

Abstract
Oncolytic herpes simplex viruses (oHSVs) have emerged as leading cancer therapeutic agents. Effective oHSV virotherapy may ultimately require both intratumoral and systemic vector administration to target the primary tumor and distant metastases. An attractive approach to enhancing oHSV tumor specificity is engineering the virus envelope glycoproteins for selective recognition of and infection via tumor-specific cell surface proteins. We previously demonstrated that oHSVs could be retargeted to EGFR-expressing cells by the incorporation of a single-chain antibody (scFv) at the N terminus of glycoprotein D (gD). Here, we compared retargeted oHSVs generated by the insertion of scFv, affibody molecule, or VHH antibody ligands at different positions within the N terminus of gD. When compared to the scFv-directed oHSVs, VHH and affibody molecules mediated enhanced EGFR-specific tumor cell entry, spread and cell killing in vitro, and enabled long-term tumor-specific virus replication following intravenous delivery in vivo. Moreover, oHSVs retargeted via a VHH ligand reduced tumor growth upon intravenous injection and achieved complete tumor destruction after intratumoral injection. Systemic oHSV delivery is important for the treatment of metastatic disease, and our enhancements in targeted oHSV design are a critical step in creating an effective tumor-specific oHSVs for safe administration via the bloodstream.
Keywords: MT: Regular Issue, HSV, oncolytic, glycoprotein, vector targeting, EGFR, GBM
Graphical abstract
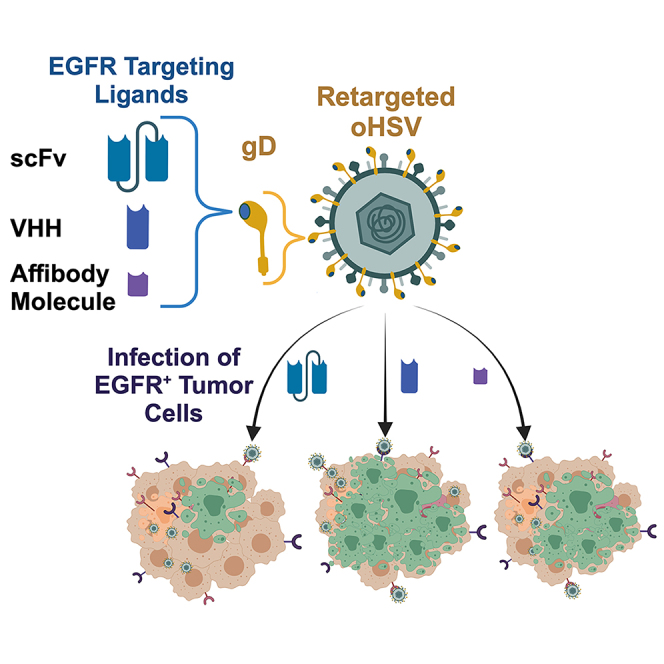
Glorioso and colleagues describe the development of oHSVs that use VHH antibodies and affibody molecules to enhance retarget virus entry and spread in EGFR-expressing tumor cells. In vivo, systemic oHSV delivery resulted in tumor-specific replication and reduced tumor growth, whereas direct intratumoral delivery resulted in complete tumor regression.
Introduction
Considerable effort has gone into creating safe and effective herpes simplex virus (HSV) vectors that demonstrate tumor cell–specific oncolysis (oHSV).1 The oHSV vector talimogene laherparepvec (T-VEC) showed durable responses in 16.3% of melanoma patients following direct intratumoral delivery,2 and was approved by the US Food and Drug Administration in 2015 for the treatment of solid tumors.3 T-VEC is attenuated by the deletion of the genes encoding γ34.5 and ICP47, and expresses the human granulocyte-macrophage colony-stimulating factor gene to stimulate an immune response.2,3 The γ34.5 protein inhibits the innate interferon (IFN)-mediated response to virus infection, and eliminating its expression inhibits virus growth in normal cells but allows growth in tumor cells that contain defects in IFN response pathways.4 Clinical trials have shown T-VEC to be well tolerated and suggest that further modifications will be required to realize effective oHSV-directed cancer therapy.5
The vectors described here use alternative approaches to accomplish tumor-restricted vector growth. First, the differential microRNA (miR) expression that exists between normal cells and tumor cells can be leveraged to restrict virus replication to tumors. In glioblastoma (GBM), miR-124 levels are low or undetectable, whereas miR-124 is highly expressed in normal neurons.6,7 Incorporation of miR-124 target sites into the 3′ UTR of the ICP4 gene blocked virus replication in miR-124-expressing cells in culture and prevented both virus replication and HSV-induced encephalitis in vivo.8 Second, vector tropism can be restricted to cells expressing epidermal growth factor receptor (EGFR) or an EGFR mutant lacking exons 2–7 (EGFRvIII),8,9 commonly upregulated in tumors such as GBM.10,11 GBM are locally invasive brain tumors, although extraneural metastases to the regional lymph nodes, lungs, bone, and liver can occur late in the course of the disease.12 EGFR is also overexpressed in cancers such as head and neck, lung, and breast, providing a cell surface marker with broad utility for targeted cancer therapy.10,13,14 To reach both the primary tumor and distant metastases, oHSV treatment will likely require combined direct intratumoral (i.t.) delivery and intravenous (i.v.) administration, making vector retargeting an ideal mechanism to ensure safety and tumor infectivity.
Vector retargeting can be accomplished by modifying the viral envelope glycoproteins involved in the process of HSV receptor binding and cell entry.15 Glycoprotein D (gD) determines vector tropism by binding to one of its cellular receptors, herpesvirus entry mediator (HVEM or HveA), nectin-1 (HveC), and 3-O-sulfated heparan sulfate. Receptor binding causes a conformational change in gD and triggers a cascade of interactions among the other essential glycoproteins, the gH/gL heterodimer, and the viral fusogen gB. gB mediates fusion between the virus envelope and the cell surface or endosomal membrane, ultimately releasing the nucleocapsid into the cytoplasm. To retarget virus infection, the natural receptor-binding properties of gD must be eliminated by the deletion or modification of specific amino acids in the N terminus of gD, and alternative protein-binding moieties that recognize the desired cell surface protein can be inserted at the site of deletion.
We previously demonstrated that a single-chain variable fragment (scFv) recognizing human EGFR and EGFRvIII (together referred to here as EGFR/vIII) inserted in place of gD residues 2–24 specifically retargeted virus infection to EGFR-expressing cells.9 In this retargeted HSV, the Δ2-24 mutation eliminated HVEM binding, and the tyrosine at gD residue 38 was replaced with cysteine to ablate nectin-1 binding (Y38C). Combining EGFR-retargeted gD with two entry-enhancing mutations in gB (gB:NT)16 significantly enhanced retargeted virus entry. This virus demonstrated EGFR/vIII-specific cell entry and enhanced survival in a mouse model of human GBM.9
We have since replaced the Y38C substitution with a complete deletion of residue 38 (Δ38) to ensure that the propagation of virus on nectin-1-expressing cell lines does not result in reversion to nectin-1-dependent entry.17 However, initial data indicated that the efficacy of the EGFR-retargeted Δ38 virus needed to be enhanced for effective tumor cell killing. Here, we explored the use of alternative EGFR/vIII ligands to mediate vector retargeting, including an affibody molecule and three variable domain from heavy-chain-only antibodies (VHH).18,19 These ligands were assessed at the original Δ2-24 position, and further downstream of the signal peptide (SP) cleavage site (Δ6/7-24). Using this approach, we identified multiple retargeted gD designs that demonstrated significantly improved entry, spread, and tumor cell killing in vitro and in vivo when compared to the original scFv-based virus.
Results
Vector engineering
Our overall goal is to optimize the safety and efficacy of our EGFR-retargeted oHSV. We have therefore explored the use of several different retargeting designs for our recombinant gD. Recombinant viruses expressing the retargeted gD variants were created in a derivative of the KG4:T124 viral backbone (strain KOS).8,20 KG4:T124 contains loxP-flanked bacterial artificial chromosome (BAC) sequences for genome manipulation in bacteria, a deletion of the internal repeat region, the EGFP gene fused in-frame to the gC gene via a 2A peptide sequence (gC-EGFP), two entry-enhancing mutations in the gB gene (gB:NT),16 and four copies of a miR-124 response element (T124) in the 3′ UTR of the ICP4 gene.8 In the present work, a Gateway (GW) cassette was introduced in place of the gD coding sequence (KG4:T124-ΔgD:GW) (Figure 1A) to facilitate the insertion of genetically modified gD genes containing alternative EGFR/vIII ligands (Figure 1B).
Figure 1.

