

Abstract
TBCK syndrome is an autosomal recessive disorder primarily characterized by global developmental delay, hypotonia, abnormal magnetic resonance imaging (MRI), and distinctive craniofacial phenotypes. High variability is observed among affected individuals and their corresponding variants, making clinical diagnosis challenging. Here, we discuss recent breakthroughs in clinical considerations, TBCK function, and therapeutic development.
Introduction
TBCK syndrome is a rare genetic disorder caused by mutations in the encoding gene, TBC1 domain-containing kinase (TBCK), and has also been referred to as TBCK encephaloneuropathy and infantile hypotonia with psychomotor retardation and characteristic facies 3 (IHPRF3). Although functional knowledge of this protein is limited, the phenotypes of individuals with TBCK syndrome have provided early insights into likely functions of TBCK in various processes, including the development and maintenance of the neuro-muscular and skeletal systems [1–7].
Clinical considerations
Over 100 affected individuals have been reported worldwide with autosomal recessive inheritance, including several demonstrating compound heterozygosity (Figure 1). Multiple organ systems are affected; major symptoms include hypotonia, progressive developmental delay, intellectual disability, seizures, skeletal abnormalities, and abnormal craniofacial features [2–6]. Brain magnetic resonance images (MRI) of affected individuals often show white matter hyperintensities and cortical atrophy [2]. Facial abnormalities, skeletal perturbations, and sleep apnea are newly recognized clinical characteristics that are consistent across multiple patients [2,4]. It has been recommended that healthcare providers consider sleep apnea in patients with TBCK mutations due to the frequency with which it has been documented in existing cases and the severity of consequences if not addressed [4]. Additionally, review of the current literature suggests early-onset osteoporosis with pathologic fractures, and ophthalmological issues may also be common features in these individuals (Figure 2).
