

Abstract
Melanocortin 4 receptor (MC4R) mutations are the most common cause of human monogenic obesity and are associated with hyperphagia and increased linear growth. While MC4R is known to activate Gsα/cAMP signaling, a substantial proportion of obesity-associated MC4R mutations do not affect MC4R/Gsα signaling. To further explore the role of specific MC4R signaling pathways in the regulation of energy balance, we examined the signaling properties of one such mutant, MC4R (F51L), as well as the metabolic consequences of MC4RF51L mutation in mice. The MC4RF51L mutation produced a specific defect in MC4R/Gq/11α signaling and led to obesity, hyperphagia, and increased linear growth in mice. The ability of a melanocortin agonist to acutely inhibit food intake when delivered to the paraventricular nucleus (PVN) was lost in MC4RF51L mice, as well as in WT mice in which a specific Gq/11α inhibitor was delivered to the PVN; this provided evidence that a Gsα-independent signaling pathway, namely Gq/11α, significantly contributes to the actions of MC4R on food intake and linear growth. These results suggest that a biased MC4R agonist that primarily activates Gq/11α may be a potential agent to treat obesity with limited untoward cardiovascular and other side effects.
Keywords: Metabolism
Keywords: G proteins, Melanocortin, Signal transduction
<p>A melanocortin 4 receptor mutation associated with human obesity leads to hyperphagia and obesity in mice and has a specific defect in Gq/11 activation.</p>
Introduction
Melanocortins are neurotransmitters that act in the CNS to promote negative energy balance via reduced food intake and increased energy expenditure and sympathetic nervous system (SNS) activity. Melanocortin 4 receptor (MC4R) is the main receptor that mediates the effects of melanocortins on food intake and energy expenditure. MC4R is an important regulator of energy balance in humans, as loss-of-function MC4R mutations constitute the most common form of monogenic obesity (1); and more than 150 pathogenic MC4R variants have been identified in patients with severe, early-onset obesity accompanied by hyperphagia, increased linear growth, impaired glucose metabolism (2) and decreased heart rate and blood pressure compared with similarly obese controls (3). MC4R deficiency also causes obesity associated with increased food intake and reduced energy expenditure in mice (4, 5). MC4R agonism is a logical therapeutic target for the treatment of obesity. However, the use of MC4R agonists has been limited by untoward cardiovascular side effects (3).
While MC4R expression in the CNS is fairly widespread (6, 7), a major site of MC4R action affecting acute inhibition of food intake (8–10) is the paraventricular nucleus of the hypothalamus (PVN), where it is activated by melanocortins released from proopiomelanocortin (POMC) neurons originating from the arcuate nucleus of the hypothalamus (ARC). PVN-specific reexpression of MC4R in MC4R-null mice reverses hyperphagia but does not correct the impaired energy expenditure (8), consistent with MC4R’s effects on food intake occurring primarily in the PVN and its effects on energy expenditure occurring at other CNS sites. MC4R activation leads to induction of the transcription factor Sim1 (11), and mutations in SIM1 and Sim1 lead to obesity, hyperphagia, and increased linear growth in humans and mice, respectively (12–15).
It is well established that activated MC4Rs stimulate the G protein Gsα, which stimulates adenylyl cyclase to increase intracellular cAMP accumulation. Heterozygous Gsα-inactivating mutations lead to obesity in patients with pseudohypoparathyroidism type 1A (PHP1A) (16) and in mice (17), but only when the mutation is on the maternal allele due to tissue-specific imprinting of GNAS/Gnas, the gene encoding Gsα. Disruption of the maternal Gsα allele limited to the CNS (mBrGsKO) produces the same obesity phenotype, which is associated with reduced energy expenditure, impaired cold-induced thermogenesis, insulin resistance, and glucose intolerance (18). However, unlike what is observed with loss of MC4R (1, 4, 19), obesity in mBrGsKO mice is not driven by hyperphagia, and the anorectic effects of a melanocortin agonist remain intact (18). Moreover, unlike Mc4r-null mice, mBrGsKO mice do not show increased linear growth (18). It appears that obesity in mBrGsKO mice is mediated by Gsα signaling defects in the dorsomedial hypothalamus (DMH), leading to reduced energy expenditure (20). Mice with Gsα deficiency limited to the PVN (mPVNGsKO) also do not show hyperphagia or an impairment in melanocortin-induced food intake inhibition (21).
The results from these mouse studies suggest that the effects of MC4R action in the PVN on food intake and linear growth are mediated by downstream signaling pathways that are independent of Gsα, which is further supported by studies showing that a significant proportion of human MC4R mutations associated with monogenic obesity do not show significant defects in activation of Gsα/cAMP signaling (22–24). One study correlating signaling properties of naturally occurring MC4R variants with BMI concluded that differences in MC4R/Gsα signaling do not account for most of the variance in BMI (25). Several alternate pathways downstream of MC4R have been implicated in mediating its effects on food intake and body weight, including β-arrestin recruitment (25, 26), closure of the inward rectifying potassium channel Kir7.1 (27), and activation of MAPK (28, 29), although there is no direct evidence that strongly links these pathways in the PVN to the regulation of energy balance. We have shown that PVN-specific deletion of both Gqα and G11α (PVNGq/11KO), 2 homologous and ubiquitously expressed G protein α-subunits that couple receptors to PLC, recapitulates the effects of MC4R deficiency on food intake and linear growth — namely, severe obesity associated with hyperphagia and loss of the anorectic response to melanocortin agonist, along with increased linear growth (11). Moreover, some MC4R mutants with normal Gsα signaling have been demonstrated to have impaired PLC activation as measured by a nuclear factor of activated T cells (NFAT) reporter system (30).
To further explore the role of MC4R signaling pathways in the regulation of energy balance, we examined in detail the physiological consequences in mice and signaling properties of one mutant MC4R that is associated with early-onset obesity in humans and has been reported to have relatively intact Gsα signaling (MC4RF51L, phenylalanine 51 to leucine) (23). Our results showed that this mutation produced a specific defect in MC4R/Gq/11α signaling and that the mutation in mice led to a phenotype closely related to that seen in PVNGq/11KO mice, providing evidence that Gq/11α signaling, rather than Gsα signaling, mediates the actions of MC4R on food intake and linear growth. This conclusion is further supported by the observation that a specific Gq/11α inhibitor delivered to the PVN of WT mice blocked the ability of a melanocortin agonist to acutely inhibit food intake. Biased MC4R agonists that primarily activate Gq/11α signaling may lead to food intake suppression and weight loss with limited untoward limited effects.
Results
MC4RF51L mice develop obesity without disruption of MC4R/Gsα/cAMP signaling.
MC4RF51L mice were generated by CRISPR/Cas9 to direct an MC4R site mutation at amino acid position 51 with phenylalanine replaced by leucine (Figure 1A) and mating to homozygosity. The human and mouse MC4R protein sequences are highly homologous (94% identical, 97% conserved), and the F51 residue is in a highly conserved region (14 amino acids upstream through 112 downstream of F51 are identical between the species).
Figure 1. MC4RF51L mice develop obesity without disruption of MC4R/Gsα signaling.

(A) DNA sequencing showing conversion of phenylalanine 51 (TTT) in WT mice to leucine (CTA) in MC4RF51L mice. (B) Quantification of pCREB/total CREB in PVN of WT, MC4RF51L (F51L), and MC4RKO mice after i.p. injection of saline or MTII (n = 3–5/group; #P < 0.05, ##P < 0.01 vs. saline; ^P < 0.05 vs. WT and MC4RF51L mice). (C) Body weights of male (left) and female (right) MC4RF51L and WT mice measured weekly from week 4 to 11 (males, n = 19–24/group; females, n = 20–24/group). (D) Total body, fat and lean mass of 8-week-old male (n = 12–14/group) and female (n = 12–16/group) MC4RF51L and WT mice. (E) Body length of 8-week-old male and female of MC4RF51L and WT mice (males, n = 5–7/group; females, n = 7–9/group). Data represent mean ± SEM. *P < 0.05, **P < 0.01 vs. WT by unpaired t test or 2-way ANOVA.
