

Abstract
Realizing the promise of prime editing for the study and treatment of genetic disorders requires efficient methods for delivering prime editors (PEs) in vivo. Here we describe the identification of bottlenecks limiting adeno-associated virus (AAV)-mediated prime editing in vivo and the development of AAV-PE vectors with increased PE expression, prime editing guide RNA stability and modulation of DNA repair. The resulting dual-AAV systems, v1em and v3em PE-AAV, enable therapeutically relevant prime editing in mouse brain (up to 42% efficiency in cortex), liver (up to 46%) and heart (up to 11%). We apply these systems to install putative protective mutations in vivo for Alzheimer’s disease in astrocytes and for coronary artery disease in hepatocytes. In vivo prime editing with v3em PE-AAV caused no detectable off-target effects or significant changes in liver enzymes or histology. Optimized PE-AAV systems support the highest unenriched levels of in vivo prime editing reported to date, facilitating the study and potential treatment of diseases with a genetic component.
Subject terms: Genetic engineering, Cell delivery, Targeted gene repair
Prime editors are efficiently delivered to mouse organs with AAV vectors.
Main
The ability to precisely correct pathogenic mutations or install protective ones in living systems has enormous therapeutic potential1. Prime editors (PEs) are precision gene editing agents that can perform virtually any substitution, small deletion and small insertion at target DNA sites in living cells2. PEs do not rely on double-strand breaks, minimizing unwanted outcomes associated with nucleases such as uncontrolled insertions and deletions (indels)3–5, large deletions6,7, chromosomal abnormalities8–13 or induction of the p53 DNA damage response14. PEs comprise a programmable nickase fused to an engineered reverse transcriptase (RT). A prime editing guide RNA (pegRNA) contains a spacer that guides the PE to the target DNA site as well as a 3′ extension that encodes the desired edit. PEs reverse transcribe the pegRNA extension directly into the genome using the nicked target DNA strand as a primer, leading to permanent changes in genomic DNA with relatively few byproducts2. PEs do not require a donor DNA template and have been used widely across many mammalian cell systems, including mitotic and post-mitotic cells3,15–21.
Before PEs can be translated into clinical settings, safe and efficient delivery methods capable of targeting therapeutically relevant tissue types are needed. Adeno-associated viruses (AAVs) are clinically validated and FDA approved for in vivo gene therapy and gene editing applications22,23. Although not without potential risks24–27, AAV remains one of the few effective and clinically validated in vivo delivery vectors for a variety of non-liver organs and tissue types28,29. At ~6.3 kilobases (kb) of encoded DNA sequence, however, PEs are currently too large to fit within the ~4.7-kb cargo size limit of AAV30,31.
Our laboratory and others have overcome the packaging capacity of AAV to accommodate large genome editing agents by splitting them into two halves, each fused to a fast-splicing intein32–34. Co-transduction of both AAVs reconstitutes the editing agent via association, or association and splicing, of the split intein-fused proteins. Existing in vivo PE delivery strategies with intein-split AAV or hydrodynamic DNA injection have thus far yielded modest in vivo editing efficiencies in postnatal animals in contexts without a survival advantage of edited cells, achieving maximum efficiencies corresponding to 1.7–13.5% editing of bulk liver or retina15–21. A recent report35 demonstrates efficient in vivo prime editing in enriched degenerating retinitis pigmentosa mouse retinas with dual-AAV delivery of PE-SpRY, but this system may not be applicable to uses that do not confer a survival advantage among edited cells. Adenoviral transduction has yielded the most efficient generalizable in vivo prime editing reported to date16 (up to 58% editing in isolated hepatocytes, corresponding to ~35–40% editing of bulk liver36) in neonatal animals at the highest dose but may be challenging to apply clinically due to the immunogenicity and toxicity of adenovirus37. The efficient in vivo delivery of PEs into therapeutically relevant cells using AAV, therefore, remains a major bottleneck to the use of prime editing for animal research and therapeutic applications.
Here we report the development, optimization and application of intein-split PE-AAVs that mediate efficient in vivo prime editing in multiple mouse organs. We systematically optimized the PE protein, pegRNA and AAV genomic elements, resulting in optimized ‘v3em PE-AAV’ delivery strategies that enabled therapeutically relevant levels of prime editing in mouse brain (42%), liver (46%) and heart (11%), representing, to our knowledge, the first prime editing in postnatal brain and heart and substantially higher AAV-mediated in vivo prime editing efficiencies than have been previously reported in the liver. We applied v3em PE3-AAV to install mutations of biomedical interest in mice that are not currently accessible with other in vivo genome editing technologies, including the rare Apolipoprotein E Christchurch (APOE3 R136S) variant that may alter Alzheimer’s disease (AD) risk and the dominant variant of proprotein convertase subtilisin/kexin type 9 (PCSK9) Q152H (mouse Pcsk9 Q155H) that is associated with a reduction in low-density lipoprotein (LDL) cholesterol levels and protection from coronary artery disease. Our results advance the potential of prime editing for basic research and therapeutic applications and establish optimized PE-AAV systems as an effective in vivo PE delivery method.
Results
Design and evaluation of split PE architectures
To deliver PE via AAV, we split the coding sequence of PE into two halves, one containing the N-terminus of the PE and the other containing the C-terminus, each fused to the N-terminal or C-terminal intein from Nostoc punctiforme32–34,38, respectively. We nominated positions 844 and 1024 as putative split sites that we hypothesized would be tolerant of structural modification (Supplementary Note 1) and would allow splitting of the PE into two halves, each of which could fit within the packaging limit of AAV while accommodating space for promoters, terminators, a pegRNA cassette and a single guide RNA (sgRNA) cassette. To maximize splicing efficiency, we also assessed mutation of the three N-terminal amino acids of the C-terminal extein from the native residues of Cas9 to the native Npu DnaB Cys-Phe-Asn sequence or to intermediate sequence variants. We transfected plasmids encoding the two candidate halves or full-length PE2 along with pegRNA and nicking sgRNA into HEK293T cells and measured editing efficiencies across three sites by high-throughput sequencing (HTS) (Fig. 1a). Among the eight split designs tested, average prime editing efficiencies ranged from 37% to 96% of full-length PE2 activity. The 1024-CFN and 844-CFN split designs yielded robust editing efficiencies (Fig. 1a and Extended Data Fig. 1), consistent with other reports showing that the 1024-NpuN split site reconstitutes prime editing activity18,21. Split designs were dependent on the presence and catalytic activity of the intein (Supplementary Note 1 and Extended Data Fig. 1).
Fig. 1. Initial development of a split PE in tissue culture and for AAV-mediated in vivo prime editing.
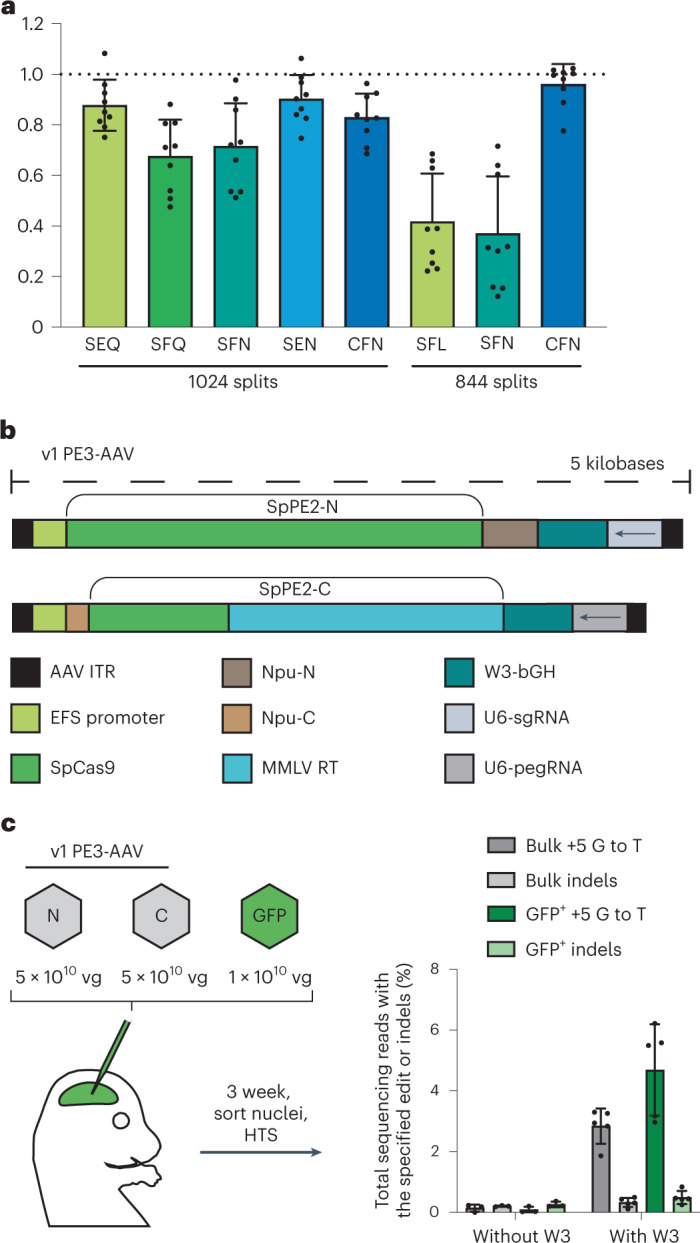
a, Editing performance at three genomic loci of intein-split PE3 variants normalized to that of unsplit, canonical PE3 when delivered by plasmid transfection in HEK293T cells. The split site in SpCas9 and the identity of the three most N-terminal residues of the C-terminal extein are indicated below each bar. A suboptimal amount of editor plasmid was used to avoid saturating editing efficiencies. Dots represent values normalized to canonical PE3 activity, and error bars represent mean ± s.e.m. of n = 3 biological replicates at three genomic loci. b, Schematic of v1 PE3-AAV architecture. c, In vivo editing activity of v1 PE3-AAV9 with pegRNA encoding the Dnmt1 +5 G-to-T edit delivered to neonatal C57BL/6 pups by P0 ICV injection at a dose of 1 × 1011 vg total (5 × 1010 vg per half). Cortex (neocortex and hippocampus) was harvested; nuclei were isolated and sorted by FACS into bulk and GFP+ populations; and genomic DNA was harvested and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3–5 mice; each condition includes both male and female mice.
Extended Data Fig. 1. Dependence on splicing of split-intein PE2 activity.

Full-length or intein-split PE2 were transfected into HEK239T cells with epegRNA and nicking sgRNA then analyzed by HTS three days after transfection. Splicing-inactive inteins contained a Cys1→Ala mutation in NpuN. A sub-optimal amount of editor plasmid was used to avoid saturating editing efficiencies. Two sites per condition are shown (HEK3 27bp insertion and RNF2 +1 C to A). Dots represent values and error bars represent mean±SEM of n = 3 biological replicates at two genomic loci.
Initial in vivo prime editing with intein-split dual PE-AAV
We chose the 1024-CFN PE split in our initial PE-AAV architecture because this split allows packaging of the PE gene across two AAV genomes, with a U6 promoter-driven pegRNA and nicking sgRNA cassettes for PE3 applications (Fig. 1b). To remain within the ~4.7-kb packaging capacity of AAV30,31, we used the small constitutive EFS promoter with a bGH polyadenylation signal39. To assess whether mRNA transcript-stabilizing cis-elements on the AAV genome would increase PE efficiency, we tested the effect of including W3, the minimized gamma portion of the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE), to the 3′ untranslated region (UTR) of the PE gene40. We tested the resulting candidate PE-AAV constructs for their ability to install a +5 G-to-T transversion in the endogenous genomic mouse Dnmt1 locus that was previously validated in cultured primary neurons2 and that is unlikely to cause a phenotypic impact in edited cells32,41.
