and cell damage. Co-treatment with UroA protected from iAs3+-induced permeability, cell damage and inflammation in all these conditions indicating benefits of UroA as a therapeutic or preventive agent.
Pro-inflammatory cytokines such as IL-8 and TNFα which are induced during oxidative stress mediated epithelial injury and are downregulated upon UroA treatment with iAs3+ exposure. Evaluation of LDH activity revealed the similar results, confirming protective effect of UroA on iAs3+ induced oxidative damage. Additionally, TJ protein analysis suggests that UroA significantly increased expression of ZO1 and protected from iAs3+-induced disruption in these 3D colon organoids. In summary, these results provide strong evidence for microbial metabolite mediated gut barrier protective activities against iAs3+−induced adverse effects. It is possible that iAs3+ -induced ROS and activation of ERK/1/2, MAPK and JNK might have significant impact on apoptosis of gut epithelial cells. The literature shows that oxidative stress induced by various ROS species including hydrogen peroxide, nitric oxide, peroxynitrite and hypochlorous acid disrupt the gut epithelial tight junctions culminating in increased permeability [106, 107]. Mechanistically, it was shown that oxidative stress-induced disruption of TJ protein includes internalization as well as protein modification such as thiol oxidation, phosphorylation, nitration and carbonylation (reviewed [107]). Treatment with UroA reduced iAs3+−induced oxidative stress and ROS production, thereby potentially blocking apoptosis and permeability (Figure 10). We acknowledge there are many complex mechanisms that might be regulating each of these physiological processes modulated by iAs3+ exposure as well as UroA. The in-depth mechanistic studies and supporting pre-clinical models are under investigation.
Figure 10:
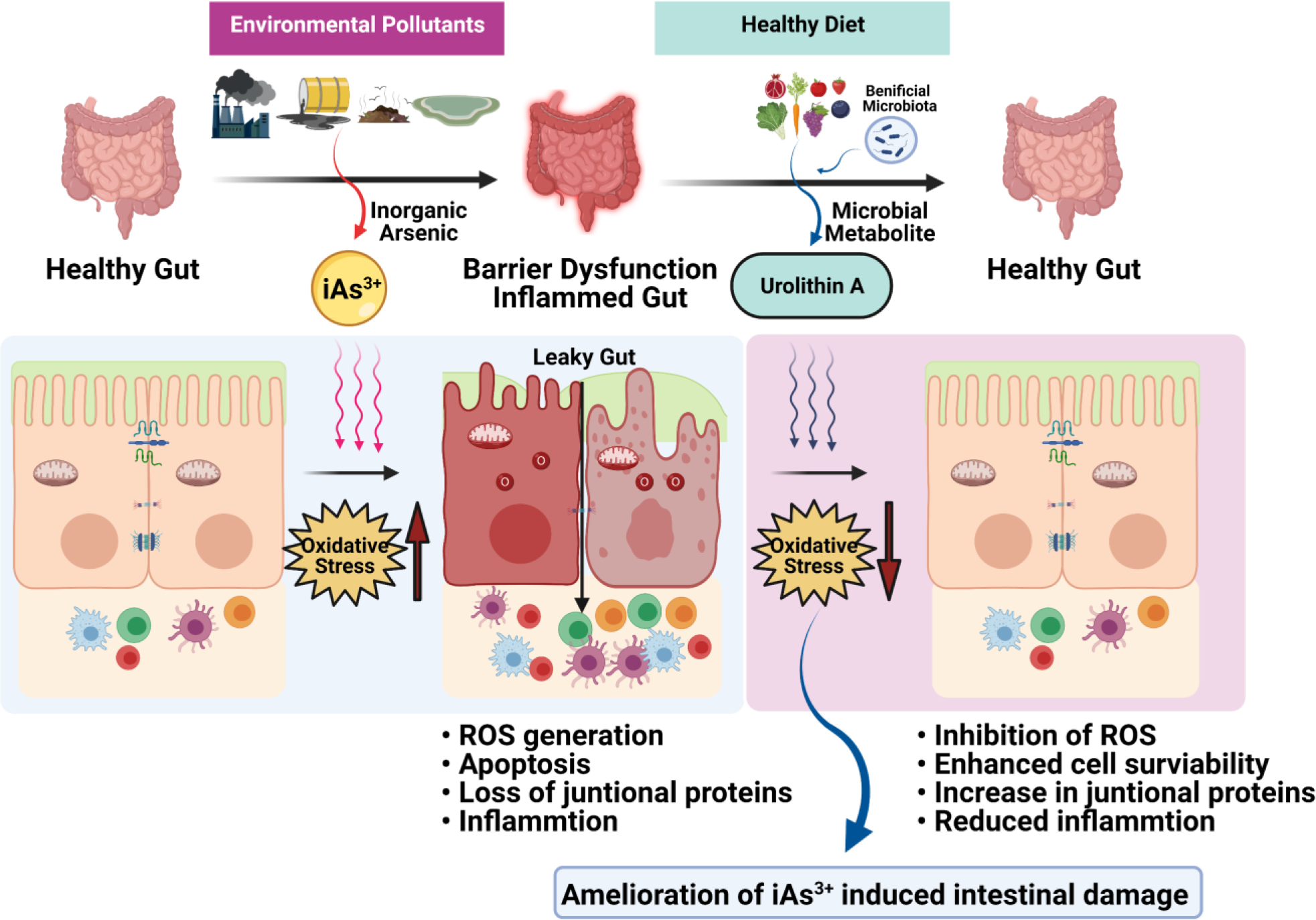
Reduction of inorganic arsenic mediated oxidative stress and enhanced barrier integrity by microbial metabolite, UroA protects the human gut from iAs3+ induced intestinal damage.