To verify that MC4R/Gsα/cAMP signaling was maintained in MC4RF51L mice in vivo, we administered the MC4R agonist melanotan II (MTII; 10 μg/g body weight) i.p. to mice and measured phosphorylation levels of CREB (pCREB) in the PVN by immunofluorescence; this served as a readout of cAMP production, as cAMP stimulates the phosphorylation of CREB via activation of PKA (Figure 1B and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI165418DS1). MC4RF51L mice and WT littermates had similar levels of total CREB protein. After administration of saline, pCREB signals were low in WT PVN, while there were significantly higher levels of pCREB in the PVN of MC4RF51L mice. This was likely due to the fact that MC4RF51L mice had increased leptinergic signaling (see Table 1), leading to greater melanocortinergic input to the PVN, which would be expected to increase CREB phosphorylation if MC4R/Gsα/cAMP signaling is intact in the mutant receptor — although other mechanisms associated with obesity may also be involved. After MTII administration, CREB phosphorylation significantly increased to similar levels in WT and MC4RF51L PVN. Concomitant administration of MTII into MC4RKO mice resulted in low pCREB levels in the PVN that were similar to those seen in WT after saline administration and significantly lower than those seen in MTII-treated WT or MC4RF51L mice, indicating that the observed pCREB responses to MTII in WT and MC4RF51L mice were primarily due to signaling through MC4R, as opposed to MC3R or other members of the melanocortin receptor family. Overall these results are consistent with the mutant MC4R receptor having intact downstream Gsα/cAMP signaling in the PVN, as was observed in the present study and by others (23) in cell-based signaling assays.
Table 1. Serum chemistry in randomly-fed 3-month-old female MC4RF51L and WT mice.
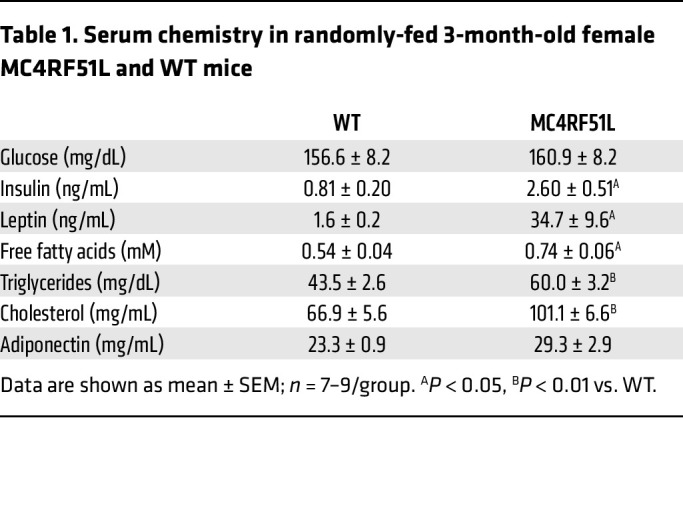
MC4RF51L mice appeared normal at birth but rapidly gained more weight than WT littermates (Figure 1C). Male mutants had significantly increased body weight at 5 weeks of age and were 21% heavier than their WT littermates at 11 weeks of age, while female mutants became significantly heavier starting at 4 weeks and gained 29% more weight than their WT littermates by 11 weeks of age. The weight gain in MC4RF51L mice was attributed to significant 2- to 3-fold increases in fat mass and small, but significant, increases in lean mass (Figure 1D). At least some of the increase in lean mass was attributable to an increase in body length (Figure 1E), a finding that was also observed in mice with germline MC4R mutations (4) and mice with PVN-specific Gq/11α deficiency (11) but not in obese mBrGsKO mice (18). Consistent with increased adiposity, MC4RF51L mice developed severe hyperleptinemia, with circulating leptin levels more than 20-fold higher in MC4RF51L mice compared with their WT littermates at 12 weeks of age (Table 1).
Obesity in MC4RF51L mice is associated with hyperphagia.
To determine the extent that differences in food intake and energy expenditure contribute to the increased weight gain in MC4RF51L mice, we measured food consumption and body composition every 7 days over a 5-week period in males starting at 5 weeks and in females starting at 4 weeks of age, and calculated energy intake and energy expenditure based on these measurements (31). During this period, both male and female MC4RF51L mice ate significantly more than their WT littermates (Figure 2A). On the other hand, energy expenditure was not decreased and in fact was increased in both males and females at various time points, reflecting their increased body mass (Figure 2B).
Figure 2. Obesity in MC4RF51L mice is associated with hyperphagia.

(A) Average daily food intake measured every 7 days for 5 weeks in male and female MC4RF51L and WT mice (n = 4–13/group). (B) Average daily energy expenditure (EE) calculated weekly for 5 weeks in male and female MC4RF51L and WT mice. (C) REE and TEE measured by indirect calorimetry and normalized to lean mass (LM) in 3-month-old male MC4RF51L and WT mice (n = 5–6/group). (D) Total and ambulatory (Amb) activity levels in male MC4RF51L and WT mice at 3 months of age (n = 5–6/group). (E) RERs (vCO2/vO2) at 22°C in 3-month-old male MC4RF51L and WT mice at 3 months of age (n = 5–6/group). (F) Cold tolerance test. Rectal temperature in 4-month-old male MC4RF51L and WT mice at room temperature (0 hours) or at the indicated time points after being placed in an environment with a 6°C ambient temperature (n = 6–9/group). Meal frequency (G) and meal size (H) measured in 3 month-old male mice during daylight and nighttime hours at 22°C and 30°C (n = 5–6/group). Meal size was significantly increased in MC4RF51L mice compared with WT mice as determined by 2-way ANOVA. Data represent mean ± SEM. *P < 0.05, **P < 0.01 vs. WT by unpaired t test.
We also studied parameters of energy metabolism in 3-month-old male mice using an Oxymax–Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments). indirect calorimetry showed no differences in resting (REE) and total energy expenditure (TEE) between the genotypes at room temperature (22°C) when normalized to lean mass (Figure 2C). The same measurements performed at thermoneutrality (30°C), a condition in which SNS activity and thermogenesis are minimized, showed increased REE and TEE in MC4RF51L mice when normalized to lean mass (Figure 2C). Physical activity tended to be lower in MC4RF51L mice than in WT mice, but these differences did not reach statistical significance (Figure 2D). Respiratory exchange ratio (RER; vCO2/vO2) was increased in MC4RF51L mice (Figure 2E), suggesting greater utilization of carbohydrates relative to fats as a fuel source in the mutant mice. Examination of food consumption patterns revealed no differences in feeding frequency between WT and MC4RF51L mice during the day or night, whether at 22°C or 30°C (Figure 2G). However, meal size was significantly larger in MC4RF51L mice as compared with WT during the night at both 22°C and 30°C (Figure 2H), consistent with a higher satiety threshold in MC4RF51L mice. These findings are consistent with previous studies showing that melanocortins working through MC4R receptors primarily affect meal size, rather than meal frequency (32–34).
MC4RF51L mice did not show evidence of acute cold intolerance, as they were able to maintain their body temperature over 5 hours at 6°C (Figure 2F), consistent with there being no overt defect in cold-induced thermogenesis, which we had previously observed in mice with whole-brain Gsα deficiency but not in mice with Gsα deficiency limited to the PVN (21). In summary, obesity in MC4RF51L mice was primarily associated with hyperphagia as a result of increased meal size. On the other hand, MC4RF51L mice showed no evidence of reduced energy expenditure or impaired cold-induced thermogenesis, which are both features of MC4R-null mice (4, 5, 8).
Melanocortin-mediated food intake inhibition is impaired in MC4RF51L mice.
We have previously shown that within the PVN, MC4R-mediated food intake inhibition appears to be mediated by Gq/11α, while MC4R-mediated cardiovascular effects are mediated by Gsα (11). To examine the effects of the MC4RF51L mutation on physiological responses to melanocortins, we delivered the melanocortin agonist MTII either systemically (i.p.) or directly into the PVN via cannula in MC4RF51L and WT mice and examined acute food intake, energy expenditure, and cardiovascular responses. While either i.p. (Figure 3A) or intra-PVN injection of MTII (Figure 3B) led to reduced food intake in WT mice, these effects on food intake were absent in MC4RF51L mice. While systemic MTII resulted in a small, but significant, increase in energy expenditure in WT mice, no increase in energy expenditure was observed in MC4RF51L mice (Figure 3C). Presumably this defect in melanocortin-stimulated energy expenditure was not occurring at the PVN, as direct delivery of MTII to the PVN had no effect on energy expenditure in WT or MC4RF51L mice (Figure 3D), consistent with prior studies showing that melanocortin actions on energy expenditure are not mediated within the PVN (8). In contrast to what we observed for food intake, MC4RF51L mice had normal heart rate and blood pressure at baseline, and the responses of both heart rate and blood pressure to intra-PVN delivery of MTII remained intact (Figure 3, E and F). Overall, the food intake and cardiovascular responses of MC4RF51L mice to intra-PVN MTII were similar to what was observed in mice with PVN-specific Gq/11α deficiency (11) and were consistent with MC4RF51L mice having a specific defect in MC4R/Gq/11α signaling.