To assess initial in vivo prime editing efficiency, we administered PE-AAVs at a total dose of 1 × 1011 viral genomes (vg) (5 × 1010 vg each of the N-terminal and C-terminal PE-AAVs) packaged in AAV9 to C57BL/6 mice on postnatal day 0 (P0) via intracerebroventricular (ICV) injection, a method of direct injection into the cerebrospinal fluid that bypasses the blood–brain barrier. To enable the analysis of transduced cells, in addition to bulk tissue, we co-injected a 1 × 1010 vg AAV9 expressing EGFP fused to a nuclear membrane-localized Klarsicht/ANC-1/Syne-1 homology (KASH) domain32,42 (Fig. 1c). Three weeks after injection, we harvested the cortex from injected and untreated control mice, isolated both FACS-sorted bulk and GFP+ nuclei, extracted genomic DNA and analyzed Dnmt1 editing by HTS. We observed 2.8% and 4.7% editing in bulk and GFP+ nuclei, respectively, in brains treated with PE-AAV containing the W3 (Fig. 1c). In mice treated with PE-AAV without W3 (bGH only), editing was very inefficient (≤0.2%; Fig. 1c). These results establish that prime editing is feasible in the mammalian brain when delivered via AAV and that addition of W3 is essential in this context to achieve even low levels of prime editing in the brain. The EFS-driven dual-AAV system with W3 (Fig. 1b) is hereafter referred to as ‘v1 PE3-AAV’.
Impact of DNA repair on in vivo prime editing efficiency
We recently discovered that DNA mismatch repair (MMR) impedes prime editing efficiency and product purity in cultured cells43,44. We sought to evaluate whether the identity of the installed edit and its susceptibility to MMR may limit prime editing in vivo. We characterized a diversity of edits at the Dnmt1 locus in mouse N2a cells (Supplementary Note 2) and found that these new edits exhibited higher editing efficiency than the Dnmt1 +5 G-to-T edit (Extended Data Fig. 2a) and were recognized to different degrees by MMR (Extended Data Fig. 2b). To assess whether MMR-recognized and MMR-evading edits show similar trends in prime editing efficiency in vivo, we performed ICV injections at P0 of 1 × 1011 vg of v1 PE3-AAV9 encoding +1 C-to-G, +1 CCC insertion or +2 G-to-C edits. We observed 6.1%, 25% and 42% prime editing for +1 C-to-G, +1 CCC insertion and +2 G-to-C edits, respectively (Fig. 2b), consistent with their relative MMR evasion and efficiencies in N2a cells (Fig. 2a and Extended Data Fig. 2). Although the activity of MMR in the mammalian brain is not well understood, Mlh1 is expressed in the mammalian brain45–47 and may limit prime editing in the central nervous system (CNS). These data suggest that MMR or related pathways likely also impede prime editing in vivo and that designing edits to natively evade MMR by adding nearby benign or silent bystander mutations43 may enhance prime editing efficiency in the mammalian brain.
Extended Data Fig. 2. Efficiencies of various prime edits at Dnmt1 and the effect of transient MMR inhibition.

a, Mouse N2A cells were transfected with plasmids encoding PE2, a +53 nicking sgRNA, and one pegRNA encoding the edit indicated below each bar. b, The impact of MMR recognition of the installed edit on editing efficiency was assessed by installing one of four edits (Dnmt1 +5 G-to-T, Dnmt1 +1 CCC insertion, Dnmt1 +1 C-to-G, or Dnmt1 +2 G-to-C) as PE2, PE3, PE4 (PE2 + MLH1dn), or PE5 (PE3 + MLH1dn). Data are shown as individual data points and mean±SEM for n = 2-3 independent biological replicates.
Fig. 2. AAV-mediated in vivo prime editing efficiency is dependent on the type of edit.
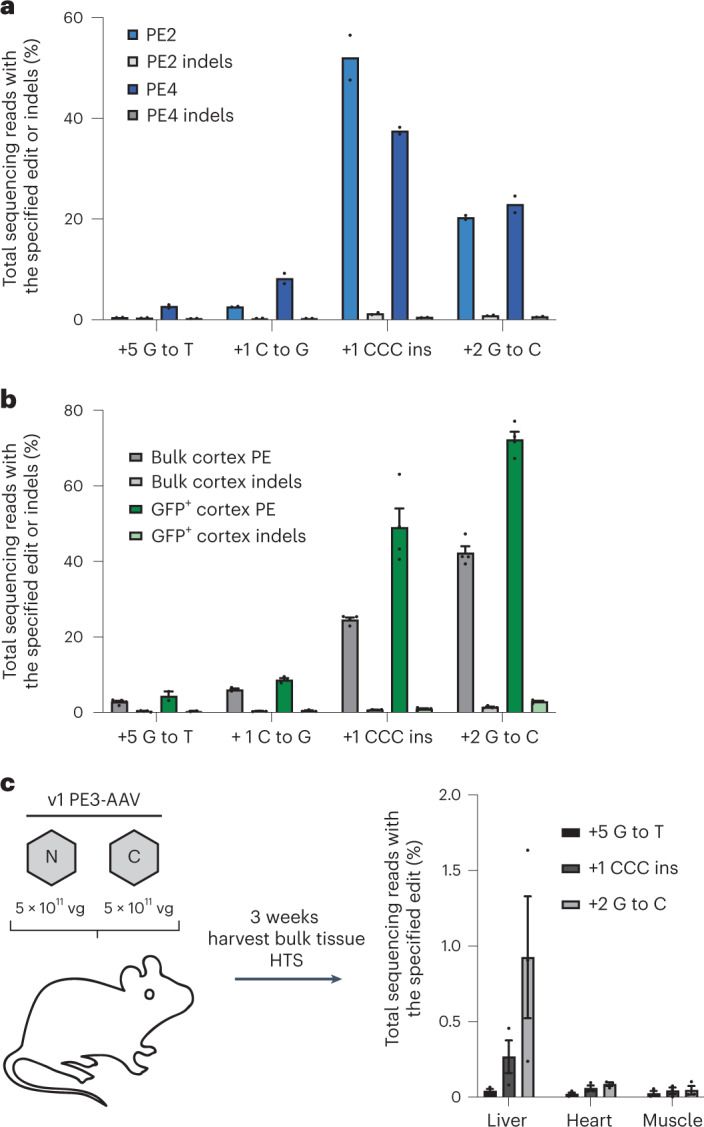
a, Editing activity of PE2 and PE4 in N2a cells by plasmid transfection for four different edits at Dnmt1. N2a cells were transfected with either PE2 and pegRNA or PE4 (PE2 + MLH1dn) and pegRNA. Three days later, gDNA was harvested and analyzed by HTS. Dots represent values, and bars represent means of n = 2 biological replicates. b, In vivo editing activity of v1 PE3-AAV9 delivered to neonatal C57BL/6 pups on P0 by ICV at a total dose of 1 × 1011 vg (5 × 1010 vg per half). Cortex (neocortex and hippocampus) was harvested, nuclei were isolated and sorted by FACS into bulk and GFP+ populations, and gDNA was analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3–4 mice; each condition includes both male and female mice. c, In vivo editing activity of PE3 delivered via v1 PE3-AAV9 by RO injection to 6–8-week-old C57BL/6 mice at a total dose of 1 × 1012 vg. Three weeks after injection, bulk tissues were harvested and gDNA was isolated and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3 mice; each condition includes both male and female mice.
Development of a PE-AAV system for systemic administration
To further explore the utility of split-intein PE-AAVs in vivo, we investigated their ability to mediate prime editing in adult animals via systemic injection. We delivered the v1 PE3-AAV9 encoding the Dnmt1 +5 G-to-T, +1 CCC insertion or +2 G-to-C edit at a dose of 1 × 1012 vg (5 × 1011 vg each of the N-terminal and C-terminal AAV) via retro-orbital (RO) injections into 6–8-week-old adult C57BL/6 mice. We harvested liver, heart, and skeletal muscle (tibialis anterior) 3 weeks after injection and analyzed editing by HTS. The +5 G-to-T edit was undetectable (≤0.1%), but we observed low prime editing in the liver for +1 CCC insertion (0.3%) and +2 G-to-C edits (0.9%), respectively (Fig. 2c). No prime editing was observed (≤0.1%) in the heart and skeletal muscle for any of the three edits (Fig. 2c). These results revealed that the PE-AAV architecture required further optimization for systemic injection.
Factors limiting systemic in vivo PE efficiency
To understand the factors limiting systemic in vivo prime editing, we first assessed the effect of time after injection on prime editing efficiencies. We administered v1 PE3-AAV9 with pegRNA encoding the Dnmt1 +2 G-to-C edit to C57BL/6 mice by RO injection and harvested tissues 3 weeks or 6 weeks after injection. We did not observe a significant increase in PE activity with increased AAV expression time, with editing for both conditions yielding ≤1.1%, indicating that time was not the main factor limiting prime editing efficiency (Extended Data Fig. 3).
Extended Data Fig. 3. Effect of incubation time on systemic in vivo prime editing efficiency.
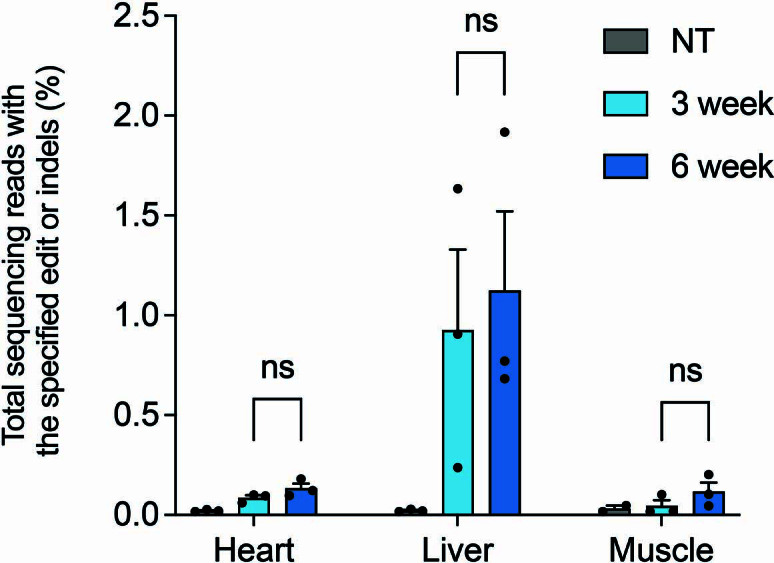
v1 PE-AAV9 encoding the Dnmt1 +2 G-to-C edit was administered at a dose of 1x1012 vg total (5x1011 vg per half) by RO injection to 6- to 8-week-old C57BL/6 mice and three weeks after, bulk tissues were harvested and isolated genomic DNA was analyzed by HTS. Dots represent individual mice and error bars represent mean±SEM of n = 3 different mice. Significance was calculated by two-tailed unpaired t-test with correction for multiple comparisons.
To assess whether the use of 3′ stabilized engineered pegRNAs (epegRNAs)48 augment prime editing in vivo, we delivered 1 × 1012 vg (5 × 1011 vg each half) of v1 PE3-AAV9 with either pegRNA or epegRNA (tevopreQ1 fused on the 3′ end of the pegRNA with an 8-nucleotide (nt) linker) via systemic RO injection. The use of an epegRNA resulted in an average of 1.7% +2 G-to-C prime editing of Dnmt1 in the liver, compared to 0.9% prime editing with unmodified pegRNAs (P = 0.20) (Fig. 3a). When assessed in the brain by P0 ICV administration of 1 × 1011 vg of v1 PE3-AAV9, epegRNAs more strongly increased Dnmt1 +1 C-to-G editing efficiencies over pegRNAs from 6.1% to 21% prime editing in bulk cortex and 8.6% to 38% prime editing in GFP+ cortex (P = 0.10 for both) (Fig. 3b). Although these findings also suggest that pegRNA stability is not the sole factor limiting editing by systemically administered PEs, we used epegRNAs for all subsequent designs and hereafter indicate use of an epegRNA with an ‘e’ (v1e PE3-AAV).