One of limitations of this study is that arsenic doses that were employed at certain conditions are in micromolar range and may not reflect chronic exposure in nanomolar ranges in humans. The pathways that are regulated by iAs3+ potentially may differ at different doses. These aspects require further investigation in the context of gut epithelium and co-treatment with UroA using pre-clinical models. However, the data from this study demonstrates from multiple cellular model systems that replicates gut barrier disruptive effects of arsenic exposure, which is rescued by treatment with microbial metabolite, UroA. We are currently extending these studies to develop a model for chronic exposure employing environmentally and toxicologically relevant dose and time of exposure using the same cell line and EpiIntestinal™ tissue model.
One of the long-standing challenges in arsenic research area is the lack of appropriate pre-clinical mouse models to investigate the adverse effects of arsenic and interventions to mitigate the effects [108]. Well documented differences exists in arsenic metabolism, efflux and detoxification between humans and rodent models [108]. iAs is methylated by arsenic methyltransferase (AS3MT) and controls detoxification and modulates risks of iAs-induced diseases. Detoxification of iAS in mice is greater than humans and attributed to higher rate of iAs methylation by mouse AS3MT compared to human AS3MT [109, 110]. Therefore, it is an uphill task to overcome this interspecies differences iAs metabolism and recapitulate human iAs exposure phenomena in mouse models. In fact, even in AS3MT knockout mice, 25–100 ppm iAs exposure (250–1000 fold higher than mean blood iAs concentration of chronically exposed human populations) was required to see adverse health effects [111]. Recently, Styblo group has developed a humanized mouse model, where mouse AS3MT gene is replaced by human AS3MT gene [112]. These humanized mice were able to successfully detoxify 400 ppb of iAs3+ for up to 4 weeks of exposure. Thus, this newly developed humanized AS3MT mice hold potential for the development of an in vivo chronic iAs exposure model in the near future, the results from which could be successfully translated to human populations. Therefore, we adopted human intestinal organoid 3D tissue model system to closely recapitulate human iAs exposure (chronic, up to 14 days) to investigate gut barrier function.
In summary, our results show that treatment with UroA efficiently protected against iAs3+-induced toxicity and loss of gut barrier homeostasis on colon epithelial barrier model system potentially through regulating apoptotic and oxidative pathways (Figure 10). Overall, this study identifies a novel role of microbial metabolite, UroA, where treatment reduces the environmental toxin (arsenic)-mediated adverse effects on gut epithelium and restores gut barrier integrity suggesting a possible new strategy to combat the toxicities of environmental toxins. Given its efficacy against arsenic toxicity together with high bioavailability through oral administration, UroA supplementation represents a promising approach towards development of a preventative and/or curative strategy to combat arsenic toxicity.
Supplementary Material
Highlights.
The gut microbial metabolite Urolithin A (UroA) enhances cell viability during inorganic arsenic (iAs3+) exposure and prevents arsenite-induced cell death in colon epithelial cells.
UroA reduced iAs3+-induced oxidative stress in intestinal epithelial cells.
UroA alleviated iAs3+-induced barrier dysfunction in both colon epithelial cell monolayers and a human 3D small intestinal tissue model.
The protective effect of UroA is associated with enhanced accumulation of tight junctional proteins including Zonula occludens-1, Occludin and Claudin-4 to repair iAs3+-induced intestinal barrier damage.
Acknowledgements:
VRJ is supported by NIH/NCI (CA191683), NIH/NIGMS CoBRE grant (P20GM125504-01), P30ES030283 (NIH/NIEHS), The Jewish Heritage Fund for Excellence Research Enhancement Grant and JGBCC. The authors thank Dr. F.M. Ausubel for poof reading the manuscript and for insightful discussions. The authors thank Dr. Xu Jason from Integrated Toxicomics and Environmental Measurement Facility Core (ITEMFC), U of L for determining intracellular arsenic levels in cell lysates.