Figure 3. Food intake, energy expenditure, heart rate, and blood pressure responses to MTII.

Food intake following i.p. injection of PBS or MTII in 3-month-old male MC4RF51L and WT mice (n = 5/group) (A) or intra-PVN injection of PBS or MTII in 4- to 5-month-old male MC4RF51L and WT mice (n = 5–6/group) (B). Energy expenditure following i.p. injection of PBS or MTII in 3-month-old male MC4RF51L and WT mice (n = 5–6/group) (C) or intra-PVN injection of PBS or MTII in 4- to 5-month-old male MC4RF51L and WT mice (n = 4–5/group) (D). Heart rate (E) and mean blood pressure (F) after intra-PVN injection of PBS or MTII in 3-month-old female MC4RF51L and WT mice (n = 8–9/group). Data represent mean ± SEM. *P < 0.05, **P < 0.01 vs. WT by 2-way ANOVA.
Glucose and lipid metabolism in MC4RF51L mice.
Adult MC4RF51L mice showed impaired glucose metabolism, with severe glucose intolerance in both male (Figure 4A) and female (Figure 4D) MC4RF51L mice at 6–7 months of age, after the establishment of obesity (Figure 4, C and F). Fasting glucose levels were unaffected in male MC4RF51L mice (Figure 4B) but significantly increased in female MC4RF51L mice (Figure 4E). Serum measurements in 3-month-old female mice in the randomly fed state showed significantly elevated insulin levels in MC4RF51L mice, with glucose levels remaining unaffected (Table 1). To determine whether abnormal glucose metabolism is independent of obesity, we assessed glucose homeostasis in young female MC4RF51L and WT mice at 4–5 weeks of age. Although body weight was only slightly, though significantly, increased in MC4RF51L mice at this age (Figure 4I), both glucose tolerance (Figure 4G) and fasting glucose (Figure 4H) were similar in MC4RF51L and WT mice. These results indicate that abnormal glucose homeostasis in MC4RF51L mice was secondary to obesity rather than to a primary defect in glucose metabolism, despite the fact that prior studies have shown that the loss of MC4R (35, 36) or Gsα in the CNS (18) leads to a primary defect in glucose metabolism. Adult female MC4RF51L mice had significantly higher serum levels of cholesterol, triglycerides, and free fatty acids in the fed state, while adiponectin levels were unaffected (Table 1). It is unclear to what extent hyperlipidemia in MC4RF51L mice results from obesity versus impaired melanocortin action, as melanocortins within the CNS have been shown to directly regulate lipid metabolism (37).
Figure 4. Glucose metabolism in MC4RF51L mice.

(A–C) Glucose metabolism in 6- to 7-month-old male MC4RF51L and WT mice (n = 8–11/group). Glucose tolerance test with AUC shown on the right (A), fasting glucose levels (B), and body weights at the time of glucose tolerance tests (C). (D–F) Glucose metabolism in 6- to 7-month-old female MC4RF51L and WT mice (n = 4–7/group). Glucose tolerance test with AUC shown to the right (D), fasting glucose levels (E), and body weights at the time of glucose tolerance tests (F). (G–I) Glucose metabolism in 4- to 5-week-old female MC4RF51L and WT mice (n = 7/group). Glucose tolerance test with AUC shown on the right (G), fasting glucose levels, (H) and body weights at the time of glucose tolerance tests (I). Data represent mean ± SEM. *P < 0.05, **P < 0.01 vs. WT by unpaired t tests.
Hyperphagia significantly contributes to the metabolic phenotype of MC4RF51L mice.
In order to examine the extent to which the metabolic phenotype of MC4RF51L mice is accounted for by excess energy intake, we performed a pair-feeding experiment in which a group of MC4RF51L mice were provided an amount of food identical to that of paired WT mice on an ad libitum diet for an 8-week period from the start of week 4 until the start of week 12 after birth. Another group of MC4RF51L mice were fed ad libitum simultaneously. Body weight curves in males showed that pair-fed MC4RF51L mice gained slightly, but significantly, more weight than WT mice, although the weight gain was significantly lower than that observed in ad libitum fed MC4RF51L mice (Figure 5, A and B). Body composition confirmed that the excess weight gain in pair-fed mice was accounted for by an increase in fat mass (Figure 5, C–E). Consistent with these findings, pair-fed MC4RF51L mice had leptin levels at the end of the study that were significantly higher than those of WT mice but significantly lower than those of ad libitum fed MC4RF51L mice (Figure 6I). The findings were similar for female mice, although the increases in body weight gain, fat mass gain, and serum leptin levels in pair-fed MC4RF51L relative to WT mice were more subtle than those observed in males (Figure 5, F–J, and Figure 6I). Energy balance studies performed during the experiments confirmed once again that both male and female ad libitum fed MC4RF51L mice ate significantly more than WT mice (Supplemental Figure 2, A and C). Reduced food intake in pair-fed MC4RF51L mice led to reduced energy expenditure compared with ad libitum fed MC4RF51L mice (Supplemental Figure 2, B and D). Overall, these results confirm that hyperphagia significantly contributes to the obesity seen in MC4RF51L mice. However, pair-fed MC4RF51L mice (particularly males) still gained more fat mass compared with WT mice with similar energy intake.
Figure 5. Effect of pair feeding on body weight and composition.

Body weight (A) and weight gain (B) during an 8 week period (weeks 4–12) in male ad libitum fed WT (WT-ad lib), ad-libitum fed MC4RF51L mice (F51L-ad lib) and MC4RF51L mice pair-fed to WT (F51L-pair-fed). (C–E) Body composition in male mice measured at weeks 6, 8, 10, and 12 during the pair-feeding study showing total body mass (C), fat mass (D), and lean mass (E) (n = 10–12/group). Body weight (F) and weight gain (G) during pair-feeding experiment in females. (H–J) Body composition showing total body mass (H), fat mass (I), and lean mass (J) in females (n = 9–16/group). Data represent mean ± SEM. Based on 2-way ANOVA, data for all 3 groups are statistically significantly different from each other in all panels except E, G, and H, in which results for ad libitum fed MC4RF51L mice are significantly different from those of the other 2 groups.
Figure 6. Glucose and lipid metabolism after pair feeding.

Body weight (A), glucose tolerance (B), and glucose tolerance AUC (C) at the end of the pair-feeding study (week 12) in male WT-ad-lib, MC4RF51L-ad-lib, and MC4RF51L pair-fed mice (n = 9–10/group). Body weight (D), glucose tolerance (E), and glucose tolerance AUC (F) at the end of the pair-feeding study in female mice (n = 8–15/group). Nonfasted serum glucose (G; n = 10–16/group for males and 9–16/group for females), insulin (H; n = 10–12/group for males and 9–16/group for females), leptin (I; n = 10–12/group for males and 9–16/group for females), triglycerides (J; n = 9–12/group for males and 8–16/group for females), and total cholesterol (K; n = 10–12/group for males and 9–16/group for females) at the end of the pair-feeding study. Data represent mean ± SEM. *P < 0.05, **P < 0.01 by 1-way ANOVA.
Examination of glucose metabolism at the end of the pair-feeding study revealed evidence that male pair-fed MC4RF51L mice — with mean body weights that were higher than those of WT but lower than those of ad libitum fed MC4RF51L mice (Figure 6A) — had impaired glucose metabolism with reduced glucose tolerance (Figure 6, B and C) and increased glucose and insulin levels (Figure 6, G and H) compared with WT mice; however, these parameters (except for random serum glucose levels) were significantly improved compared with ad libitum fed MC4RF51L mice. In contrast, female pair-fed MC4RF51L mice, which also had a significant increase in body weight compared with WT mice (Figure 6D), showed no differences in glucose tolerance or random glucose or insulin levels compared with WT mice, while ad libitum fed MC4RF51L mice showed significant glucose intolerance and elevations in serum glucose and insulin levels (Figure 6, E–H). There were no differences in triglyceride levels between groups (Figure 6J), while pair-fed and ad libitum fed MC4RF51L mice had similarly elevated total cholesterol levels compared with WT mice (Figure 6K). Overall, these results confirm that impaired glucose metabolism in female MC4RF51L mice was completely accounted for by increased energy intake and obesity. In male mice, the partial impairment in glucose metabolism that remains after pair feeding may be secondary to the fact that these mice still have increased adiposity.
