

To the Editor:
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrosing interstitial lung disease (ILD) with a poor prognosis (1). In 2014, two antifibrotic agents, nintedanib and pirfenidone, were approved by the U.S. Food and Drug Administration to slow the progression of the disease (2–4). Subsequently, studies among patients enrolled in IPF registries reported antifibrotic agent uptake rates among eligible patients with IPF as high as 70% (5, 6), whereas adoption among insured patients in the general population and veterans was lower (approximately 17–25%) (7, 8). The uptake of and factors associated with antifibrotic agent use in socioeconomically diverse populations is unclear. We evaluated the adoption of antifibrotic medications for patients with IPF in a large, multicenter, all-payer database.
Methods
We used the TriNetX Analytics Platform Research Network (https://trinetx.com/), an electronic health record and claims database with inpatient and outpatient data on all patients treated at more than 76 healthcare organizations across the United States (regardless of insurance/payer), including academic and community facilities. We defined incident cases of IPF from January 1, 2016, to December 22, 2022, as patients ⩾50 years of age (1) with at least one ambulatory visit without an IPF diagnosis (International Classification of Diseases, Tenth Revision code J84.112 [9]) followed by at least two subsequent ambulatory visits with an IPF diagnosis. Patients with another ILD or connective tissue disease before the first IPF code or medication initiation more than 30 days before the first IPF visit were excluded; our cohort definition is intentionally more specific than previously published cohort definitions (7, 8).
We calculated the annual incidence rate of antifibrotic agents by assessing the number of antifibrotic prescriptions prescribed divided by the total person-time accumulated within each calendar year. Patients contributed person-time to only the year in which they were first diagnosed with IPF. Person-time was censored at death, new connective tissue disease or interstitial lung disease diagnosis code, or the last encounter in the database before the end of the calendar year of the diagnosis.
We used multivariable regression analysis to assess factors associated with the receipt of antifibrotic agents within 1 year of diagnosis. The included factors were demographic characteristics (age, sex, race [White/non-Hispanic versus non-White/Hispanic/unknown]), adverse social determinants of health, comorbidities (modified individual components of the combined comorbidity score [10, 11]), prior testing (pulmonary function testing, echocardiography, oximetry), and prior healthcare encounters (emergency visit, inpatient admission). The presence of prior testing and prior healthcare encounters were assessed in the year before the first IPF code. Codes used to define the variables can be found at https://osf.io/x6dw7/?view_only=08c77e52720c46729b007ccef57fb6b2. Statistical testing was two-tailed, with an α of 0.05, and was conducted using R (version 4.0.2; R Foundation for Statistical Computing). This study was deemed exempt by the Boston University Institutional Review Board (H-43331).
Results
We identified 5,111 patients with IPF who met the inclusion criteria (Figure 1) (mean age, 71.0 yr; 61.8% male; 62.0% White/non-Hispanic); 839 patients (16.4%) received an antifibrotic agent after an initial IPF diagnosis. Among patients who received an antifibrotic agent, the median time to initiation was 43 days (interquartile range, 0–224.5 d). Patient characteristics stratified by antifibrotic treatment are depicted in Table 1.
Figure 1.