Footnotes
Conflict of Interest: VRJ is one of the scientific co-founders of Artus Therapeutics. SG, MB and BH have no conflicts of interest to declare.
Availability of data and material:
The data, analytic methods, and study materials will be made available to other researchers.
References:
- [1].Ghosh S, Whitley CS, Haribabu B, Jala VR, Regulation of Intestinal Barrier Function by Microbial Metabolites, Cell Mol Gastroenterol Hepatol 11(5) (2021) 1463–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kho ZY, Lal SK, The Human Gut Microbiome - A Potential Controller of Wellness and Disease, Front Microbiol 9 (2018) 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Claus SP, Guillou H, Ellero-Simatos S, The gut microbiota: a major player in the toxicity of environmental pollutants?, NPJ Biofilms Microbiomes 2 (2016) 16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Claus SP, Guillou H, Ellero-Simatos S, The gut microbiota: a major player in the toxicity of environmental pollutants?, npj Biofilms and Microbiomes 2(1) (2016) 16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tu P, Chi L, Bodnar W, Zhang Z, Gao B, Bian X, Stewart J, Fry R, Lu K, Gut Microbiome Toxicity: Connecting the Environment and Gut Microbiome-Associated Diseases, Toxics 8(1) (2020) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chi L, Bian X, Gao B, Tu P, Ru H, Lu K, The Effects of an Environmentally Relevant Level of Arsenic on the Gut Microbiome and Its Functional Metagenome, Toxicol Sci 160(2) (2017) 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cervantes C, Ji G, Ramirez JL, Silver S, Resistance to arsenic compounds in microorganisms, FEMS Microbiol Rev 15(4) (1994) 355–67. [DOI] [PubMed] [Google Scholar]
- [8].IARC., Special Report: Policy, A review of Human Carcinogens—Part C: Metals, Arsenic, Dusts, and Fibres., IARC Monogr Eval Carcinog Risks Hum, 2012, pp. 11–465.23189751 [Google Scholar]
- [9].Garza-Lombo C, Pappa A, Panayiotidis MI, Gonsebatt ME, Franco R, Arsenic-induced neurotoxicity: a mechanistic appraisal, J Biol Inorg Chem 24(8) (2019) 1305–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hunt KM, Srivastava RK, Elmets CA, Athar M, The mechanistic basis of arsenicosis: pathogenesis of skin cancer, Cancer Lett 354(2) (2014) 211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Coryell M, Roggenbeck BA, Walk ST, The Human Gut Microbiome’s Influence on Arsenic Toxicity, Curr Pharmacol Rep 5(6) (2019) 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Faita F, Cori L, Bianchi F, Andreassi MG, Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies, Int J Environ Res Public Health 10(4) (2013) 1527–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schuhmacher-Wolz U, Dieter HH, Klein D, Schneider K, Oral exposure to inorganic arsenic: evaluation of its carcinogenic and non-carcinogenic effects, Crit Rev Toxicol 39(4) (2009) 271–98. [DOI] [PubMed] [Google Scholar]
- [14].Hong YS, Song KH, Chung JY, Health effects of chronic arsenic exposure, J Prev Med Public Health 47(5) (2014) 245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA, The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem, Environ Health Perspect 121(3) (2013) 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Podgorski J, Berg M, Global threat of arsenic in groundwater, Science 368(6493) (2020) 845–850. [DOI] [PubMed] [Google Scholar]
- [17].Hong Y-S, Song K-H, Chung J-Y, Health effects of chronic arsenic exposure, Journal of preventive medicine and public health = Yebang Uihakhoe chi 47(5) (2014) 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Antonelli R, Shao K, Thomas DJ, Sams R 2nd, Cowden J, AS3MT, GSTO, and PNP polymorphisms: impact on arsenic methylation and implications for disease susceptibility, Environ Res 132 (2014) 156–67. [DOI] [PubMed] [Google Scholar]
- [19].Mazumder DN, Das Gupta J, Santra A, Pal A, Ghose A, Sarkar S, Chronic arsenic toxicity in west Bengal--the worst calamity in the world, J Indian Med Assoc 96(1) (1998) 4–7, 18. [PubMed] [Google Scholar]
- [20].Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M, Arsenic: toxicity, oxidative stress and human disease, J Appl Toxicol 31(2) (2011) 95–107. [DOI] [PubMed] [Google Scholar]