Vector engineering
(A) Schematic representation of the KG4:T124-ΔgD:GW vector, illustrating the unique long (UL), unique short (US), terminal repeat long (TRL), and terminal repeat short (TRS) segments of the HSV genome. KG4:T124-ΔgD:GW contains BAC sequence between UL37 and UL38, two viral entry-enhancing mutations in the gB glycoprotein gene (gB:NT), a deletion encompassing the adjacent internal repeat sequences IRL and IRS (ΔJOINT), a GFP marker gene linked to the gC open reading frame via a T2A sequence (gC-T2A-GFP), and 4 copies of a miR-124 recognition sequence in the 3′ UTR of the ICP4 gene (ICP4:T124). A GW cassette replaces the coding sequence for the gD glycoprotein (ΔgD:GW). (B) Schematic of gD showing the locations of the SP, immunoglobulin-like fold (Ig fold), profusion domain (PFD), and transmembrane domain (TM). Residue 38 is deleted in all of the retargeted gD glycoproteins to ablate nectin-1 binding. The black box (Δx-24) indicates the residues removed to eliminate HVEM binding (the x position varies per construct); this is the insertion site for the targeting ligands shown below. (C) Glycoprotein incorporation into purified virus particles was assessed by western blot. 1 × 108 gc of purified virus was loaded per lane and probed with antibodies recognizing the envelope glycoproteins gD and gB and the tegument protein VP16. Relative gD and gB band intensities normalized to VP16 and set to gD:scEΔ38 = 1× are shown below the lanes. Western blots were performed in duplicate and representative images and band intensities are shown.
We assessed several different criteria in the construction of the retargeted gD proteins, including different EGFR binding ligands and alternative insertion positions within the recombinant gD molecule. The original EGFR-targeted gD used an scFv (248 amino acids [aa]) in place of residues 2–24 of gD. The alternative EGFR/vIII ligands tested here included an affibody molecule (58 aa; ZEGFR)19 and 3 VHH antibodies (referred to here as single domain [SD] 1–3; 124–130 aa)18 (Figure 1B). We tested insertion sites at the original Δ2-24 position and further downstream of the SP cleavage site (Δ6/7-24). To create the recombinant gDs, we genetically replaced residues 2–24, 6–24, or 7–24 in the HVEM binding N-terminal region with the appropriate EGFR ligands (Figure 1B), and deleted residue 38 (Δ38) to ablate nectin-1 recognition. The modified gDs were introduced into the KG4:T124-ΔgD:GW BAC by LR-Clonase reaction. Viruses were produced by transfection of BAC DNA into Vero cells that naturally express both nectin-1 and EGFR (Table 1).
Table 1.
Viral recombinants
| Virusa | Genome copy titer (gc/mL)b | Plaque-forming unit titer (pfu/mL)c | gc/pfu |
|---|---|---|---|
| gD:WT | 1 × 1012 | 1.9 × 109 | 526 |
| gD:scEGFRΔ2-24 | 5.5 × 1011 | 7.5 × 107 | 7,000 |
| gD:scEGFRΔ7-24 | 3.5 × 1011 | 1.1 × 107 | 31,800 |
| gD:SD1Δ6-24 | 6.6 × 1011 | 5.8 × 108 | 1,100 |
| gD:SD2Δ6-24 | 6.2 × 1011 | 1.6 × 108 | 3,900 |
| gD:SD3Δ6-24 | 8 × 1011 | 1.4 × 108 | 5,700 |
| gD:SD1Δ2-24 | ND | ND | |
| gD:SD2Δ2-24 | 8.5 × 1010 | 1.2 × 107 | 7,100 |
| gD:SD3Δ2-24 | ND | ND | |
| gD:ZEGFRΔ2-24 | 6.4 × 1011 | 3 × 108 | 2,000 |
| gD:ZEGFRΔ7-24 | 5.5 × 1011 | 1.6 × 108 | 3,400 |
| gD:ZdimΔ2-24 | 1.8 × 1011 | 5.6 × 108 | 321 |
| gD:ZdimΔ7-24 | 1 × 1011 | 2 × 107 | 5,000 |
| Fluc-gD:scE | 3.8 × 1012 | 5 × 109 | 760 |
| Fluc-gD:ZE | 2 × 1012 | 3 × 109 | 660 |
| Fluc-gD:SD2 | 2.5 × 1012 | 2.5 × 109 | 1,000 |
Virus names describe the corresponding recombinant gD construct design. Deletion notations Δ2-24, Δ6-24, or Δ7-24 specify the insertion site of the noted targeting ligand: scEGFR and scE (scFv), SD (VHH), ZEGFR/ZE, and Zdim (affibody monomer and dimer molecules). All contain the Δ38 nectin-1 detargeting mutation. Fluc, firefly luciferase added to the viral backbone.
Genome copy titers were determined by qPCR for UL5; DNA from purified virus particles. ND, not determined.
Plaque-forming unit titers were determined by standard plaque assay on Vero cells. ND, not determined.
Characterization of recombinant virus production and retargeted gD expression
The majority of gD derivatives produced functional virus, able to form plaques on Vero cells; however, the efficiency of virus production varied significantly between constructs (Table 1). ZEGFR was tested as a monomer or dimer at positions Δ2-24 and Δ7-24. Based on virus replication and stock production, the ZEGFR ligand functioned similarly in all cases, yielding high titer stocks of replicating virus. When positioned at Δ2-24, SD1, SD2, and SD3 yielded viruses that formed small plaques that did not grow well on Vero cells. Virus containing gD:SD2Δ2-24 grew well enough to obtain a low titer stock of virus, whereas the SD1 and SD3 variants were discontinued due to poor growth. The same SD ligands inserted at position Δ6-24 generated viruses that grew robustly, and these were selected for further study. For the original scFv ligand, the two positions tested (Δ2-24 and Δ7-24) produced viruses with comparable growth. These data suggested that ligand position may be adjusted to improve the function of specific ligands, such as the SD antibodies, when designing retargeted gD proteins.
For each recombinant virus, biological titers in plaque-forming units (pfu) per milliliter were determined by plaque assay on Vero cells and physical titers in genome copies (gc) per milliliter were established by real-time qPCR for the HSV-1 early gene unique long 5 (UL5) (Table 1). Because gD modification significantly influences virus entry and plaque formation, gc titers were used for viral backbone comparisons. Purified virus particles were analyzed by western blot for the envelope glycoproteins gD and gB and the tegument protein VP16. When compared at an equivalent gc input, similar VP16 levels were seen for each virus stock, indicating that gc titers provide a good estimate of viral particles for comparison of the recombinant viruses. The viruses also demonstrated comparable levels of the gB glycoprotein. Each virus contained a modified gD glycoprotein at the predicted molecular weight. However, unlike the levels of VP16 and gB, recombinant gD levels varied considerably between constructs, falling well below wild-type (wt) gD (gD:wt) levels (Figure 1C).
Entry specificity of the retargeted recombinant viruses
To assess the ability of purified KG4:T124-gD recombinant viruses to enter cells via interaction with full-length human (h)EGFR, we infected J1.1-2 cells that are resistant to HSV infection due to the absence of gD receptors21 and derivatives expressing human nectin-1 (J-C)22 or hEGFR (J-EGFR).23 Cells were infected at an MOI of 1,000 gc/cell and virus entry was assessed 6 h postinfection (hpi) by immunostaining for the immediate-early (IE) HSV protein ICP4. As shown in Figure 2, the gD:scEGFRΔ2-24, gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 viruses were able to enter J-EGFR cells but were unable to enter J-C or J1.1-2 cells. In contrast, the control virus expressing gD:wt from the same backbone was able to enter J-C cells, but not J1.1-2 or J-EGFR cells.
Figure 2.

The retargeted viruses enter cells via human EGFR
Human EGFR-specific cell entry was assessed on HSV receptor-deficient J1.1-2 cells, J-C (nectin-1), and J-EGFR (hEGFR) cells. Cells were infected for 6 h at an MOI of 1,000 gc/cell and immunostained with antibody recognizing the IE protein ICP4 as a marker of virus entry (green) and counterstained with DAPI as a positive control (blue). Scale bars, 200 μM.
To assess virus entry via EGFRvIII, we infected B78H1 cells that do not express the natural HSV-1 receptors24 and B78H1 derivatives transduced with EGFRvIII (B78-vIII)17 or nectin-1 (B78-C).25 At 6 hpi, ICP4 staining was observed on B78-vIII cells for the gD:scEGFRΔ2-24, gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 viruses, whereas no staining was observed on either B78H1 or B78-C cells. As expected, the gD:wt control virus was able to enter the B78-C cell line, but not B78H1 and B78-vIII (Figure 3). These results indicated that entry of the retargeted viruses was no longer supported by the cellular expression of nectin-1, and that the scFv, affibody molecule, and VHH domains mediated entry via both EGFR and EGFRvIII.
Figure 3.