MC4RF51L has a specific defect in Gq/11α signaling.
Melanocortin receptor accessory protein 2 (MRAP2) is expressed in the hypothalamus, interacts with MC4R, and is required for normal energy balance and MC4R signaling (38–41). We therefore generated a cell line in which MRAP2 was stably transfected into HEK293 cells (HEK293MRAP2) to examine the signaling properties of WT MC4R and MC4RF51L. MRAP2 expression in HEK293MRAP2 cells and its absence in parental HEK293 cells was confirmed by both quantitative real-time PCR (qRT-PCR) and immunoblotting (Supplemental Figure 3). We first examined the ability of α-melanocyte–stimulating hormone (α-MSH) to stimulate cAMP accumulation in parental HEK293 or HEK293MRAP2 cells that were transiently transfected with either WT MC4R or MC4RF51L plasmid in the presence of the phosphodiesterase inhibitor IBMX. For both WT and mutant MC4R, the maximal response was doubled and the EC50 decreased by at least 1 order of magnitude in the presence of MRAP2 (Figure 7A), indicating that MRAP2 is required for optimal MC4R/Gsα/cAMP signaling. Maximal cAMP accumulation was similar in cells transfected with WT MC4R or MC4RF51L, in both the presence and absence of MRAP2 (Figure 7A). This finding was consistent with prior results (22, 23) and our observation that CREB phosphorylation in response to MTII was unaffected in the PVNs of MC4RF51L mice (Figure 1B). Consistent with prior results (22), α-MSH was somewhat less potent in stimulating cAMP accumulation via MC4RF51L (EC50: 22.6 nM [95% CI, 11.5–41.2 nM] for WT MC4R vs. 363 nM [95% CI, 219–619 nM] for MC4RF51L in HEK293MRAP2 cells). Whether the decreased potency was due to reduced abundance of the mutant receptor at the plasma membrane is unclear, as one study reported reduced MC4RF51L abundance at the plasma membrane (26), while another study showed no differences (22).
Figure 7. MC4RF51L has impaired Gq/11α signaling.

Dose response curves for cAMP (n = 3) (A) and IP1 (n = 5) (B) in parental HEK293 or HEK293MRAP2 cells that were transfected with WT or mutant (F51L) MC4R plasmids. (C and D) Representative experiment showing immunoblots probed with antibodies for phospho-ERK1/2 (p-ERK; top row) and total ERK1/2 (bottom row) in lysates from HEK293MRAP2 cells transfected with WT, mutant (F51L) MC4R or empty vector (EV), pretreated with the Gq/11α inhibitor FR900359 (FR) or vehicle and incubated with 10–5 M α-MSH at the indicated times in minutes (C), with quantification of pERK/total ERK ratios normalized to WT at time 0 (n = 8–9/group; except for EV, n = 3–5/group) (D). (E) IP1 generation in response to 10–4 M α-MSH in HEK293 cells transfected with empty vector or HEK293 or HEK293MRAP2 cells transfected with WT MC4R after pretreatment with either FR or vehicle (n = 2–6/group). (F) cAMP generation in response to 0 or 10–6 M α-MSH in HEK293MRAP2 cells transfected with WT or mutant MC4R that were pretreated with either FR or vehicle (n = 3–7/group). (G) Food intake (normalized to PBS injection) in response to intra-PVN administration of vehicle alone, MTII, or MTII + FR in 4- to 6-month-old male WT mice (n = 4–8/group). (H) Food intake (normalized to PBS injection) in response to intra-PVN administration of FR alone (n = 8/group). (I) Effect of intra-PVN FR injection on heart rate response to intra-PVN MTII injection (n = 3/group). Data represent mean ± SEM. *P < 0.05, **P < 0.01 by unpaired t test or 1-way ANOVA.
We next examined the ability of α-MSH to activate MC4R/Gq/11α signaling by measuring the accumulation of inositol phosphate (IP1) in the presence of lithium, which blocks IP1 degradation, as a readout of PLC activity (Figure 7B). Similar to what we observed for cAMP accumulation, the presence of MRAP2 significantly increased the ability of α-MSH to maximally stimulate IP1 accumulation via the WT MC4R, as well as its potency. However, α-MSH was less potent for IP1 accumulation as compared with cAMP accumulation (EC50: 1,049 nM for IP1 vs. 22.6 nM for cAMP in HEK293MRAP2 cells). As compared with WT MC4R, MC4RF51L showed no IP1 response in the absence of MRAP2 and only a small increase in IP1 at high α-MSH concentrations (~10–4 M) in the presence of MRAP2. Accumulation of IP1 by WT MC4R in response to 10–4 M α-MSH was completely blocked by FR900359 (FR; formerly UBO-QIC), a specific Gq/11α inhibitor (42–45), confirming that the IP1 responses observed in our experiments reflect effects on MC4R/Gq/11α signaling (Figure 7E). We also observed a partial (~25%) decrease in cAMP accumulation in the presence of FR (Figure 7F). However, studies in HEK293 cells have confirmed that this effect of FR on cAMP accumulation is the direct result of Gq/11α inhibition, rather than Gsα inhibition (42), and other studies have found that FR does not affect Gsα activation (43–45).
Finally, we examined MC4R/MAPK/ERK signaling by measuring the time course of ERK1/2 phosphorylation over the first 10 minutes after addition of 10–5 M α-MSH in HEK293MRAP2 cells (Figure 7, C and D). Overall, there were no statistical differences in the responses in cells expressing WT MC4R or MC4RF51L (Figure 7D). At 3 minutes the responses were similar, while at 5 and 10 minutes there were small, but statistically insignificant, decreases in responses to α-MSH in cells expressing MC4RF51L. However, FR-treated cells expressing WT MC4R showed virtually the same response as cells expressing MC4RF51L, indicating that the extent of any decrease in ERK phosphorylation observed with MC4RF51L would be due to downstream effects resulting from loss of Gq/11α signaling. Our results showing lack of effect of the F51L mutation on MAPK signaling is consistent with prior results (28). In summary, our results show that MRAP2 is critical for both the MC4R/Gsα and MC4R/Gq/11α signaling pathways and that MC4RF51L has a specific defect in Gq/11α signaling.
MC4R-mediated food intake inhibition in PVN is blocked by Gq/11α inhibition.
To examine the effect of Gq/11α inhibition on the ability of intra-PVN delivery of MC4R agonist to acutely inhibit food intake, we repeated the experiment examining the ability of intra-PVN delivery of MTII to acutely inhibit food intake in WT mice, but injected FR or vehicle into the intra-PVN cannula 5 minutes prior to injection of MTII. In this experiment injection of MTII alone resulted in an approximately 70% inhibition of food intake, while FR almost completely blocked the effect of MTII on food intake (Figure 7G). Intra-PVN administration of FR alone had no effect on food intake (Figure 7H). In contrast to the effect of intra-PVN administration on food intake, intra-PVN FR injection failed to block the rise in heart rate after intra-PVN delivery of MTII (Figure 7I; compare with Figure 3E). This provides direct in vivo evidence for the role of MC4R/Gq/11α signaling in the PVN in food intake regulation.
Discussion
MC4R mutations are the most common cause of monogenic obesity and are associated with hyperphagia, increased body length, reduced energy expenditure, abnormal glucose and lipid metabolism, and decreased heart rate and blood pressure (1, 4, 35–37, 46). While some studies have suggested that cAMP in MC4R-expressing PVN neurons plays a role in the regulation of food intake and body weight (47, 48), many MC4R mutations linked to human obesity do not show defects in Gsα/cAMP signaling (22, 23, 49), suggesting that alternative MC4R signaling pathways may also be involved in the normal maintenance of energy balance. Consistent with this notion, a study correlating the signaling properties of MC4R variants to BMI concluded that Gsα/cAMP explains only up to 12% of the variance in BMI (25). In the present study, we further investigated the signaling properties and physiological consequences of MC4RF51L, which was reported to maintain its ability to activate the Gsα/cAMP pathway (23). We confirmed that stimulation of CREB phosphorylation by a melanocortin agonist was maintained in the PVN of MC4RF51L mice, consistent with intact MC4R/Gsα signaling, and that MC4RF51L maintained the ability to stimulate cAMP accumulation in response to α-MSH, with a Vmax similar to that of WT MC4R, although α-MSH potency was somewhat reduced.