Fig. 3. Evaluation of factors limiting systemic prime editing efficiency.

a, Comparison of unmodified pegRNA or epegRNA installing a +2 G-to-C edit in bulk liver. v1 PE3-AAV9 was delivered by RO injection to 6–8-week-old C57BL/6 mice at a dose of 1 × 1012 vg (5 × 1011 vg per N-terminal and C-terminal AAVs). Bulk tissue was harvested 3 weeks after injection, and gDNA was isolated and analyzed by HTS. Significance was calculated by unpaired t-test. b, Comparison of unmodified pegRNA or epegRNA installing a +1 C-to-G edit in cortical tissue. v1 PE3-AAV9 was delivered via P0 ICV injection at a total dose of 1 × 1011 vg. Cortex was harvested 3 weeks after injection, nuclei were isolated, and bulk and GFP+ populations were isolated by FACS. gDNA was extracted and analyzed by HTS. c, Schematic of v2 PE3-AAV. d, C57BL/6 mice were RO injected with 1.25 × 1012 vg total of either v2 PE3-AAV9 (5 × 1011 vg each N-terminal and C-terminal AAVs encoding PEmax plus 2.5 × 1011 vg epegRNA/sgRNA AAV encoding the Dnmt1 +2 G-to-C edit) or architecture-matched v2 Cas9 nuclease AAV9 (5 × 1011 vg each N-terminal and C-terminal AAVs encoding Cas9 nuclease plus 2.5 × 1011 vg sgRNA AAV). Bulk liver, heart, and muscle tissues were harvested 3 weeks after injection, and gDNA was analyzed by HTS. e, Comparison of PE3 and PE3max by v2 AAV9 PE at Dnmt1 installing the +1 C-to-G edit. For a, b, d and e, dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3 mice; each condition includes both male and female mice.
Next, we considered whether insufficient PE protein production driven by the EFS promoter may be a major limiting factor for efficient systemic in vivo prime editing. To improve PE expression, we tested the Cbh promoter, a strong and ubiquitous promoter that mediates efficient AAV-mediated base editing in mice when delivered systemically32,49. To accommodate the 0.7-kb Cbh promoter, we moved the pegRNA and sgRNA cassettes to a third AAV vector containing a human U6 promoter that drives epegRNA expression and a mouse U6 promoter that drives nicking sgRNA expression to avoid long stretches of homology on the AAV genome that can lead to recombination50 (Fig. 3c). We designated this triple-AAV system ‘v2e PE3-AAV’.
We co-delivered all three v2e PE3-AAV9 PE components at a 1:1:0.5 ratio (5 × 1011 vg each of the N-terminal and C-terminal PE-AAVs and 2.5 × 1011 vg epegRNA/sgRNA AAV) to 6–8-week-old C57BL/6 mice via systemic RO injection and evaluated prime editing of Dnmt1 +2 G-to-C edit (Fig. 3d). This strategy yielded 48% prime editing in the bulk liver and 7.2% prime editing in the bulk heart, 27- and 70-fold higher editing compared to v1e PE3-AAV9 that uses the EFS promoter (Fig. 3d). Prime editing in heart was 7.2%, and prime editing in muscle was less than 1%, whereas Cas9 delivered at the same dose yielded 18% and 8.7% indels in heart and muscle, respectively (Fig. 3d), suggesting that cell-type-specific factors or insufficient PE expression may still limit prime editing in some tissues, which can potentially be overcome by increasing AAV dose or by changing delivery route or timing of injection51. These data suggest that PE expression level is a major determinant of in vivo prime editing efficiency, consistent with a report of prime editing in the retina18.
To further improve efficiency, we tested architectural improvements from PEmax43, including codon optimization of MMLV RT for improved expression, mutations in Cas9 for enhanced nickase activity and optimized nuclear localization signals (NLS) into the v2e PE3-AAV design, resulting in v2em PE3-AAV. We delivered all three v2e or v2em PE3-AAV9 vectors in a 1:1:0.5 ratio (5 × 1011 vg each of the N-terminal and C-terminal PE-AAVs and 2.5 × 1011 vg epegRNA/sgRNA AAV) to 6–8-week-old C57BL/6 mice via systemic RO injection and evaluated the efficiency of the Dnmt1 +1 C-to-G edit. These improvements from PEmax resulted in a similar level of editing efficiency in the well-edited liver but 2.4-fold higher prime editing efficiency in the heart (P = 0.017; Fig. 3e), suggesting that the PEmax improvements improve in vivo editing in tissues that do not reach high editing levels with older PE architectures. In the brain, incorporation of epegRNA and PEmax together resulted in 41% prime editing, corresponding to a 6.8-fold increase in prime editing in bulk cortex compared to PE3 with pegRNA (Extended Data Fig. 4; P < 0.0001). We used the PEmax architecture for all subsequent experiments.
Extended Data Fig. 4. Incorporation of epegRNAs and PEmax together substantially enhance prime editing in CNS.

a-b, PE3-AAVs encoding a Dnmt1 +1 C-to-G edit were delivered via P0 ICV injection as v1, v1e, or v1em architecture at a dose of 1x1011 vg (5x1010 vg each N- and C-terminal AAVs) with 1x1010 vg promoter-matched AAV9 EGFP:KASH. At three weeks, nuclei from cortex were isolated and sorted by FACS to either all nuclei (bulk) or GFP+ populations. Genomic DNA was extracted and analyzed by HTS. While improvement from incorporation of epegRNA alone did not reach statistical significance (P=0.17), incorporation of epegRNA and PEmax (v1em) yielded highly statistically significant improvement in prime editing over v1 (for both bulk and GFP+ populations, P<0.0001). Significance was calculated by two-tailed unpaired t-test with correction for multiple comparisons. Dots represent individual data points and error bars represent mean±SEM for n = 3 different mice.
The observed liver editing efficiencies, which suggest prime editing in the majority of hepatocytes, would likely be relevant for the study or potential treatment of a variety of genetic diseases in the liver16,52,53 and are similar to bulk liver editing efficiencies achieved with optimized AAV-delivered base editors and Cas9 nuclease systems32,33,39. These data also represent the highest AAV-mediated in vivo liver prime editing efficiencies reported thus far, to our knowledge15–21.
Highly active dual-AAV PE for systemic editing in adult mice
We next sought to design a dual PE3-AAV system to simplify delivery and reduce the total AAV dose while preserving the improvements that enhanced in vivo prime editing efficiency of the triple-AAV v2em PE3 system. To reduce the size of the prime editing system to fit into two AAV, we considered using smaller Cas variants, such as SaCas9 (Supplementary Note 3 and Extended Data Fig. 5a), but chose to focus on SpCas9-based PEs owing to their more robust editing activity. To minimize the size of SpCas9-based PE, we assessed truncated MMLV RT variants that lack the RNaseH domain (Supplementary Note 3 and Extended Data Fig. 5b). To directly assess whether PE ΔRNaseH maintains its full-length activity in vivo after systemic injection, we used the v2em PE3-AAV system to compare full-length PE programmed to edit Dnmt1 +1 C-to-G with two variants of RNaseH-deleted PEs. The first ΔRNaseH variant uses a previously published truncation at residue 497 of MMLV RT that appends six additional amino acids54. The second RT ΔRNaseH variant is a clean truncation at residue 497 of MMLV RT without additional amino acids. All three PEmax variants (PEmax, PEmax ΔRNaseH+6AA, and PEmax ΔRNaseH) delivered systemically as v2em PE3-AAVs injected at 1.25 × 1012 vg total with AAV9 (5 × 1011 vg each of the N-terminal and C-terminal PE-AAVs plus 2.5 × 1011 vg epegRNA/sgRNA AAV) into 6–8-week-old mice were similarly active in the liver and heart (Fig. 4a), indicating that PE3max ΔRNaseH maintains full-length prime editing efficiency in vivo at the target site assessed. Similarly, when PEmax or PEmax ΔRNaseH v2em PE3-AAVs were injected by P0 ICV injection at 5 × 1010 vg total with AAV9 (2.5 × 1010 vg each N-terminal and C-terminal PE3-AAV plus 1.1 × 1010 vg epegRNA/sgRNA AAV), PEmax ΔRNaseH performed similarly to PEmax (Fig. 4b), indicating that the RNaseH domain is also not necessary for prime editing in the brain.
Extended Data Fig. 5. Efforts to minimize the size of the prime editor protein.

Activity of full-length and intein-split SaPE2 by plasmid transfection in HEK293T cells with a pegRNA encoding the HEK3 +6 G-to-T edit. An intein split at previously validated position 7401 was assessed to maintain on-target editing efficiency compared to full-length (SaPE2). The three N-terminal residues of the C-terminal extein are indicated, with ‘SMP’ being the native residues to SaCas9, and CFN as the consensus residues of Npu intein. b. PE ∆RNaseH maintains prime editing activity in cultured cells across a variety of edits. HEK293T cells were transfected with plasmids encoding PE2 or PE2 ∆RNaseH and pegRNA (PE2) or pegRNA and nicking sgRNA (PE3). Genomic DNA was harvested after three days and analyzed by HTS. Dots represent individual data points and error bars represent mean±SEM for n = 3 independent biological replicates.
Fig. 4. Development of a dual-AAV system for efficient in vivo prime editing.

The RNaseH domain of PE3 is not essential in the liver, heart, or brain. a, C57BL/6 mice were RO injected with 1.25 × 1012 vg total of v2em PE3-AAV9 (5 × 1011 vg each N-terminal and C-terminal AAVs plus 2.5 × 1011 vg epegRNA/sgRNA AAV) encoding PE3max with its native full-length RT or PE3max with one of two truncated ΔRNaseH RTs to install a Dnmt1 +1 C-to-G edit. Three weeks after injection, liver, heart, and muscle tissues were harvested and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 1–3 mice; each condition includes both male and female mice. b, C57BL/6 pups were injected on P0 by ICV injection of a total of 5.7 × 1010 vg total of v2em PE3-AAV (2.3 × 1010 vg each N-terminal and C-terminal AAVs plus 1.1 × 1010 vg epegRNA/sgRNA AAV) encoding PE3max with full-length RT or PE3max with a truncated ΔRNaseH RT to install the Dnmt1 +2 G-to-C edit. Three weeks after injection, mice were harvested; cortex was dissected; and nuclei were isolated and sorted by FACS. The bulk population (all nuclei) and a GFP+ subpopulation were lysed, and gDNA was analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3–4 mice; each condition includes both male and female mice. c, Schematic of v3em PE3max AAV. d, C57BL/6 mice were RO injected with either 1.25 × 1012 vg total of v2em PE3 ΔRNaseH AAV9 (5 × 1011 vg each N-terminal and C-terminal AAVs plus 2.5 × 1011 vg epegRNA/sgRNA AAV) or 1 × 1012 vg total of v3em PE3-AAV9 (5 × 1011 vg each N-terminal and C-terminal AAVs), both installing the Dnmt1 +1 C-to-G edit. Three weeks after injection, liver, heart, and muscle tissues were harvested and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3–5 mice; each condition includes both male and female mice. e, Prime editing efficiency in bulk liver of v1em or v3em PE3-AAV9 after a single injection of AAV at two different doses. C57BL/6 mice were RO injected with 1 × 1011 vg or 1 × 1012 vg total of either v1em or v3em PE3-AAV9 containing an epegRNA encoding the Dnmt1 +2 G-to-C edit. Three weeks after injection, liver tissue was harvested and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 4 mice; each condition includes both male and female mice.