The retargeted viruses enter cells via the tumor-associated EGFRvIII mutant
EGFRvIII-specific cell entry was assessed on HSV receptor-deficient B78H1 cells, B78-C (nectin-1), and B78-vIII (mutant EGFRvIII) cells. Cells were infected for 6 h (MOI of 1,000 gc/cell) and immunostained with antibody recognizing the IE protein ICP4 (green) and counterstained with DAPI as a positive control (blue). Scale bars, 200 μM.
Lateral spread of EGFR retargeted gD
Effective oncolytic activity is influenced by both entry efficiency and subsequent lateral spread. The infectious center (IC) assay specifically examines viral cell-to-cell spread by eliminating the first stage of virus entry into the cells as a variable. Using the IC assay, we compared different acceptor cells for plaque formation induced by lateral spread from a single source of intracellular virus (donor cell).25 Vero donor cells were infected at an MOI of 10,000 gc/cell to achieve 100% infection and were washed with acidic glycine at 2 hpi to remove the extracellular virus. Equal numbers of infected donor cells were overlaid on monolayers of uninfected acceptor cells, and plaque areas were quantified 2 days later. We assessed plaque formation on Vero cells and on three tumor cell lines that express EGFR.26,27,28
As shown in Figure 4A, each of the viruses formed plaques on Vero cells, and plaque size varied significantly among the viruses. The gD:wt virus showed an average plaque size of 0.25 mm2. The gD:scEGFRΔ2-24 and gD:scEGFRΔ7-24 viruses, targeted to EGFR via the same scFv ligand located at different positions within the recombinant gD molecule, demonstrated similar plaque sizes of 0.04 and 0.03 mm2, respectively. Likewise, the viruses retargeted via a monomer affibody molecule, gD:ZEGFRΔ2-24 and gD:ZEGFRΔ7-24, yielded similar plaque sizes (0.13 mm2) regardless of the position of the ligand within recombinant gD.Viruses targeted via an affibody dimer molecule demonstrated smaller plaques that also differed in size between the two ligand positions in gD; the average plaque size for gD:ZdimΔ2-24 was 0.09 mm2 and the average plaque size for gD:ZdimΔ7-24 was 0.02 mm2. The gD:SD1Δ6-24, gD:SD2Δ6-24, and gD:SD3Δ6-24 viruses, targeted to EGFR via 3 distinct SD antibodies, demonstrated plaque sizes of 0.13 mm2, 0.36 mm2, and 0.13 mm2, respectively (Figure 4A).
Figure 4.

Viruses retargeted by the SD antibodies or affibody molecules demonstrate enhanced cell-to-cell spread
Vero donor cells were infected at an MOI of 10,000 gc/cell, extracellular virus was inactivated, and equal numbers of infected donor cells were added onto monolayers of uninfected (A) Vero, (B) U251, (C) SNB19, or (D) A549 acceptor cells. Plaques were imaged at 2 dpi and plaque size was quantified using ImageJ software. Averages were calculated ±SEM, and 1-way ANOVA analyses were used to determine differences observed between groups (n = 5–15; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001).
Similar data were obtained using U251 acceptor cells (Figure 4B). The average plaque sizes observed for gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, gD:ZEGFRΔ2-24, gD:ZEGFRΔ7-24, gD:ZdimΔ2-24, and gD:ZdimΔ7-24 were significantly larger than those recorded for gD:scEGFRΔ2-24, and we detected no significant differences in plaque formation between the gD:scEGFRΔ2-24 and gD:scEGFRΔ7-24 viruses. Since we observed no significant difference between gD:scEGFRΔ2-24 and gD:scEGFRΔ7-24 on both Vero and U251 cells, we omitted the latter from further analyses. For similar reasons, we selected the gD:ZEGFRΔ7-24 virus to represent the viruses targeted via affibody molecules.
We tested two more human tumor cell lines to evaluate lateral spread, the SNB19 and A549 cell lines, respectively a GBM and an adenocarcinoma cell line. When infected Vero cells were seeded on either SNB19 cells (Figure 4C) or A549 cells (Figure 4D), the gD:SD1Δ6-24, gD:SD2Δ6-24, and gD:SD3Δ6-24 viruses consistently showed significantly larger plaque sizes than the gD:scEGFRΔ2-24 virus (p < 0.0001). The gD:ZEGFRΔ7-24 virus also showed significantly improved lateral spread relative to the gD:scEGFRΔ2-24 virus on both the SNB19 (p < 0.05) and A549 (p < 0.0001) cell lines. Together, these results indicated that cell-to-cell spread through EGFR was enhanced in the gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 viruses relative to that observed for the gD:scEGFRΔ2-24 virus. Interestingly, the gD:SD2Δ6-24 and gD:SD3Δ6-24 viruses exhibited enhanced lateral spread also compared to the gD:wt virus on both the SNB19 and A549 cell lines (p < 0.0001), and the gD:SD1Δ6-24 virus demonstrated enhanced lateral spread compared to the gD:wt virus on the A549 cells (p < 0.0001).
Entry efficiency for EGFR retargeted gD
To quantify viral entry, Vero (Figure 5A), U251 (Figure 5B), SNB19 (Figure 5C), and A549 (Figure 5D) cells were infected at an MOI of 1,000 gc/cell, and the percentage of ICP4+ cells relative to the total number of DAPI+ cells was determined at 6 hpi. On the Vero cell line, comparable numbers of ICP4+ cells were observed for all viruses (Figure 5A). The gD:scEGFRΔ2-24 virus entered 63% of Vero cells, and the gD:wt, gD:SD1Δ6-24, gD:SD2Δ6-24, and gD:SD3Δ6-24 viruses entered 76%, 55%, 69%, and 58% of Vero cells, respectively, with no observable statistical difference. The gD:ZEGFRΔ7-24 virus entered 82% of Vero cells, representing a significant but moderate increase relative to the gD:scEGFRΔ2-24 virus. On the U251 cell line, the percentage of ICP4+ cells was 13% for gD:scEGFRΔ2-24, 52% for gD:SD1Δ6-24, 85% for gD:SD2Δ6-24, 64% for gD:SD3Δ6-24, 39% for gD:ZEGFRΔ7-24, and 85% for gD:wt (Figure 5B). These data showed that both the VHH antibody–based and the affibody molecule–based retargeted viruses entered significantly more U251 tumor cells at the same gc input than the gD:scEGFRΔ2-24 virus.
Figure 5.

Viruses retargeted via the SD antibodies or affibody molecule demonstrate enhanced cell entry
(A) Vero, (B) U251, (C) SNB19, and (D) A549 cells were infected at 1,000 gc/cell, fixed at 6 hpi, and stained for ICP4 and DAPI. Fluorescent images were captured and quantified with ImageJ software. Data are presented as the percentage of ICP4+ cells relative to the total DAPI+ cells. Averages were calculated ±SEMs (n = 5). One-way ANOVA analyses were used to determine differences observed between groups. Statistical significance (∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001) is shown for gD:scEGFRΔ2-24 relative to the other viruses. Statically significant differences (p < 0.0001) were also observed between gD:wt and gD:SD2Δ6-24(SNB19 and A549); gD:wt and gD:SD3Δ6-24 (SNB19 and A549); gD:wt and gD:SD1Δ6-24 (A549) but are not illustrated.
Similar to what we observed for the U251 cell line, on the SNB19 cell line, the gD:wt virus entered 88% of the cells compared to only 18% for gD:scEGFRΔ2-24. Relative to gD:scEGFRΔ2-24, gD:SD1Δ6-24 (54%), gD:SD2Δ6-24 (73%), gD:SD3Δ6-24 (47%), and gD:ZEGFRΔ7-24 (36%) demonstrated significantly improved entry (Figure 5C). On the A549 cell line, the ICP4+ percentages observed for gD:wt and gD:scEGFRΔ2-24 were not as disparate (93% for gD:wt and 50% for gD:scEGFRΔ2-24) (Figure 5D). Still, significant increases in entry were observed for gD:SD1Δ6-24 (85%), gD:SD2Δ6-24 (82%), and gD:ZEGFRΔ7-24 (76%). At 64% ICP4+ cells, the gD:SD3Δ6-24 virus did not demonstrate a significant increase in entry relative to gD:scEGFRΔ2-24. Together, these data showed that entry varied in a cell type–dependent manner, with the gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 viruses generally demonstrating improvement relative to gD:scEGFRΔ2-24 virus and often approaching the entry values observed for gD:wt.
Tumor cell line killing
We next assessed virus-mediated killing using the alamarBlue cell viability assay. The U251, SNB19, and A549 cells were infected at an MOI of 1,000 gc/cell and followed for 72 h. The gD:wt, gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, gD:ZEGFRΔ7-24, and gD:scEGFRΔ2-24 viruses were cytotoxic in every cell line, resulting in a substantial reduction in cell viability over the 72 h time course (Figures 6A–6C). On all three cell lines, the gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 viruses resulted in a significant improvement in cell killing when compared to the gD:scEGFRΔ2-24 virus. These data were consistent with the overall improvement in both entry and spread observed for the viruses retargeted via SD antibodies or affibody molecules. Given the variability in entry observed among the viruses, we compared cell killing on the U251 cell line by first infecting donor Vero cells at high MOI with gD:scEGFRΔ2-24, gD:SD2Δ6-24, or gD:ZEGFRΔ7-24. As in the IC assay, infected Vero cells were then seeded on the U251 acceptor cells, and cell viability was evaluated by the alamarBlue assay. The results confirmed that even once the initial entry differences were removed, the gD:SD2Δ6-24 and gD:ZEGFRΔ7-24 viruses resulted in a significant improvement in cell killing relative to the gD:scEGFRΔ2-24 virus (Figure 6D).
