

Abstract
Rationale
Chronic obstructive pulmonary disease (COPD) is associated with high morbidity, mortality, and healthcare costs. Cigarette smoke is a causative factor; however, not all heavy smokers develop COPD. Microbial colonization and infections are contributing factors to disease progression in advanced stages.
Objectives
We investigated whether lower airway dysbiosis occurs in mild-to-moderate COPD and analyzed possible mechanistic contributions to COPD pathogenesis.
Methods
We recruited 57 patients with a >10 pack-year smoking history: 26 had physiological evidence of COPD, and 31 had normal lung function (smoker control subjects). Bronchoscopy sampled the upper airways, lower airways, and environmental background. Samples were analyzed by 16S rRNA gene sequencing, whole genome, RNA metatranscriptome, and host RNA transcriptome. A preclinical mouse model was used to evaluate the contributions of cigarette smoke and dysbiosis on lower airway inflammatory injury.
Measurements and Main Results
Compared with smoker control subjects, microbiome analyses showed that the lower airways of subjects with COPD were enriched with common oral commensals. The lower airway host transcriptomics demonstrated differences in markers of inflammation and tumorigenesis, such as upregulation of IL-17, IL-6, ERK/MAPK, PI3K, MUC1, and MUC4 in mild-to-moderate COPD. Finally, in a preclinical murine model exposed to cigarette smoke, lower airway dysbiosis with common oral commensals augments the inflammatory injury, revealing transcriptomic signatures similar to those observed in human subjects with COPD.
Conclusions
Lower airway dysbiosis in the setting of smoke exposure contributes to inflammatory injury early in COPD. Targeting the lower airway microbiome in combination with smoking cessation may be of potential therapeutic relevance.
Keywords: microbiome, transcriptomics, metatranscriptomics, COPD, lung inflammation
At a Glance Commentary
Scientific Knowledge on the Subject
Not all heavy smokers develop physiological evidence of chronic obstructive pulmonary disease. The lower airway microbiome may play a role in disease pathogenesis.
What This Study Adds to the Field
Lower airway dysbiosis contributes to the inflammatory signatures seen in the lungs of patients with mild-to-moderate chronic obstructive pulmonary disease.
Chronic obstructive pulmonary disease (COPD) is a multifaceted and heterogeneous disease characterized by lower airway inflammation, airway narrowing, parenchymal destruction with alveolar damage, and irreversible airflow obstruction. Patients with COPD are at a higher risk of infections, malignancy, and other comorbid conditions, such as cardiovascular disease (1). Many inciting factors are linked to the pathogenesis of COPD, including cigarette smoking, air pollution, childhood respiratory illnesses, and genetic predisposition (2). However, many smokers do not develop the airflow obstruction required for a COPD diagnosis, and other environmental factors are likely at play in the pathogenesis of this disease. In moderate COPD, subjects with different types of inflammation (endotypes) defined by sputum neutrophilia, eosinophilia, or a pauci-immune profile have a distinct microbial composition of sputum microbiomes and serum inflammatory biomarkers (3). In patients with late-stage COPD, an increased abundance of Moraxella and Hemophilus is associated with an increased FEV1 decline and increased concentrations of inflammatory markers in sputum (4). However, patients with moderate to advanced COPD may have architectural destruction and chronic inflammation, leading to frequent administration of antimicrobial and systemic steroids during exacerbations, confounding the assessment of the lung microbiome as a possible driver in lung injury. Thus, studying the microbial host interaction in the lower airways of patients with mild disease may help elucidate the role of the lung microbiome in this disease process. Using culture-independent techniques, we have previously analyzed BAL samples from “healthy” smokers and healthy control subjects, demonstrating a direct correlation between lower airway enrichment with common oral commensals, such as Prevotella, Veillonella, Streptococcus, and Rothia, with biomarkers of lower airway inflammation (5). Recent cross-sectional investigations have identified similar microbial signals associated with mild-stage COPD (6, 7). As part of the SubPopulations and InteRmediate Outcome Measures in COPD Study (SPIROMICS) cohort, Opron and colleagues sampled the lower airways of 181 subjects and studied their lung microbiome (7). There, in comparing smokers without COPD to smokers with mild or moderate COPD, Streptococcus, Prevotella, Staphylococcus, and Pseudomonas were all associated with impaired lung function. Importantly, Streptococcus, Lactobacillales, and Veillonella were all differentially enriched in subjects with mild or moderate COPD. In a follow-up study of 137 subjects with COPD Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 1–2, enrichment with Streptococcus, Neisseria, and Veillonella was also associated with impaired lung function, COPD diagnosis, and increased symptom burden (6). Adding to these observations, dissecting the interaction between the microbiome and lung immunity may require a preclinical model. We previously showed in mice that lower airway aspiration with a mixture of oral commensal bacteria led to lower airway inflammation and altered susceptibility to Streptococcus pneumoniae (8). In this study, we tested whether lower airway dysbiosis identified among patients with mild-to-moderate COPD can cause lower airway inflammatory injury contributing to the pathogenesis of COPD. To do this, we examined a human smoking cohort, including patients with mild-to-moderate COPD and smoker control (SC) subjects, and conducted parallel investigations in a preclinical mouse model of smoke-induced lung injury.
Methods
Cohort
Smoking volunteers (>10 pack-year smoking history, based on self-report) were recruited to evaluate their lung function and undergo research bronchoscopy. Subjects underwent an initial screening visit that included blood sample collection, 12-lead ECG, chest X-ray, and pulmonary function testing. Subjects met the inclusion criteria for mild-to-moderate COPD if there was evidence of airflow obstruction (FEV1/FVC < 70) but with an FEV1 > 50% predicted. The SC cohort included subjects with no evidence of obstruction on spirometry (FEV1/FVC > 70% and/or FEV1 > 70%). The study protocol was approved by the Institutional Review Board of New York University (IRB#S14-01546), and all patients gave written consent.
Lower Airway Bronchoscopic Sampling Procedure
Background and supraglottic samples were obtained before the procedure, as previously described (5). Background samples were obtained by passing sterile saline through the suctioning channel of the bronchoscope before the procedure. BAL samples were obtained from the right middle lobe and lingula. Samples were transported on ice to the laboratory, where aliquots of whole BAL and BAL cell pellets were generated (for microbiome and host transcriptome, respectively) and stored at −80°C until further processing.
DNA/RNA Isolation, Library Preparation, and Sequencing
DNA and RNA were isolated from BAL samples, upper airway (UA), and environmental background controls (BKG) in parallel using zymoBIOMICS DNA/RNA Miniprep Kit (Cat: R2002) as per the manufacturer’s instructions. To ensure sufficient taxonomic data were generated from background samples, we included a higher amount of template from these samples compared with the amount from upper and lower airway samples. This allowed us to have sufficient depth of sequencing from background samples and to explore for potential background contaminants. DNA obtained from whole BAL was targeted sequenced (16S rRNA gene encoding the V4 region) (9) on a MiSeq platform (Illumina) and untargeted sequenced (whole-genome sequencing [WGS]) on a NovaSeq platform (Illumina). RNA obtained from whole BAL underwent RNA Sequencing for metatranscriptome, and RNA obtained from BAL cell pellets underwent RNA Sequencing for host transcriptome, both on a NovaSeq platform (Illumina). The obtained 16S rRNA gene sequences were analyzed using the Quantitative Insights into Microbial Ecology 2 package (10). Metagenomic and metatranscriptomic data were analyzed using Trimmomatic v0.36 (11), Bowtie2 v2.3.4.1 (12), Kraken v2.0.7 (13), Bracken v2.5 (14), and FMAP v0.15 (15), as previously described (16). Host transcriptomic reads were analyzed using Rsubread v2.10.5 (17). Figure E1 in the online supplement shows a summary of the depth achieved with the parallel 16S rRNA gene sequencing, metagenome, and metatranscriptome approaches across sample types and the number of reads assigned to different microbial subfractions (bacteria, fungi, DNA viruses, RNA viruses, and phages). Further analyses were performed to identify possible contaminants in the datasets; a description of this is in the online supplement, and possible contaminants are listed in Table E1. Taxa identified as possible contaminants were tagged as such but not removed from any subsequent analyses.