Although MC4RF51L had relatively intact coupling to Gsα/cAMP signaling, mice with homozygous MC4RF51L mutation developed severe obesity associated with hyperphagia and impaired anorectic responses to melanocortin agonist, as well as increased linear growth, without primary effects on cold tolerance, glucose metabolism, or cardiovascular responses to melanocortin administration. Overall the phenotypes observed in MC4RF51L mice were remarkably similar to those observed with PVNGq/11KO mice (11) (Table 2) and, along with the severe defect in Gq/11α signaling that we observed in vitro, they implicate defective MC4R/Gq/11α signaling in the PVN as the likely cause of hyperphagia and increased linear growth in MC4RF51L mice. Loss of Sim1, a transcription factor highly expressed in the PVN and shown to be regulated by MC4R (11), produces similar effects on food intake and linear growth and leads to loss of the anorectic response to melanocortin agonist (12–15). The major role of Gq/11α signaling in mediating the inhibition of food intake by melanocortins in the PVN is further supported by the observation that the inhibition of food intake in response to intra-PVN delivery of MTII in WT mice was blocked by prior intra-PVN delivery of FR, a specific inhibitor of Gq/11α. Moreover, an MC4R variant associated with protection from human obesity and lower BMI (V103I) (50–52), which has been shown to have increased ligand-induced β-arrestin recruitment (25), has also been shown to lead to increased stimulation of Gq/11α signaling in response to α- and β-MSH (53). Finally, loss of the K+-dependent Na+/Ca2+ exchanger NCKX4 led to an MC4R- and Ca2+-dependent increase in PVN neuron activation and severe hypophagia and weight loss (54).
Table 2. Phenotypic comparison of different mouse models.

In contrast to what we observed in PVNGq/11KO (11) and MC4RF51L mice, Gsα deficiency in the CNS (18) or specifically limited to the PVN (Sim1 neurons) (21) had no impact on food intake or linear growth, nor the ability of melanocortin agonist to inhibit food intake (Table 2). CNS-specific Gsα deficiency (mBrGsKO) does lead to obesity, but this is associated with reduced energy expenditure rather than hyperphagia, and is also associated with impaired glucose metabolism, impaired cold tolerance, and reduced blood pressure (18), none of which were observed in PVNGq/11KO or MC4RF51L mice (Table 2). In contrast to mBrGsKO mice, mPVNGsKO mice do not show a significant metabolic phenotype nor impaired melanocortin action on food intake or energy expenditure, indicating that the metabolic defects observed in mBrGsKO mice do not result from loss of MC4R action in the PVN (21) (Table 2). However, like mBrGsKO mice, mPVNGsKO mice have lower blood pressure and heart rate, and intra-PVN delivery of melanocortin agonist fails to stimulate cardiovascular responses (11), consistent with Gsα mediating the cardiovascular actions of melanocortins in the PVN. Several lines of evidence show that Gq/11α is not involved in the cardiovascular actions of melanocortins in the PVN, including intact cardiovascular responses to intra-PVN delivery of melanocortin agonist in PVNGq/11KO mice (11) and MC4RF51L mice (the present study), as well as the inability of FR to block the cardiovascular responses to melanocortins in the PVN of WT mice (the present study).
One aspect in which the MC4RF51L mouse phenotype differed from that of PVNGq/11KO mice is that the ability of peripherally administered (i.p.) MTII to stimulate energy expenditure was absent in MC4RF51L mice, while it was maintained in PVNGq/11KO mice (Table 2). Unlike other models in which we observed the same deficit (18, 20), MC4RF51L mice did not show a reduction in baseline energy expenditure, and therefore reduced energy expenditure was not a major driver of the obesity phenotype observed under basal conditions (room temperature, ad libitum feeding). It is possible that we missed a small but significant response to i.p. MTII due to the limited numbers of mice examined. The apparent defect observed with i.p. MTII does not appear to be due to defective MC4R action in the PVN, as MTII delivery to the PVN had no effect on energy expenditure in either MC4RF51L or WT mice, consistent with the finding that reexpression of MC4R in the PVN of MC4R-null mice did not reverse the impaired ability of melanocortin agonist to stimulate energy expenditure (8). Our pair-feeding study also indicates that energy expenditure may be reduced in MC4RF51L mice when energy intake is restricted. While we have shown that the loss of Gq/11α in the DMH leads to reduced energy expenditure, stimulation of energy expenditure in response to i.p. MTII was unaffected in these mice (55). These subtle changes in energy expenditure observed in MC4RF51L mice may reflect the loss of MC4R/Gq/11α signaling at another site, moderately reduced ability of melanocortins to stimulate MC4R/Gsα signaling, or impaired activation of other signaling pathways downstream of MC4R.
We performed several experiments examining the effects of the MC4RF51L mutation on downstream signaling pathways in parental HEK293 cells and HEK293 cells stably transfected with MRAP2 (HEK293MRAP2 cells). Consistent with prior studies, the presence of MRAP2 led to a large increase in the ability of the WT MC4R to increase cAMP accumulation in the presence of melanocortin agonist, with increases in both Vmax and potency (38, 56). It is unlikely that MC4RF51L has a defect in MRAP2 interaction, as the mutant receptor showed improvements in Vmax and agonist potency for cAMP accumulation similar to those observed for the WT receptor. WT MC4R also showed a dose-dependent increase in IP1 accumulation in response to melanocortin agonist, which was confirmed to reflect MC4R coupling to Gq/11α, as the response was completely blocked by the Gq/11α inhibitor FR. As was observed for the cAMP response, WT MC4R demonstrated a large increase in Vmax and agonist potency for the IP1 response in the presence of MRAP2. In contrast, MC4RF51L demonstrated essentially no IP1 response to agonist in the absence of MRAP2 and a minimal response at high agonist doses in the presence of MRAP2, confirming a major defect in activation of the Gq/11α pathway. Finally, we examined MAPK activation by examining acute changes in ERK1/2 phosphorylation in response to agonist in the presence of MRAP2 and found no significant defect in the ability of MC4RF51L to stimulate ERK phosphorylation.
Recently MC4R/β-arrestin signaling has been implicated as a potentially important pathway in the regulation of body weight by a study correlating β-arrestin recruitment to MC4R variants with BMI (25). It is difficult to interpret this study, however, as β-arrestin recruitment to receptor is a general response to receptor activation, and therefore, β-arrestin recruitment may reflect the receptor being in the active conformation rather than activation of downstream β-arrestin signaling per se. The study did show a correlation between β-arrestin recruitment and ERK1/2 phosphorylation, which suggested that MAPK is activated by MC4R in a β-arrestin–dependent manner. While we did not directly measure β-arrestin recruitment to MC4RF51L, we did not see a defect in ERK1/2 phosphorylation, which might be expected if β-arrestin recruitment and/or signaling were impaired in this mutant receptor. Our study does not rule out the possibility that MC4R/β-arrestin interaction or signaling is involved in energy balance. MC4R-mediated closure of the inwardly rectifying potassium channel Kir7.1 by a G protein–independent mechanism has also recently been implicated as a regulator of energy balance and food intake (27). However, the role of Kir7.1 in body weight regulation is unclear, as Kir7.1-null mice only develop mild obesity at a very late age (57), which does not mimic the phenotype of MC4R-null (4) or MC4RF51L mice.
This study has several limitations. Although the PVN is a critical site for regulation of food intake by melanocortins, several studies have shown that the MC4R receptor is highly expressed in the dorsal vagal complex and that melanocortins delivered to this region can affect food intake and cardiovascular responses (58–60). We did not directly examine how the F51L mutation affects melanocortin actions in the dorsal vagal complex but would predict that MC4RF51L mice have altered melanocortin actions at this site that contribute to the overall phenotype. Although the results herein and in prior work (11) provide evidence that the phenotype of MC4RF51L mice is due to impaired Gq/11α signaling, we did not rule out the possibility that other defects in MC4R function (e.g., abnormal cellular localization or protein stability; changes in potency for Gsα/cAMP signaling; impaired β-arrestin signaling; protein-protein interactions) did not contribute to the phenotype. We also did not directly compare the phenotype of MC4RF51L mice with that of other relevant mouse lines (e.g., MC4R-null mice) in the same experiment. Finally, the results of pair-feeding experiments may be affected by the fact that the pair-fed mice do not have a normal feeding pattern, as they consume food soon after it is provided each day.