Figure 6.

Virus-mediated cell death in vitro
(A) U251, (B) SNB19, or (C) A549 cells were infected at 1,000 gc/cell, and cell viability was assessed at 24, 48, and 72 hpi by alamarBlue assay. Data are presented as the percentage of viable cells relative to uninfected cells at each time point; average ±SEM (n = 5–8). Statistics were determined by 2-way ANOVA. The viability of U251, SNB19, and A549 cells infected with gD:wt, gD:SD1Δ6-24, gD:SD2Δ6-24, gD:SD3Δ6-24, and gD:ZEGFRΔ7-24 was significantly reduced compared to gD:scEGFRΔ2-24. For U251 cells, p < 0.0001 for all time points. For SNB19 cells, p < 0.0001 at 24 and 48 hpi for each comparison; at 72 hpi, p < 0.001 for gD:SD1Δ6-24, gD:SD2Δ6-24, and gD:wt, p < 0.01 for gD:SD3Δ6-24, and p < 0.05 for gD:ZEΔ7-24. For A549 cells, at 24 hpi, p < 0.0001 for gD:SD2Δ6-24, p < 0.01 for gD:wt and gD:SD1Δ6-24, and p < 0.05 for gD:SD3Δ6-24; at 48 hpi, p < 0.0001 for gD:SD2Δ6-24, p < 0.001 for gD:wt, and p < 0.01 for gD:SD1Δ6-24 and gD:ZEΔ7-24; at 72 hpi, p < 0.01 for gD:SD1Δ6-24, gD:wt, and gD:SD2Δ6-24. (D) Infected Vero donor cells were mixed with U251 acceptor cells (1 Vero cell per 10 U251 cells), and cell viability was evaluated by alamarBlue assay. The gD:ZEGFRΔ7-24 virus significantly reduced cell viability when compared to gD:scEGFRΔ2-24 at 24, 48 (p < 0.05), and 72 hpi (p < 0.001). gD:SD2Δ6-24 significantly reduced cell viability at 48 and 72 hpi when compared to gD:scEGFRΔ2-24 (p < 0.001).
EGFR retargeted HSV-induced tumor regression in a nude mouse model
To assess the in vivo oncolytic activity of the EGFR-retargeted viruses, we introduced a firefly luciferase (fLuc) reporter gene under the control of the cytomegalovirus (CMV) promoter into the KG4:T124-ΔgD:GW BAC genome at the intergenic region between UL50 and UL51 (KG4:T124_fLuc-ΔgD:GW). The modified gD glycoprotein genes, gD:scEGFRΔ2-24, gD:SD2Δ6-24, and gD:ZEGFRΔ7-24, were introduced into the KG4:T124_fLuc-ΔgD:GW BAC, and the Fluc-gD:scE, Fluc-gD:SD2, and Fluc-gD:ZE viruses were produced as described above (Table 1). It was previously reported that the affibody molecule showed high affinity for both murine (m)EGFR and hEGFR.19 We therefore generated a B78H1 cell line that expresses mEGFR (B78-mEGFR; Figure S1) and demonstrated that Fluc-gD:ZE but not Fluc-gD:scE or Fluc-gD:SD2 entered cells via mEGFR (Figure S1). In murine tumor models, the Fluc-D:ZE virus offers the utility of evaluating both virus replication in the targeted tumor and off-target infection in nontumor tissue.
We treated U251 subcutaneous flank tumors with a single dose of oHSV by i.v. administration and monitored tumor volume and virus biodistribution by bioluminescent imaging (BLI). Animals received 1 × 1010 gc of Fluc-gD:scE, Fluc-gD:SD2, Fluc-gD:ZE, or vehicle control by tail vein delivery when tumors reached an average volume of 100 mm3. At 1 day postvirus delivery, luciferase signal was detected for all three viruses in the liver, which cleared by 48 h. We also observed that all three viruses accumulated in the tumor with no further off-target detection. The BLI signal for Fluc-gD:SD2 peaked at 7 days (average of 1.7 × 108 photons per second [p/s]) and stayed within 1 log of the peak value over the remaining observation period. Similarly, the Fluc-gD:ZE signal peaked at 5 days (average of 1.7 × 108 p/s) and stayed within 1–1.5 log of the peak value for the remaining observation period. The luciferase signal for Fluc-gD:scE also peaked at 5 days (average of 4.4 × 107 p/s), but in contrast to the other two viruses, the BLI signal declined progressively (Figure 7A). BLI signals were significantly higher at 7 and 10 days for Fluc-gD:SD2 (p < 0.0.0001 and p < 0.001, respectively) and at 5 days for Fluc-gD:ZE virus (p < 0.001) when compared to Fluc-gD:scE. Following treatment, tumor volume in the vehicle and Fluc-gD:scE-treated groups increased steadily, reaching in average ∼1,300 mm3 at 18 days (Figure 7B). At 18 days, the average tumor size in the Fluc-gD:SD2- and Fluc-gD:ZE-treated groups was ∼730 and ∼890 mm3, respectively. Fluc-gD:SD2-treated tumors were significantly smaller than PBS-treated tumors at 18 days postvector delivery (p < 0.01). We compared the area under the curve (AUC)29 for each treatment group to evaluate the trajectory of tumor growth (Figure 7C), and observed a significant difference between Fluc-gD:SD2-treated tumors and PBS-treated controls (p < 0.05). Although these data indicated that a single i.v. dose of oHSV was not sufficient to cause tumor regression, they provided compelling evidence of tumoricidal activity.
Figure 7.

Treatment of U251 subcutaneous tumors by systemic or intratumoral vector delivery
U251 cells were implanted in the right hind flank of BALB/c athymic nude mice, and when tumors reached a volume of ∼100 mm3, the mice were treated with 1 × 1010 gc of the indicated virus or vehicle control (PBS). Black arrows indicate the days of treatment. (A–C) The i.v. delivery of a single dose of Fluc_gD:scE (scE), Fluc_gD:SD2 (SD2), or Fluc_gD:ZE (ZE) (n = 5 mice per group). (D–F) Four doses of SD2 delivered i.t. or i.v. (n = 3 mice per group). (A and F) Fluc expression from the viral backbone was quantified by BLI in a time course beginning 1 day after vector delivery and expressed as p/s (mean ± SEM). (B and D) Tumor growth was assessed by creating a growth curve of tumor volume (mm3; mean ± SEM) and by (C and E) determining the AUC for the growth curves generated in (B) and (D). Statistical differences were determined by 2-way ANOVA (A, B, D, and F) and by 2-tailed nonparametric Mann-Whitney test (C and E) (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
To determine whether multiple injections of oHSV delivered by either i.v. administration or direct i.t. injection could improve oHSV activity, we treated U251 tumor-bearing mice with gD:SD2 virus by i.v. or i.t. delivery every 2 days for a total of 4 doses. The vehicle-treated tumors increased steadily in size over the 18-day time course to ∼1,000 mm3. Multiple i.t. injections resulted in a decrease in the average tumor size by 5 days postvector delivery, and tumors were completely cleared by 18 days (Figure 7D). For animals treated with multiple i.v. injections of Fluc-gD:SD2, the tumor size remained significantly smaller than controls for 9 days after treatment, followed by a progressive increase (Figure 7D). Based on the AUC, multiple i.v. doses significantly reduced tumor growth when compared to PBS-treated controls (Figure 7E). The BLI signal in i.t.-treated animals was higher than in i.v.-treated animals at 1 day postdelivery (3 × 107 p/s i.t. compared to 3 × 104 p/s i.v.; p < 0.05) and 3 days postdelivery (1 × 108 p/s i.t. compared to 5 × 107 p/s i.v.; p < 0.001), remained high for 7 days, and then decreased in a manner consistent with the decrease in tumor size, reaching background levels when tumor regression was complete (Figure 7F). In the i.v.-treated animals, the BLI signal peaked at 3 days (5 × 107 p/s) and remained largely constant within the tumor over the observation period, declining between 13 and 18 dpi (Figure 7F). These data show that by repeated i.t. delivery of 1 × 1010 viral gc, retargeted oHSV was able to eradicate the tumor. Following i.v. delivery with the same variables, the retargeted vector was able to transduce and replicate in the tumor cells, reducing tumor growth, but did not completely lead to tumor regression.