Mouse Dysbiosis and Cigarette Smoke Exposure
Female 12-week-old C57/BL6J mice obtained from Jackson Laboratories were used for the experiments described below, including tobacco smoke exposure and aspiration with a representative mixture of oral commensals (MOC). All animal studies were performed with the approval from the Institutional Animal Care and Use Committee of the Columbia University School of Medicine and New York University Grossman School of Medicine. Further detailed description of the mouse model methods can be found in the online supplement and have been previously published (18). Lungs from killed mice were harvested for histological assessment, including measurement of mean linear intercept (MLI), cytokine measurement, flow cytometry, fluorescence-activated cell sorting (FACS), multispectral immunohistochemistry (IHC), and host transcriptome.
Data Availability
Sequencing data are available in National Center for Biotechnology Information (NCBI) Sequence Read Archive under project number PRJNA870929 for human microbiome data, PRJNA936182 for murine microbiome data, and PRJNA870929 for host transcriptomic data. Codes used for the analyses presented in the current manuscript are available at https://github.com/segalmicrobiomelab/Mild_Moderate_COPD_microbiome. See the online supplement for more details on the methods used.
Results
We recruited 57 subjects with >10 pack-years of smoking for a research bronchoscopy. Among them, 26 met spirometry criteria for mild-to-moderate COPD (8 [30%] GOLD I and 18 [70%] GOLD II), and 31 did not (SC). Comparing the two groups, although overall age was similar, there were more subjects between the ages of 61 and 70 in the COPD cohort. Table 1 further lists demographics and clinical characteristics of these two groups. All subjects underwent research bronchoscopy. Samples collected included BAL, UA, and BKG.
Table 1.
Basic Demographics of Patient Cohort
| Subjects with COPD | Smoker Control Subjects | P Value | |
|---|---|---|---|
| N | 26 (45.6) | 31 (54.4) | — |
| Age, yr | 57.5 (51.5–61) | 54 (49.5–58) | 0.089 |
| Age category, yr | |||
| <51 | 6 (23.1) | 9 (29) | 0.257 |
| 51–60 | 12 (46.2) | 18 (58.1) | |
| 61–70 | 8 (30.8) | 4 (12.9) | |
| Sex (male) | 20 (76.9) | 22 (71) | 0.622 |
| BMI | 24.6 (21.9–29.8) | 28.4 (25.6–32.1) | 0.025 |
| BMI category | |||
| <30 | 19 (73.1) | 18 (58.1) | 0.237 |
| ⩾30 | 7 (26.9) | 13 (41.9) | |
| Race | |||
| Asian | 1 (3.8) | 1 (3.2) | 0.690 |
| Black or African American | 18 (69.2) | 17 (54.8) | |
| Caucasian | 5 (19.2) | 10 (32.3) | |
| Hispanic | 2 (7.7) | 3 (9.7) | |
| Smoking status | |||
| Former | 3 (11.5) | 2 (6.5) | 0.499 |
| Current | 23 (88.5) | 29 (93.5) | |
| Pack-years (average) | 21 (16–35) | 22 (12–34) | 0.798 |
| Inhaled corticosteroid | 5 (19.2) | 2 (6.5) | 0.143 |
| Inhaled β-agonist | 8 (30.8) | 3 (9.7) | 0.045 |
| FEV1 | |||
| Pre, L | 2.345 (2.025–2.76) | 2.99 (2.615–3.305) | <0.001 |
| Pre, % | 70.5 (63.3–75.8) | 89.5 (75.5–99) | <0.001 |
| Post, L | 2.44 (2.19–2.75) | 3.13 (2.66–3.45) | <0.001 |
| Post, % | 72 (68–84) | 93 (80–102.5) | <0.001 |
| FVC | |||
| Pre, L | 3.61 (3.19–4.27) | 3.72 (3.31–4.36) | 0.724 |
| Pre, % | 93 (85.3–94.8) | 93.5 (82.3–107.3) | 0.576 |
| Post, L | 3.63 (3.26–4.35) | 3.78 (3.26–4.35) | 0.656 |
| Post, % | 92 (85–101) | 95 (85–103.5) | 0.609 |
| FEV1:FVC | |||
| Pre, % | 65 (56–68) | 79 (72.5–82) | <0.001 |
| Post, % | 67 (61–69) | 81 (74.5–83.5) | <0.001 |
| R5 | |||
| Pre | 5.365 (4.37–6.66) | 4.77 (3.92–5.66) | 0.180 |
| Post | 4.61 (3.43–5.92) | 3.73 (3.48–4.54) | 0.106 |
| SGRQ | |||
| Median | 9.33 (3.28–25.93) | 16.56 (5.5–33.37) | 0.392 |
| High (>25) | 7 (28) | 12 (42.9) | 0.260 |
Definition of abbreviations: BMI = body mass index; COPD = chronic obstructive pulmonary disease; R5 = resistance 5; SGRQ = St. George’s Respiratory Questionnaire.
Values are presented as median (interquartile range) or n (proportion). Bold value indicates a P value < 0.05.
Topographical Analysis of the Microbiome
16S rRNA gene sequencing (16S) was performed on all collected samples (sequence depth shown in Figure E1). UA samples had significantly lower α-diversity, as measured by Shannon diversity index, compared with BAL or BKG samples (Figure E2A; Kruskal-Wallis test P < 0.01). Similarly, β-diversity analysis based on Bray-Curtis dissimilarity showed significant compositional differences between BKG, UA, and BAL samples (Figure E2B; permutational multivariate ANOVA [PERMANOVA] P < 0.01). We next evaluated differences between lower airway samples (BAL) from different sites (right middle lobe and lingula). Overall microbial composition (β-diversity) of the two lower airway samples from the same subject at different locations was more similar than two lower airway samples from different subjects in the same location (Figure E2C; Kruskal-Wallis test P < 0.01). These data support that respiratory microbial communities are less heterogeneous topographically within the same subject than between different subjects. We next compared the relative abundance of taxa in negative controls (BKG) to UA and/or BAL samples to identify potential contaminants. The top identified potential contaminants (by Wilcoxon rank-sum test) included Flavobacterium succinicans, Candidatus Aquiluna rubra, and Methylotenera mobilis (Figure E2D). Importantly, given the compositional nature of the data, potential contaminants were not removed from further analyses but kept labeled for identification.
Given the topographical similarity noted within each subject in the 16S data, we then selected one BAL sample for every subject plus 10 UA samples for metagenome and metatranscriptome sequencing with 7 and 10 BKG samples to evaluate the metagenome and metatranscriptome by WGS and RNA sequencing, respectively (sequence depth shown in Figure E1). Although the top taxa identified in the lower airways differed among the three datasets, similar to the 16S data, the lower airway metagenome and metatranscriptome showed significant differences in both α- and β-diversity by sample type (Figures E3A and E3B and E4A and E4B, respectively). Furthermore, the top potential environmental contaminants identified in the metagenome dataset were different from those identified in the 16S data and included Burkholderia dolosa, Xanthomonas citri, and Pseudoalteromonas sp. (Figure E3C and Table E1). The top potential contaminants identified in the metatranscriptome data were also different and included Malassezia restricta, Cutibacterium acnes, and Talaromyces rugulosus (Figure E4C and Table E1). Similar to the 16S data, we did not remove potential contaminants from subsequent analyses.
Microbial Differences between Subjects with COPD and SC Subjects
We tested for the identification of microbial signatures in the lower airways of subjects with physiological evidence of COPD. Bacterial burden measured by digital droplet PCR (ddPCR) was similar in the lower airways of subjects with COPD and SC subjects (Figure E5A; Kruskal-Wallis test P = 0.27). Microbiota analyses of the 16S data showed that within-sample diversity (Shannon diversity index) was significantly lower for COPD when compared with SC (Figure E5B; Kruskal-Wallis test P = 0.003). Lower airway microbial composition evaluated by β-diversity metrics (Bray-Curtis) showed nonsignificant differences between subjects with COPD and SC subjects (PERMANOVA P = 0.06). Differential enrichment analysis comparing the lower airway microbiota in subjects with COPD versus SC subjects identified several taxa as enriched in COPD, including many common oral commensals, such as those belonging to the genera Prevotella, Veillonella, and Streptococcus (Figure 1A). Taxa significantly enriched in SC included some from the genus Corynebacterium and Pseudomonas (Table E2).