- [21].Vantroyen B, Heilier JF, Meulemans A, Michels A, Buchet JP, Vanderschueren S, Haufroid V, Sabbe M, Survival after a lethal dose of arsenic trioxide, J Toxicol Clin Toxicol 42(6) (2004) 889–95. [DOI] [PubMed] [Google Scholar]
- [22].Au WY, Kwong YL, Arsenic trioxide: safety issues and their management, Acta Pharmacol Sin 29(3) (2008) 296–304. [DOI] [PubMed] [Google Scholar]
- [23].Ohnishi K, Yoshida H, Shigeno K, Nakamura S, Fujisawa S, Naito K, Shinjo K, Fujita Y, Matsui H, Sahara N, Takeshita A, Satoh H, Terada H, Ohno R, Arsenic trioxide therapy for relapsed or refractory Japanese patients with acute promyelocytic leukemia: need for careful electrocardiogram monitoring, Leukemia 16(4) (2002) 617–22. [DOI] [PubMed] [Google Scholar]
- [24].McDermott TR, Stolz JF, Oremland RS, Arsenic and the gastrointestinal tract microbiome, Environ Microbiol Rep 12(2) (2020) 136–159. [DOI] [PubMed] [Google Scholar]
- [25].Coryell M, McAlpine M, Pinkham NV, McDermott TR, Walk ST, The gut microbiome is required for full protection against acute arsenic toxicity in mouse models, Nat Commun 9(1) (2018) 5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li D, Yang Y, Li Y, Li Z, Zhu X, Zeng X, Changes induced by chronic exposure to high arsenic concentrations in the intestine and its microenvironment, Toxicology (2021) 152767. [DOI] [PubMed] [Google Scholar]
- [27].Brabec JL, Wright J, Ly T, Wong HT, McClimans CJ, Tokarev V, Lamendella R, Sherchand S, Shrestha D, Uprety S, Dangol B, Tandukar S, Sherchand JB, Sherchan SP, Arsenic disturbs the gut microbiome of individuals in a disadvantaged community in Nepal, Heliyon 6(1) (2020) e03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shao M, Zhu Y, Long-term metal exposure changes gut microbiota of residents surrounding a mining and smelting area, Sci Rep 10(1) (2020) 4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu W, Zhang S, Jiang W, Xu S, Jin P, Arsenic Accumulation of Realgar Altered by Disruption of Gut Microbiota in Mice, Evid Based Complement Alternat Med 2020 (2020) 8380473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mazumder P, Sharma SK, Taki K, Kalamdhad AS, Kumar M, Microbes involved in arsenic mobilization and respiration: a review on isolation, identification, isolates and implications, Environ Geochem Health 42(10) (2020) 3443–3469. [DOI] [PubMed] [Google Scholar]
- [31].Van de Wiele T, Gallawa CM, Kubachka KM, Creed JT, Basta N, Dayton EA, Whitacre S, Du Laing G, Bradham K, Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils, Environ Health Perspect 118(7) (2010) 1004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].SS DCR, Alava P, Zekker I, Du Laing G, Van de Wiele T, Arsenic thiolation and the role of sulfate-reducing bacteria from the human intestinal tract, Environ Health Perspect 122(8) (2014) 817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Choiniere J, Wang L, Exposure to inorganic arsenic can lead to gut microbe perturbations and hepatocellular carcinoma, Acta Pharm Sin B 6(5) (2016) 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fernández Fernández N, Estevez Boullosa P, Gómez Rodríguez A, Rodríguez Prada JI, A Rare Cause of Gastric Injury: Arsenic Intake, Am J Gastroenterol 114(8) (2019) 1193. [DOI] [PubMed] [Google Scholar]
- [35].Chiocchetti GM, Vélez D, Devesa V, Inorganic arsenic causes intestinal barrier disruption, Metallomics 11(8) (2019) 1411–1418. [DOI] [PubMed] [Google Scholar]
- [36].Chiocchetti GM, Domene A, Kühl AA, Zúñiga M, Vélez D, Devesa V, Monedero V, In vivo evaluation of the effect of arsenite on the intestinal epithelium and associated microbiota in mice, Arch Toxicol 93(8) (2019) 2127–2139. [DOI] [PubMed] [Google Scholar]
- [37].Flora SJS, Pachauri V, Arsenic, Free Radical and Oxidative Stress, in: Kretsinger RH, Uversky VN, Permyakov EA (Eds.), Encyclopedia of Metalloproteins, Springer New York, New York, NY, 2013, pp. 149–159. [Google Scholar]
- [38].Hu Y, Li J, Lou B, Wu R, Wang G, Lu C, Wang H, Pi J, Xu Y, The Role of Reactive Oxygen Species in Arsenic Toxicity, Biomolecules 10(2) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE, Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases, Physiol Rev 94(2) (2014) 329–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chiocchetti GM, Vélez D, Devesa V, Effect of subchronic exposure to inorganic arsenic on the structure and function of the intestinal epithelium, Toxicol Lett 286 (2018) 80–88. [DOI] [PubMed] [Google Scholar]