We next optimized the AAV genome architecture to enable packaging of intein-split PE3max ΔRNaseH into two AAV genomes by retaining the Cbh promoter and substituting with SV40 late poly(A), the size of which allows the pegRNA and nicking sgRNA cassettes to both fit in the C-terminal PE-AAV, resulting in v3em PE3-AAV (Fig. 4c). We compared the performance of v2em PE3-AAV9 with v3em PE3-AAV9, each with an epegRNA programmed to install the +1 C-to-G edit at Dnmt1 in 6–8-week-old C57BL/6 mice via systemic RO injection. Doses were 1.25 × 1012 vg total v2em PE3-AAV (5 × 1011 vg each N-terminal and C-terminal PE-AAV and 2.5 × 1011 vg epegRNA/sgRNA AAV) or 1 × 1012 vg of total v3em PE3-AAV (5 × 1011 vg each of the N-terminal and C-terminal v3em PE3-AAVs). Three weeks after injection, we harvested tissues for analysis by HTS. In bulk liver tissue, v3em PE3-AAV9 yielded efficiencies similar to v2em PE3-AAV9 (35% for v3em PE3-AAV versus 36% for v2em PE3-AAV). In bulk heart and skeletal muscle, prime editing increased 2.1-fold (9.1%, P = 0.026) and 4.9-fold (1.3%, not significant by unpaired t-test), respectively, for v3em compared to v2em PE3-AAV (Fig. 4d). The largely unchanged editing efficiency in liver but strong improvement in extra-hepatic tissues upon reduction of the co-transduction requirement likely reflects that transduction efficiency in liver is only modestly limiting for either AAV system39, in contrast with less efficiently transduced tissues in which AAV delivery remains a key bottleneck.
Next, we directly compared v1em and v3em PE3-AAV architectures. We RO injected 6–8-week-old C57BL/6 mice with v1em or v3em PE3-AAV9, each at a high dose of 1 × 1012 vg total (5 × 1011 vg N-terminal and C-terminal PE-AAVs) or a 10-fold lower dose of 1 × 1011 vg total (5 × 1010 vg N-terminal and C-terminal PE-AAVs) and with each epegRNA encoding the Dnmt1 +2 G-to-C edit. Three weeks after injection, DNA and RNA were isolated from bulk liver tissue. We observed 46% and 14% prime editing with v3em PE3-AAV9 at the high dose and low dose, respectively (Fig. 4e), and 5.7% and 0.1% prime editing with v1em PE3-AAV9 in bulk liver, consistent with the observed improved PE mRNA expression from the Cbh promoter (Supplementary Note 4 and Extended Data Fig. 6).
Extended Data Fig. 6. Transduction, RNA expression, and assessment of higher N:C ratio of PE-AAVs in liver.

a, In vivo comparison of viral genomes per diploid genome by promoter and dose in liver tissue. Prime editors were expressed from either the v3em (Cbh promoter) or v1em (EFS promoter) architecture at the doses indicated. Isolated DNA was quantified by ddPCR using primers and a probe specific to either the N- or C-terminal half of SpCas9. Viral genome concentrations were normalized to Gapdh. b, In vivo comparison of PE transcript levels by promoter and dose in liver tissue. Prime editors were expressed from either the v3em (Cbh promoter) or v1em (EFS promoter) architecture at the doses indicated. Mature mRNA transcripts were reverse transcribed, and PE transcript quantities from the resulting cDNA were measured by droplet digital PCR (ddPCR) using primers and a probe specific to either the N- or C-terminal half of SpCas9 (Supplementary Table 4). Transcript concentrations were normalized to Gapdh. c, In vivo comparison of higher ratio of N-terminal v3em PE-AAV to C-terminal half v3em PE-AAV. v3em PE3-AAV9 encoding the Dnmt1 +2 G-to-C edit was delivered at a total dose of 1×1012 vg at the ratio indicated: either a ratio of 1:1 (5×1011 vg each N- and C-terminal halves) or a ratio of 2.5:1 (7.1×1011 vg N-terminal half and 2.9×1011 vg C-terminal half) and bulk tissues were analyzed three weeks post injection by HTS. Dots represent individual mice and error bars represent mean±SEM of n = 3-4 different mice.
Finally, we assessed v3em PE3-AAVs using the 844-CFN intein split architecture that also yielded high-efficiency prime editing in vitro (Fig. 1a and Extended Data Fig. 7). We delivered 1 × 1012 vg of total 1024-CFN or 844-CFN v3em PE3-AAV (5 × 1011 vg each of the N-terminal and C-terminal PE-AAVs) to 6–8-week-old C57BL/6 mice via systemic RO injection and analyzed prime editing 3 weeks after injection. Editing efficiency differences were statistically insignificant by unpaired two-tailed t-test (Extended Data Fig. 7). Thus, 884-CFN offers an alternate PE protein split that can accommodate other elements on the AAV genome that may be necessary for different applications. We continued to use the 1024-CFN (v3em PE-AAV) for further characterization.
Extended Data Fig. 7. Comparison of v3em PE3-AAV split at two different positions.

6- to 8-week-old C57BL/6 mice were injected with 1x1012 vg total of v3em PE3-AAV9 (5x1011 vg each N & C terminal AAVs) using the intein-split indicated (residue 1024 or 844, in both cases with extein residues mutated to CFN) installing a Dnmt1 +1 C-to-G edit. Dots represent individual mice and error bars represent mean±SEM of n = 3-5 mice.
Improved PE-AAV prime editing in the CNS
Although v1 PE3-AAV supported robust CNS prime editing when injected directly to the brain in neonatal mice (Figs. 2b and 3b), improvements in v3em PE3-AAV design might offer increased CNS editing efficiency over v1em PE3-AAV. To test this possibility, we delivered PE3max with epegRNA installing the Dnmt1 +1 C-to-G edit via P0 ICV injection using the v1em or v3em PE3-AAV9 architecture at a dose of 1 × 1011 vg (5 × 1010 vg per half) with 1 × 1010 vg capsid-matched, promoter-matched and terminator-matched EGFP:KASH to facilitate sorting of transduced cells. The capsid-, promoter-, and terminator-matched GFP AAVs yielded 50% and 61% GFP+ nuclei for v1 and v3, respectively (Fig. 5a), indicating that the Cbh promoter may be active in more cells across the CNS than the EFS promoter. Although both approaches led to similarly high levels of editing in bulk cortex (41% and 42% for v1em and v3em, respectively), v1em PE3-AAV yielded higher editing among GFP+ nuclei (81% for v1em and 69% for v3em, P = 0.0087) (Fig. 5b). We also assessed the importance of inclusion of a nicking sgRNA in vivo (the PE3 strategy) and found that PE3 increased editing over PE2 by 9.8-fold and 2.8-fold in bulk cortex for the Dnmt1 +1 C-to-G edit and +2 G-to-C edit, respectively (Supplementary Note 5 and Extended Data Fig. 8).
Fig. 5. Improved prime editing in the mouse CNS.

Comparison of prime editing from v1em and v3em PEmax AAVs in the CNS via direct injection in neonatal mice or systemic injection in adult mice. a, Percent GFP+ nuclei of capsid-matched, promoter-matched and terminator-matched EGFP:KASH used for enrichment. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3 mice; each condition includes both male and female mice. b, Either v1em or v3em PE3-AAV9 encoding the +1 C-to-G edit at Dnmt1 was delivered via P0 ICV injection at a dose of 1 × 1011 vg. Cortex was harvested after 3 weeks, and nuclei were isolated. GFP+ subpopulations were sorted by FACS. gDNA from bulk or GFP+ nuclei was extracted and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3 mice; each condition includes both male and female mice. c, Adult 6–8-week-old mice were RO injected with a total dose of 1 × 1012 vg AAV PHP.eB with either v1em or v3em PE3-AAV encoding the +2 G-to-C edit at Dnmt1. Cortex was harvested after 3 weeks, and nuclei were isolated. GFP+ subpopulations were sorted by FACS. gDNA from bulk or GFP+ nuclei was extracted and analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 4–5 mice; each condition includes both male and female mice. d, Installation of APOE3 R136S (APOE Christchurch) in humanized APOE3 mice. 1 × 1011 vg v3em PE3-AAV9 was administered by ICV injection to mice on P1 or P3. gDNA or total RNA was isolated from neocortex and hippocampus (matched hemispheres). RNA was converted to cDNA, and both gDNA and cDNA were analyzed by HTS. Dots represent individual mice, and error bars represent mean ± s.e.m. of n = 3–4 mice; each condition includes both male and female mice.
Extended Data Fig. 8. Comparison of PE2 and PE3 in vivo.

v1em AAV9 with an epegRNA encoding a +1 C-to-G edit or +2 G-to-C edit at Dnmt1 was delivered via P0 ICV injection at a dose of 1x1011 vg (5x1010 vg each N- and C-terminal AAVs) with 1x1010 vg promoter-matched AAV9 EGFP:KASH. At three weeks, nuclei from cortex were isolated and sorted by FACS to either all nuclei (bulk) or GFP+ populations. Genomic DNA was extracted and analyzed by HTS. Dots represent individual mice (n = 2-4) and error bars represent mean±SEM of n = 3-4 mice.
To test whether delivery of PEs in adult animals via systemic injection with a blood–brain-barrier-crossing capsid might enable efficient CNS editing, we injected C57BL/6 mice with a total dose of 1 × 1012 vg of either v1em or v3em PE3-AAV PHP.eB55–57 encoding an epegRNA to install the Dnmt1 +2 G-to-C edit (5 × 1011 vg each half) with promoter- and terminator-matched 1 × 1011 vg AAV PHP.eB EGFP:KASH to facilitate enrichment of transduced nuclei (Fig. 5c). With v1em PE3-AAV PHP.eB, we observed 14% prime editing in bulk cortex and 30% prime editing among GFP+ nuclei, with few indels (0.5%), whereas v3em PE3-AAV PHP.eB yielded 11% prime editing and 0.4% indels in bulk cortex and 22% prime editing and 0.5% indels in the GFP+ population (Fig. 5c). Taken together, these results demonstrate prime editing of the CNS in adult mice after a systemic injection of a blood–brain-barrier-crossing AAV capsid.
Installation of a disease-relevant mutation in the CNS
To test the capabilities of PE-AAV systems to edit mutations of biomedical interest in vivo, we first used PE-AAVs to install the putatively protective APOE Christchurch (APOE3 R136S) coding variant, a G-to-T transversion mutation that cannot be installed via base editing and that would be difficult to install in post-mitotic cells via homology-directed repair (HDR). This mutation is of biological and therapeutic interest, as it has been observed in an individual who carried the risk-associated PSEN1 (presenilin 1) E280A mutation but who did not develop cognitive impairment until three decades after the expected age of onset of AD among PSEN1 E280A carriers58. A subsequent study suggests that the APOE Christchurch variant may be deleterious in other genetic contexts59. The ability to precisely install the APOE Christchurch allele in relevant cells in vivo could help illuminate the mutation’s influence on AD pathology.