In summary, we have characterized the signaling properties and the physiological consequences of a human obesity-associated MC4R mutation and conclude that this mutation had a defect in the activation of Gq/11α and when introduced into mice led to obesity, hyperphagia, and increased linear growth, with no significant direct effects on glucose metabolism or cardiac function, the latter of which are mediated by Gsα (61). While melanocortin agonists have potential as a therapeutic option for obesity, its use has been limited by the presence of significant side effects, particularly increased heart rate and blood pressure and untoward sexual responses. Recent evidence shows that setmelanotide, an MC4R agonist with few reported side effects, has increased potency for activation of both Gsα and PLC (30). Clarifying the divergent MC4R signals that mediate the diverse physiological effects of melanocortins may allow the development of more specific MC4R agonists to treat obesity. Specifically, our findings suggest that a biased MC4R agonist that preferentially activates Gq/11α versus Gsα may reduce food intake by increasing meal-based satiety (mediated by Gq/11α), with lower cardiovascular side effects, which are mediated by Gsα.
Methods
In vivo experiments in mice
Sex as a biological variable.
Our study examined male and female animals, and similar findings are reported for both sexes.
Generation of MC4RF51L mice.
Mice having an Mc4r gene mutation replacing the phenylalanine (F) at position 51 with leucine (L) (MC4RF51L mice) were generated by CRISPR/Cas9 using MC4R guide RNA with sequence CCCGAGGTGTTTGTGACTCT; repair donor DNA sequence after cutting, CCCGAGGTGTTTGTGACTCT; repair donor DNA sequence after cutting, TACGGGCTGCACGGCAATGCCAGCGAGTCGCTGGGGAAGGGCCACCCGGACGGAGGATGCTATGAGCAACTTTTTGTTTCCCCCGAGGTGCTAGTGACTCTGGGTGTCATAAGCCTGTTGGAGAACATTCTAGTGATCGTGGCGATAGCCAAGAACAAGAACCTGCACTCACCCATGTACTTT; and spCas9 mRNA (System Biosciences). Mice were maintained on a C57BL/6J background and bred to homozygosity to generate MC4RF51L mice. Mice were housed on a 12-hour light/12-hour dark cycle (with light from 0600 hours to 1800 hours) and fed a standard chow diet (NIH-07, 5% fat by weight, Envigo).
Body composition, food intake, energy expenditure, and physical activity measurements.
Body weight was measured weekly starting at 4 weeks of age. Body composition was measured in nonanesthetized mice using the EchoMRI 3-in-1 NMR analyzer (Echo Medical Systems). Energy expenditure, food intake, and activity levels were measured in 3-month-old mice using a 12-chamber CLAMS/Oxymax system (Columbus Instruments) after a 48-hour acclimation period, with each chamber sampled every 13 minutes. TEE was determined over a 24-hour period at 22°C, followed by a 24-hour period at 30°C. Total and ambulatory activity was determined by infrared beam interruption (Opto-Varimex Mini; Columbus Instruments). Resting energy expenditure (REE) was determined as the means of points measured when mice were not ambulating. Feeding frequency was calculated based upon the number of 13-minute intervals in which mice were eating over a 24-hour period, while meal size was calculated by the amount of food eaten per 13-minute interval when mice were eating. For energy balance analysis, body weight, body composition, and food consumption were measured every 7 days in young mice starting at 4 weeks of age. Energy expenditure was calculated based on a previously validated formula (31) that includes coefficients for change in food intake, lean mass, and fat mass over a given period, all expressed in kcal.
Cannula implantation into the PVN.
Male MC4RF51L and WT mice at age 2–3 months underwent unilateral stereotaxic implantation of a guide cannula (26G, Plastics One) at a position above the PVN at bregma — anterioposterior –0.8 mm, mediolateral +0.3 mm, dorsoventral –3.8 mm — using a stereotaxic apparatus. A dummy cannula was placed above the guide cannula to prevent leaks and blockage. After mice recovered at 7–10 days after surgery, the dummy cannula was replaced by an internal cannula (30G) with a 1.2 mm projection; thus, the PVN injection position was at dorsoventral –4.9 to –5.0 mm. Surgery was performed under anesthesia with isoflurane, and after surgery mice received subcutaneous injections of banamine (2.2 mg/kg; MWI) to minimize postoperative discomfort.
CREB phosphorylation in the PVN.
Mice were injected with the MC3R/MC4R agonist MTII (10 mg/g i.p., 4039778; Bachem) or saline (100 μL) and 45 minutes later were anesthetized and perfused with cold 4% paraformaldehyde by intracardiac injection. Brain sections (40 mm) containing the PVN were confirmed under the microscope and collected and pretreated with heat-mediated antigen retrieval by incubation in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) at 95˚C for 30 minutes. The brain sections were then blocked in 2.5% horse serum (Vector Laboratories) plus 0.3% Triton X-100 at room temperature for 2 hours and incubated with anti-CREB (4820, Cell Signaling Technology), and their consecutive sections were incubated with anti-pCREB (Ser133, 9191; Cell Signaling Technology) in blocking solution overnight at 4˚C, followed by incubation with Alexa Fluor–conjugated secondary antibody (Alexa Fluor 555, A21428; MilliporeSigma). The sections were mounted with mounting medium with DAPI (Vector Laboratories). The fluorescent signals were captured and visualized with a fluorescence microscope (BZ-X800; Keyence). For quantification of pCREB and CREB signals, images were processed using haze reduction and black balance tools to ensure consistency between slides (2–4 sections/mouse). By use of the hybrid cell count and macro cell count tools, the integrated brightness was measured within a 200 μm diameter circle placed over the PVN on both sides of the third ventricle to ensure only signal from the PVN was counted.
Responses to MTII.
After stereotaxic cannula placement, mice were single-caged for 2 weeks. After 24 hours of fasting, mice received 200 nL PBS into the PVN unilaterally at 30 minutes prior to lights out, and food intake was measured over the first 3.5 hours after injection. After a 1-week recovery, the same procedure was followed, except mice received intra-PVN MTII (150 pmol/200 nL PBS). To examine the effect of FR900359 (formerly UBO-QIC) (42) on MTII-mediated inhibition of food intake, we pretreated mice with FR intra-PVN (150 pmol/150 nL) or vehicle 5 minutes prior to administration of MTII. For measurement of energy expenditure, mice were acclimated for 48 hours in an indirect calorimetric chamber. Nonanesthetized mice were injected with PBS (intra-PVN, 200 nL) prior to a 24-hour measurement period at 30°C, and then mice received 150 pmol/200 nL intra-PVN MTII after a 1-week recovery period. For food intake and energy expenditure responses to systemically delivered MTII, mice underwent the same procedures except receiving either i.p. PBS (200 μL) or MTII (200 μg for food intake; 10 μg/g for energy expenditure). To measure heart rate and blood pressure responses to intra-PVN MTII with or without FR, mice were acclimated to the BP-2000 specimen platform (Visitech) for 2 days prior to measurement. Blood pressure and heart rate were measured 5 minutes after PBS or drug administration following the same paradigm as described for food intake responses above.
Glucose tolerance test.
Overnight-fasted mice were given glucose (2 mg/g i.p.), and tail blood was collected before (time 0) and at the indicated times after glucose injection for blood glucose measurement using a Glucometer Contour (Bayer).
Cold tolerance test.
Rectal temperature was measured with a TH-5 rectal probe (Thermalet) inserted 1 cm deep. Before cold tolerance testing, mice were acclimated to experimental conditions at room temperature for a minimum of 48 hours, with daily measurement of rectal temperature. During cold tolerance tests, mice were housed singly without bedding, but provided food and water ad libitum, and exposed to 6°C conditions for 5 hours. Rectal temperature was measured before (time 0) and at the indicated times after exposure to 6°C conditions.
Biochemical assays.
ELISA kits were used for the measurement of serum insulin (Crystal Chem), leptin (R&D Systems), and adiponectin (ALPCO). Serum free fatty acids were measured using a kit from FUJIFILM (Healthcare Solutions), and triglyceride and cholesterol levels were measured using reagents from Thermo Fisher Scientific.
In vitro signaling assays
Generation of a stable MRAP2 cell line (HEK293MRAP2).