- [41].Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang HG, Haribabu B, Vemula PK, Jala VR, Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway, Nat Commun 10(1) (2019) 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Agus A, Clement K, Sokol H, Gut microbiota-derived metabolites as central regulators in metabolic disorders, Gut (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saha P, Yeoh BS, Singh R, Chandrasekar B, Vemula PK, Haribabu B, Vijay-Kumar M, Jala VR, Gut Microbiota Conversion of Dietary Ellagic Acid into Bioactive Phytoceutical Urolithin A Inhibits Heme Peroxidases, PLoS One 11(6) (2016) e0156811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tomás-Barberán FA, González-Sarrías A, García-Villalba R, Núñez-Sánchez MA, Selma MV, García-Conesa MT, Espín JC, Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status, Molecular Nutrition & Food Research 61(1) (2017) 1500901. [DOI] [PubMed] [Google Scholar]
- [45].Espin JC, Gonzalez-Barrio R, Cerda B, Lopez-Bote C, Rey AI, Tomas-Barberan FA, Iberian pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans, J Agric Food Chem 55(25) (2007) 10476–85. [DOI] [PubMed] [Google Scholar]
- [46].Espin JC, Larrosa M, Garcia-Conesa MT, Tomas-Barberan F, Biological significance of urolithins, the gut microbial ellagic Acid-derived metabolites: the evidence so far, Evid Based Complement Alternat Med 2013 (2013) 270418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tomas-Barberan FA, Gonzalez-Sarrias A, Garcia-Villalba R, Nunez-Sanchez MA, Selma MV, Garcia-Conesa MT, Espin JC, Urolithins, the rescue of ‘old’ metabolites to understand a ‘new’ concept: metabotypes as a nexus between phenolic metabolism, microbiota dysbiosis and host health status, Mol Nutr Food Res (2016). [DOI] [PubMed] [Google Scholar]
- [48].Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J, Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents, Nat Med 22(8) (2016) 879–88. [DOI] [PubMed] [Google Scholar]
- [49].D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J, Impact of the Natural Compound Urolithin A on Health, Disease, and Aging, Trends Mol Med 27(7) (2021) 687–699. [DOI] [PubMed] [Google Scholar]
- [50].Banerjee M, Ferragut Cardoso AP, Lykoudi A, Wilkey DW, Pan J, Watson WH, Garbett NC, Rai SN, Merchant ML, States JC, Arsenite Exposure Displaces Zinc from ZRANB2 Leading to Altered Splicing, Chem Res Toxicol 33(6) (2020) 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat Methods 9(7) (2012) 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jala VR, Bodduluri SR, Ghosh S, Chheda Z, Singh R, Smith ME, Chilton PM, Fleming CJ, Mathis SP, Sharma RK, Knight R, Yan J, Haribabu B, Absence of CCR2 reduces spontaneous intestinal tumorigenesis in the Apc(Min) (/+) mouse model, Int J Cancer (2021). [DOI] [PubMed] [Google Scholar]
- [53].Larrosa M, Gonzalez-Sarrias A, Yanez-Gascon MJ, Selma MV, Azorin-Ortuno M, Toti S, Tomas-Barberan F, Dolara P, Espin JC, Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism, The Journal of nutritional biochemistry 21(8) (2010) 717–25. [DOI] [PubMed] [Google Scholar]
- [54].Gonzalez-Sarrias A, Gimenez-Bastida JA, Garcia-Conesa MT, Gomez-Sanchez MB, Garcia-Talavera NV, Gil-Izquierdo A, Sanchez-Alvarez C, Fontana-Compiano LO, Morga-Egea JP, Pastor-Quirante FA, Martinez-Diaz F, Tomas-Barberan FA, Espin JC, Occurrence of urolithins, gut microbiota ellagic acid metabolites and proliferation markers expression response in the human prostate gland upon consumption of walnuts and pomegranate juice, Mol Nutr Food Res 54(3) (2010) 311–22. [DOI] [PubMed] [Google Scholar]
- [55].Heilman J, Andreux P, Tran N, Rinsch C, Blanco-Bose W, Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid, Food Chem Toxicol 108(Pt A) (2017) 289–297. [DOI] [PubMed] [Google Scholar]
- [56].Tomas-Barberan FA, Gonzalez-Sarrias A, Garcia-Villalba R, Nunez-Sanchez MA, Selma MV, Garcia-Conesa MT, Espin JC, Urolithins, the rescue of “old” metabolites to understand a “new” concept: Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status, Mol Nutr Food Res 61(1) (2017). [DOI] [PubMed] [Google Scholar]