Discussion
Despite significant therapeutic advancement, oHSV therapy still faces significant challenges, such as improving viral delivery to cancer cells, augmenting oHSV replication and spread in the tumor microenvironment (TME), and ultimately stimulating an efficacious antitumor immune response. In this study, our overall goal was to create a safe and effective vector that retargets virus infection to the tumor-associated surface marker EGFR. Tumor-selective vector infection allows for high-dose i.v. delivery that will expand the utility of oHSV therapies and may prove important for the treatment of metastatic disease where direct i.t. delivery is not possible.
During WT virus entry, gD–receptor interaction results in conformational changes30,31,32 that are transmitted via the gH/gL heterodimer to the fusogen gB to induce fusion of the viral envelope with the cell membrane.33 The aa residues necessary for cognate receptor binding have been well defined.32,34,35,36,37 Vector retargeting may be accomplished by the modification of gD to eliminate its cognate receptor interactions, introduce sequences for targeted receptor recognition, and preserve its required downstream interactions. oHSV vectors targeting tumor-associated human epidermal growth factor receptor 2 (HER2)38,39,40; epithelial cell adhesion molecule (EpCAM)41; carcinoembryonic antigen (CEA)9; chemokine receptor 4 (CXCR4)42; and glial-derived neurotrophic factor (GDNF)43 have achieved positive results in preclinical studies and illustrated the relative plasticity of the envelope glycoproteins to tolerate insertions of different ligands.37
Although scFv ligands have been used successfully in multiple oHSV retargeting studies,9,41,44,45 it has been suggested that the insertion of large sequences, such as scFv, into gD is not well tolerated.37 Our initial retargeted viruses used an scFv ligand specific for EGFR and EGFRvIII8,9,16,17; this targeting ligand introduces 248 aa residues into the Δ2-24 position of gD. scFv directed viruses mediated tumor cell–specific oncolysis but did not achieve WT levels of infection. Here, we have demonstrated that viruses retargeted via SD antibody ligands, approximately 120 aa residues smaller, achieved more robust tumor-specific entry and lateral cell-to-cell spread. In addition, viruses retargeted via an affibody molecule (58 aa) yielded a significant improvement over those retargeted via the scFv ligand but did not typically reach the entry, spread, or cell killing capability of the SD-based viruses. These data are consistent with the suggestion that the scFv-based virus could have been restricted in its infection capabilities based on the overall size of the ligand positioned within the N terminus of gD. However, the enhanced infectivity observed for the viruses retargeted via SD antibodies and affibody molecules was also likely influenced by differences between the ligands in overall affinity for EGFR.18,19,46
Insertion site also plays a pivotal role in retargeted oHSV design. The sites explored for ligand insertion include 6–38, 61–218,44,47 35–39,48 214–223, 219–223,49 and 2–24.8,9 These studies suggested that not all insertion sites are equivalent and that ligand position can greatly alter oHSV yields and gD function. For instance, the N terminus of gD seems to tolerate insertions better than internal positions in the gD ectodomain.37,50 Here, we engineered recombinant gD molecules with ligands at different positions within the N terminus of gD and demonstrated that the relative contribution of the insertion site to gD function varied with respect to the ligand used. The function of the recombinant gD glycoproteins was not significantly affected by ligand position when scFv or affibody molecules were used as targeting ligands. However, recombinant gD function varied greatly with respect to VHH position. The SD1-3 ligands grew poorly when inserted at position 2-24 of gD, forming small plaques and yielding low titer virus stocks (Table 1). In contrast, insertion of the SD1-3 ligands at position 6-24 resulted in a functional gD glycoprotein capable of supporting robust virus growth and high titer stock production. These findings suggest that ligand position may be adjusted to improve function when designing retargeted gD proteins and that no single rule can be defined that applies to all targeting ligands.
Our results suggest that oHSV retargeted to EGFR via the VHH and affibody molecules are more efficient in both entry and spread than the scFv ligand. Recently, a tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-armed oHSV was targeted to GBM stem-like cells (GSC) via insertion of an SD (nanobody) ligand and demonstrated promising results in preclinical studies,42 supporting the use of these smaller targeting moieties. Among the oHSVs retargeted to EGFR via VHH, the gD:SD2Δ6-24 virus outperformed the gD:SD1Δ6-24 and gD:SD3Δ6-24 viruses in entry efficiency, lateral spread, and tumor cell killing.
We compared retargeted vector activity in vivo in subcutaneous U251 xenografts in immunodeficient mice. Although not made up of a characteristic GBM brain TME, subcutaneous tumor models created with established human GBM cell lines are frequently used for initial assessments of HSV vector oncolytic activity following both i.t. and i.v. delivery.9,51,52 In comparison to gD:scE, the gD:SD2 virus showed improved oncolytic activity upon a single i.v. injection, significantly limiting overall tumor progression, although regression was not accomplished. Studies have shown that repeat administration of oHSV was more effective at inhibiting tumor growth and significantly increased the proportion of tumor-free mice.47,51 Consistent with this observation, we observed that multiple doses of gD:SD2 virus delivered i.v. significantly decreased tumor growth and multiple doses of gD:SD2 injected i.t. led to complete tumor clearance. These data suggest that we have significantly improved the oncolytic activity of our oHSV backbone but that additional steps must be taken to achieve complete responses by i.v. administration.
Several options exist to enhance retargeted HSV oncolysis. For example, we have recently shown that lateral spread of retargeted HSV can be enhanced by the addition of complementing mutations in other viral proteins.53 It was also demonstrated that i.v. delivery of retargeted oHSV-bearing syncytial mutations resulted in complete responses in immunodeficient mice bearing human xenografts.51 As mentioned above, subcutaneous xenografts in immunocompromised animals, such as the U251 assessed here, do not represent the complex and typically immunosuppressive TME that would be encountered by oHSVs in a clinical setting. Current evidence suggests that optimal oHSV candidates for cancer therapy will provide both efficient oncolysis and the induction of antitumor immunity. A recent clinical trial has linked oncolytic immunoactivation to survival in GBM,54 showing that the median overall survival was increased in individuals who had preexisting antibodies to HSV-1 relative to seronegative patients. In preclinical studies, promising results in immunocompetent mice have shown that arming a HER2-retargeted oHSV with interleukin-12 (IL-12) induced a proinflammatory TME and enhanced therapeutic activity.55
Although the majority of retargeted oHSVs specifically recognize the human protein, oHSVs retargeted via the ZEGFR affibody molecule mediated entry into both human and murine EGFR-expressing cells, offering the potential to study retargeted oHSV treatment in a completely murine syngeneic system without the introduction of exogenous human EGFR. Our future studies will assess the ability of systemically administered, retargeted oHSVs to achieve lytic infection of multiple tumor sites along with the induction of antitumor immunity in syngeneic EGFR-expressing tumor models. The retargeted oHSV backbones defined here will be improved by the addition of vector modifications to enhance lateral spread within the tumor mass and transgenes to induce potent antitumor immunity. Tumor targeting will likely play an essential role in realizing the full potential of oHSVs.
Materials and methods
Cells
Baby hamster kidney J1.1-2 cells were provided by Gabriella Campadelli-Fiume (University of Bologna) and murine melanoma B78H1 cells were provided by Gary Cohen (University of Pennsylvania); both cell lines were cultured in DMEM (Corning, Durham, NC) supplemented with 5% fetal bovine serum (FBS; MilliporeSigma, Burlington, MA) and 1% penicillin/streptomycin (Corning-Mediatech, Manassas, VA). Nectin-1 transduced J-C cells and B78-C cultures, hEGFR transduced J-EGFR cells, and EGFRvIII transduced B78-vIII were grown in the presence of antibiotic, as previously described.17,22,25,56 Human GBMs SNB19 and U251,57 osteosarcoma U2OS (American Type Culture Collection [ATCC], Manassas, VA), African green monkey kidney Vero cells (ATCC), and A549 cells (ATCC) were cultured in DMEM supplemented with 5% FBS and 1% penicillin/streptomycin. B78-mEGFR cells were established by the transfection of B78H1 cells with pFLAG-mEgfr(wt) followed by selection with 0.8 mg/mL G418. Resistant clones were screened by immunofluorescent staining for the FLAG tag present at the N terminus of mEGFR (Figure S1).