The HEK293MRAP2 cell line was generated by Acrogenic Technologies. In brief, HEK293 cells (ATCC) were cultured in DMEM (high glucose, ATCC) supplemented with 10% FBS and 1% antibiotic-antimycotic (Thermo Fisher Scientific, Gibco) and transfected with Myc-DDK–tagged human MRAP2-expressing plasmid (RC203668, OriGene) using Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, Invitrogen) according to the manufacturer’s instructions. At 48 hours after transfection, cells were cultured in a selection medium containing 500 μg/mL Geneticin (Gibco), which efficiently eliminated 90% of nontransfected parental cells within 72 hours and 100% within 96 hours. The transfected cells were cultured in the selection medium for 4 weeks, and when 90% confluency was reached cells were split at a subcultivation ratio of 1:3. The exogenous expression of DDK-Myc–tagged human MRAP2 in the stable cell pool was evaluated by immunoblotting membrane fractions with mouse anti-DDK (FLAG) monoclonal antibody (1:250; TA50011-100, OriGene) and anti–α-tubulin antibody (CP06, MilliporeSigma) to control for loading. MRAP2 expression was verified with qRT-PCR (forward primer: 5′-ATTTTCTCGCCAAGGCAACG-3′, reverse primer: 5′-TGCTTCTGATGGCTTCCTGG-3′) normalized by β-actin (Supplemental Figure 3). HEK293MRAP2 cells were maintained in DMEM supplemented with 10% FBS and 1 mg/mL Geneticin at 37°C in 5% CO2.
Cell culture.
Parental HEK293 and HEK293MRAP2 cells were seeded in 96-well plates (5 × 104 cells/100 μL per well) or 24-well plates (2.5 × 105 cells/500 μL per well) and cultured overnight. On the experiment days, cells were transiently transfected with human WT-MC4R plasmid (cDNA Resource Center, catalog MC4R0400000) or F51L-MC4R plasmid that was generated via site-directed mutagenesis (Quintara Biosciences). All transfections were performed using Lipofectamine 3000 in Opti-MEM I reduced serum medium, according to the manufacturer’s protocol. At 24 hours after transfection, parental HEK293 and HEK293MRAP2 cells were treated with different concentrations of α-MSH (4008476, Bachem), ranging from 10–8 to 10–4 M, diluted in serum-free, antibiotic-free DMEM. For stimulation of intracellular cAMP levels, cells were treated with α-MSH plus 100 μM 3-isobutyl-1-methylxanthine (IBMX; I5879, MilliporeSigma) for 2 hours at 37°C in 5% CO2, and cAMP was measured using the DetectX Direct Cyclic AMP (cAMP) Enzyme Immunoassay Kit (K019-H1, Arbor Assays). For stimulation of intracellular IP1 production, cells were treated with α-MSH at various doses in lithium chloride–containing buffer provided in the IP-One Gq ELISA Kit (72IP1PEA, PerkinElmer Cisbio) for 1 hour at 37°C in 5% CO2, and IP1 was measured using the same kit. For experiments in which FR was used, cells were pretreated with 1 μM FR or DMSO vehicle for 3 minutes and then treated with α-MSH. Each treatment was performed in triplicate and repeated with 3 independent experiments.
ERK phosphorylation.
For measurement of α-MSH–stimulated ERK1/2 phosphorylation, HEK293MRAP2 cells that were transfected with WT or mutant MC4RF51L plasmid and pretreated with FR or DMSO vehicle were treated with 10–5 M α-MSH for 3, 5, or 10 minutes at 37°C and then were lysed in RIPA buffer (20-188, MilliporeSigma) with a protease inhibitor cocktail (cOmplete Protease Inhibitor Cocktail, Roche). After measurement of protein concentrations using the Pierce BCA protein assay kit (23225, Thermo Fisher Scientific), cell extracts were prepared using NuPAGE sample reducing agent (NP0009, Invitrogen) and NuPAGE LDS sample buffer (NP0007, Invitrogen), heated at 70°C for 10 minutes, and run on NuPAGE 4-12% Bis-Tris gels (NP0335BOX, Invitrogen); proteins were transferred onto PVDF membranes with iBlot (IB24001, Invitrogen). Blots were probed with rabbit anti–phospho-ERK1/2 polyclonal antibody (1:1,000, 9101S, Cell Signaling Technology) or rabbit anti-ERK1/2 monoclonal antibody (1:1,000, 4695, Cell Signaling Technology) at 4°C overnight. Membranes were then incubated in HRP-linked secondary antibodies (NA934V, MilliporeSigma) and developed with ECL substrate. ERK1/2 signals were analyzed as a ratio of phospho-ERK1/2 to ERK1/2 with ImageJ software (NIH).
Statistics
Data are presented as mean ± SEM and were analyzed using Prism v7.00 (GraphPad). Statistical significance was determined using 2-tailed paired or unpaired Student’s t test or 1- or 2-way ANOVA, with correction for multiple comparisons when appropriate. Differences were considered significant at P < 0.05.
Study approval
All animal studies were approved by the NIDDK Animal Care and Use Committee.
Data availability
All data are available in the main text or the supplemental materials. Values for all data points in graphs are reported in the Supporting Data Values file.
Author contributions
PJM, AZ, BAC, MC, and LSW conceived the project and directed the study with input from all authors. PJM, AZ, BAC, MC, OG, EK, and LSW were involved in design of experiments. PJM, AZ, BAC, HS, YL, MTJ, ZC, DRL, MBG, NL, MC, and OG performed the experiments. PJM, AZ, BAC, MC, OG, and LSW were involved in data analysis. PJM, AZ, MC, and LSW wrote the manuscript, and all authors were involved in review and editing of the manuscript.
Supplementary Material
Acknowledgments
We would like to thank Huiyan Lu for assistance with making the mutant mouse line; Roger Cone for providing MC4RKO mice; and Yinyan Ma, Tamar Demby and Emily Shapland for technical assistance. This study was supported by the Intramural Research Program of the National Institute for Diabetes and Digestive and Kidney Diseases (NIDDK); the National Institutes of Health (NIH), Bethesda, Maryland, USA; and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) — 290847012/FOR2372 to EK.
Version 1. 01/04/2024
In-Press Preview
Version 2. 02/15/2024
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2024, Metzger et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2024;134(4):e165418. https://doi.org/10.1172/JCI165418.
Contributor Information
Peter J. Metzger, Email: pmetzger@princeton.edu.
Aileen Zhang, Email: aileen.zhang2000@gmail.com.
Bradley A. Carlson, Email: Bradley-carlson@northwestern.edu.
Hui Sun, Email: bluebird14325@yahoo.com.
Zhenzhong Cui, Email: cuiz@mail.nih.gov.
Yongqi Li, Email: yong-qi.li@nih.gov.
Marshal T. Jahnke, Email: mtjahnke@uiowa.edu.
Daniel R. Layton, Email: daniel.layton@nih.gov.
Meenakshi B. Gupta, Email: meenakshi.gupta@nih.gov.
Naili Liu, Email: naili.liu@nih.gov.
Evi Kostenis, Email: kostenis@uni-bonn.de.
Oksana Gavrilova, Email: oksanag@INTRA.NIDDK.nih.gov.
Min Chen, Email: MinC@niddk.nih.gov.
Lee S. Weinstein, Email: leew@mail.nih.gov.