Figure 1. Schematic of reported TBCK variants.
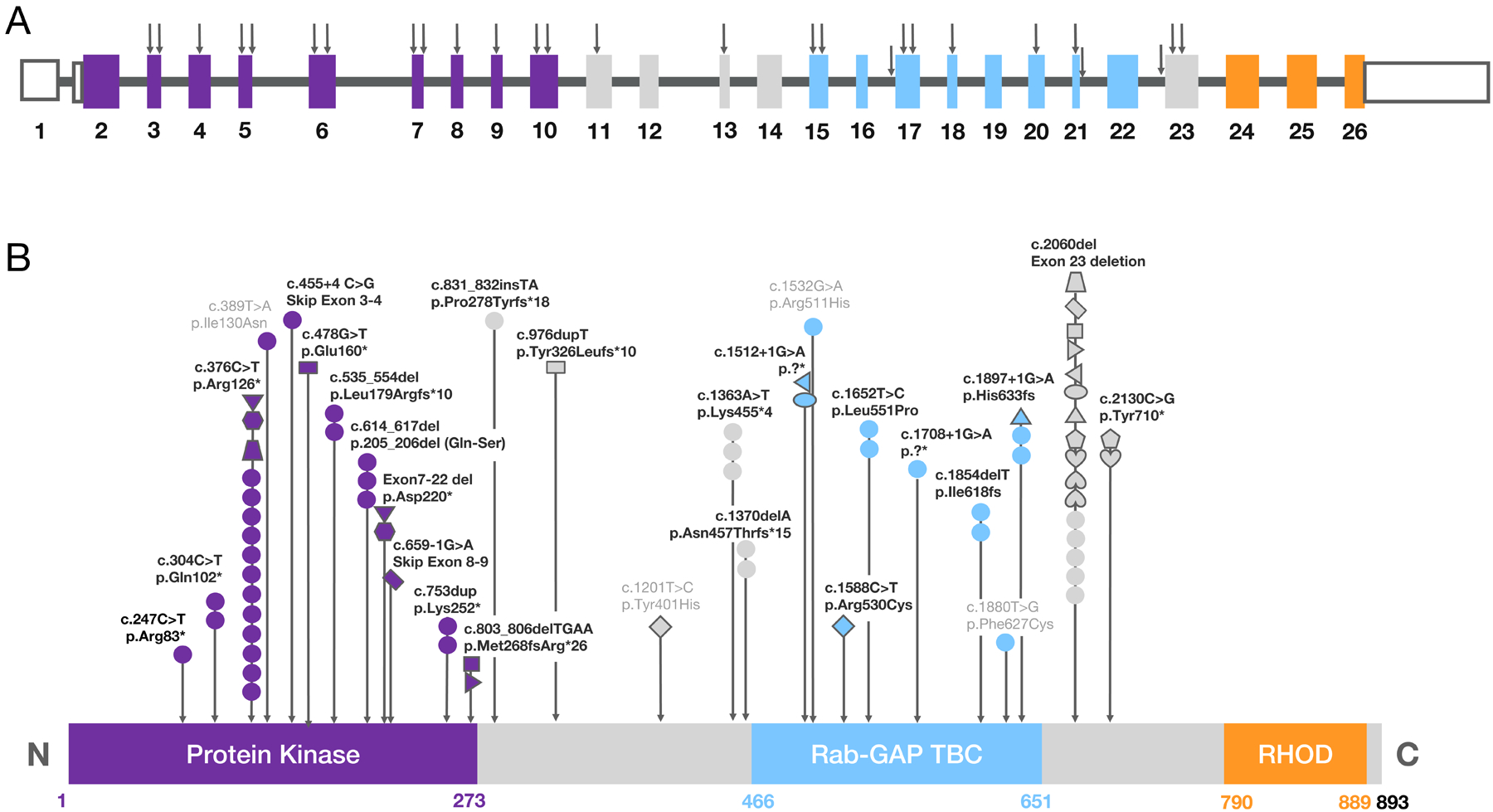
(A) Diagram representing the location of reported variants within the genomic DNA for the canonical TBCK transcript. Black arrows correspond to the genomic loci for each of the reported variants. Color coding of the exons (rectangles) correspond to their contributing protein domain in panel B: exon 1 encodes the 5’UTR, dark purple exons (2–10) encode the protein kinase domain, light blue exons (15–22) encode the Rab-GAP TBC domain, and the orange exons (24–26) encode the rhodanese homology domain (RHOD). (B) Diagram of TBCK protein domains illustrating the reported variants and their frequencies. Each circle represents one individual with a homozygous mutation. For an individual with a compound heterozygous mutation, a distinct shape is given to match the two variants. Matching shapes correspond to the variants reported in the same individual. Some individuals with the same mutations are from the same family. Color coding of the domains corresponds to the color-coded exons in panel A. Mutation p. Arg126* is referred to as the founder mutation or the Boricua mutation associated with a patient population with severe phenotype [5]. The numbers under each domain denote the amino acid range for that domain, with the canonical protein consisting of 893 amino acids total in length. Abbreviations: 5’UTR, 5’ untranslated region; fs, frameshift; *, nonsense mutation; del, deletion; light gray letters, missense mutation.
Figure 2. Symptoms and complications observed in individuals with TBCK syndrome.
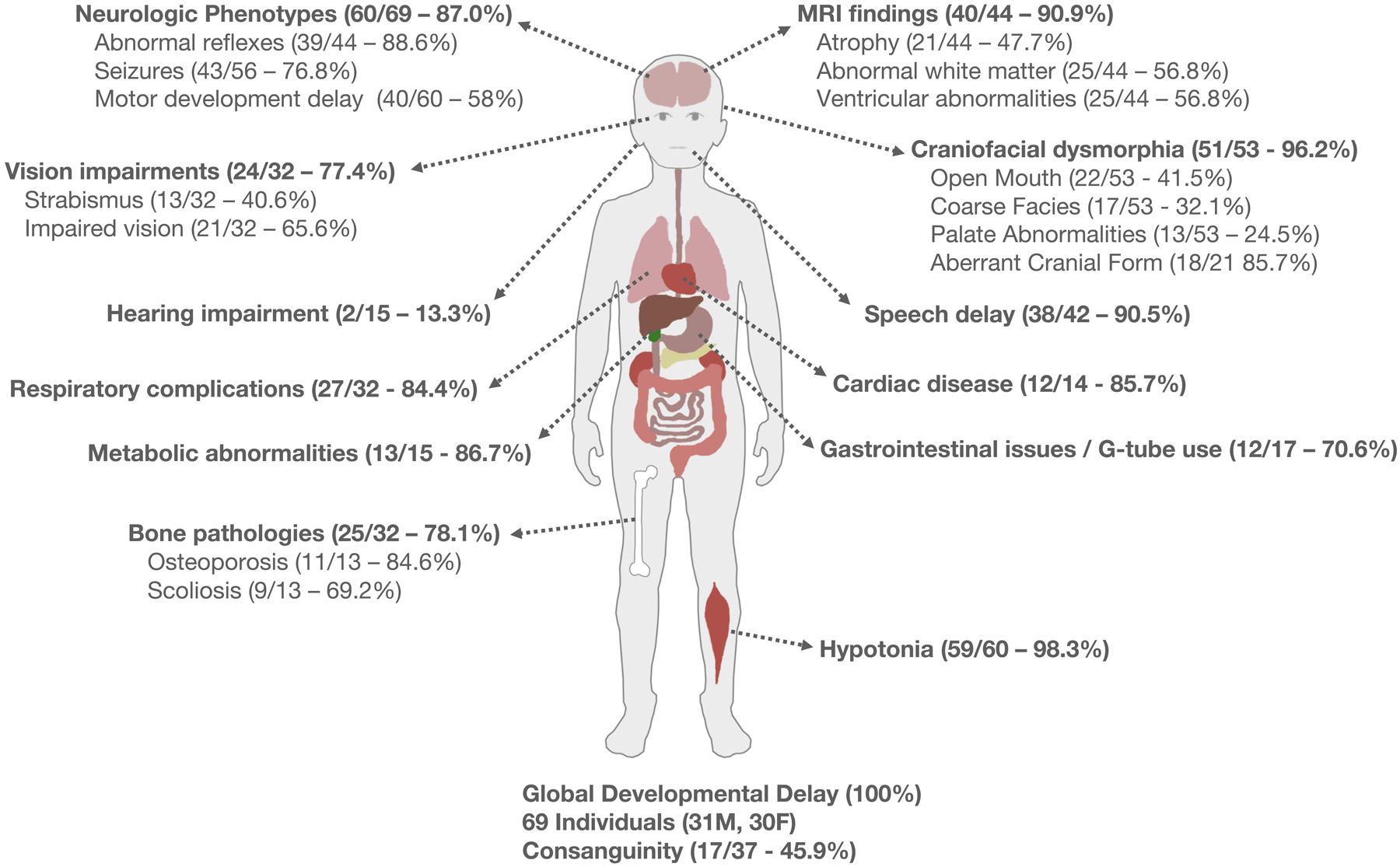