- [57].Stevens JJ, Graham B, Walker AM, Tchounwou PB, Rogers C, The effects of arsenic trioxide on DNA synthesis and genotoxicity in human colon cancer cells, Int J Environ Res Public Health 7(5) (2010) 2018–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Keim A, Rossler OG, Rothhaar TL, Thiel G, Arsenite-induced apoptosis of human neuroblastoma cells requires p53 but occurs independently of c-Jun, Neuroscience 206 (2012) 25–38. [DOI] [PubMed] [Google Scholar]
- [59].Pan X, Jiang L, Zhong L, Geng C, Jia L, Liu S, Guan H, Yang G, Yao X, Piao F, Sun X, Arsenic induces apoptosis by the lysosomal-mitochondrial pathway in INS-1 cells, Environ Toxicol 31(2) (2016) 133–41. [DOI] [PubMed] [Google Scholar]
- [60].Satoh T, Enokido Y, Aoshima H, Uchiyama Y, Hatanaka H, Changes in mitochondrial membrane potential during oxidative stress-induced apoptosis in PC12 cells, J Neurosci Res 50(3) (1997) 413–20. [DOI] [PubMed] [Google Scholar]
- [61].Zhao RZ, Jiang S, Zhang L, Yu ZB, Mitochondrial electron transport chain, ROS generation and uncoupling (Review), Int J Mol Med 44(1) (2019) 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kim KB, Lee S, Kim JH, Neuroprotective effects of urolithin A on H2O2-induced oxidative stress-mediated apoptosis in SK-N-MC cells, Nutr Res Pract 14(1) (2020) 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Karim MR, Salam KA, Hossain E, Islam K, Ali N, Haque A, Saud ZA, Yeasmin T, Hossain M, Miyataka H, Himeno S, Hossain K, Interaction between chronic arsenic exposure via drinking water and plasma lactate dehydrogenase activity, Sci Total Environ 409(2) (2010) 278–83. [DOI] [PubMed] [Google Scholar]
- [64].Jovanovic P, Zoric L, Stefanovic I, Dzunic B, Djordjevic-Jocic J, Radenkovic M, Jovanovic M, Lactate dehydrogenase and oxidative stress activity in primary open-angle glaucoma aqueous humour, Bosn J Basic Med Sci 10(1) (2010) 83–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Devriese S, Van den Bossche L, Van Welden S, Holvoet T, Pinheiro I, Hindryckx P, De Vos M, Laukens D, T84 monolayers are superior to Caco-2 as a model system of colonocytes, Histochem Cell Biol 148(1) (2017) 85–93. [DOI] [PubMed] [Google Scholar]
- [66].Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB, Reactive oxygen species in inflammation and tissue injury, Antioxid Redox Signal 20(7) (2014) 1126–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ayehunie S, Stevens Z, Landry T, Taimi M, Klausner M, Hayden P, Novel 3 D Human Small Intestinal Tissue Model (EpiInestinal TM) to Assess Drug Permeation & Inflammation, 2014.
- [68].Cui Y, Claus S, Schnell D, Runge F, MacLean C, In-Depth Characterization of EpiIntestinal Microtissue as a Model for Intestinal Drug Absorption and Metabolism in Human, Pharmaceutics 12(5) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saha JC, Dikshit AK, Bandyopadhyay M, Saha KC, A Review of Arsenic Poisoning and its Effects on Human Health, Critical Reviews in Environmental Science and Technology 29(3) (1999) 281–313. [Google Scholar]
- [70].Guha Mazumder D, Dasgupta UB, Chronic arsenic toxicity: studies in West Bengal, India, Kaohsiung J Med Sci 27(9) (2011) 360–70. [DOI] [PubMed] [Google Scholar]
- [71].Calatayud M, Devesa V, Velez D, Differential toxicity and gene expression in Caco-2 cells exposed to arsenic species, Toxicol Lett 218(1) (2013) 70–80. [DOI] [PubMed] [Google Scholar]
- [72].Sanyal T, Bhattacharjee P, Paul S, Bhattacharjee P, Recent Advances in Arsenic Research: Significance of Differential Susceptibility and Sustainable Strategies for Mitigation, Front Public Health 8 (2020) 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zheng Y, Lessons Learned from Arsenic Mitigation among Private Well Households, Curr Environ Health Rep 4(3) (2017) 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Weerasundara L, Ok YS, Bundschuh J, Selective removal of arsenic in water: A critical review, Environ Pollut 268(Pt B) (2021) 115668. [DOI] [PubMed] [Google Scholar]
- [75].Hossain M, Rahman SN, Bhattacharya P, Jacks G, Saha R, Rahman M, Sustainability of arsenic mitigation interventions—an evaluation of different alternative safe drinking water options provided in Matlab, an arsenic hot spot in Bangladesh, Frontiers in Environmental Science 3(30) (2015). [Google Scholar]