Figure 1.
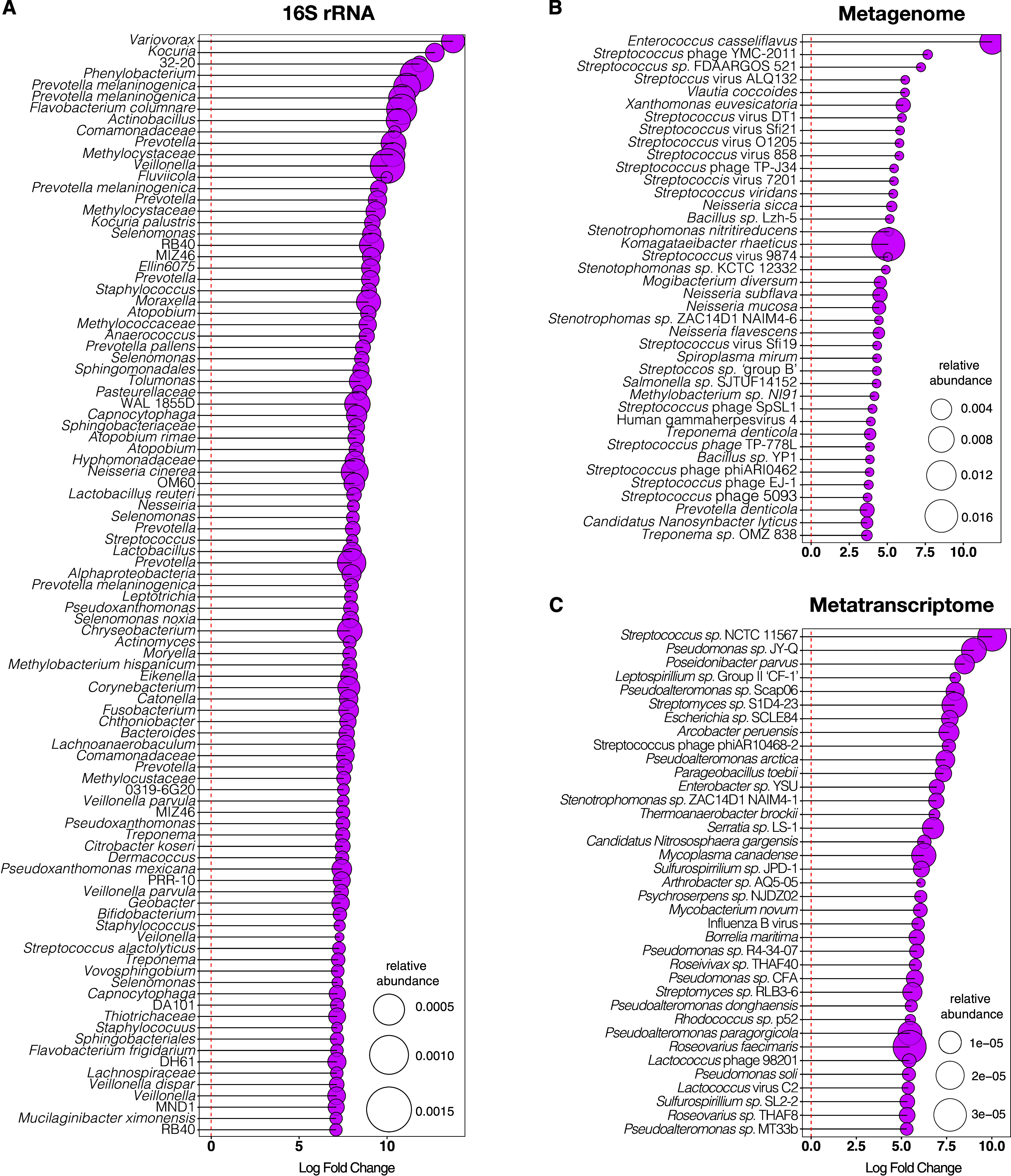
Lower airway dysbiotic signatures in mild-to-moderate chronic obstructive pulmonary disease as compared with smoker control subjects. Differential enrichment analysis based on edgeR (with multiple comparison adjustment based on false discovery rate [FDR]) on (A) 16S rRNA gene sequencing, displaying genera amplicon sequence variant; (B) whole-genome sequencing; and (C) RNA metatranscriptome sequencing. Only the top differentially enriched (based on fold change) and significant data with multiple comparisons based on FDR are displayed. Bubble size is based on relative abundance for each dataset separately. WGS = whole-genome sequencing.
Within the metagenome dataset, there were no significant differences in α- or β-diversity of lower airway samples between the COPD and SC groups (Figures E6A and E6B). Compared with SC, the lower airways of COPD were enriched with taxa belonging to the genera Enterococcus and Streptococcus as well as with several Streptococcus phages (Figure 1B and Table E2). Similar to the metagenome data, in the metatranscriptome dataset there were no significant differences in α- or β-diversity in lower airway samples between the COPD and SC groups (Figures E6C and E6D), but taxa annotated to the genera Streptococcus and a Streptococcus phage were among those taxa enriched in the lower airways of subjects with COPD (Figure 1C and Table E2). Thus, with the data presented in Figure 1, we identified that although some taxonomic differences were noted across datasets, a common pattern was the enrichment with common oral commensals in the lower airways in COPD. We then evaluated the cooccurrence of these taxa identified as oral commensals belonging to the genera Streptococcus, Prevotella, and Veillonella. Correlation analysis of these taxa showed that in all three datasets (16S rRNA gene sequencing, metagenome, and metatranscriptome) their relative abundance in the lower airways have, for the most part, a positive correlation, indicating that they tend to cooccur (Figures E7–E9 and Table E3). Thus, despite differences in which taxa were identified as differentially enriched across the three datasets, we conclude that enrichment with a mixture of oral commensals (MOC) frequently cooccurs in the lower airways of COPD.
Host Transcriptomic Differences between Subjects with COPD and SC Subjects
We next evaluated for differences in host transcriptomic signatures in lower airway samples. Principal component analysis of the lower airway host transcriptome showed significant compositional differences between subjects with COPD and SC subjects (Figure 2A; PERMANOVA P = 0.02). Differential analysis revealed several genes as significantly enriched in COPD versus SC (Figure 2B; edgeR false discovery rate < 0.2). Top genes, based on P value and fold change, identified as enriched in COPD include KRT6A (keratin), FFAR3 (short-chain fatty acid receptor), and ARNT2 (Aryl Hydrocarbon Receptor Nuclear Translocator 2). A list of all differentially enriched genes is in Table E4. The lower airway transcriptomic signatures from this cohort were similar to prior reports (19) comparing subjects with COPD with SC subjects (Figure E10; Gene Set Enrichment Analysis P < 0.001). Ingenuity pathway analysis (IPA) of consistent transcriptomic signatures associated with COPD identified activation of STAT3, IL-1B, VEGFA, IFN-γ, and IL-6 as potential upstream transcriptional regulators in COPD, which are associated with tumor cell adhesions and epithelial tissue branching (20, 21) (Figures 2C and E11). Importantly, many of these transcriptional pathways are key to mucosal immune responses to microbes. Thus, these data, presented in Figure 2, demonstrate significant differences in the host transcriptional profile of mild-to-moderate COPD, concurrent with the changes in the lower airway microbial environment.
Figure 2.

Changes in lower airway host transcriptome in chronic obstructive pulmonary disease (COPD). (A) PC analysis comparing subjects with COPD and smoker control (SC) subjects with permutational multivariate ANOVA P value. (B) Volcano plot showing differentially enriched genes based on fold change versus log10 adjusted P value (false discovery rate [FDR]) using edgeR comparing COPD and SC subjects. Genes with a log fold change >0 are enriched in COPD and <0 are enriched in SC. With an FDR cutoff of 0.2, only genes with an FDR < 0.2 are colored (green or red). (C) Ingenuity pathway analysis identifying cascade of upstream transcriptional regulators and associated disease or function relating to overlapping gene sets found using Gene Set Enrichment Analysis of our cohort and the publicly available GSE37147 dataset. PC = principal component.