An array of phenotypes are identified in 69 documented TBCK syndrome patients encompassing 23 ethnicities. Ratio and percentages of symptom frequency identified in the literature are reported. (Clockwise from top left) Neurologic phenotypes included areflexia (18 patients), reduced reflexes (19), hyperreflexia (1), seizures, and motor developmental delay (mild 18, moderate/severe 20). MRI commonly indicated abnormal white matter including hyperintensities and ventricular abnormalities. A lesser percent of patients (30.9%) had abnormal findings in the corpus collosum. Interrogation of the published literature indicates high variability in reporting of craniofacial anomalies. An open mouth phenotype including a tented upper lip and/or macroglossia (17.3%) was common, as was aberrant cranial form (microcephaly 3, macrocephaly 7, brachycephaly 5, turricephaly 3). Most patients had nonverbal speech delay. Cardiac abnormalities were reported in 12 patients. Eleven patients were reported to use G-tubes. Almost all patients reported hypotonia. Bone pathologies included delayed bone age, osteoporosis, and scoliosis reported in over half of the patients. Metabolic symptoms including high cholesterol, dyslipidemia, and hypertriglyceridemia were common. Respiratory maladies included ventilator dependency (13/30), tracheostomy (4/30), use of BiPAP (4/30), and apnea (2/30). Hearing impairment was relatively uncommon while vision impairment occurred in more than half of the patients. Vision-related phenotypes included ptosis (1), strabismus (12), impaired vision (20), nystagmus (8), and nocturnal lagophthalmos (1). All patients reported global developmental delay. The variability identified with these phenotypes and available data highlights the need for a more systematic diagnosis process. Abbreviation: MRI, magnetic resonance imaging.