To develop a strategy for the installation of APOE3 R136S, we first optimized a prime editing strategy for this mutation in HEK293T cells, achieving 40% prime editing by plasmid transfection (Supplementary Fig. 1a), and verified prime editing in cultured humanized mouse astrocytes, observing 15% prime editing with PE3 and 27% prime editing with PE5 (Supplementary Fig. 1d). These results indicate that prime editing is feasible in cultured astrocytes, the major apoE-expressing cells in the CNS and the primary cell type of interest for this mutation60,61. To assess installation of APOE3 R136S in vivo, we performed ICV injection of 1 × 1011 vg (5 × 1010 vg per half) of v3em PE3-AAV9 carrying the optimized epegRNA and nicking sgRNA to humanized APOE3 mice via ICV injection. We also assessed the impact of injection timing by injecting via ICV at P1 or P3, as the extent of non-neuronal cell transduction is known to increase with age62–64. Three weeks after injection, we analyzed prime editing efficiencies in bulk nuclei from neocortex and hippocampus, two brain regions relevant to AD pathology. DNA editing efficiencies in bulk neocortical and hippocampal tissues for P1 injected mice were 12% prime editing with 5.0% indels and 14% prime editing with 3.1% indels, respectively, whereas P3 injections resulted in 8.2% prime editing with 4.6% indels and 7.1% prime editing with 3.8% indels in the bulk neocortex and hippocampus tissues, respectively (Fig. 5d). To measure installation of APOE3 R136S in apoE-expressing cells, we isolated total RNA from AAV-injected and control brain tissues 3 weeks after injection, generated cDNA and observed 9.4% prime editing with 3.5% indels in neocortex APOE cDNA and 11% prime editing with 2.8% indels in hippocampal APOE cDNA. Together, these data reveal that prime editing can install mutations of therapeutic interest in relevant CNS cell types in vivo.
Installation of a protective mutation in adult mouse liver
To further assess in vivo editing activity of PE-AAV systems, we tested their ability to mediate therapeutically relevant prime edits in adult animals. We targeted Pcsk9, a gene involved in cholesterol homeostasis65–70. Although the use of genome editing to knock down PCSK9 has yielded encouraging results39,71–73, and base editing of PCSK9 to lower LDL cholesterol levels has recently entered clinical trials74, the ability of prime editing to precisely introduce virtually any small substitution, insertion or deletion provides access to PCSK9 variants that could offer unique strengths.
To test whether PE-AAV could edit the liver of an adult animal to confer a protective phenotypic change, we designed and produced PE-AAV to install the mouse homolog of PCSK9 Q152H, a G-to-C substitution that blocks autocatalytic processing of PCSK9 (ref. 75) with retention of both the mutated protein as well as autocatalytically competent wild-type PCSK9 in the endoplasmic reticulum (ER) without inducing ER stress or the unfolded protein response in humans or in mice overexpressing PCSK9 Q152H76. Individuals homozygous for PCSK9 Q152H show a marked reduction in LDL cholesterol levels, and heterozygous individuals also show a reduction in levels of circulating PCSK9 and LDL75,77. The development of an in vivo prime editing strategy to install this mutation in postnatal animals may help illuminate its physiological consequences and test a potential therapeutic strategy to lower coronary heart disease risk.
We first optimized prime editing in cultured cells to install the mouse homolog of PCSK9 Q152H (Pcsk9 Q155H) (Supplementary Fig. 2a–c). We transfected plasmids encoding PEmax, an epegRNA and a nicking sgRNA, resulting in up to 17% prime editing in mouse N2a cells (Supplementary Fig. 2a–c). We next assessed whether the inclusion of MMR-evading silent mutations near the intended edit43 could improve installation of Pcsk9 Q155H and found a 2.8-fold improvement when the silent mutation was incorporated with PE2max and 1.3-fold with PE3max (Supplementary Fig. 2d), achieving up to 25% prime editing with 5.5% indels in N2a cells.
We then delivered v3em PE3-AAV9 into 6–8-week-old mice via systemic RO injection at a total dose of 1 × 1012 vg per mouse and compared epegRNAs encoding Pcsk9 Q155H with a PAM-disrupting edit or with MMR-evading silent edits (Supplementary Fig. 2e), achieving 31% and 38% prime editing, respectively. Inclusion of MMR-evading silent edits modestly increased average liver editing efficiency 8 weeks after injection (P = 0.06, unpaired t-test) (Supplementary Fig. 2e). Together, these findings demonstrate that MMR-evading silent edits can increase prime editing efficiency in vitro and in vivo and should be considered for the design of prime editing experiments in animals when possible.
In a separate cohort of mice, we assessed whether v3em PE3-AAV-mediated Pcsk9 prime editing with this MMR-evading strategy in liver reduces circulating lipid levels. We delivered v3em PE3-AAV9 into 6–8-week-old mice via systemic RO injection at a total dose of 1 × 1012 vg per mouse and achieved 39% editing 8 weeks after injection (Fig. 6b and Extended Data Fig. 9a). Total plasma cholesterol in Pcsk9 Q155H-edited mice decreased by 20% on average compared to age-matched untreated mice 2 weeks after injection, and this effect persisted until the end of the study at 8 weeks (P = 0.022 by two-way ANOVA at 8 weeks) (Fig. 6c). We observed 27% reduction in plasma LDL cholesterol at 2 weeks after treatment compared to age-matched untreated controls, and this effect also persisted until the end of the study at 8 weeks (Fig. 6d) (P = 0.023 by two-way ANOVA). Male mice showed a larger reduction in both total plasma cholesterol and LDL cholesterol than age- and condition-matched female mice (Supplementary Note 6, Extended Data Fig. 9 and Supplementary Fig. 3). Although the effect of PCSK9 Q152H transgene overexpression has been reported76,77, the installation of this mutation into the genome in vivo and the examination of the resulting physiological effect has not, to our knowledge, been previously described. Collectively, these results demonstrate that the optimized v3em PE3-AAV architecture can achieve robust and therapeutically relevant prime editing in vivo to install genomic mutations that cannot readily be installed in relevant tissues by other methods.
Fig. 6. In vivo prime editing to install Pcsk9 Q155H and its effect on plasma cholesterol levels.

a, Timing of injections of v3em PE3-AAV9, evaluation of prime editing and plasma analysis. b, Bulk liver editing efficiencies for installation of Pcsk9 Q155H with an epegRNA that includes additional MMR-evading silent edits. c,d, Plasma total cholesterol and plasma LDL cholesterol in mice treated with prime editing AAV normalized to untreated mice at the same age. Data are shown as mean ± s.e.m. for n = 8 mice (four male and four female), *P = 0.0064 and P = 0.023, respectively. Significance was calculated using two-way repeated-measures ANOVA with Sidak multiple comparisons and is shown for the 8-week timepoint. e,f, Off-target prime editing for 10 CIRCLE-seq-nominated off-target loci (Off-target1–Off-target10) for pegRNA and nicking sgRNA from the livers of v3em PE3-AAV-treated and untreated mice. For b, e and f, dots represent individual mice, and error bars represent mean ± s.e.m. of n = 8 mice (four male and four female).
Extended Data Fig. 9. Prime editing and plasma analytes (normalized to untreated) data of either untreated or v3em PE3-AAV9 treated male and female C57BL/6 mice used to plot Fig. 6.

a, Installation of Pcsk9 Q155H in mouse liver using v3em PE-AAV9 with epegRNA encoding MMR-evading silent edits used to plot Fig. 6b. Dots represent individual mice and error bars represent mean±SEM for n = 4 mice. b-c, Total plasma cholesterol in C57BL/6 male mice (b) and female mice (c) normalized to untreated used to plot Fig. 6c. Data are shown as mean±SEM for n = 4 mice. d-e, Plasma LDL cholesterol levels from C57BL/6 male mice d and female mice e normalized to untreated used to plot Fig. 6d. Data are shown as mean±SEM for n = 4 mice.
Characterization of in vivo off-target editing and toxicity
To assess in vivo off-target prime editing in liver tissue from v3em PE3-AAV treatment, we performed circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq)78 to nominate potential off-target loci that might be engaged by either the Pcsk9 Q155H pegRNA or nicking sgRNA. From the nominated loci, we then selected 10 candidate off-target sites based on the highest read counts from CIRCLE-seq to examine by HTS (Supplementary Tables 5 and 6). No detected off-target editing above background levels was present at any of these loci in mice treated with v3em PE3-AAV9, indicating that prime editing at this site maintains a high degree of sequence specificity, consistent with several other reports2,79–88 (Fig. 6e,f).
Finally, we investigated toxicity from v3em PE3-AAV delivery by measuring serum alanine aminotransferase (ALT) and aspartate transaminase (AST) levels, biomarkers of hepatocellular and kidney injury89. We used v3em PE3-AAV8, a serotype that efficiently and specifically transduces murine hepatocytes90, and delivered into a separate cohort of mice dose- and serotype-matched AAV8 encoding EGFP or Cas9 nickase and a fourth cohort with saline alone. Although we observed a slight elevation of ALT and AST in AAV-injected mice at 5 weeks and 7 weeks after injection compared to saline controls, the levels were within the normal physiological range for all conditions91,92, and there were no statistically significant differences between saline-treated and PE-AAV-treated mice (P = 0.28 by two-way ANOVA) (Extended Data Fig. 10b,c). Additionally, we performed liver histology 8 weeks after injection and found no evident histological or morphological changes compared to saline-treated mice (Extended Data Fig. 10a). Together, these results demonstrate that our optimized v3em PE3-AAV can mediate efficient prime editing in the mouse liver with no detected off-target editing and without evident toxicity.
Extended Data Fig. 10. Assessment of liver toxicity following systemic v3em PE-AAV8 injection.

Adult mice were systemically injected v3em PE-AAV8 along with dose- and serotype-matched AAV8 encoding EGFP to control for toxicity induced by the AAV vector, dose- and serotype-matched Cas9 nickase to control for toxicity caused by Cas9 nickase and sgRNA portion of the prime editor, and a fourth cohort injected with saline alone. a, Histopathological assessment by hematoxylin and eosin staining of livers at eight weeks post-injection of saline or 1x1012 vg total AAV8 encoding either EGFP, Cas9 nuclease, or prime editor. Scale bars, 50 μm, representative images from two separate mice shown per condition. b-c, Plasma aspartate transaminase (AST) and alanine transaminase (ALT) levels. Data are shown as mean±SEM for n = 4 mice. The dotted lines mark the upper limit of normal physiological range for AST and ALT in adult C57BL/6 mice. d, Installation of Pcsk9 Q155H in vivo for liver toxicity study. v3em PE3-AAV8 was injected into 6- to 8-week old adult C57BL/6 mice at a dose of 1x1012 vg total by retro-orbital injection and liver was harvested eight weeks post injection to assess bulk editing. Dots represent individual mice and error bars represent mean±SEM for n = 4 mice.
Discussion
By systematically identifying and addressing the factors limiting in vivo prime editing via AAV, we developed a platform for the delivery of PEs to mouse CNS, liver, and heart. Our findings highlight promoter choice and PE expression as key bottlenecks of PE performance in vivo that can be overcome through vector engineering. We also show that recent improvements in prime editing, such as epegRNAs, PEmax and the use of MMR-evading bystander edits, can each improve prime editing in vivo. In the brain and liver, v3em PE-AAVs can achieve ≥40% precise prime editing, substantially outperforming previously reported platforms for in vivo PE delivery in these systems with a clinically validated vector and establishing prime editing in the mammalian brain. PE-AAVs can facilitate the installation of various therapeutic edits, including protective alleles associated with AD and coronary artery disease. We anticipate that properties of optimized v1em and v3em PE-AAV systems will advance the study and potential treatment of a wide variety of diseases with a genetic component.
We limited the doses of systemically delivered AAV in this study to a maximum of 1 × 1012 vg per animal (4–5 × 1013 vg kg−1 for a 20–25-g mouse), a dose considered to be tolerated in clinical trials involving AAV22,24,93,94. Prime editing efficiency in extra-hepatic tissues may be further increased by improvements in PE expression using tissue-specific optimization, such as delivery routes that increase AAV transduction in relevant tissues51 and the use of capsids with enhanced tropism for a given tissue.
In vivo AAV-mediated prime editing currently requires the use of two or more AAVs. For prime editing applications that require additional proteins, such as installing site-specific recombinase landing sites for gene-sized genomic insertions2,95,96, further size reduction of the components could facilitate a dual-AAV strategy. Substantial size reduction of components may enable single-AAV prime editing, which would further simplify use and enhance therapeutic relevance, similar to the improvements that we recently observed from single-AAV base editor delivery39.