Effect of Dysbiosis on Cigarette Smoke–related Lung Injury in a Preclinical Model
To better understand the interactions between lower airway dysbiosis due to common oral commensals and smoke-related lung inflammatory injury, we next used an established smoke mouse model (18), in which we induced dysbiosis as previously described (8), with a set of oral taxa identified as differentially enriched and cooccurring in the lower airways of the human cohort (22). C57/B6J mice were exposed to phosphate-buffered saline (PBS) (control), whole-body cigarette smoke (smoke group), intratracheal weekly aspiration with a representative MOC (including Streptococcus mitis, Veillonella parvula, and Prevotella melaninogenica), and MOC + smoke for 12 weeks (Figure 3A). In a subset of mice recently exposed to MOC or PBS, we documented the presence of the aspirated microbes in the lower airways using RNAscope with strain-specific probes (Figure E12). Evaluation of the microbial burden by ddPCR and microbial composition by 16S rRNA gene sequencing of lower airway samples obtained 5 days after the last experimental exposure showed no significant differences between the PBS, smoke, MOC, and MOC + smoke groups (Figures E13A and E13B). Indeed, the relative abundance of the taxa introduced with MOC exposure were only identified in a very small subset of lower airway samples from MOC-related groups (Figure E13C). These data support the transient nature of the exposure to this MOC that are likely being cleared from the lower airways within a few days, consistent with our prior publications (8). We then assessed the MLI in all groups as a surrogate for parenchymal lung destruction as described in preclinical models of emphysema (23–25). Figures 3B and 3C show that although there is some degree of overlap between the MLIs calculated for each histological field, smoke alone led to statistically higher MLIs than PBS, whereas MOC alone did not increase the MLIs. Similarly, the combination of MOC + smoke exposure led to statistically higher MLIs when compared with PBS.
Figure 3.
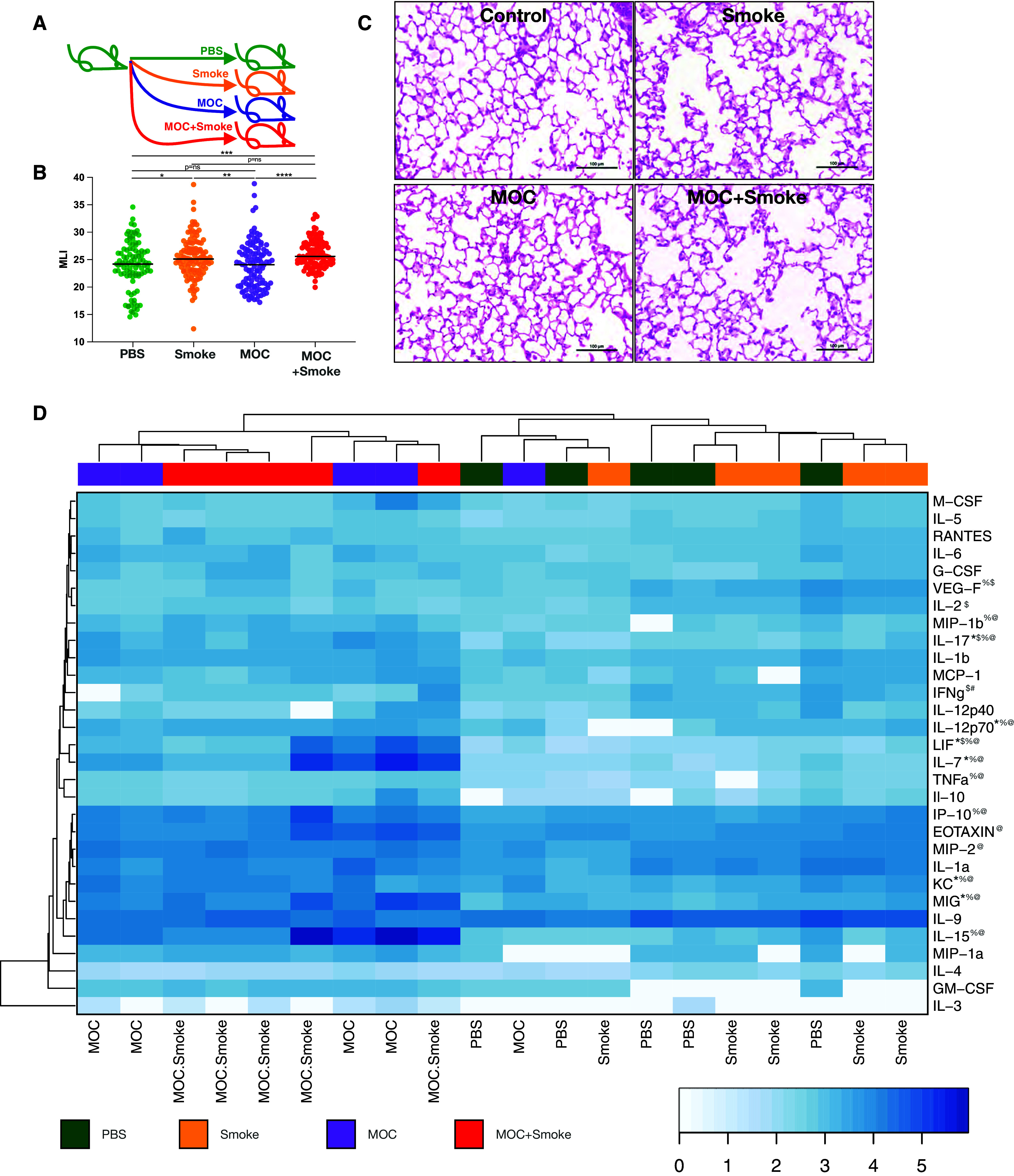
Effects of lower airway dysbiosis in a preclinical model of smoke-induced lower airway injury. (A) Murine model and exposures. (B) MLI calculated in (C) 20 different lung sections per mouse (P value, Mann-Whitney *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). (D) Heat map of log-transformed cytokines or chemokines (pg/ml) normalized by grams of lung weight (n = 5 per group). Mann-Whitney comparisons demonstrate statistically significant comparisons (+phosphate-buffered saline [PBS] vs. smoke, P < 0.05; $smoke vs. mixture of oral commensals [MOC], P < 0.05; #MOC vs. MOC + smoke, P < 0.05; *PBS vs. MOC, P < 0.05; %smoke vs. MOC + smoke, P < 0.05; and @PBS vs. MOC + smoke, P < 0.05; n = 5 mice per group). MLI = mean linear intercept.
Next, we measured inflammatory cytokines, chemokines, and growth factors in lung homogenates with a multiplexed fluorescent bead assay. Clustering analyses showed that cytokine concentrations in MOC- and MOC + smoke–exposed samples clustered together and separately from PBS and smoke alone (Figure 3D). The MOC-exposed groups (MOC and MOC + smoke) had higher concentrations of inflammatory markers, whereas VEGF, IFNγ, and IL-2 were significantly higher in the smoke-alone group than the MOC challenge. To evaluate overall changes and evaluation of cytokine differences, see Table E5. When MOC alone was compared with PBS control–exposed mice, IL-17, LIF, IL-7, KC, and MIG were increased among the MOC-exposed mice compared with PBS control mice (Figure 3D). The inflammatory profile between smoke-alone compared with MOC + smoke demonstrated that the only cytokine we measured elevated in smoke exposure was VEGF (Figure 3D); all other cytokines found to be different between MOC + smoke versus smoke alone were elevated in the MOC + smoke group. The MOC challenge drove many of the inflammatory patterns seen in our murine model. Important differences between the groups demonstrate that MOC + smoke exposure increased IFN-γ versus MOC alone and also increased IL-17 versus PBS or smoke alone. Some of the inflammatory cytokines within the murine experiments experienced high heterogeneity (Figure 3D). Thus, it would be helpful for a greater sample size to ensure trends and results are consistent. FACS analyses showed that MOC exposure increased the CD4/CD8 ratio and activation of CD4 and CD8 T cells (Figures 4A and E14). MOC exposure greatly increased lower airway IL-17–producing T cells, consistent with prior observations (8). The expression of exhaustion markers (PD-1) among CD4 and CD8 T cells was increased in the MOC group regardless of smoke exposure (Figure 4A). We next used IHC and multispectral labeling to detect and quantify key immune cell types in the airways and lung (Figures 4B and E15). In airway sections, a lower frequency of CD3+ and CD4+ T cells were found in the smoke-exposed mice than in the PBS group. The lungs of MOC + smoke–exposed mice had increased CD3+ and CD4+ frequency compared with smoke alone, supporting that the combined exposure augments lower airway inflammation. Airway macrophages and neutrophils were also increased in MOC + smoke versus PBS or smoke alone (Figure 4B). Parenchymal changes showed that CD3, CD4, and CD8 frequency increased with MOC exposure (Figure 4B). However, in contrast to the airway, MOC + smoke tended to have a lower number of CD4+ and CD8+ cells than MOC alone (representative images shown in Figure 4C). Therefore, with the data presented in Figures 3 and 4, in a preclinical model, we identified both an inflammatory and histological change associated with smoke exposure as well as with induced lower airways dysbiosis, as seen in COPD.