Plasmids
pFLAG-mEgfr(wt) used for the generation of B78-mEGFR cells was created from pFLAG-Egfr(Velvet) (Addgene plasmid no. 18788).58 We first corrected the A833G substitution in the mEGFR coding region of Egfr(Velvet) by designing primers to introduce the WT aa sequence at position 833 between two EcoR1 sites in the pFLAG-Egfr(Velvet) plasmid (Table S1). First, overlapping PCR products were generated with primer pairs (1) mEGFR F1 (velvet) and mEGFR R1 (velvet), and (2) mEGFR F2 (velvet) and mEGFR R2 (velvet). The final DNA fragment containing the corrected WT sequence flanked by EcoRI sites was produced by overlap PCR with products 1 and 2. Finally, the corrected DNA fragment was subcloned into pFLAG-Egfr(Velvet) between the indicated EcoR1 sites and confirmed by DNA sequencing across the mEGFR coding sequence.
pENTR-gD:scEGFRΔ2-24/Δ38 was derived from pgD:Δ224/38C-scEGFR9 by deleting the mutant codon at position 38 (Y38C) using flanking BstBI and BspEI restriction sites, as described.17 The gD:scEGFRΔ224/Δ38 coding sequence was then transferred to the pENTR1A plasmid (Thermo Fisher Scientific, Pittsburgh, PA) to facilitate LR-Clonase-mediated recombination into the GW containing vector. pENTR-gD:scEGFRΔ7-24/Δ38 was derived from pENTR-gD:scEGFRΔ2-24/Δ38 by modification of the N-terminal NcoI-BglII fragment using primers Bgl2EGFRd7R and Nco1EGFRd7F.
pENTR-gD:SD1Δ6-24/Δ38, pENTR-gD:SD2Δ6-24/Δ38, and pENTR-gD:SD3Δ6-24/Δ38 were generated from a pENTR-gD:wt plasmid that contains the wt gD coding sequence (strain KOS), modified to contain a BstB1 site between aa 34 and 36, positioned between the attL1 and attL2 recombination sites in pENTR1A.17 First, the NcoI-BstBI fragment at the 5′ end of gD was modified to contain a unique BamHI restriction site in place of aa 6–24 in the gD coding sequence to generate pENTR-gDΔ6-24. The modified NcoI-BamHI sequence was created by annealing and extension of the complementary oligonucleotides gDd6-24_NcoI-BstBI R and gDd6-24_NcoI-BstBI F (Table S1). Codon-optimized DNA fragments encoding VHH SD1, SD2, and SD318 followed by a glycine-serine spacer (G4S x 2) were obtained from GeneScript (Piscataway, NJ) in pUC57 shuttle vectors. The VHH sequences were PCR amplified to introduce 5′ and 3′ BamHI restriction sites (EGFR SD1-3 BamH1 F and R; Table S1) and subcloned into the BamHI site of pENTR-gDΔ6-24 to generate pENTR-gD:SD1Δ6-24, pENTR-gD:SD2Δ6-24, and pENTR-gD:SD3Δ6-24. We deleted aa Y38 from the internal BstBI-BspEI fragment, as described previously,17 to obtain pENTR-gD:SD1Δ6-24/Δ38, pENTR-gD:SD2Δ6-24/Δ38, and pENTR-gD:SD3Δ6-24/Δ38. All of the constructs were confirmed by DNA sequencing.
pENTR-gD:SD1Δ2-24/Δ38 was generated by modifying the 5′ Nco1-BstB1 fragment of pENTR-gD:SD1Δ6-24/Δ38 using primers gDSD1d2-24 Ncol F and gD BstBI R (Table S1) to remove aa 2–5 of gD and retain the BamHI-flanked SD1 VHH. pENTR-gD:SD2Δ2-24/Δ38 and pENTR-gD:SD3Δ2-24/Δ38 were then obtained by replacement of the SD1 VHH in pENTR-gD:SD1Δ2-24/Δ38 with the BamH1-flanked insert sequences described above.
Codon-optimized sequences encoding the affibody molecule (Affibody AB; Solna, Sweden) as either a monomer (ZEGFR) or dimer (Zdim) followed by a glycine-serine linker (G4S x 2) were obtained from GeneScript in pUC57 shuttle vectors with NcoI and BamHI sites for cloning. For the affibody dimer molecule, the monomer subunits were separated by a G4S glycine-serine linker. pENTR-gD:ZegfrΔ7-24/Δ38 and pENTR-gD:ZdimΔ2-24/Δ38 were generated from pENTR-gD:scEGFRΔ2-24/Δ38 by replacing a NcoI-BamHI fragment containing the scEGFR sequence with the affibody sequence. pENTR-gD:ZEGFRΔ2-24/Δ38 was generated from pENTR-gD:ZEGFRΔ7-24/Δ38 using primers gD:ZEGFR d2-24F and gD:ZEGFR d2-24R to delete aa 2–6 from gD:ZEGFRΔ7-24/Δ38. We replaced aa 2–6 in pENTR-gD:ZdimΔ2-24/Δ38 by replacing the NcoI-BglII fragment with an oligonucleotide generated by annealing primers gDZdimd7-24top and gDZdimd7-24bottom.
Viruses
The GW-compatible gD null BAC, KG4:T124-ΔgD:GW, was derived from KG4:T124 BAC8 by Red-mediated replacement59 of the gD coding sequence with a GW cassette, GW-Zeo, amplified with primers targeting the proximal 5′ and 3′ gD untranslated sequences, as described.17,43,60 The KG4:T124_fLuc-ΔgD:GW backbone was generated by PCR amplification of a CMV-Fluc reporter gene8 to introduce homology arms targeting the intergenic region between UL50 and UL51 (primers UL50FlucF and UL51FlucR; Table S1).
All of the viruses tested here were derived from either the KG4:T124-ΔgD:GW or KG4:T124_fLuc-ΔgD:GW BAC by LR-Clonase II-mediated recombination between the GW cassette in the BAC and the pENTR-based plasmids containing either the WT gD or retargeted gD sequences. Recombinants were confirmed by field inversion gel electrophoresis of restriction enzyme digested BAC DNAs and DNA sequencing across the gD genomic region. Infectious viruses were produced by transfection of BAC DNA into Vero cells with Lipofectamine LTX (Thermo Fisher Scientific). Virus was propagated by infecting Vero cells at an MOI of 0.005 and purified essentially as described.25 Briefly, supernatants were harvested when 100% of the cell monolayer had undergone cytopathic effects, incubated with 0.45 M NaCl for 30 min at room temperature (RT), centrifuged at 1,000 × g for 10 min to remove cell debris, and filtered through a 0.8-μm CN membrane filter. Virus was purified by centrifugation at 38,000 × g for 45 min (4°C), washed with 1X PBS, and resuspended in 1X PBS with 10% glycerol as a cryoprotectant.
Viral titer determination
Viral titers in genome copies were determined as previously described.17 Briefly, viral DNA was isolated using the QIAGEN blood and tissue DNA extraction kit (QIAGEN, Germantown, MD). Titers were determined by qPCR using a custom FAM-MGB primer probe set (Thermo Fisher Scientific) and a standard curve generated using a 10-fold dilution series of plasmid pUL5.17 Samples, standard curve, and negative controls were run together in triplicate in MicroAmp optical 96-well reaction plates. Viral titers in plaque-forming units were determined on Vero cells. Vero cells were plated at 90% confluency in 48-well tissue culture–treated plates. The following day, cells were infected with serial 1:10 dilutions of the virus stock in serum-free media. Two hours later, cells were overlaid with 1% methylcellulose and resulting viral plaques were counted using an inverted fluorescent microscope.