In vivo prime editing resulted in no detected off-target editing and no significant increase in markers of liver damage compared to control treatments, suggesting that expression of the PE in vivo does not induce apparent hepatotoxicity. Measures to minimize inflammation and immune response to PE components16 should be investigated for therapeutic applications. Additional improvements that offer transient delivery, such as lipid nanoparticles (LNPs) or virus-like particles (VLPs), temporally controlled expression of PEs, and further enhanced potency may improve the safety profile of prime editing in vivo.
Methods
Molecular biology
Editor plasmids used for mammalian cell transfection were generated using Gibson assembly or USER assembly using pCMV-PE2 (Addgene, 132775) and pCMV-PEmax (Addgene, 174820) plasmids. pegRNA and epegRNA constructs were cloned by Golden Gate assembly using custom pU6-pegRNA-GG-acceptor (Addgene,132777) and pU6-tevopreQ1-GG-acceptor (Addgene, 174038) plasmids. All nicking sgRNA plasmids were generated by KLD assembly or Golden Gate assembly using pFYF1320 (Addgene, 47511) as a template plasmid. rAAV vector plasmids were cloned by restriction digestion of v5 AAV CBE (Addgene, 137176) or v5 AAV ABE (Addgene, 137177) followed by Gibson assembly with eBlocks or polymerase chain reaction (PCR) amplicons. All plasmids used for mammalian tissue culture were purified from MACH1, DH5alpha or NEBstable Escherichia coli using Plasmid Plus Maxiprep or Midiprep kits (Qiagen), ZymoPURE II Midiprep Kit (Zymo Research) or PureYield Plasmid Miniprep kit (Promega).
Cell culture
HEK293T cells (American Type Culture Collection (ATCC), CRL-3216) and Neuro-2A cells (ATCC, CCL-131) were grown in DMEM plus GlutaMAX (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS at 37 °C with 5% CO2. Immortalized mouse astrocytes containing the APOE4 isoform of the human APOE gene (Taconic Biosciences) were grown in DMEM plus GlutaMAX (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS and 200 μg ml−1 Geneticin (Thermo Fisher Scientific). Cell lines were authenticated by their suppliers and were verified to be mycoplasma negative during the study.
Transfections of HEK293T and Neuro-2A cells
Sixteen to twenty-four hours before transfection, HEK293T and Neuro-2A cells at more than 90% viability were seeded on 48-well poly-d-lysine-coated plates (BioCoat plates, Corning) at a density of 30,000–40,000 cells per well. Cells were transfected with 1 μl of Lipofectamine 2000 (Thermo Fisher Scientific) and 750 ng of PE plasmid, 250 ng of pegRNA plasmid and 83 ng of sgRNA plasmid. For conditions delivering MLH1dn, 100 ng of additional plasmid was transfected. For 96-well plate (Corning) transfections, 15,000–20,000 cells were plated 16–24 h before transfection, and 200 ng of PE, 40 ng of pegRNA and 13.3 ng of nicking guide with 0.5 μl of Lipofectamine 2000 (Thermo Fisher Scientific) were used. For intein-split PE plasmid transfections, total PE plasmid was reduced to 50 ng. Seventy-two hours after transfection, genomic DNA (gDNA) was isolated with 75–150 μl of lysis buffer (10 mM Tris-HCl pH 8.0, 9.05% SDS, 25 μg ml−1 proteinase K (Thermo Fisher Scientific)) at 37 °C for 1 h, followed by 80 °C for 30 min.
In vitro transcription of PE2 and MLH1dn mRNA
Plasmid templates for in vitro transcription carry an inactivated T7 promoter, 5′ UTR, Kozak sequence, coding sequences and 3′ UTR. Transcription templates were PCR amplified from these plasmids using Phusion U Green Multiplex Master Mix (Thermo Fisher Scientific) with primers that correct the T7 promoter and add a 119-nt poly(A) tail to the 3′ UTR. After purification of the product with QIAquick PCR Purification Kit (Thermo Fisher Scientific), PE2 and hMLH1dn mRNAs were transcribed from these templates using HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs) with full replacement of UTP with N1-methylpseudouridine-5′-triphosphate (TriLink Biotechnologies) and co-transcriptional capping by CleanCap Reagant AG (TriLink Biotechnologies). mRNA products were precipitated in 2.5 M lithium chloride, washed twice with 70% ethanol, dissolved in nuclease-free water and stored at −80 °C.
Nucleofection of APOE4 murine astrocytes
Astrocytes containing APOE4 isoform of the APOE gene (originated from Taconic Biosciences) were nucleofected using program EN-150 with SF Cell Line 4D-Nucleofector X Kit (Lonza). In brief, 200,000 astrocytes were resuspended in 20 μl of buffer with 1 μg of PE2 mRNA, 90 pmol synthetic pegRNA (Integrated DNA Technologies) and 60 pmol synthetic nicking sgRNA (Synthego). For PE5 experiments, 2 μg of hMLH1dn mRNA was also included in the nucleofection. After nucleofection, the cells were diluted to 100 μl of pre-warmed media and recovered for 10 min at 37 °C, followed by plating in 12-well plates. Seventy-two hours after nucleofection, gDNA was harvested.
HTS and data analysis
Genomic loci of interest were amplified from isolated gDNA via two rounds of PCR with PhusionU or PhusionHS polymerase (Thermo Fisher Scientific). The initial PCR step (PCR1) was done using primers with Illumina adapter overhangs (Supplementary Table 1) with the following conditions: 95 °C for 3 min; 27–30 cycles of 95 °C for 15 s, 61–70 °C (corresponding to the experimentally optimized Tm) for 20 s and 72 °C for 30 s, followed by 72 °C for 1 min. Unique Illumina sequencing barcodes were added in subsequent PCR2 step, using 1–2 μl of PCR1 as a template with the following conditions: 95 °C for 3 min; nine cycles of 95 °C for 15 s, 61 °C for 20 s and 72 °C for 30 s, followed by 72 °C for 1 min. After PCR2, samples were pooled according to amplicon size and gel purified in a 1% agarose gel using a Qiaquick Gel Extraction Kit (Qiagen). Pooled library concentration was quantified (Qubit dsDNA HS assay kit, Thermo Fisher Scientific) and run on an Illumina MiSeq 300 v2 Kit with 280–300 cycles.
Sequencing reads were demultiplexed using MiSeq Reporter (Illumina). For single base changes, alignment of amplicon sequences to reference sequence was performed using CRISPResso2 (ref. 97) in batch mode with ‘-q30’, ‘discard indel reads TRUE’ and ‘qwc’ coordinates spanning the sequence between pegRNA-directed and nicking sgRNA-directed Cas9 cut sites. Indels were calculated as percentage of (discarded reads) / (total aligned reads). Prime editing at a given position was calculated explicitly as: (frequency of specified point mutation in non-discarded reads) × 100 × (100 – (indel reads)) / 100)). For insertions, CRISPResso2 was executed in HDR mode using identical parameters as described above but with an additional parameter ‘e’ specifying sequence of the edited ‘desired’ amplicon. Indels were calculated as percentage of (discarded reads from the reference-aligned sequences and HDR-aligned sequences) / (total aligned reads). In HDR mode, prime editing efficiency was quantified as (HDR-aligned reads without indels) / (number of total reads aligned to the reference amplicon) × 100 × (100 – (indel reads)) / 100)).
AAV production
HEK293T clone 17 cells (ATCC, CRL-11268) were maintained in DMEM plus GlutaMAX (Thermo Fisher Scientific) with 10% (v/v) heat-inactivated FBS without antibiotic in 150-mm2 dishes (Thermo Fisher Scientific) at 37 °C with 5% CO2 and passaged every 2–3 days. Cells were split 1:3 18–22 h before polyethyleneimine transfection (PEI MAX, Polysciences) with 5.7 μg of AAV genome plasmid, 11.4 μg of pHelper (Clontech) and 22.8 μg of rep-cap plasmid per plate. Four days after transfection, cells were harvested using a cell scraper (Corning), pelleted by centrifugation at 2,000g for 10 min, resuspended in 500 µl of hypertonic lysis buffer per plate (40 mM Tris base, 500 mM NaCl, 2 mM MgCl2 and 100 U ml−1 salt active nuclease (ArcticZymes)) and incubated at 37 °C for 1 h. Media were then decanted to a new bottle and a 5× solution of poly(ethylene glycol) (PEG) 8000 (Sigma-Aldrich) and NaCl was added to a final concentration of 8% PEG and 500 mM NaCl, incubated on ice for 2 h or overnight and then centrifuged at 3,200g for 30 min. The pellet was resuspended in 500 μl of hypertonic lysis buffer per plate and added to the cell lysate. Cell lysates were either stored at 4 °C overnight or taken immediately to ultracentrifugation.
Cell lysates were centrifuged at 3,400g for 10 min and added to Beckman Coulter Quick-Seal tubes via 16-gauge, 5-inch needles (Air-Tite N165) in a discontinuous gradient of iodixanol in sequentially floating layers: 9 ml of 15% iodixanol in 500 mM NaCl and 1× PBS-MK (1× PBS plus 1 mM MgCl2 and 2.5 mM KCl), 6 ml of 25% iodixanol in 1× PBS-MK and 5 ml each of 40% and 60% iodixanol in 1× PBS-MK with phenol red at a concentration of 1 μg ml−1 in the 15%, 25% and 60% layers to visualize layers. Ultracentrifugation was performed using a fixed-angle Ti 70 rotor in a Optima XPN-100 Ultracentrifuge (Beckman Coulter) at 68,000 r.p.m. (340,000g for an rav of 65.7 mm) for 1 h or 58,600 r.p.m. (253,000g for an rav of 65.7 mm) for 2 h and 15 min at 18 °C. Immediately after centrifugation, 3 ml of solution was removed from the 40–60% iodixanol interface via an 18-gauge needle. Buffer was exchanged for cold PBS with 0.001% F-68 using PES 100 kD MWCO columns (Thermo Fisher Scientific) and concentrated. The AAV solution was sterile filtered using a 0.22-μm filter and then quantified by quantitative PCR (qPCR) (AAVpro Titration Kit version 2, Clontech) and stored at 4 °C until use.
Animal care
All experiments involving live animals were approved by the Broad Institute Institutional Animal Care and Use Committee (D16-00903; 0048-04-15-2) and were consistent with local, state and federal regulations as applicable, including the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6 mice were purchased as adults from The Jackson Laboratory (000664) for all adult RO injections. For neonatal injections, pregnant C57BL/6 females were purchased from Charles River Laboratories (027), and their pups were injected; or, for APOE4 experiments, mice were originally purchased from Taconic Biosciences (1549) and were bred and maintained in the Broad Institute vivarium. All mice were housed on a 12-h light/dark cycle between 68 °F and 79 °F and 30–70% humidity. Mice had ad libitum access to standard rodent diet and water except for 6-h fasts just before submandibular bleeding for plasma analysis when access to water was still maintained.
RO injections
AAV was diluted into 100 μl of sterile 0.9% NaCl USP (Fresenius Kabi) before injection. Anesthesia was induced with 3–4% isoflurane. Under anesthesia, the right eye was protruded; the needle of the loaded insulin syringe was inserted into the RO sinus; bevel faced away from the eye; and the syringe was slowly advanced. Directly after injection, a drop of proparacaine hydrochloride ophthalmic solution (Patterson Veterinary) was applied to the eye as an analgesic.
Neonatal ventricle injections
For neonatal ICV injections, Drummond PCR pipettes were pulled at the ramp test value of a Sutter P1000 micropipette puller for a tip diameter size of approximately 100 μm. The injection solution of 4 μl including a small amount of Fast Green dye was front-loaded. Pups were anesthetized by cryoanesthesia. Then, 2 μl was then freehand injected into each ventricle. Transillumination of the head was then used to verify successful ventricle targeting.