Figure 4.
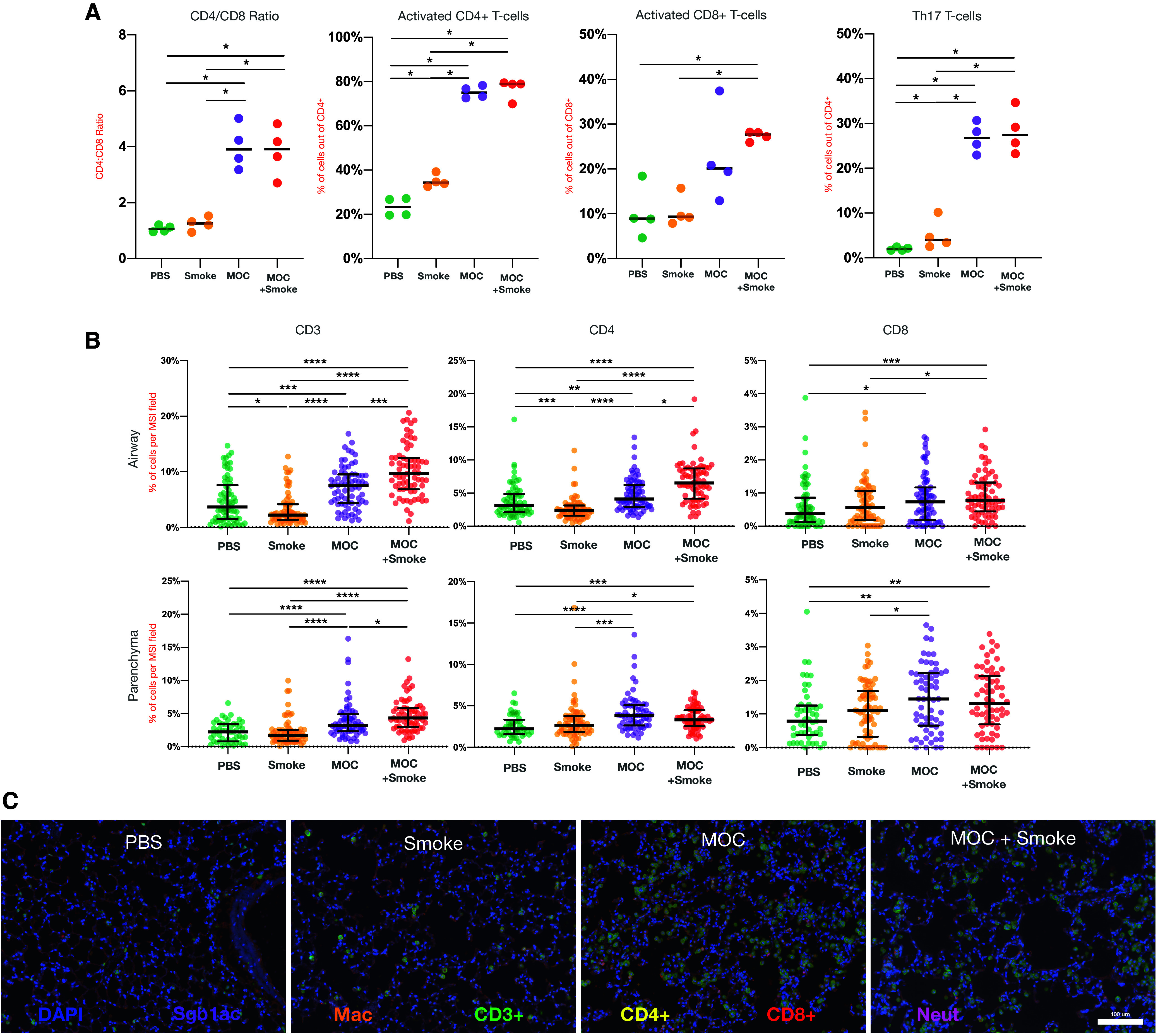
Effects of lower airway dysbiosis in a preclinical model of smoke-induced lower airway inflammation. (A) Flow cytometry of single-cell suspensions of lung homogenates with differences between PBS, smoke, mixture of oral commensals (MOC), and MOC + smoke groups. (B) Differences in cell composition in the airways and parenchyma identified using immunohistochemistry (IHC) performed on the lung sections of five mice per group and obtained from 20 different sections per mouse (P value Mann-Whitney, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). (C) Representative IHC images (DAPI = nuclei; orange = Mac; green = CD3+; yellow = CD4+; red = CD8+; and pink = Neu]). Mac = macrophage; Neut = neutrophils; PBS = phosphate-buffered saline.
We then compared transcriptomic differences in the murine lungs. Principal component analysis of host transcriptomic data showed significant differences between the four exposures (Figure 5A). Differential analysis of each paired group was used to uncover transcriptional differences; the highest number of differentially enriched genes was seen with any MOC-containing group, whereas the lowest number of significant genes was seen when smoke alone was compared with PBS (Figure 5B and Table E6). The degree of overlap between transcriptomic changes seen in human and mouse under different experimental conditions was assessed using Gene Set Enrichment Analysis. Figure 5C shows that there is a greater overlap between the human signatures (COPD vs. SC) and the mouse lower airway dysbiotic signatures (MOC alone or MOC + smoke), whereas the lowest overlap was with the smoke-alone signature. We then used IPA to summarize lower airway host transcriptomic signatures between different experimental conditions in mice with host transcriptomic differences found in COPD versus SC (Figure 5D and Table E7). Transcriptomic data in mice demonstrated that dysbiosis (MOC alone or MOC + smoke) leads to significant upregulation of various inflammatory mediators, tryptophan metabolism, ferroptosis, matrix metalloproteinases, and notch signaling, although there is downregulation of oxidative phosphorylation, PPARs (peroxisome proliferator–activated receptors), and fatty acid β-oxidation pathways (Figure 5D). Parallel analyses of lower airway transcriptome data from subjects with COPD compared with SC showed consistent signatures compared with those found among mice exposed to MOC, even within the subgroup subjected to both MOC + smoke. All together, these data shown in Figure 5 support that in a preclinical model, lower airway dysbiosis in combination with cigarette smoke exposure contributes to airway injury with molecular signatures similar to those seen in mild-to-moderate COPD.
Figure 5.

Effects of lower airway dysbiosis on the lung transcriptome. (A) PC analysis based on Bray-Curtis dissimilarity index on host lung transcriptome comparing all four exposures, permutational multivariate ANOVA P < 0.01. (B) Volcano plots showing differentially enriched genes [DEGs] identified by fold change versus log10 adjusted (false discovery rate) P value based on edgeR analyses between different exposures. (C) Gene Set Enrichment Analysis using DEGs identified significant and concordant pathways for experimental conditions in the preclinical mouse model with those identified in the human cohort (subjects with chronic obstructive pulmonary disease [COPD] vs. smoker control [SC] subjects). First comparing the mouse smoke versus phosphate-buffered saline (PBS) analysis to the human COPD versus SC analysis, then the mouse mixture of oral commensals (MOC) versus PBS analysis to the human COPD versus SC analysis, followed by the mouse MOC + smoke versus PBS analysis to the human COPD versus SC analysis, and finally the mouse MOC + smoke versus smoke analysis to the human COPD versus SC analysis. (D) Ingenuity pathway analysis comparing regulation of canonical pathways between different conditions in the preclinical mouse model and those identified in the human COPD versus SC cohort. The values for the heatmap represent a z-score. PC = principal component.