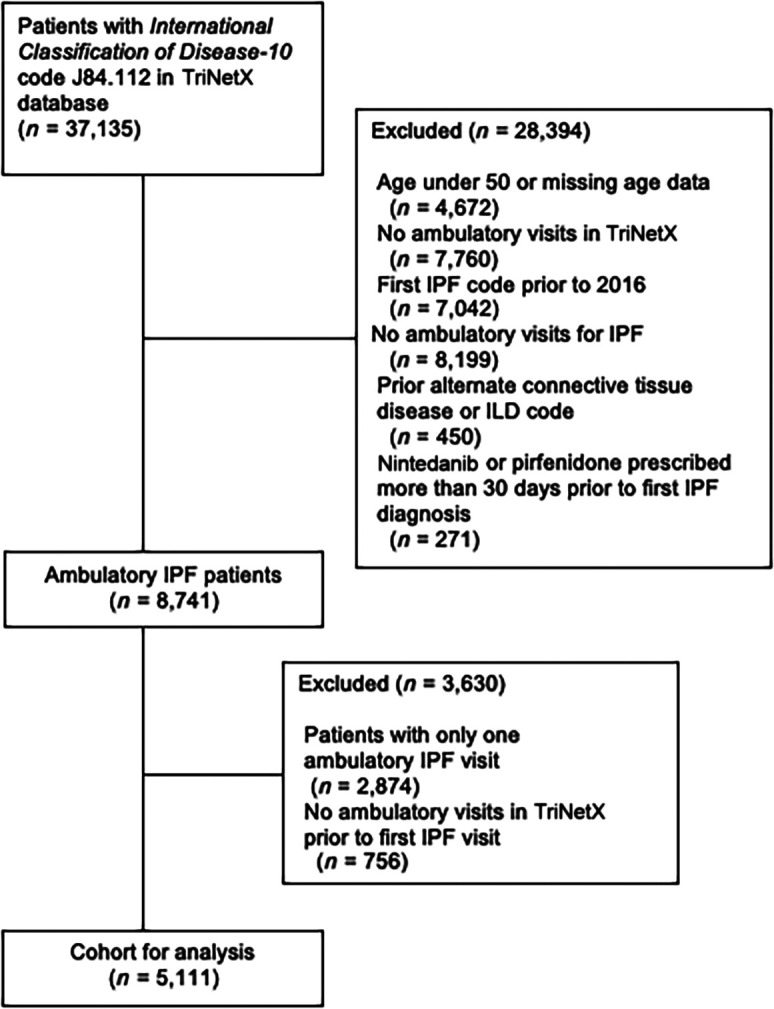
Flow diagram of cohort selection. IPF = idiopathic pulmonary fibrosis; ILD = interstitial lung disease.
Table 1.
Baseline characteristics and factors associated with receipt of antifibrotic agents within 1 year of IPF diagnosis
| Patient Characteristic | No Antifibrotic (n = 4,272) | Antifibrotic (n = 839) | aOR (95% CI) |
|---|---|---|---|
| Mean age ± SD, yr | 71.1 ± 8.35 | 70.9 ± SD 8.04 | 0.94 (0.86–1.03) |
| Sex | |||
| Male (reference) | 2,581 (60.4%) | 579 (69.0%) | – |
| Female | 1,518 (35.5%) | 249 (29.7%) | 0.71 (0.59–0.85)* |
| Unknown | 173 (4.0%) | 11 (1.3%) | 0.12 (0.04–0.28)* |
| Race and ethnicity | |||
| White/non-Hispanic (reference) | 2,549 (59.7%) | 619 (73.8%) | – |
| Non-white, Hispanic, and/or unknown race/ethnicity | 1,723 (40.3%) | 220 (26.2%) | 0.65 (0.54–0.79)* |
| Adverse social determinants of health† | 142 (3.3%) | 13 (1.5%) | 0.60 (0.28–1.14) |
| Geographic region | |||
| Northeast (reference) | 580 (13.6%) | 209 (24.9%) | – |
| Midwest | 247 (5.8%) | 57 (6.8%) | 0.47 (0.31–0.68)* |
| South | 2,152 (50.4%) | 301 (35.9%) | 0.33 (0.26–0.43)* |
| West | 1,144 (26.8%) | 270 (32.2%) | 0.68 (0.53–0.87)* |
| Unknown | 149 (3.5%) | 2 (0.2%) | 0.03 (0.00–0.13)* |
| Ischemic heart disease‡ | 1,226 (28.7%) | 211 (25.1%) | 0.85 (0.68–1.06) |
| Congestive heart failure‡ | 550 (12.9%) | 88 (10.5%) | 0.74 (0.54–1.00) |
| Liver disease‡ | 384 (9.0%) | 55 (6.6%) | 0.78 (0.55–1.08) |
| Gastrointestinal composite§ | 1,715 (40.1%) | 343 (40.9%) | 0.98 (0.81–1.18) |
| Renal failure | 500 (11.7%) | 79 (9.4%) | 0.99 (0.73–1.35) |
| Alcohol abuse | 121 (2.8%) | 23 (2.7%) | 0.96 (0.55–1.61) |
| Any tumor | 384 (9.0%) | 67 (8.0%) | 0.82 (0.59–1.12) |
| Cardiac arrythmias | 1,002 (23.5%) | 179 (21.3%) | 0.85 (0.67–1.07) |
| Chronic pulmonary disease | 1,765 (41.3%) | 282 (33.6%) | 0.68 (0.56–0.82)* |
| Coagulopathy | 275 (6.4%) | 49 (5.8%) | 0.76 (0.51–1.11) |
| Complicated diabetes | 505 (11.8%) | 89 (10.6%) | 0.91 (0.67–1.22) |
| Anemia | 237 (5.5%) | 36 (4.3%) | 0.86 (0.56–1.29) |
| Fluid and electrolyte disorder | 586 (13.7%) | 120 (14.3%) | 1.10 (0.83–1.45) |
| HIV/AIDS | 27 (0.6%) | 3 (0.4%) | 1.13 (0.26–3.49) |
| Hypertension | 2,012 (47.1%) | 401 (47.8%) | 0.98 (0.81–1.20) |
| Metastatic cancer | 190 (4.4%) | 19 (2.3%) | 0.58 (0.32–0.99)* |
| Peripheral vascular disease | 664 (15.5%) | 106 (12.6%) | 0.89 (0.68–1.16) |