Mice tissue collection
Mice in this study were sacrificed by CO2 asphyxiation, and unperfused tissues were immediately dissected. Bulk tissue was harvested unless otherwise noted. For harvest of brain tissue, hemispheres were split sagittally by razor blade, and then neocortex and hippocampus were isolated with a micro spatula. Tissues were harvested in DNAdvance lysis buffer (Beckman Coulter), and the gDNA was purified.
Nuclear isolation and sorting
Dissected brain tissue was homogenized in 2 ml of ice-cold EZ-PREP buffer (Sigma-Aldrich) using a glass Dounce homogenizer (Sigma-Aldrich) with 20 strokes of pestle A followed by 20 strokes of pestle B. Homogenized samples were then decanted into a new tube containing an additional 2 ml of EZ-PREP buffer on ice. After 5-min incubation, samples were centrifuged for 5 min at 500g at 4 °C. The nuclei pellet was resuspended in 4 ml of ice-cold nuclei suspension buffer (NSB) consisting of 100 µg ml−1 BSA (New England Biolabs) and 3.33 µM Vybrant DyeCycle Ruby (Thermo Fisher Scientific) in cold PBS. Nuclei were then centrifuged at 500g for 5 min at 4 °C. The nuclei were resuspended in 1–2 ml of NSB, passed through a 35-µm cell strainer and then sorted using a MA900 Cell Sorter (Sony Biotechnology) at the Broad Institute Flow Cytometry Core. See Supplementary Fig. 17 for FACS gating strategy. Nuclei were sorted into DNAdvance lysis buffer (Beckman Coulter), and gDNA was purified.
Off-target analysis
CIRCLE-seq was performed and analyzed as described previously78 save for the following modifications. For the Cas9 cleavage step, guide denaturation, incubation and proteinase K treatment were conducted using the more efficient method described in the CHANGE-seq protocol98. Specifically, the sgRNAs with the spacer sequences ‘GCAUGGCUGUCUGGUUCUGU’ (the PCKS9 nicking sgRNA) and ‘GCCAGGUUCCAUGGGAUGCUC’ (used in the PCKS9 pegRNA) were ordered from Synthego with their standard chemical modifications—2′-O-methyl for the first three and last three bases and phosphonothioate bonds between the first three and last two bases. A 5′ ‘G’ nucleotide was included with the 20-nt pegRNA spacer sequence to recapitulate the sequence expressed from AAVs. The sgRNAs were diluted to 9 µM in nuclease-free water and re-folded by incubation at 90 °C for 5 min, followed by a slow annealing down to 25 °C at a ramp rate of 0.1 °C per second. sgRNA was complexed with Cas9 nuclease (New England Biolabs) via a 10-min room temperature incubation after mixing 5 µl of 10× Cas9 Nuclease Reaction Buffer provided with the nuclease, 4.5 µl of 1 µM Cas9 nuclease (diluted from the 20 µM stock in 1× Cas9 Nuclease Reaction Buffer) and 1.5 µl of 9 µM annealed sgRNA. Circular DNA from mouse N2a cells was added to a total mass of 125 ng and diluted to a final volume of 50 µl. After 1 h of incubation at 37 °C, proteinase K (New England Biolabs) was diluted four-fold in water, and 5 µl of the diluted mixture was added to the cleavage reaction. After a 15-min proteinase K treatment at 37 °C, DNA was A-tailed, adapter ligated, USER treated, and PCR amplified as described in the CIRCLE-seq protocol78. After PCR, samples were loaded on a preparative 1% agarose gel, and DNA was extracted between the 300-bp and 1-kb range to eliminate primer dimers before sequencing on an Illumina MiSeq. Data were processed using the CIRCLE-seq analysis pipeline and aligned to the mouse genome ‘mm10’ with parameters: ‘read_threshold: 4; window_size: 3; mapq_threshold: 50; start_threshold: 1; gap_threshold: 3; mismatch_threshold: 6; search_radius: 30; PAM: NGG; merged_analysis: True’.
Analysis of PE activity at Cas9 off-target sites
For detailed off-target analysis, the top 10 sites each for pegRNA and sgRNA spacer (20 total sites) with highest read counts were deep sequenced from liver tissues of untreated or v3em PE3-AAV-treated mice. The sequencing reads were then aligned to reference off-target amplicons using CRISPResso2 (ref. 97) in batch mode with ‘-q30’, ‘discard indel reads TRUE’, ‘plot_window_size 80’, ‘-w25’, ‘min_alleles_around_cut_to_plot 0.1’ and ‘max_rows_alleles_around_cut_to_plot 600’. Off-target reads were called as leniently as possible to capture all potential reverse transcription product including point mutations, insertions or deletions at the Cas9 nick site. For off-target sites nominated by pegRNA spacer, the 6-nt sequences 3′ of Cas9 nick site (prime-editable target) were compared to the 3′ DNA flap sequence encoded by pegRNA reverse transcription. Any aligned reads with nucleotide sequence within prime editing target window that matches to the nucleotide encoded by pegRNA reverse transcription were noted as off-target reads. Off-target editing efficiencies were, thus, quantified as a percentage of (number of off-target reads) / (number of reference-aligned reads). For off-target sites nominated by nicking sgRNA spacer, insertions or deletions at the Cas9 nick site were quantified as a percentage of (discarded reads) / (total aligned reads).
ALT and AST assay
Blood was collected via submandibular bleeding in a serum separator tube. The serum was then separated by centrifugation at 2,000g for 15 min and stored at −80 °C until the end of the experiment. The samples were sent to IDEXX BioAnalytics for analysis.
Plasma measurements for total cholesterol and LDL cholesterol
To track total cholesterol and LDL cholesterol levels in plasma, blood was collected using a submandibular bleed in heparin-coated tubes. Plasma was recovered by centrifugation at 2,000g for 15 min and stored at −80 °C in one-time-use aliquots until the end of the experiment. Total cholesterol levels were determined using the Total Cholesterol Reagent (Thermo Fisher Scientific) with cholesterol standards from Pointe Scientific. Plasma LDL cholesterol levels were measured using the LDL Cholesterol Kit (WakoChemical).
Western blot of liver tissues
For western blot analysis, liver samples were lysed in a homogenization buffer (Cell Signaling Technology) containing protease inhibitor cocktail (Sigma-Aldrich). Samples were run on Mini-Protean TGX Gel, 4–15% gradient gels (Bio-Rad), followed by transfer of proteins to nitrocellulose. Antibodies against LDLR (1:1,000) (Proteintech, 10785-1-AP) and β-actin (1:3,000) (Cell Signaling Technology, 8H10D10) were used with LI-COR 926-68070 and LI-COR 926-32211 secondaries. Precision Plus Protein All Blue Prestained Protein Standards (Bio-Rad, 1610373) ladder was used. Secondaries used were LI-COR 926-68070 and LI-COR 926-32211, both at 1:20,000. The western blot data were quantified through densitometry of the bands using LI-COR Odyssey analyzer software.
Tissue fixation and histology
Mice were sacrificed by CO2 asphyxiation, and tissues were immediately harvested. For liver histology, the left medial lobe was dissected and fixed in freshly prepared 4% paraformaldehyde in 1× PBS. Tissues were rocked at 4 °C for 24 h, washed with PBS with 10 mM glycine and then rocked again at 4 °C in PBS with 10 mM glycine for another 24 h. Fixed tissues were then stored at 4 °C until further analysis.
Liver histopathology was performed by the Rodent Histopathology Core of Harvard Medical School. Fixed liver tissue was embedded in paraffin, cut into 5-μm sections and stained with hematoxylin and eosin. Samples were analyzed by a blinded mouse histopathologist.
Digital droplet PCR
Total DNA was isolated from liver tissue (DNeasy Blood and Tissue Kit, Qiagen). For quantification of viral genomes, digital droplet PCR (ddPCR) was carried out using ddPCR Supermix for Probes (Bio-Rad) with 10 ng of isolated DNA used as template and 6.25 U of HindIII-HF (New England Biolabs) per 25-µl reaction. Droplets were autogenerated, and PCR was performed with an annealing temperature of 57 °C for 2 min for a total of 40 cycles. Droplets were analyzed on a QX200 droplet analyzer, and fluorescence was quantified using QuantaSoft (Bio-Rad). Total RNA was isolated from flash-frozen liver tissue (RNeasy Plus Mini Kit, Qiagen). Isolated RNA was reverse transcribed into cDNA with SuperScript III first-strand synthesis mix with oligo dT primer (Invitrogen). RNA expression analysis was carried out in the same manner but with Gapdh expression reference assay kit (Bio-Rad). Numbers reported are normalized to reference probes in the same reaction. Primer and probe sequences are available in Supplementary Table 4. To ensure minimal DNA contamination in RNA expression analysis, RT negative controls were verified to yield only background positive droplets.
Statistical analysis
Data are presented as mean and s.e.m. Sample size and the statistical tests used for each experiment are described in the figure legends. No statistical methods were used to pre-determine sample size. Statistical analysis was performed using GraphPad Prism software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41587-023-01758-z.
Supplementary information
Supplementary Figs. 1–6, Supplementary Notes 1–6, Supplementary Sequences and Table of Contents also describing supplementary tables (which are provided in a separate Excel file).
Acknowledgements
This work was supported by US National Institutes of Health grants UG3AI150551, U01AI142756, R35GM118062 and RM1HG009490 (to D.R.L.); the MassCATS New Target Grant (to S.B., J.R.D. and D.R.L); HL163805 (to G.A.N.); R35HL145203 and R01HL148769 (to K.M.); the Bill and Melinda Gates Foundation; and the Howard Hughes Medical Institute. P.J.C. and P.B.R were supported by National Science Foundation graduate research fellowships. We thank M. Howard, S. Graffam and the Broad vivarium staff for advice and assistance with mouse husbandry and bleeding; K. Kormier and R. Bronson for histology services and analysis; S. Vallabh, E. Vallabh-Minikel, K. Chan, and B. Deverman for helpful discussions; B. Hyman and R. Jackson for APOE4 astrocytes and helpful discussions; and A. Vieira for manuscript editing assistance.
Extended data
Author contributions
J.R.D. and S.B. designed and executed in vitro and in vivo experiments, cloned plasmids, prepared AAV, injected mice, performed HTS and analyzed data. J.M.L. also designed initial experiments, cloned plasmids, prepared AAV, injected mice, performed analysis and analyzed data. G.A.N. performed CIRCLE-seq. X.W. performed western blots on prime-edited livers. A.V.A. designed and assessed PE ΔRNaseH in cell culture. A.T.N. performed ddPCR for quantification of viral genomes and PE transcript expression. A.H., M.A. and H.R. designed and cloned intein-split plasmids. P.J.C. provided PEmax constructs and performed astrocyte editing experiments. P.B.R. provided epegRNA motifs and cloned plasmids. K.M. and X.W. advised on Pcsk9 editing experiments. J.R.D., S.B. and D.R.L. designed the research and drafted the manuscript, with input from all authors.
Peer review
Peer review information
Nature Biotechnology thanks Yong-Sam Kim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
All pegRNA, nicking sgRNA and HTS primer sequences used for this study are provided in supplementary tables. High-throughput DNA sequencing data files are available from the National Center for Biotechnology Informationʼs Sequence Read Archive under accession code PRJNA898625 (ref. 99). DNA sequences of AAV genomes are provided in the Supplementary Information. Key plasmids from this work are available from Addgene, and other plasmids and raw data are available from the corresponding author upon reasonable request.
Code availability
The code used for analysis of HTS data is available at https://github.com/pinellolab/CRISPResso2.