One of the challenges in diagnosing rare genetic disorders like TBCK syndrome is the lack of awareness and familiarity among healthcare providers. Variability in clinical manifestations and overlapping features with similar conditions compound this issue. However, advances in diagnostic technologies, such as exome sequencing and transcriptome-directed approaches, have improved the accuracy and rate of TBCK syndrome diagnosis. Recent studies have demonstrated the usefulness of RNA sequencing in identifying TBCK mutations that were missed by exome sequencing [8], highlighting the importance of considering multiple diagnostic modalities when TBCK syndrome is suspected. We recognize that worldwide not all individuals have access to these technologies, leaving many undiagnosed.
Several studies have touched on a genotype-phenotype correlation of TBCK syndrome; however, more data are required for statistically significant conclusions. While all individuals exhibit developmental delay, shared facial features are beginning to emerge. Further investigations are needed to characterize a unique facial gestalt, which may include bitemporal narrowing, arched eyebrows, high-arched palate, and an open mouth posture [2,4–6]. In addition, it is unclear if the systemic disturbances are directly due to TBCK loss in each tissue type or if they are secondary to neurologic dysfunction, pointing to an unmet need to understand the underlying mechanisms of TBCK function in different tissues.
TBCK molecular function
Recent molecular studies using patient fibroblasts suggest TBCK function affects multiple organelles that may vary depending on tissue type, possibly through the autophagy-lysosomal axis. Originally it was thought that mTORC1 dysregulation from TBCK loss led to a storage-like disorder. Comprehensive biochemical analyses showed individuals with TBCK syndrome having widespread accumulation of vesicles positive for sialylated oligosaccharides, lipofuscin, and glycogen in neurons, astrocytes, hepatocytes, spleen, and lymphocytes [7,9]. Patient-derived fibroblasts also exhibit mitochondrial dysfunction and oxidative phosphorylation defects [3], and these pathways appear to be regulated by lysosomes, since treatment with acidifying nanoparticles cleared auto-phagosomes and improved mitochondrial function. A recent study of patient-derived pluripotent stem cells (iPSCs) reprogrammed to neural progenitor cells (NPCs) described defects in early secretory pathways due to impaired endoplasmic reticulum to Golgi vesicle transport affecting proliferation and differentiation [10]. Evidence also suggests that the Rhodanese domain (Figure 1) is essential for cellular localization of the TBCK protein and interacts with Golgi proteins in zebrafish [11]. While it is well established that the mTOR pathway regulates autophagy and lysosomal functions, the link between TBCK and mTORC1 activity is still unclear. Deeper investigations in different cell models are necessary to determine how TBCK function may differ in distinct cell types.
A significant breakthrough in understanding the TBCK protein function was the identification of its structure as part of a multi-protein complex called Five-subunit Early endosome RNA and Ribosome intermediarY (FERRY). The FERRY complex interacts with RAB5 (an early endosome regulator) and nascent mRNA to play an essential role in local protein translation through early endosomes [12,13]. Local protein translation is essential for subcellular homeostasis, particularly in neuronal cells that contain elongated processes. Defects in another FERRY complex protein, PPP1R21, leads to a disease phenotype like TBCK syndrome. Structurally, TBCK is a high molecular weight protein (99kD) with Serine/Threonine kinase, Rab-GAP-TBC, and Rho domains (Figure 1). Understanding TBCK function in the FERRY complex will reveal regulatory pathways involved in TBCK syndrome, which will guide treatment strategies and targets for disease management.
Potential therapeutics
Treatment for TBCK syndrome is currently limited to palliative care for specific symptoms. This includes noninvasive and invasive ventilation for hypotonia-associated respiratory dysfunction and treatment with anticonvulsants to reduce seizures. There are no treatments which directly ameliorate the effects of TBCK deficiency, nor are there any which slow disease progression. Thus, there is a pressing need for new therapeutics aimed at reducing the effects of this fatal pediatric neurodegenerative disease.