References
- 1.Farooqi IS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348(12):1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 2.Tan K, et al. Functional characterization and structural modeling of obesity associated mutations in the melanocortin 4 receptor. Endocrinology. 2009;150(1):114–125. doi: 10.1210/en.2008-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenfield JR, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 4.Huszar D, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 5.Ste Marie L, et al. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci U S A. 2000;97(22):12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mountjoy KG, et al. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8(10):1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 7.Kishi T, et al. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457(3):213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 8.Balthasar N, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 9.Shah BP, et al. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc Natl Acad Sci U S A. 2014;111(36):13193–13198. doi: 10.1073/pnas.1407843111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garfield AS, et al. A neural basis for melanocortin-4 receptor-regulated appetite. Nat Neurosci. 2015;18(6):863–871. doi: 10.1038/nn.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YQ, et al. Gq/11α and Gsα mediate distinct physiological responses to central melanocortins. J Clin Invest. 2015;126(1):40–49. doi: 10.1172/JCI76348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holder JL, Jr, et al. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9(1):101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 13.Michaud JL, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10(14):1465–1473. doi: 10.1093/hmg/10.14.1465. [DOI] [PubMed] [Google Scholar]
- 14.Kublaoui BM, et al. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol. 2006;20(10):2483–2492. doi: 10.1210/me.2005-0483. [DOI] [PubMed] [Google Scholar]
- 15.Tolson KP, et al. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J Neurosci. 2010;30(10):3803–3812. doi: 10.1523/JNEUROSCI.5444-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long DN, et al. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Gαs in the development of human obesity. J Clin Endocrinol Metab. 2007;92(3):1073–1079. doi: 10.1210/jc.2006-1497. [DOI] [PubMed] [Google Scholar]
- 17.Chen M, et al. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci U S A. 2005;102(20):7386–7391. doi: 10.1073/pnas.0408268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen M, et al. Central nervous system imprinting of the G protein Gsα and its role in metabolic regulation. Cell Metab. 2009;9(6):548–555. doi: 10.1016/j.cmet.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsh DJ, et al. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21(1):119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, et al. Gsα deficiency in the dorsomedial hypothalamus underlies obesity associated with Gsα mutations. J Clin Invest. 2017;127(2):500–510. doi: 10.1172/JCI88622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, et al. Gsα deficiency in the paraventricular nucleus of the hypothalamus partially contributes to obesity associated with Gsα mutations. Endocrinology. 2012;153(9):4256–4265. doi: 10.1210/en.2012-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang Z, et al. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry. 2010;49(22):4583–4600. doi: 10.1021/bi100068u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao YX, Segaloff DL. Functional analyses of melanocortin-4 receptor mutations identified from patients with binge eating disorder and nonobese or obese subjects. J Clin Endocrinol Metab. 2005;90(10):5632–5638. doi: 10.1210/jc.2005-0519. [DOI] [PubMed] [Google Scholar]
- 24.Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–543. doi: 10.1210/er.2009-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lotta LA, et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell. 2019;177(3):597–607. doi: 10.1016/j.cell.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouwers B, et al. Human MC4R variants affect endocytosis, trafficking and dimerization revealing multiple cellular mechanisms involved in weight regulation. Cell Rep. 2021;34(12):108862. doi: 10.1016/j.celrep.2021.108862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghamari-Langroudi M, et al. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015;520(7545):94–98. doi: 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He S, Tao YX. Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene. Int J Biol Sci. 2014;10(10):1128–1137. doi: 10.7150/ijbs.10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniels D, et al. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003;986(1–2):1–11. doi: 10.1016/S0006-8993(03)03162-7. [DOI] [PubMed] [Google Scholar]
- 30.Clement K, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24(5):551–555. doi: 10.1038/s41591-018-0015-9. [DOI] [PubMed] [Google Scholar]
- 31.Ravussin Y, et al. Estimating energy expenditure in mice using an energy balance technique. Int J Obes (Lond) 2013;37(3):399–403. doi: 10.1038/ijo.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adan RA, et al. The MC4 receptor and control of appetite. Br J Pharmacol. 2006;149(7):815–827. doi: 10.1038/sj.bjp.0706929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atalayer D, et al. Food demand and meal size in mice with single or combined disruption of melanocortin type 3 and 4 receptors. Am J Physiol Regul Integr Comp Physiol. 2010;298(6):R1667–R1674. doi: 10.1152/ajpregu.00562.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richard CD, et al. Meal pattern analysis in neural-specific proopiomelanocortin-deficient mice. Eur J Pharmacol. 2011;660(1):131–138. doi: 10.1016/j.ejphar.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan W, et al. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141(9):3072–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- 36.Obici S, et al. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108(7):1079–1085. doi: 10.1172/JCI200112954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nogueiras R, et al. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest. 2007;117(11):3475–3488. doi: 10.1172/JCI31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asai M, et al. Loss of function of the melanocortin 2 receptor accessory protein 2 is associated with mammalian obesity. Science. 2013;341(6143):275–278. doi: 10.1126/science.1233000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novoselova TV, et al. Loss of Mrap2 is associated with Sim1 deficiency and increased circulating cholesterol. J Endocrinol. 2016;230(1):13–26. doi: 10.1530/JOE-16-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang J, et al. Pharmacological effect of human melanocortin-2 receptor accessory protein 2 variants on hypothalamic melanocortin receptors. Endocrine. 2018;61(1):94–104. doi: 10.1007/s12020-018-1596-2. [DOI] [PubMed] [Google Scholar]
- 41.Berruien NNA, Smith CL. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene. 2020;757:144949. doi: 10.1016/j.gene.2020.144949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patt J, et al. An experimental strategy to probe Gq contribution to signal transduction in living cells. J Biol Chem. 2021;296:100472. doi: 10.1016/j.jbc.2021.100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inamdar V, et al. Characterization of UBO-QIC as a Gαq inhibitor in platelets. Platelets. 2015;26(8):771–778. doi: 10.3109/09537104.2014.998993. [DOI] [PubMed] [Google Scholar]
- 44.Gao ZG, Jacobson KA. On the selectivity of the Gαq inhibitor UBO-QIC: a comparison with the Gαi inhibitor pertussis toxin. Biochem Pharmacol. 2016;107:59–66. doi: 10.1016/j.bcp.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kukkonen JP. G-protein inhibition profile of the reported Gq/11 inhibitor UBO-QIC. Biochem Biophys Res Commun. 2016;469(1):101–107. doi: 10.1016/j.bbrc.2015.11.078. [DOI] [PubMed] [Google Scholar]
- 46.Fan W, et al. Thermogenesis activated by central melanocortin signaling is dependent on neurons in the rostral raphe pallidus (rRPa) area. Brain Res. 2007;1179:61–69. doi: 10.1016/j.brainres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 47. Zhang SX, et al. Competition between stochastic neuropeptide signals calibrates the rate of satiation [preprint]. Posted on bioRxiv July 12, 2023. [DOI]
- 48.Wang Y, et al. Melanocortin 4 receptor signals at the neuronal primary cilium to control food intake and body weight. J Clin Invest. 2021;131(9):142064. doi: 10.1172/JCI142064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinney A, et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88(9):4258–4267. doi: 10.1210/jc.2003-030233. [DOI] [PubMed] [Google Scholar]
- 50.Young EH, et al. The V103I polymorphism of the MC4R gene and obesity: population based studies and meta-analysis of 29 563 individuals. Int J Obes (Lond) 2007;31(9):1437–1441. doi: 10.1038/sj.ijo.0803609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heid IM, et al. Association of the 103I MC4R allele with decreased body mass in 7937 participants of two population based surveys. J Med Genet. 2005;42(4):e21. doi: 10.1136/jmg.2004.027011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geller F, et al. Melanocortin-4 receptor gene variant I103 is negatively associated with obesity. Am J Hum Genet. 2004;74(3):572–581. doi: 10.1086/382490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paisdzior S, et al. Differential signaling profiles of MC4R mutations with three different ligands. Int J Mol Sci. 2020;21(4):1224. doi: 10.3390/ijms21041224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li XF, Lytton J. An essential role for the K+-dependent Na+/Ca2+-exchanger, NCKX4, in melanocortin-4-receptor-dependent satiety. J Biol Chem. 2014;289(37):25445–25459. doi: 10.1074/jbc.M114.564450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson EA, et al. Gqα/G11α deficiency in dorsomedial hypothalamus leads to obesity resulting from decreased energy expenditure and impaired sympathetic nerve activity. Am J Physiol Endocrinol Metab. 2021;320(2):E270–E280. doi: 10.1152/ajpendo.00059.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Habara M, et al. Molecular characterization of feline melanocortin 4 receptor and melanocortin 2 receptor accessory protein 2. Gen Comp Endocrinol. 2018;261:31–39. doi: 10.1016/j.ygcen.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 57.Anderson EJP, et al. Late onset obesity in mice with targeted deletion of potassium inward rectifier Kir7.1 from cells expressing the melanocortin-4 receptor. J Neuroendocrinol. 2019;31(1):e12670. doi: 10.1111/jne.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li SJ, et al. Melanocortin antagonists define two distinct pathways of cardiovascular control by alpha- and gamma-melanocyte-stimulating hormones. J Neurosci. 1996;16(16):5182–5188. doi: 10.1523/JNEUROSCI.16-16-05182.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng H, et al. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. Am J Physiol Regul Integr Comp Physiol. 2005;289(1):R247–R258. doi: 10.1152/ajpregu.00869.2004. [DOI] [PubMed] [Google Scholar]
- 60.Williams DL, et al. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology. 2000;141(4):1332–1337. doi: 10.1210/endo.141.4.7410. [DOI] [PubMed] [Google Scholar]
- 61.Li P, et al. Melanocortin 3/4 receptors in paraventricular nucleus modulate sympathetic outflow and blood pressure. Exp Physiol. 2013;98(2):435–443. doi: 10.1113/expphysiol.2012.067256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplemental materials. Values for all data points in graphs are reported in the Supporting Data Values file.