Discussion
Here, we used complementary techniques to evaluate the lower airway microbiome in subjects with mild-to-moderate COPD compared with SC subjects without physiological evidence of COPD. In this investigation, we used multiple sequencing datasets that provide different orthogonal views of the microbiome: 1) 16S rRNA gene sequencing with bacterial taxonomic characterization; 2) WGS that provides better species resolution plus data on fungi, viruses, and phages; and 3) RNA metatranscriptome that expands with profiling functionally active taxa. Although several differences can be noted among these different datasets, a common finding of taxonomic differences associated with COPD was the enrichment of the lower airway microbiome with some common oral commensals. Although we found taxa from the genera Streptococcus consistently associated with COPD, we noted that this taxon commonly cooccurs with other oral commensals, such as taxa from the genus Prevotella and Veillonella. This difference in the lower airway microbiome was associated with differences in the host immune response in patients with COPD with increased expression of IL-17 and IL-6. To assess for potential causal relationships underlying these associations, we used a cigarette smoke–exposed mouse model to show that lower airway dysbiosis produced by aspiration of a representative mixture of common oral commensals, more than smoke exposure alone, leads to an increase in inflammatory signatures, as shown in our cohort data of subjects with COPD and previously published data (26). Thus, these data support the hypothesis that lower airway dysbiosis contributes to the inflammatory injury seen in mild-to-moderate COPD.
Cigarette smoke exposure and ambient air pollution exposure, such as particulate matter ⩽2.5 μm in aerodynamic diameter, are well-accepted causes of lung injury resulting in the development and progression of COPD (27, 28). However, not all patients with significant smoking history (29) or significant exposure to air pollution develop physiological evidence of COPD. Other inciting factor(s), in combination with smoke and/or air pollution, likely contribute to lung injury and airway remodeling that lead to COPD. Here, we explored the role of the lower airway microbial microenvironment in lung inflammation and remodeling in the setting of exposure to tobacco smoke.
Most research investigating the lung microbiome and COPD has focused on advanced-stage COPD. Indeed, a recent investigation involving a large number of subjects from three different cohorts (two in the United Kingdom and one from China) with a large percentage of patients with advanced-stage COPD identified several taxa differentially enriched in sputum samples of patients with progressive disease (based on lung function decline and exacerbations) (30). Among those taxa identified, the authors conducted experiments in an LPS/elastase preclinical model of COPD, in which induction of dysbiosis with Staphylococcus aureus contributed to an increase in homocysteine in the lower airways and inflammatory injury leading to a COPD-like phenotype. Although these microbial signatures were identified in sputum rather than in BAL or lung tissue samples, the experiments shown in that publication provide a proof-of-concept for the potential contribution of microbes to the lower airway damage in COPD. The above-mentioned investigation takes a reductionist approach, singling out one taxon and one molecule to study its contribution to the inflammatory injury. However, multiple taxa and molecules are part of the dysbiotic signatures commonly identified in different studies as associated with COPD (7, 31, 32). Furthermore, a disadvantage of research on patients with moderate-to-severe COPD is that the lungs of these patients have suffered extensive physiological changes (structural and immunological). These changes in the lower airway microenvironment could lead to changes in the microbiome, thus limiting interpretation of cross-sectional studies. Furthermore, in advanced COPD there may be other confounders, such as antibiotic and systemic corticosteroid use, which further limit the interpretation of these studies (3, 32, 33). Another knowledge gap is that few publications reported results of assays that define bacterial gene expression in COPD. One group used metagenomic and metatranscriptomic sequencing to evaluate acute exacerbations of COPD (34); however, exacerbations are a late-stage phenomenon that may be distinct from the original pathogenic mechanism that produced COPD. Thus, there is increasing interest in exploring the microbial signatures of mild COPD. Similar to prior observations from the SPIROMICS investigators (7), we used bronchoscopy samples to evaluate the lower airway microbiome in patients with a significant smoking history (>10 pack-years). 16S rRNA target gene sequencing demonstrated enrichment of common oral commensals in subjects with mild-to-moderate COPD when compared with SC subjects, such as taxa from the genera Prevotella, Veillonella, and Streptococcus, as previously described (22, 26). However, 16S rRNA target gene sequencing data can only define taxonomic community structure with poor strain resolution. The metagenome and metatranscriptome data generated here also identified significant differences in microbial composition, particularly involving similar oral commensals such as Streptococcus and Prevotella between subjects with mild-to-moderate COPD versus SC subjects. Here, inconsistencies in taxonomic signatures identified between different approaches might be explained on the basis of technical differences (e.g., sample processing, amplification approaches, sequencing techniques) or biological differences (e.g., DNA vs. RNA focused). For example, we have shown, using paired DNA and RNA genomic methods, that identification of signals from aspirated microbes to the lower airways of mice vary for different aspirated oral commensal, but, overall, microbial DNA persists longer than microbial RNA in the lower airways (22). Thus, it is not surprising that in cross-sectional samples, some degree of discrepancy related to the presence and absence of specific taxa among DNA and RNA methods is observed. However, the similarity in the identification of oral commensals, which can come from aspiration and/or decreased clearance, as enriched in the lower airways of patients with COPD across different orthogonal sequencing approaches, increases the confidence of its relevance as a microbial signature present in milder stages of this disease. Furthermore, a consistent signature with Streptococcus species and associated phages seen in the metatranscriptome further supports at least some transient viability of these common oral commensals in the lower airways in mild-to-moderate COPD.
Changes in the lower airway microbiome have been associated with host immunity. A recent analysis used sputum from subjects with COPD (mostly from advanced stages) and healthy control subjects in a multiomic analysis of the metagenome, metabolome, and host transcriptome (35). This identified significant associations between the host transcriptome and some microorganisms in COPD, particularly Prevotella and Streptococcus. In our analyses, we also explored the host transcriptome and identified several genes differentially enriched between the two groups, with changes consistent with transcriptomic differences found in other patient cohorts (36–44) as well as in COPD cohorts (30, 45–47). Furthermore, IPA identified several pathways that were upregulated in COPD, such as MUC1 and MUC4, pathways important to the production of mucin, as well as inflammatory pathways associated with Th-17. Interestingly, we have previously shown that lower airway enrichment with common oral commensals leads to increased lower airway inflammatory tone, characterized by a Th-17–like inflammatory phenotype (5).
To establish the possible causal nature of these associations, we explored the effects of lower airway dysbiosis in a preclinical mouse model. In an established tobacco smoke exposure model, we superimposed lower airway dysbiosis by intratracheal aspiration with a representative mixture of common cooccurring human oral commensals identified as dysbiotic signatures in multiple independent cohorts (including ours): taxa from the genus Streptococcus, Prevotella, and Veillonella (5, 48–52). Aspiration of this combination of human oral commensals affects the lower airway inflammatory tone (8), including in preclinical models of lung cancer (53) and acute bacterial pneumonia (8). The combination of lower airway exposure to common oral commensals and cigarette smoke produced greater transcriptomic changes than those produced by smoking alone. However, histological worsening of MLI was not evident when the MOC + smoke group was compared with smoke alone. Whether these results support a causal role for dysbiosis in lung injury that may contribute to airflow obstruction may need to be further tested in more chronic smoke exposures. Molecular profiling of immune signals using FACS, IHC, and transcriptomics determined that experimental lower airway dysbiosis causes increased inflammatory injury, driven by neutrophilic and lymphocytic inflammation, with IL-17 playing a central role. This is not a total surprise, as LPS has been used before in combination with elastase to induce emphysema and inflammation in mice (54). Although traditionally LPS from well-recognized pathogens is used for those experiments, here we identify the “pathobiont” potential of oral commensals in the lower airways contributing to the development of COPD. Interestingly, a single episode of exposure to oral commensals reduces susceptibility to a respiratory pathogen (S. pneumoniae) in a preclinical mouse model. This may be counterintuitive, because patients with COPD have increased susceptibility to respiratory pathogens. It is possible that the effects of aspiration of oral commensals (or the repetitive and increased frequency of microaspiration events) are different in the setting of smoke-induced lung injury and may warrant further investigation. Conversely, the key molecular features present in the setting of exposure to these oral commensals might be multifaceted and require further investigation to decipher effects over time and to different exposures.