As a monogenetic loss-of-function (LOF) disorder, TBCK syndrome is potentially amenable to treatment via gene replacement with a functional copy of TBCK. Even low levels of exogenous functional TBCK are likely to be therapeutic. For example, we reported on two sisters who are compound heterozygous with a LOF and a canonical splice variant who have not yet reported neurologic regression even in the second decade of life [2]. This is strikingly different from the many patients with homozygous variants who become ventilator dependent in infancy or early childhood. Such variability indicates that adding even a small amount of exogenous functional TBCK protein to patient cells is likely to have powerful therapeutic effects, although this still needs to be confirmed in cell and animal models.
With an approximately 2.7 kb cDNA, TBCK can be readily packaged into all commonly used viral gene therapy vectors, including recombinant adeno-associated virus (rAAV). rAAV-based gene therapy vectors have exhibited safe delivery with minimal side effects and have already shown promising results in animal studies and human trials for neurodegenerative diseases. Furthermore, there is evidence that TBCK syndrome’s respiratory insufficiency is driven by motor neuron dysfunction in the anterior gray column of the spinal cord [7]. A recent Novartis-led clinical trial for spinal muscular atrophy identified the anterior gray column of the spinal cord as a viable target for gene therapy, bolstering the argument that gene therapy-based treatment will be effective for TBCK syndrome patients, especially with early intervention. As the field of gene therapy progresses and begins to fulfill its potential, individuals with TBCK syndrome can become a gene therapy success story.
Concluding remarks
Overall, clinical, genetic, and molecular advances of TBCK syndrome are enhancing our ability to diagnose and manage affected individuals. We have highlighted the need for continued research and collaboration to understand the pathophysiology of this rare genetic disorder and offered hope for effective therapies for this vulnerable population.
Footnotes
Declaration of interests
No interests are declared.
References
- 1.Wu J and Lu G (2021) Multiple functions of TBCK protein in neurodevelopment disorders and tumors (Review). Oncol. Lett 21, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhoj EJ et al. (2016) Mutations in TBCK, encoding TBC1-domain-containing kinase, lead to a recognizable syndrome of intellectual disability and hypotonia. Am. J. Hum. Genet 98, 782–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tintos-Hernández JA et al. (2021) Lysosomal dysfunction impairs mitochondrial quality control and is associated with neurodegeneration in TBCK encephaloneuronopathy. Brain Commun. 3, fcab215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabanathan S et al. (2023) Expanding the phenotype of children presenting with hypoventilation with biallelic TBCK pathogenic variants and literature review. Neuromuscul. Disord 33, 50–57 [DOI] [PubMed] [Google Scholar]
- 5.Ortiz-González XR et al. (2018) Homozygous boricua TBCK mutation causes neurodegeneration and aberrant autophagy. Ann. Neurol 83, 153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chong JX et al. (2016) Recessive inactivating mutations in TBCK, encoding a Rab GTPase-activating protein, cause severe infantile syndromic encephalopathy. Am. J. Hum. Genet 98, 772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck-Wödl S et al. (2018) Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease - a new type of neuronal ceroid lipofuscinosis (CLN15)? Acta Neuropathol. Commun 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murdock DR et al. (2021) Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J. Clin. Invest 131, e141500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sumathipala D et al. (2019) TBCK encephaloneuropathy with abnormal lysosomal storage: use of a structural variant bioinformatics pipeline on whole-genome sequencing data unravels a 20-year-old clinical mystery. Pediatr. Neurol 96, 74–75 [DOI] [PubMed] [Google Scholar]
- 10.Moreira D.de.P. et al. (2021) Neuroprogenitor cells from patients with TBCK encephalopathy suggest deregulation of early secretory vesicle transport. Front. Cell. Neurosci 15, 803302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D et al. (2020) Structure of TBC1D23 N-terminus reveals a novel role for rhodanese domain. PLoS Biol. 18, e3000746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quentin D et al. (2023) Structural basis of mRNA binding by the human FERRY Rab5 effector complex. Mol. Cell 83, 1856–1871.e9 [DOI] [PubMed] [Google Scholar]
- 13.Schuhmacher JS et al. (2023) The Rab5 effector FERRY links early endosomes with mRNA localization. Mol. Cell 83, 1839–1855.e13 [DOI] [PubMed] [Google Scholar]