Western blot
Viruses were diluted in 1X Laemmli sample buffer to 1 × 108 gc/well. Lysates were heated for 5 min at 100°C, and proteins were separated by electrophoresis on precast 4%–15% SDS-PAGE gels (BioRad, Hercules, CA) and transferred to polyvinylidene fluoride membranes (MilliporeSigma). The filter was cut horizontally to detect both the intended glycoproteins and the VP16 loading control from the same lane. The membrane was blocked for 1 h in 5% nonfat dry milk in PBS + 0.05% Tween (PBS-T) and incubated sequentially with primary antibody and horseradish peroxidase–conjugated secondary antibody (anti-mouse immunoglobulin G; Abcam, Cambridge, UK) diluted 1:50,000 in 5% nonfat milk/PBS-T. The primary antibodies were gB (10B7; Virusys Corporation, Taneytown, MD) 1:5,000 in 5% nonfat dry milk/PBS-T; gD (DL6; Santa Cruz Biotechnology, Dallas, TX) 1:1,000 in 5% nonfat dry milk/PBS-T; and VP16 (1–21; Santa Cruz Biotechnology) 1:2,000 in PBS-T. Membranes were developed with ECL Plus (Thermo Fisher Scientific). Signal intensities were measured with ImageJ software version 1.53a (NIH).61
Virus entry assay
Cells were infected at the indicated MOI for 6 h at 37°C and fixed in 4% paraformaldehyde (Thermo Fisher Scientific) at RT for 30 min. Immunofluorescence was performed by permeabilization with 0.1% Triton 100X at RT for 5 min. The cells were incubated with 10% horse serum in PBS (HS-PBS) at RT for 1 h, followed by an overnight incubation at 4°C with an ICP4 mouse monoclonal antibody (sc-69809; Santa Cruz Biotechnology) diluted 1:400 in 1% HS-PBS. The cell nuclei were stained by incubation with 0.0001% DAPI (MilliporeSigma) at RT for 10 min. Images were obtained with a Nikon Diaphot fluorescence microscope (Nikon, Melville, NY) and MetaMorph imaging software (Molecular Devices, San Jose, CA), and cells were counted using ImageJ software version 1.53a.61
IC assay
The IC assay was performed as described,25 with minor modifications. Donor Vero cells were infected at an MOI of 10,000 gc/cell at 37°C for 2 h, treated with 0.1 M glycine (pH 3.0) at RT for 3 min, and washed 3 times with 1X PBS. The cells were incubated at 37°C for 1 h, dispersed with trypsin, and resuspended in serum-free culture medium. Equal numbers of donor cells were seeded onto monolayers of acceptor cells to create well-separated single plaques in a 48-well plate. The plate was incubated for 2 h at 37°C, and the cells were overlaid with medium containing 1% methylcellulose (MilliporeSigma). Two days later, plaques were visualized by fluorescence microscopy for the EGFP marker gene with the Nikon Diaphot fluorescence microscope, and images were captured with MetaMorph imaging software (Molecular Devices). Plaque size was quantified using ImageJ software version 1.53a.61
AlamarBlue cell viability assay
Indicated cell types were seeded at 70%–80% confluency in 48-well tissue culture–treated plates 24 h before infection. The following day, cells were infected with an MOI of 1,000 gc/cell in 120 μL serum-free media at 37°C for 1.5 h and overlaid with 130 μL media containing 10% FBS. For assays performed using Vero donor cells, donor cells were prepared as described for the IC assay and added to uninfected U251 cells at a ratio of 1 donor cell per 10 U251 cells. At 24, 48, and 72 hpi, 25 μL alamarBlue reagent (Thermo Fisher Scientific) was added to the cells, and the plates were incubated for a further 3–5 h at 37°C. Wells without cells and uninfected cells were used as controls. Supernatant was monitored for color change, transferred to opaque black 96-well plates, and measured for fluorescence using a Biotek (Winooski, VT) plate reader (560 nm excitation/590 nm emission).
Animal studies
We established subcutaneous U251 flank tumors in athymic nude mice (Jackson Laboratories, Bar Harbor, ME) by implanting 3 × 106 cells. Tumors were treated with either 1 × 1010 gc of virus or vehicle control (PBS) when they reached an average volume of 100 mm3. For experiments using multiple treatment doses, repeat injections were delivered every other day for a total of 4 doses. For i.v. injections, virus was resuspended in 100μL PBS and delivered via the lateral tail vein. For intratumoral injection, virus was resuspended in 25μL PBS. Tumor diameters were measured by calipers by an animal technician who was blinded to the experimental details, and volumes were calculated as (L × W2) × 0.52. Mice were sacrificed when the tumor ulcerated, reached a maximum diameter of 20 mm or when mice showed any sign of discomfort (e.g., unable to ambulate, eat, or drink, lost >10% body weight). If the cell or virus injections induced redness and inflammation at the injection site, then a topical antibiotic was administered to minimize the risk of topical pathogen infection. All of the animal studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee in accordance with the requirements and recommendations in the NIH Guide for the Care and the Use of Laboratory Animals.62
In vivo imaging
Mice were injected intraperitoneally (i.p.) with d-luciferin (Promega, Madison, WI) at 150 mg/kg in 100 μL saline solution (PBS, pH 7.4). Five minutes after d-luciferin injection, mice were anesthetized in an induction chamber with 2%–3% isoflurane in 100% oxygen and imaged with the 2D Lumina S5 (PerkinElmer, Waltham, MA) in vivo imaging system. Consecutive 1-min frames were acquired around the maximum signal at 10–20 min after d-luciferin injection. Photon emission was quantified using Living Image software and reported as photon flux (p/s) from a 1.5-cm2 circular region of interest (ROI) on the tumor. The limit of detection was 104 p/s measured by the signal emitted by the fur from a 1.5-cm2 circular ROI. Statistical analyses were performed using GraphPad Prism 10 (GraphPad Software, La Jolla, CA) software.
Statistical analysis
GraphPad Prism 8 software for MacOS was used for all of the statistical analyses. Averages for each experiment were calculated ±SEMs. One-way ANOVA was used to determine entry efficiency and lateral spread. For in vitro and in vivo studies, two-way ANOVAs were used to determine the statistical significance of differences observed between groups. The two-tailed nonparametric Mann-Whitney test was used to determine the differences in the tumor growth profile among groups.
Data and code availability
Data and materials described in this article will be available upon reasonable request to the corresponding author.
Acknowledgments
This work was supported by grant R01-CA222804 (to J.C.G.) from the NIH. We thank Lisa Bailey at the University of Pittsburgh for technical support. We thank Affibody AB (Solna, Sweden) and Stefan Ståhl for the sequence of the ZEGFR:2377 affibody molecule.
Author contributions
Conceptualization, B.L.H., J.B.C., and J.C.G.; methodology, B.L.H., J.B.C., and J.C.G.; investigation, S.I. and B.L.H.; writing – original draft, S.I. and B.L.H.; writing – review & editing, B.L.H., J.B.C., and J.C.G.; funding acquisition, J.C.G.; supervision, J.B.C. and J.C.G.
Declaration of interests
J.B.C. and J.C.G. are inventors of intellectual property licensed to Oncorus Inc. (Cambridge, MA). J.C.G. is a consultant to and Chair of the Scientific Advisory Board of Oncorus Inc. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200761.
Supplemental information
References
- 1.Glorioso J.C., Cohen J.B., Goins W.F., Hall B., Jackson J.W., Kohanbash G., Amankulor N., Kaur B., Caligiuri M.A., Chiocca E.A., et al. Oncolytic HSV Vectors and Anti-Tumor Immunity. Curr. Issues Mol. Biol. 2021;41:381–468. doi: 10.21775/cimb.041.381. [DOI] [PubMed] [Google Scholar]
- 2.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 3.Pol J., Kroemer G., Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5 doi: 10.1080/2162402X.2015.1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dambach M.J., Trecki J., Martin N., Markovitz N.S. Oncolytic viruses derived from the gamma34.5-deleted herpes simplex virus recombinant R3616 encode a truncated UL3 protein. Mol. Ther. 2006;13:891–898. doi: 10.1016/j.ymthe.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Kaufman H.L., Shalhout S.Z., Iodice G. Talimogene Laherparepvec: Moving From First-In-Class to Best-In-Class. Front. Mol. Biosci. 2022;9 doi: 10.3389/fmolb.2022.834841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaur A., Jewell D.A., Liang Y., Ridzon D., Moore J.H., Chen C., Ambros V.R., Israel M.A. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 7.Silber J., Lim D.A., Petritsch C., Persson A.I., Maunakea A.K., Yu M., Vandenberg S.R., Ginzinger D.G., James C.D., Costello J.F., et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzacurati L., Marzulli M., Reinhart B., Miyagawa Y., Uchida H., Goins W.F., Li A., Kaur B., Caligiuri M., Cripe T., et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol. Ther. 2015;23:99–107. doi: 10.1038/mt.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchida H., Marzulli M., Nakano K., Goins W.F., Chan J., Hong C.-S., Mazzacurati L., Yoo J.Y., Haseley A., Nakashima H., et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. 2013;21:561–569. doi: 10.1038/mt.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halder S., Basu S., Lall S.P., Ganti A.K., Batra S.K., Seshacharyulu P. Targeting the EGFR signaling pathway in cancer therapy: What’s new in 2023? Expert Opin. Ther. Targets. 2023;27:305–324. doi: 10.1080/14728222.2023.2218613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An Z., Aksoy O., Zheng T., Fan Q.-W., Weiss W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37:1561–1575. doi: 10.1038/s41388-017-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beauchesne P. Extra-neural metastases of malignant gliomas: myth or reality? Cancers. 2011;3:461–477. doi: 10.3390/cancers3010461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholson R.I., Gee J.M., Harper M.E. EGFR and cancer prognosis. Eur. J. Cancer. 2001;37(Suppl 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 14.Sigismund S., Avanzato D., Lanzetti L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilterbrand A.T., Heldwein E.E. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida H., Chan J., Goins W.F., Grandi P., Kumagai I., Cohen J.B., Glorioso J.C. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J. Virol. 2010;84:12200–12209. doi: 10.1128/JVI.01633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuzmen C., Cairns T.M., Atanasiu D., Lou H., Saw W.T., Hall B.L., Cohen J.B., Cohen G.H., Glorioso J.C. Point Mutations in Retargeted gD Eliminate the Sensitivity of EGFR/EGFRvIII-Targeted HSV to Key Neutralizing Antibodies. Mol. Ther. Methods Clin. Dev. 2020;16:145–154. doi: 10.1016/j.omtm.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz K.R., Bagchi A., Roovers R.C., van Bergen en Henegouwen P.M.P., Ferguson K.M. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013;21:1214–1224. doi: 10.1016/j.str.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolmachev V., Rosik D., Wållberg H., Sjöberg A., Sandström M., Hansson M., Wennborg A., Orlova A. Imaging of EGFR expression in murine xenografts using site-specifically labelled anti-EGFR 111In-DOTA-Z EGFR:2377 Affibody molecule: aspect of the injected tracer amount. Eur. J. Nucl. Med. Mol. Imag. 2010;37:613–622. doi: 10.1007/s00259-009-1283-x. [DOI] [PubMed] [Google Scholar]