There are some limitations to this study. We focused exclusively on mild-to-moderate COPD, defined by spirometry. However, some studies show the importance of respiratory symptoms irrespective of significant airflow obstruction (29). Although we were able to identify several significant differences between our two cohorts, our sample size was small, and a larger study would be helpful in validating these findings. Furthermore, the results presented here come mostly from active smokers, and our small sample size prevents us from conducting further subgroup analyses within former and current smokers. Also, the cross-sectional design compares current smokers with and without airway obstruction, and, therefore, future longitudinal studies would be needed to further elucidate the pathogenic importance of these microbial signatures and their association with symptom burden, progressive disease, and frequent exacerbations. Importantly, smoking quantification and alcohol use were based on self-report, which, paired with the relatively small cohort size, prevents us from performing further subgroup analyses. Here, we conducted the preclinical mouse experiments with a defined set of microbes to induce lower airway dysbiosis; however, we recognize that there are multiple different forms of dysbiosis, including many different oral commensals. In addition, one possible confounder was that murine exposure to tobacco smoke occurred within a cage. However, we attempted to control for the cage effect by cohousing MOC exposure and PBS control mice within the same cage. Furthermore, the dynamic change in the lung microbiome seen in our preclinical data, exemplified by the early detection of microbial DNA using RNAscope, but loss of most of this microbial DNA signal 5 days after exposure, exposes a challenge ensuring that a mouse model fully replicates conditions that occur in the lower airways of humans, which we can only study under cross-sectional design. The investigation presented here was done as a proof-of-concept to test the potential role of lower airway dysbiosis in the pathogenesis of COPD-related lung inflammation, and it is possible that other forms of dysbiosis could contribute to this process as well. Finally, experimental interventions in clinical trials are required to evaluate targeted interventions that may target specific molecular derangements triggered by lower airway dysbiosis in COPD.
In summary, data from this cohort support the conclusion that patients with mild-to-moderate COPD have a distinct lower airway microenvironment and that the combination of cigarette smoke exposure with lower airway dysbiosis might result in continued inflammatory injury and eventually lead to altered lung structure and physiological impairments. Importantly, these findings highlight potential novel targets in conjunction with smoking cessation that could aid in preventing COPD or progression of disease.
Acknowledgments
Acknowledgment
The authors thank the NYU Genome Technology Center for expert library preparation and sequencing and the Applied Bioinformatics Laboratories for providing bioinformatics support and helping with the analysis and interpretation of the data. This study also used the Office of Cyber Infrastructure and Computational Biology High-Performance Computing cluster at the National Institute of Allergy and Infectious Diseases, Bethesda, MD. The authors thank the Experimental Pathology Research Laboratory for histopathology services and imaging. The Genome Technology Center and Applied Bioinformatics Laboratories are shared resources partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. This work has used computing resources at the NYU School of Medicine High-Performance Computing Facility.
Footnotes
Supported by National Cancer Institute/NIH grant R37 CA244775 (L.N.S.), NHLBI/NIH grant R01 HL125816 (L.N.S. and S.B.K.), National Cancer Institute/NIH grants U2C CA271890 (L.N.S.) and R56 HL151700 (M.C.K.), Foundation for the NIH Partnership for Accelerating Cancer Therapies (PACT) grant (L.N.S.), National Center for Advancing Translational Science grant UL1 TR001445 (L.N.S.), NIH/NIGMS (National Institute of General Medical Sciences) grant R21GM147800 (L.N.S.), Veterans Affairs grant IK2BX005309-01A2 (B.G.W.), NIH grant KL2TR001446 (B.G.W.), CHEST Foundation Research Grant in Chronic Obstructive Pulmonary Disease (B.G.W.), and an American Association for Cancer Research grant (L.N.S.). The Genome Technology Center is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center, a Flight Attendant Medical Research Institute Young Clinical Scientist Award (B.G.W.), Stony Wold-Herbert Fund Grant-in-Aid/Fellowships (I.S., C.R.B., and M.C.K.), and a Will Rogers Research Fellowship (M.C.K.). This work was supported in part by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases (NIAID)/NIH (E.G. and M.C.); National Health and Medical Research Council Australia fellowship and grants 1059238, 1175134, and 2010287; and the Cancer Council New South Whales and University of Technology Sidney (P.M.H.). The PACT project is supported by funding provided to the Foundation for the NIH by: AbbVie Inc., Amgen Inc., Boehringer-Ingelheim Pharma GmbH & Co. KG, Bristol-Myers Squibb, Celgene Corporation, Genentech Inc., Gilead, GlaxoSmithKline plc, Janssen Pharmaceutical Companies of Johnson & Johnson, Novartis Institutes for Biomedical Research, Pfizer Inc., and Sanofi. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC. The project described was supported by the National Center for Advancing Translational Sciences, NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author Contributions: Study conception and design: I.S., B.G.W., B.I., J.-C.J.T., M. Chung., E.G., and L.N.S. Data were obtained by I.S., B.G.W., M.C., Y.K., B.I., C.R.B., M.H., J.-C.J.T., Y.L., R.S., J.C., D.C., L.P., B.W.O., K.I.B., R.M.G., S.B.K., R.X., J.D’A., E.G., and L.N.S. Data were analyzed by I.S., B.G.W., M. Chung., B.K., Y.K., B.I., C.R.B., M.H., J.-C.J.T., M.C.K., S.S., J.G.N., Y.L., D.C., C.D.C., P.M.H., M.D.W., J.D’A., J.C.C., E.G., and L.N.S. The first draft of the manuscript was written by I.S., B.G.W., B.I., J.-C.J.T., E.G., and L.N.S. All authors read, critically revised, and approved the final manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202210-1865OC on September 7, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Patel AR, Patel AR, Singh S, Singh S, Khawaja I. Global Initiative for Chronic Obstructive Lung Disease: the changes made. Cureus. 2019;11:e4985. doi: 10.7759/cureus.4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med . 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 3. Wang Z, Locantore N, Haldar K, Ramsheh MY, Beech AS, Ma W, et al. Inflammatory endotype-associated airway microbiome in chronic obstructive pulmonary disease clinical stability and exacerbations: a multicohort longitudinal analysis. Am J Respir Crit Care Med . 2021;203:1488–1502. doi: 10.1164/rccm.202009-3448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilkinson TM, Patel IS, Wilks M, Donaldson GC, Wedzicha JA. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2003;167:1090–1095. doi: 10.1164/rccm.200210-1179OC. [DOI] [PubMed] [Google Scholar]
- 5. Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol . 2016;1:16031. doi: 10.1038/nmicrobiol.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Madapoosi SS, Cruickshank-Quinn C, Opron K, Erb-Downward JR, Begley LA, Li G, et al. Lung microbiota and metabolites collectively associate with clinical outcomes in milder stage COPD. Am J Respir Crit Care Med . 2022;206:427–439. doi: 10.1164/rccm.202110-2241OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Opron K, Begley LA, Erb-Downward JR, Freeman C, Madapoosi S, Alexis NE, et al. Lung microbiota associations with clinical features of COPD in the SPIROMICS cohort. NPJ Biofilms Microbiomes . 2021;7:14. doi: 10.1038/s41522-021-00185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu BG, Sulaiman I, Tsay JJ, Perez L, Franca B, Li Y, et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med . 2021;203:1099–1111. doi: 10.1164/rccm.202005-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J . 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol . 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics . 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods . 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol . 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu JBF, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci . 2017;3:e104. [Google Scholar]