| Psychosis | 24 (0.6%) | 5 (0.6%) | 1.08 (0.24–3.35) |
| Pulmonary circulation disorder | 696 (16.3%) | 117 (13.9%) | 0.93 (0.72–1.20) |
| Weight loss | 336 (7.9%) | 44 (5.2%) | 0.81 (0.54–1.19) |
| Dementia | 60 (1.4%) | 6 (0.7%) | 0.83 (0.31–1.90) |
| Hemiplegia | 58 (1.4%) | 6 (0.7%) | 0.79 (0.26–1.94) |
| Pulmonary function testsǁ | 1,907 (44.6%) | 462 (55.1%) | 1.42 (1.18–1.70)* |
| Echocardiogramǁ | 765 (17.9%) | 195 (23.2%) | 1.21 (0.96–1.53) |
| Oximetryǁ | 400 (9.4%) | 131 (15.6%) | 1.48 (1.15–1.90)* |
| Emergency department visitǁ | 466 (10.9%) | 145 (17.3%) | 1.89 (1.47–2.41)* |
| Inpatient visitǁ | 725 (17.0%) | 196 (23.4%) | 1.68 (1.34–2.10)* |
Definition of abbreviations: AIDS = acquired immunodeficiency syndrome; aOR = adjusted odds ratio; HIV = human immunodeficiency virus; SD = standard deviation.
The value signifies that the adjusted odds ratio does not cross 1.
Social determinants of health identified by International Classification of Diseases, 10th Revision coding.
Gastrointestinal composite comprising International Classification of Diseases codes for gastrointestinal reflux disease, irritable bowel syndrome, aphagia, and nausea.
Prior testing and visits occurring as long as 1 year before the presence of the first idiopathic pulmonary fibrosis code.
Annual rates of antifibrotic agent uptake remained stable to slightly increasing until 2021, after which nintedanib use increased and pirfenidone use remained stable (Figure 2). The factors associated with the receipt of antifibrotic agents after full model adjustment are shown in Table 1. The odds of receiving antifibrotic agents within 1 year of a IPF diagnosis was lower in patients who were female (odds ratio [OR], 0.71; 95% confidence interval [CI], 0.59–0.85; reference category, male), were of non-White/Hispanic/unknown race or ethnicity (OR, 0.65; 95% CI, 0.54–0.79; reference category, White/non-Hispanic), had chronic pulmonary disease (OR, 0.68; 95% CI, 0.56–0.82), or had metastatic cancer (OR, 0.58; 95% CI, 0.32–0.99). Pulmonary testing and healthcare encounters in the 1 year before the IPF diagnosis were associated with increased odds of receiving an antifibrotic agent within 1 year of diagnosis.
Figure 2.

Antifibrotic prescriptions per annual person-year among incident cases of IPF. IPF = idiopathic pulmonary fibrosis.