- [76].Carson RT, Koundouri P, Nauges C, Arsenic Mitigation in Bangladesh: A Household Labor Market Approach, American Journal of Agricultural Economics 93(2) (2010) 407–414. [Google Scholar]
- [77].Cerda B, Periago P, Espin JC, Tomas-Barberan FA, Identification of urolithin a as a metabolite produced by human colon microflora from ellagic acid and related compounds, J Agric Food Chem 53(14) (2005) 5571–6. [DOI] [PubMed] [Google Scholar]
- [78].Cerda B, Espin JC, Parra S, Martinez P, Tomas-Barberan FA, The potent in vitro antioxidant ellagitannins from pomegranate juice are metabolised into bioavailable but poor antioxidant hydroxy-6H-dibenzopyran-6-one derivatives by the colonic microflora of healthy humans, Eur J Nutr 43(4) (2004) 205–20. [DOI] [PubMed] [Google Scholar]
- [79].Garcia-Villalba R, Beltran D, Espin JC, Selma MV, Tomas-Barberan FA, Time course production of urolithins from ellagic acid by human gut microbiota, J Agric Food Chem 61(37) (2013) 8797–806. [DOI] [PubMed] [Google Scholar]
- [80].Tomas-Barberan FA, Garcia-Villalba R, Gonzalez-Sarrias A, Selma MV, Espin JC, Ellagic acid metabolism by human gut microbiota: consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status, J Agric Food Chem 62(28) (2014) 6535–8. [DOI] [PubMed] [Google Scholar]
- [81].Cerda B, Tomas-Barberan FA, Espin JC, Metabolism of antioxidant and chemopreventive ellagitannins from strawberries, raspberries, walnuts, and oak-aged wine in humans: identification of biomarkers and individual variability, J Agric Food Chem 53(2) (2005) 227–35. [DOI] [PubMed] [Google Scholar]
- [82].Seeram NP, Zhang Y, McKeever R, Henning SM, Lee RP, Suchard MA, Li Z, Chen S, Thames G, Zerlin A, Nguyen M, Wang D, Dreher M, Heber D, Pomegranate juice and extracts provide similar levels of plasma and urinary ellagitannin metabolites in human subjects, J Med Food 11(2) (2008) 390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Seeram NP, Henning SM, Zhang Y, Suchard M, Li Z, Heber D, Pomegranate juice ellagitannin metabolites are present in human plasma and some persist in urine for up to 48 hours, J Nutr 136(10) (2006) 2481–5. [DOI] [PubMed] [Google Scholar]
- [84].Nunez-Sanchez MA, Garcia-Villalba R, Monedero-Saiz T, Garcia-Talavera NV, Gomez-Sanchez MB, Sanchez-Alvarez C, Garcia-Albert AM, Rodriguez-Gil FJ, Ruiz-Marin M, Pastor-Quirante FA, Martinez-Diaz F, Yanez-Gascon MJ, Gonzalez-Sarrias A, Tomas-Barberan FA, Espin JC, Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients, Mol Nutr Food Res 58(6) (2014) 1199–211. [DOI] [PubMed] [Google Scholar]
- [85].Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C, The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans, Nature Metabolism 1(6) (2019) 595–603. [DOI] [PubMed] [Google Scholar]
- [86].Singh A, D’Amico D, Andreux PA, Dunngalvin G, Kern T, Blanco-Bose W, Auwerx J, Aebischer P, Rinsch C, Direct supplementation with Urolithin A overcomes limitations of dietary exposure and gut microbiome variability in healthy adults to achieve consistent levels across the population, Eur J Clin Nutr (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Huang C, Ma WY, Li J, Dong Z, Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway, Cancer Res 59(13) (1999) 3053–8. [PubMed] [Google Scholar]
- [88].Banerjee N, Banerjee M, Ganguly S, Bandyopadhyay S, Das JK, Bandyopadhay A, Chatterjee M, Giri AK, Arsenic-induced mitochondrial instability leading to programmed cell death in the exposed individuals, Toxicology 246(2–3) (2008) 101–11. [DOI] [PubMed] [Google Scholar]
- [89].Prakash C, Chhikara S, Kumar V, Mitochondrial Dysfunction in Arsenic-Induced Hepatotoxicity: Pathogenic and Therapeutic Implications, Biol Trace Elem Res (2021). [DOI] [PubMed] [Google Scholar]
- [90].Biswas R, Ghosh P, Banerjee N, Das JK, Sau T, Banerjee A, Roy S, Ganguly S, Chatterjee M, Mukherjee A, Giri AK, Analysis of T-cell proliferation and cytokine secretion in the individuals exposed to arsenic, Hum Exp Toxicol 27(5) (2008) 381–6. [DOI] [PubMed] [Google Scholar]
- [91].Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY, Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients, Blood 89(9) (1997) 3354–60. [PubMed] [Google Scholar]