Competing interests
J.R.D., S.B., J.M.L., A.V.A., G.A.N. and D.R.L. have filed patent applications on systems for PE delivery. J.R.D., J.M.L., A.V.A. and P.J.C. are currently employees of Prime Medicine. S.B. is currently an employee of Nvelop Therapeutics. K.M. is a consultant and equity holder of Verve Therapeutics and Variant Bio and a consultant for LEXEO Therapeutics. D.R.L. is a consultant for Prime Medicine, Beam Therapeutics, Pairwise Plants, Chroma Medicine and Nvelop Therapeutics, companies that use or deliver genome editing or epigenome engineering agents, and owns equity in these companies. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jessie R. Davis, Samagya Banskota.
Extended data
is available for this paper at 10.1038/s41587-023-01758-z.
Supplementary information
The online version contains supplementary material available at 10.1038/s41587-023-01758-z.
References
- 1.Korf, B. R., Pyeritz, R. E. & Grody, W. W. Nature and frequency of genetic disease. In Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics 7th edn (eds Pyeritz, R. E., Korf, B. R. & Grody, W. W.) 47–51 (Academic Press, 2019).
- 2.Anzalone AV, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 4.Newby GA, Liu DR. In vivo somatic cell base editing and prime editing. Mol. Ther. 2021;29:3107–3124. doi: 10.1016/j.ymthe.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song C-Q, et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020;4:125–130. doi: 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giannoukos G, et al. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stadtmauer EA, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turchiano G, et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Webber BR, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10:5222. doi: 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leibowitz ML, et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alanis-Lobato G, et al. Frequent loss of heterozygosity in CRISPR–Cas9-edited early human embryos. Proc. Natl Acad. Sci. USA. 2021;118:e2004832117. doi: 10.1073/pnas.2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 15.Liu P, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Böck, D. et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med.14, eabl9238 (2022). [DOI] [PMC free article] [PubMed]
- 17.Zheng C, et al. A flexible split prime editor using truncated reverse transcriptase improves dual-AAV delivery in mouse liver. Mol. Ther. 2022;30:1343–1351. doi: 10.1016/j.ymthe.2022.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhi S, et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2022;30:283–294. doi: 10.1016/j.ymthe.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Z, et al. A truncated reverse transcriptase enhances prime editing by split AAV vectors. Mol. Ther. 2022;30:2942–2951. doi: 10.1016/j.ymthe.2022.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jang H, et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 2022;6:181–194. doi: 10.1038/s41551-021-00788-9. [DOI] [PubMed] [Google Scholar]
- 21.She K, et al. Dual-AAV split prime editor corrects the mutation and phenotype in mice with inherited retinal degeneration. Signal Transduct. Target. Ther. 2023;8:57. doi: 10.1038/s41392-022-01234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mendell JR, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 23.Russell S, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulk N. Gene therapy: it is time to talk about high-dose AAV: the deaths of two children with X-linked myotubular myopathy in the ASPIRO trial prompts a reexamination of vector safety. Genetic Engineering & Biotechnology News. 2020;40:14–16. [Google Scholar]
- 25.Chand DH, et al. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy: a case series. J. Pediatr. 2021;231:265–268. doi: 10.1016/j.jpeds.2020.11.054. [DOI] [PubMed] [Google Scholar]
- 26.Morales L, Gambhir Y, Bennett J, Stedman HH. Broader implications of progressive liver dysfunction and lethal sepsis in two boys following systemic high-dose AAV. Mol. Ther. 2020;28:1753–1755. doi: 10.1016/j.ymthe.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinderer C, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzmin DA, et al. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021;20:173–174. doi: 10.1038/d41573-021-00017-7. [DOI] [PubMed] [Google Scholar]
- 29.Au HKE, Isalan M, Mielcarek M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Front. Med. 2022;8:809118. doi: 10.3389/fmed.2021.809118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. Biodrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy JM, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villiger L, et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 34.Truong DJ, et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin H, et al. Vision rescue via unconstrained in vivo prime editing in degenerating neural retinas. J. Exp. Med. 2023;220(5):e20220776. doi: 10.1084/jem.20220776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 37.Varnavski AN, Calcedo R, Bove M, Gao G, Wilson JM. Evaluation of toxicity from high-dose systemic administration of recombinant adenovirus vector in vector-naive and pre-immunized mice. Gene Ther. 2005;12:427–436. doi: 10.1038/sj.gt.3302347. [DOI] [PubMed] [Google Scholar]
- 38.Zettler J, Schutz V, Mootz HD. The naturally split Npu DnaE intein exhibits an extraordinarily high rate in the protein trans-splicing reaction. FEBS Lett. 2009;583:909–914. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Davis JR, et al. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022;6:1272–1283. doi: 10.1038/s41551-022-00911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi JH, et al. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol. Brain. 2014;7:17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swiech L, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR–Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen PJ, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreira da Silva J, et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat. Commun. 2022;13:760. doi: 10.1038/s41467-022-28442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uhlén M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 46.Brooks PJ. DNA repair in neural cells: basic science and clinical implications. Mutat. Res. 2002;509:93–108. doi: 10.1016/s0027-5107(02)00222-1. [DOI] [PubMed] [Google Scholar]
- 47.Pinto RM, et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 2013;9:e1003930. doi: 10.1371/journal.pgen.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson JW, et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022;40:402–410. doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gray SJ, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidigal JA, Ventura A. Rapid and efficient one-step generation of paired gRNA CRISPR–Cas9 libraries. Nat. Commun. 2015;6:8083. doi: 10.1038/ncomms9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koblan LW, et al. In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature. 2021;589:608–614. doi: 10.1038/s41586-020-03086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Packer MS, et al. Evaluation of cytosine base editing and adenine base editing as a potential treatment for alpha-1 antitrypsin deficiency. Mol. Ther. 2022;30:1396–1406. doi: 10.1016/j.ymthe.2022.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murillo O, et al. Long-term metabolic correction of Wilsonʼs disease in a murine model by gene therapy. J. Hepatol. 2016;64:419–426. doi: 10.1016/j.jhep.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 54.Kotewicz ML, Sampson CM, D’Alessio JM, Gerard GF. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. Nucleic Acids Res. 1988;16:265–277. doi: 10.1093/nar/16.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan KY, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hordeaux J, et al. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood–brain barrier. Mol. Ther. 2019;27:912–921. doi: 10.1016/j.ymthe.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang Q, et al. Delivering genes across the blood–brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE. 2019;14:e0225206. doi: 10.1371/journal.pone.0225206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arboleda-Velasquez JF, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 2019;25:1680–1683. doi: 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hernandez I, et al. Heterozygous APOE Christchurch in familial Alzheimer’s disease without mutations in other Mendelian genes. Neuropathol. Appl. Neurobiol. 2021;47:579–582. doi: 10.1111/nan.12670. [DOI] [PubMed] [Google Scholar]
- 60.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol. Brain. Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 62.Kim JY, et al. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. Eur. J. Neurosci. 2013;37:1203–1220. doi: 10.1111/ejn.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foust KD, et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang H, et al. Several rAAV vectors efficiently cross the blood–brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011;19:1440–1448. doi: 10.1038/mt.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cohen J, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 66.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 67.Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–448. doi: 10.1016/j.atherosclerosis.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 68.Rao AS, et al. Large-scale phenome-wide association study of PCSK9 variants demonstrates protection against ischemic stroke. Circ. Genom. Precis. Med. 2018;11:e002162. doi: 10.1161/CIRCGEN.118.002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abifadel M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 70.Fitzgerald K, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Musunuru K, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 72.Rothgangl T, et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Banskota S, et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185:250–265. doi: 10.1016/j.cell.2021.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verve takes base editors into humans. Nat. Biotechnol.40, 1159 (2022). [DOI] [PubMed]
- 75.Mayne J, et al. Novel loss-of-function PCSK9 variant is associated with low plasma LDL cholesterol in a French-Canadian family and with impaired processing and secretion in cell culture. Clin. Chem. 2011;57:1415–1423. doi: 10.1373/clinchem.2011.165191. [DOI] [PubMed] [Google Scholar]
- 76.Lebeau P, et al. Loss-of-function PCSK9 mutants evade the unfolded protein response sensor GRP78 and fail to induce endoplasmic reticulum stress when retained. J. Biol. Chem. 2018;293:7329–7343. doi: 10.1074/jbc.RA117.001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lebeau PF, et al. The loss-of-function PCSK9Q152H variant increases ER chaperones GRP78 and GRP94 and protects against liver injury. J. Clin. Invest. 2021;131:e128650. doi: 10.1172/JCI128650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lazzarotto CR, et al. Defining CRISPR–Cas9 genome-wide nuclease activities with CIRCLE-seq. Nat. Protoc. 2018;13:2615–2642. doi: 10.1038/s41596-018-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim DY, Moon SB, Ko J-H, Kim Y-S, Kim D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020;48:10576–10589. doi: 10.1093/nar/gkaa764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, et al. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schene IF, et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020;11:5352. doi: 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin S, et al. Genome-wide specificity of prime editors in plants. Nat. Biotechnol. 2021;39:1292–1299. doi: 10.1038/s41587-021-00891-x. [DOI] [PubMed] [Google Scholar]
- 83.Geurts MH, et al. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci Alliance. 2021;4:e202000940. doi: 10.26508/lsa.202000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park SJ, et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime editor. Genome Biol. 2021;22:170. doi: 10.1186/s13059-021-02389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lin J, et al. Modeling a cataract disorder in mice with prime editing. Mol. Ther. Nucleic Acids. 2021;25:494–501. doi: 10.1016/j.omtn.2021.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao P, et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol. 2021;22:83. doi: 10.1186/s13059-021-02304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Habib O, Habib G, Hwang GH, Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 2022;50:1187–1197. doi: 10.1093/nar/gkab1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao R, et al. Genomic and transcriptomic analyses of prime editing guide RNA–independent off-target effects by prime editors. CRISPR J. 2022;5:276–293. doi: 10.1089/crispr.2021.0080. [DOI] [PubMed] [Google Scholar]
- 89.Meunier L, Larrey D. Drug-induced liver injury: biomarkers, requirements, candidates, and validation. Front. Pharmacol. 2019;10:1482. doi: 10.3389/fphar.2019.01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao G-P, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thakore PI, et al. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR–Cas9 repressors. Nat. Commun. 2018;9:1674. doi: 10.1038/s41467-018-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meyer K, et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose–response study in mice and nonhuman primates. Mol. Ther. 2015;23:477–487. doi: 10.1038/mt.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kishimoto TK, Samulski RJ. Addressing high dose AAV toxicity—‘one and done’ or ‘slower and lower’? Expert Opin. Biol. Ther. 2022;22:1067–1071. doi: 10.1080/14712598.2022.2060737. [DOI] [PubMed] [Google Scholar]
- 95.Anzalone AV, et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022;40:731–740. doi: 10.1038/s41587-021-01133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yarnall, M. T. N. et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol.10.1038/s41587-022-01527-4 (2022). [DOI] [PMC free article] [PubMed]
- 97.Clement K, et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lazzarotto CR, et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davis, J. R. et al. High throughput sequencing data for the efficient delivery of prime editors in vivo. NCBI, PRJNA898625 (2023). https://www.ncbi.nlm.nih.gov/bioproject/PRJNA898625
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–6, Supplementary Notes 1–6, Supplementary Sequences and Table of Contents also describing supplementary tables (which are provided in a separate Excel file).
Data Availability Statement
All pegRNA, nicking sgRNA and HTS primer sequences used for this study are provided in supplementary tables. High-throughput DNA sequencing data files are available from the National Center for Biotechnology Informationʼs Sequence Read Archive under accession code PRJNA898625 (ref. 99). DNA sequences of AAV genomes are provided in the Supplementary Information. Key plasmids from this work are available from Addgene, and other plasmids and raw data are available from the corresponding author upon reasonable request.
The code used for analysis of HTS data is available at https://github.com/pinellolab/CRISPResso2.