Discussion
Using a large, multicenter, all-payer claims-based database, from 2016 to 2022, we found that the uptake of antifibrotic medications was lower than prior registry-based estimates but has increased over time. We observed disparities in prescription rates along gender and racial lines.
Society guidelines recommend the use of antifibrotic therapy in patients with mild to moderate IPF (12). The adoption of antifibrotic agents in our study (16%) is lower than previously observed in registry-based studies (58–70%), a study using private insurance/Medicare Advantage administrative data (25%), and a study in the U.S. Veterans Affairs Health Care System (17%) (5–8). Registry-based studies are susceptible to selection bias because participants are often carefully selected patients at ILD centers, and adoption of new practices at ILD centers may differ widely from those at nonspecialty centers. Our slightly lower adoption rate compared with insured patients or veterans (7, 8) may be due to our inclusion of uninsured/underinsured patients without universal medication coverage, suggesting that costs may be a barrier to the receipt of antifibrotic agents. For example, out-of-pocket costs for a 30-day supply of an antifibrotic agent are $5–$11 for veterans but may be as high as $400 for the general population (2, 7). Despite generally low prescribing rates for antifibrotic agents, rates of nintedanib use increased after approval expanded to the treatment of progressive pulmonary fibrosis in 2019 (13); the expanded indication may have increased provider familiarity with nintedanib. Nintedanib is also dosed twice daily, compared with pirfenidone’s thrice-daily dosing, possibly leading some providers and patients to favor nintedanib (14).
Our cohort was more diverse than prior registry-based or population-specific administrative studies (i.e., 62% male and 62% White/non-Hispanic vs. 75–95% male and 90–95% White [5, 6, 8]). Additionally, we intentionally used narrow, highly specific inclusion criteria to best isolate a population of patients with true and incident IPF who were receiving ambulatory care (and therefore eligible for antifibrotic agent initiation) within the healthcare systems captured in TriNetX; even though our resulting cohort is smaller, there is a decreased risk for misclassification than in previously published work using the same dataset (15). Similar to prior studies, we found that women and participants of minority race had lower odds of receiving antifibrotic therapy even after adjusting for social determinants of health, a pattern that has also been noted in the initiation of immunomodulatory and antifibrotic treatments in a larger cohort of patients with ILD (16). Further studies are needed to identify the reasons for the large disparities in antifibrotic agent prescribing.
Our study has limitations. First, there is the possibility of misclassification bias in the identification of our cohort within administrative data, and there is a possibility that patients with non-IPF fibrotic ILDs were misclassified as patients with IPF. However, we chose to use a validated definition of IPF to increase the probability of identifying true cases of IPF (17). Second, we were unable to completely adjust for the severity of IPF. We were also unable to assess for practice pattern clusters because healthcare organizations were deidentified. Finally, even though TriNetX represents a highly socioeconomically diverse cohort, the majority of included health organizations are academic centers, and antifibrotic agent uptake may be even lower in other settings.
Conclusions
Antifibrotic agent uptake in an incident IPF population is low but increasing in recent years. Future work should focus on addressing potential gender bias, structural racism, and cost barriers to improve the equitable uptake of antifibrotic agents in eligible populations.
Footnotes
Supported by National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute grants T32 HL007035 and 1F32HL168959-01 (D.A.S.), Cystic Fibrosis Foundation Regenerating Airway Epithelium with Basal Cells Derived from Human iPSCs (F.H.), NIH grants 5R01HL139799-04 (F.H.) and K08HL163494 (K.-D.A.), Boston University School of Medicine Department of Medicine Career Investment Awards (K.-D.A., A.J.W., and A.C.L.), NIH grants R01HL139751, R01HL151607, R01HL136660, and OT2HL156812-01 (A.J.W.), NIH/National Center for Advancing Translational Sciences grant 1KL2TR001411 (N.A.B.), NIH grants 1UL1TR001430 (N.A.B.) and K23HL153482 (A.C.L.), and a Doris Duke Charitable Foundation Fund to Retain Clinician Scientists (A.C.L.).