- [92].Gonsebatt ME, Vega L, Montero R, Garcia-Vargas G, Del Razo LM, Albores A, Cebrian ME, Ostrosky-Wegman P, Lymphocyte replicating ability in individuals exposed to arsenic via drinking water, Mutat Res 313(2–3) (1994) 293–9. [DOI] [PubMed] [Google Scholar]
- [93].Gonsebatt ME, Vega L, Salazar AM, Montero R, Guzmán P, Blas J, Del Razo LM, García-Vargas G, Albores A, Cebrián ME, Kelsh M, Ostrosky-Wegman P, Cytogenetic effects in human exposure to arsenic, Mutat Res 386(3) (1997) 219–28. [DOI] [PubMed] [Google Scholar]
- [94].Pi J, Kumagai Y, Sun G, Yamauchi H, Yoshida T, Iso H, Endo A, Yu L, Yuki K, Miyauchi T, Shimojo N, Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia, Free Radic Biol Med 28(7) (2000) 1137–42. [DOI] [PubMed] [Google Scholar]
- [95].Aviello G, Knaus UG, ROS in gastrointestinal inflammation: Rescue Or Sabotage?, Br J Pharmacol 174(12) (2017) 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zuo L, Kuo WT, Turner JR, Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis, Cell Mol Gastroenterol Hepatol 10(2) (2020) 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Buckley A, Turner JR, Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease, Cold Spring Harb Perspect Biol 10(1) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].France MM, Turner JR, The mucosal barrier at a glance, J Cell Sci 130(2) (2017) 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Odenwald MA, Turner JR, Intestinal permeability defects: is it time to treat?, Clin Gastroenterol Hepatol 11(9) (2013) 1075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zeisel MB, Dhawan P, Baumert TF, Tight junction proteins in gastrointestinal and liver disease, Gut 68(3) (2019) 547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Caricilli AM, Castoldi A, Câmara NO, Intestinal barrier: A gentlemen’s agreement between microbiota and immunity, World J Gastrointest Pathophysiol 5(1) (2014) 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Rao RK, Seth A, Sheth P, Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease, Am J Physiol Gastrointest Liver Physiol 286(6) (2004) G881–4. [DOI] [PubMed] [Google Scholar]
- [103].Li B, Lee C, Chuslip S, Lee D, Biouss G, Wu R, Koike Y, Miyake H, Ip W, Gonska T, Pierro A, Intestinal epithelial tight junctions and permeability can be rescued through the regulation of endoplasmic reticulum stress by amniotic fluid stem cells during necrotizing enterocolitis, Faseb j 35(1) (2021) e21265. [DOI] [PubMed] [Google Scholar]
- [104].Garbinski LD, Rosen BP, Chen J, Pathways of arsenic uptake and efflux, Environ Int 126 (2019) 585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Roggenbeck BA, Banerjee M, Leslie EM, Cellular arsenic transport pathways in mammals, J Environ Sci (China) 49 (2016) 38–58. [DOI] [PubMed] [Google Scholar]
- [106].Aviello G, Knaus UG, NADPH oxidases and ROS signaling in the gastrointestinal tract, Mucosal Immunology 11(4) (2018) 1011–1023. [DOI] [PubMed] [Google Scholar]
- [107].Rao R, Oxidative stress-induced disruption of epithelial and endothelial tight junctions, Front Biosci 13 (2008) 7210–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].States JC, Barchowsky A, Cartwright IL, Reichard JF, Futscher BW, Lantz RC, Arsenic toxicology: translating between experimental models and human pathology, Environ Health Perspect 119(10) (2011) 1356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vahter M, Methylation of inorganic arsenic in different mammalian species and population groups, Sci Prog 82 (Pt 1) (1999) 69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Vahter M, Mechanisms of arsenic biotransformation, Toxicology 181–182 (2002) 211–7. [DOI] [PubMed] [Google Scholar]
- [111].Coryell M, McAlpine M, Pinkham NV, McDermott TR, Walk ST, The gut microbiome is required for full protection against acute arsenic toxicity in mouse models, Nature Communications 9(1) (2018) 5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Koller BH, Snouwaert JN, Douillet C, Jania LA, El-Masri H, Thomas DJ, Stýblo M, Arsenic Metabolism in Mice Carrying a BORCS7/AS3MT Locus Humanized by Syntenic Replacement, Environ Health Perspect 128(8) (2020) 87003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, analytic methods, and study materials will be made available to other researchers.