- 15. Kim J, Kim MS, Koh AY, Xie Y, Zhan X. FMAP: Functional Mapping and Analysis Pipeline for metagenomics and metatranscriptomics studies. BMC Bioinformatics . 2016;17:420. doi: 10.1186/s12859-016-1278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sulaiman I, Chung M, Angel L, Tsay JJ, Wu BG, Yeung ST, et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat Microbiol . 2021;6:1245–1258. doi: 10.1038/s41564-021-00961-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res . 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao R, Goldklang MP, D’Armiento JM. Parenchymal airspace profiling: sensitive quantification and characterization of lung structure evaluating parenchymal destruction. Am J Respir Cell Mol Biol . 2016;55:708–715. doi: 10.1165/rcmb.2016-0143OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steiling K, van den Berge M, Hijazi K, Florido R, Campbell J, Liu G, et al. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med . 2013;187:933–942. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Varner VD, Nelson CM. Cellular and physical mechanisms of branching morphogenesis. Development . 2014;141:2750–2759. doi: 10.1242/dev.104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol . 2019;10:1078. doi: 10.3389/fimmu.2019.01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sulaiman I, Wu BG, Li Y, Tsay JC, Sauthoff M, Scott AS, et al. Functional lower airways genomic profiling of the microbiome to capture active microbial metabolism. Eur Respir J . 2021;58:2003434. doi: 10.1183/13993003.03434-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knudsen L, Weibel ER, Gundersen HJ, Weinstein FV, Ochs M. Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J Appl Physiol (1985) . 2010;108:412–421. doi: 10.1152/japplphysiol.01100.2009. [DOI] [PubMed] [Google Scholar]
- 24. Kugler MC, Loomis CA, Zhao Z, Cushman JC, Liu L, Munger JS. Sonic Hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am J Respir Cell Mol Biol . 2017;57:280–293. doi: 10.1165/rcmb.2016-0268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beckett EL, Stevens RL, Jarnicki AG, Kim RY, Hanish I, Hansbro NG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol . 2013;131:752–762. doi: 10.1016/j.jaci.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bowerman KL, Rehman SF, Vaughan A, Lachner N, Budden KF, Kim RY, et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat Commun . 2020;11:5886. doi: 10.1038/s41467-020-19701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao J, Li M, Wang Z, Chen J, Zhao J, Xu Y, et al. Role of PM2.5 in the development and progression of COPD and its mechanisms. Respir Res . 2019;20:120. doi: 10.1186/s12931-019-1081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song C, He J, Wu L, Jin T, Chen X, Li R, et al. Health burden attributable to ambient PM2.5 in China. Environ Pollut . 2017;223:575–586. doi: 10.1016/j.envpol.2017.01.060. [DOI] [PubMed] [Google Scholar]
- 29.Lamprecht B, McBurnie MA, Vollmer WM, Gudmundsson G, Welte T, Nizankowska-Mogilnicka E, et al. BOLD Collaborative Research Group COPD in never smokers: results from the population-based burden of obstructive lung disease study. Chest. 2011;139:752–763. doi: 10.1378/chest.10-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang W, Yang Y, Gong S, Wei M, Ma Y, Feng R, et al. Airway dysbiosis accelerates lung function decline in chronic obstructive pulmonary disease. Cell Host Microbe . 2023;31:1054–1070.e9. doi: 10.1016/j.chom.2023.04.018. [DOI] [PubMed] [Google Scholar]
- 31. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One . 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One . 2012;7:e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2015;192:438–445. doi: 10.1164/rccm.201502-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang J, Zhang Q, Zhang J, Ouyang Y, Sun Z, Liu X, et al. Exploring the change of host and microorganism in chronic obstructive pulmonary disease patients based on metagenomic and metatranscriptomic sequencing. Front Microbiol . 2022;13:818281. doi: 10.3389/fmicb.2022.818281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z, Yang Y, Yan Z, Liu H, Chen B, Liang Z, et al. Multi-omic meta-analysis identifies functional signatures of airway microbiome in chronic obstructive pulmonary disease. ISME J . 2020;14:2748–2765. doi: 10.1038/s41396-020-0727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krokidis MG, Vlamos P. Transcriptomics in amyotrophic lateral sclerosis. Front Biosci (Elite Ed) . 2018;10:103–121. doi: 10.2741/e811. [DOI] [PubMed] [Google Scholar]
- 37. Krokidis MG. Transcriptomics and metabolomics in amyotrophic lateral sclerosis. Adv Exp Med Biol . 2020;1195:205–212. doi: 10.1007/978-3-030-32633-3_29. [DOI] [PubMed] [Google Scholar]
- 38. Burel JG, Singhania A, Dubelko P, Muller J, Tanner R, Parizotto E, et al. Distinct blood transcriptomic signature of treatment in latent tuberculosis infected individuals at risk of developing active disease. Tuberculosis (Edinb) . 2021;131:102127. doi: 10.1016/j.tube.2021.102127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong HS, Guo CL, Lin GH, Lee KY, Okada Y, Chang WC. Transcriptome network analyses in human coronavirus infections suggest a rational use of immunomodulatory drugs for COVID-19 therapy. Genomics . 2021;113:564–575. doi: 10.1016/j.ygeno.2020.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bagyinszky E, Giau VV, An SA. Transcriptomics in Alzheimer’s disease: aspects and challenges. Int J Mol Sci . 2020;21:3517. doi: 10.3390/ijms21103517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kadara H, Fujimoto J, Yoo SY, Maki Y, Gower AC, Kabbout M, et al. Transcriptomic architecture of the adjacent airway field cancerization in non-small cell lung cancer. J Natl Cancer Inst . 2014;106:dju004. doi: 10.1093/jnci/dju004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beane J, Vick J, Schembri F, Anderlind C, Gower A, Campbell J, et al. Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res (Phila) . 2011;4:803–817. doi: 10.1158/1940-6207.CAPR-11-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature . 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 44. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods . 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 45. Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Müller-Quernheim J, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe . 2021;2:e300–e310. doi: 10.1016/S2666-5247(21)00035-5. [DOI] [PubMed] [Google Scholar]
- 46. Titz B, Sewer A, Schneider T, Elamin A, Martin F, Dijon S, et al. Alterations in the sputum proteome and transcriptome in smokers and early-stage COPD subjects. J Proteomics . 2015;128:306–320. doi: 10.1016/j.jprot.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 47. Yan Z, Chen B, Yang Y, Yi X, Wei M, Ecklu-Mensah G, et al. Multi-omics analyses of airway host-microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat Microbiol . 2022;7:1361–1375. doi: 10.1038/s41564-022-01196-8. [DOI] [PubMed] [Google Scholar]
- 48. Segal LN, Alekseyenko AV, Clemente JC, Kulkarni R, Wu B, Gao Z, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome . 2013;1:19. doi: 10.1186/2049-2618-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Charlson ES, Bittinger K, Chen J, Diamond JM, Li H, Collman RG, et al. Assessing bacterial populations in the lung by replicate analysis of samples from the upper and lower respiratory tracts. PLoS One . 2012;7:e42786. doi: 10.1371/journal.pone.0042786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio . 2015;6:e00037. doi: 10.1128/mBio.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Lung HIV Microbiome Project Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med . 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsay JJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, et al. Lower airway dysbiosis affects lung cancer progression. Cancer Discov . 2021;11:293–307. doi: 10.1158/2159-8290.CD-20-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yadava K, Pattaroni C, Sichelstiel AK, Trompette A, Gollwitzer ES, Salami O, et al. Microbiota promotes chronic pulmonary inflammation by enhancing IL-17A and autoantibodies. Am J Respir Crit Care Med . 2016;193:975–987. doi: 10.1164/rccm.201504-0779OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data are available in National Center for Biotechnology Information (NCBI) Sequence Read Archive under project number PRJNA870929 for human microbiome data, PRJNA936182 for murine microbiome data, and PRJNA870929 for host transcriptomic data. Codes used for the analyses presented in the current manuscript are available at https://github.com/segalmicrobiomelab/Mild_Moderate_COPD_microbiome. See the online supplement for more details on the methods used.