Author Contributions: D.A.S., F.H., K.-D.A., K.C.W., A.J.W., N.A.B., and A.C.L. were involved in the planning of this study. D.A.S., A.J.W., N.A.B., and A.C.L. were involved in the data collection and data analysis of this study. D.A.S., F.H., K.-D.A., K.C.W., A.J.W., N.A.B., and A.C.L. were involved in writing the manuscript for this study. D.A.S. submitted the study.
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med . 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assayag D. Broad adoption of antifibrotics in idiopathic pulmonary fibrosis: still a long way to go. Ann Am Thorac Soc. 2021;18:1115–1116. doi: 10.1513/AnnalsATS.202102-123ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med . 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 4.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 5. Salisbury ML, Conoscenti CS, Culver DA, Yow E, Neely ML, Bender S, et al. IPF-PRO Registry principal investigators as follows Antifibrotic drug use in patients with idiopathic pulmonary fibrosis. data from the IPF-PRO Registry. Ann Am Thorac Soc . 2020;17:1413–1423. doi: 10.1513/AnnalsATS.201912-880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holtze CH, Freiheit EA, Limb SL, Stauffer JL, Raimundo K, Pan WT, et al. Patient and site characteristics associated with pirfenidone and nintedanib use in the United States; an analysis of idiopathic pulmonary fibrosis patients enrolled in the Pulmonary Fibrosis Foundation Patient Registry. Respir Res . 2020;21:48. doi: 10.1186/s12931-020-1315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dempsey TM, Payne S, Sangaralingham L, Yao X, Shah ND, Limper AH. Adoption of the antifibrotic medications pirfenidone and nintedanib for patients with idiopathic pulmonary fibrosis. Ann Am Thorac Soc . 2021;18:1121–1128. doi: 10.1513/AnnalsATS.202007-901OC. [DOI] [PubMed] [Google Scholar]
- 8. Kaul B, Lee JS, Petersen LA, McCulloch C, Rosas IO, Bandi VD, et al. Disparities in antifibrotic medication utilization among veterans with idiopathic pulmonary fibrosis. Chest . 2023;164:441–449. doi: 10.1016/j.chest.2023.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ley B, Urbania T, Husson G, Vittinghoff E, Brush DR, Eisner MD, et al. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis. Case validation and improvement. Ann Am Thorac Soc . 2017;14:880–887. doi: 10.1513/AnnalsATS.201610-764OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quan H, Sundararajan V, Halfon P, Fong A, Burnand B, Luthi JC, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care . 2005;43:1130–1139. doi: 10.1097/01.mlr.0000182534.19832.83. [DOI] [PubMed] [Google Scholar]
- 11. Sun JW, Rogers JR, Her Q, Welch EC, Panozzo CA, Toh S, et al. Adaptation and validation of the combined comorbidity score for ICD-10-CM. Med Care . 2017;55:1046–1051. doi: 10.1097/MLR.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 12. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, et al. American Thoracic Society European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT Clinical Practice Guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med . 2015;192:e3–e19. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 13. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. INBUILD Trial Investigators Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med . 2019;381:1718–1727. doi: 10.1056/NEJMoa1908681. [DOI] [PubMed] [Google Scholar]
- 14. Coleman CI, Limone B, Sobieraj DM, Lee S, Roberts MS, Kaur R, et al. Dosing frequency and medication adherence in chronic disease. J Manag Care Pharm . 2012;18:527–539. doi: 10.18553/jmcp.2012.18.7.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao J, Fares J, George G, Maheu A, Loizidis G, Roman J, et al. Racial and ethnic disparities in antifibrotic therapy in idiopathic pulmonary fibrosis. Respirology . 2023;28:1036–1042. doi: 10.1111/resp.14563. [DOI] [PubMed] [Google Scholar]
- 16. Assayag D, Adegunsoye A, Sheehy R, Morisset J, Khalil N, Johannson KA, et al. Sex- and race-based differences in the treatment of interstitial lung diseases in North America and Australasia. Chest . 2023;163:1156–1165. doi: 10.1016/j.chest.2022.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herberts MB, Teague TT, Thao V, Sangaralingham LR, Henk HJ, Hovde KT, et al. Idiopathic pulmonary fibrosis in the United States: time to diagnosis and treatment. BMC Pulm Med . 2023;23:281. doi: 10.1186/s12890-023-02565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]