- 20.Gierasch W.W., Zimmerman D.L., Ward S.L., Vanheyningen T.K., Romine J.D., Leib D.A. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods. 2006;135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 21.Cocchi F., Menotti L., Mirandola P., Lopez M., Campadelli-Fiume G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998;72:9992–10002. doi: 10.1128/jvi.72.12.9992-10002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frampton A.R., Stolz D.B., Uchida H., Goins W.F., Cohen J.B., Glorioso J.C. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J. Virol. 2007;81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano K., Kobayashi M., Nakamura K.i., Nakanishi T., Asano R., Kumagai I., Tahara H., Kuwano M., Cohen J.B., Glorioso J.C. Mechanism of HSV infection through soluble adapter-mediated virus bridging to the EGF receptor. Virology. 2011;413:12–18. doi: 10.1016/j.virol.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasamura Y., Tashjian A.H., Sato G.H. Establishment of four functional, clonal strains of animal cells in culture. Science. 1966;154:1186–1189. doi: 10.1126/science.154.3753.1186. [DOI] [PubMed] [Google Scholar]
- 25.Uchida H., Shah W.A., Ozuer A., Frampton A.R., Goins W.F., Grandi P., Cohen J.B., Glorioso J.C. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J. Virol. 2009;83:2951–2961. doi: 10.1128/JVI.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramis G., Thomàs-Moyà E., Fernández de Mattos S., Rodríguez J., Villalonga P. EGFR inhibition in glioma cells modulates Rho signaling to inhibit cell motility and invasion and cooperates with temozolomide to reduce cell growth. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038770. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Zhou P., Hu J., Wang X., Wang J., Zhang Y., Wang C. Epidermal growth factor receptor expression affects proliferation and apoptosis in non-small cell lung cancer cells via the extracellular signal-regulated kinase/microRNA 200a signaling pathway. Oncol. Lett. 2018;15:5201–5207. doi: 10.3892/ol.2018.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L., Chen L., Zhu H., Li Y., Chen C.C., Li M. FHL1 promotes glioblastoma aggressiveness through regulating EGFR expression. FEBS Lett. 2021;595:85–98. doi: 10.1002/1873-3468.13955. [DOI] [PubMed] [Google Scholar]
- 29.Duan F., Simeone S., Wu R., Grady J., Mandoiu I., Srivastava P.K. Area under the curve as a tool to measure kinetics of tumor growth in experimental animals. J. Immunol. Methods. 2012;382:224–228. doi: 10.1016/j.jim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Di Giovine P., Settembre E.C., Bhargava A.K., Luftig M.A., Lou H., Cohen G.H., Eisenberg R.J., Krummenacher C., Carfi A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krummenacher C., Supekar V.M., Whitbeck J.C., Lazear E., Connolly S.A., Eisenberg R.J., Cohen G.H., Wiley D.C., Carfí A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carfí A., Willis S.H., Whitbeck J.C., Krummenacher C., Cohen G.H., Eisenberg R.J., Wiley D.C. Herpes simplex virus glycoprotein D bound to the human receptor. Mol. Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 33.Cooper R.S., Heldwein E.E. Herpesvirus gb: A finely tuned fusion machine. Viruses. 2015;7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Connolly S.A., Landsburg D.J., Carfi A., Wiley D.C., Cohen G.H., Eisenberg R.J. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 2003;77:8127–8140. doi: 10.1128/JVI.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon M., Zago A., Shukla D., Spear P.G. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 2003;77:9221–9231. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou G., Roizman B. Separation of receptor-binding and profusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc. Natl. Acad. Sci. USA. 2007;104:4142–4146. doi: 10.1073/pnas.0611565104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan Q., Kopp S., Connolly S.A., Muller W.J., Longnecker R. Mapping sites of herpes simplex virus type 1 glycoprotein D that permit insertions and impact gD and gB receptors usage. Sci. Rep. 2017;7 doi: 10.1038/srep43712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gambini E., Reisoli E., Appolloni I., Gatta V., Campadelli-Fiume G., Menotti L., Malatesta P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012;20:994–1001. doi: 10.1038/mt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reisoli E., Gambini E., Appolloni I., Gatta V., Barilari M., Menotti L., Malatesta P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012;19:788–795. doi: 10.1038/cgt.2012.62. [DOI] [PubMed] [Google Scholar]
- 40.Menotti L., Cerretani A., Campadelli-Fiume G. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 2006;80:5531–5539. doi: 10.1128/JVI.02725-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata T., Uchida H., Shiroyama T., Okubo Y., Suzuki T., Ikeda H., Yamaguchi M., Miyagawa Y., Fukuhara T., Cohen J.B., et al. Development of an oncolytic HSV vector fully retargeted specifically to cellular EpCAM for virus entry and cell-to-cell spread. Gene Ther. 2016;23:479–488. doi: 10.1038/gt.2016.17. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez Gil J., Dubois M., Neirinckx V., Lombard A., Coppieters N., D’Arrigo P., Isci D., Aldenhoff T., Brouwers B., Lassence C., et al. Nanobody-based retargeting of an oncolytic herpesvirus for eliminating CXCR4+ GBM cells: A proof of principle. Mol. Ther. Oncolytics. 2022;26:35–48. doi: 10.1016/j.omto.2022.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall B.L., Leronni D., Miyagawa Y., Goins W.F., Glorioso J.C., Cohen J.B. Generation of an oncolytic herpes simplex viral vector completely retargeted to the GDNF receptor gfrα1 for specific infection of breast cancer cells. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21228815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menotti L., Cerretani A., Hengel H., Campadelli-Fiume G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008;82:10153–10161. doi: 10.1128/JVI.01133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appolloni I., Alessandrini F., Menotti L., Avitabile E., Marubbi D., Piga N., Ceresa D., Piaggio F., Campadelli-Fiume G., Malatesta P. Specificity, Safety, Efficacy of EGFRvIII-Retargeted Oncolytic HSV for Xenotransplanted Human Glioblastoma. Viruses. 2021;13:1677. doi: 10.3390/v13091677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asano R., Sone Y., Makabe K., Tsumoto K., Hayashi H., Katayose Y., Unno M., Kudo T., Kumagai I. Humanization of the bispecific epidermal growth factor receptor x CD3 diabody and its efficacy as a potential clinical reagent. Clin. Cancer Res. 2006;12:4036–4042. doi: 10.1158/1078-0432.CCR-06-0059. [DOI] [PubMed] [Google Scholar]
- 47.Menotti L., Nicoletti G., Gatta V., Croci S., Landuzzi L., De Giovanni C., Nanni P., Lollini P.-L., Campadelli-Fiume G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA. 2009;106:9039–9044. doi: 10.1073/pnas.0812268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrovic B., Leoni V., Gatta V., Zaghini A., Vannini A., Campadelli-Fiume G. Dual ligand insertion in gb and gd of oncolytic herpes simplex viruses for retargeting to a producer vero cell line and to cancer cells. J. Virol. 2018;92 doi: 10.1128/JVI.02122-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leoni V., Gatta V., Casiraghi C., Nicosia A., Petrovic B., Campadelli-Fiume G. A Strategy for Cultivation of Retargeted Oncolytic Herpes Simplex Viruses in Non-cancer Cells. J. Virol. 2017;91 doi: 10.1128/JVI.00067-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leoni V., Petrovic B., Gianni T., Gatta V., Campadelli-Fiume G. Simultaneous insertion of two ligands in gd for cultivation of oncolytic herpes simplex viruses in noncancer cells and retargeting to cancer receptors. J. Virol. 2018;92 doi: 10.1128/JVI.02132-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki T., Uchida H., Shibata T., Sasaki Y., Ikeda H., Hamada-Uematsu M., Hamasaki R., Okuda K., Yanagi S., Tahara H. Potent anti-tumor effects of receptor-retargeted syncytial oncolytic herpes simplex virus. Mol. Ther. Oncolytics. 2021;22:265–276. doi: 10.1016/j.omto.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Speranza M.-C., Kasai K., Lawler S.E. Preclinical mouse models for analysis of the therapeutic potential of engineered oncolytic herpes viruses. ILAR J. 2016;57:63–72. doi: 10.1093/ilar/ilw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marzulli M., Hall B.L., Zhang M., Goins W.F., Cohen J.B., Glorioso J.C. Novel mutations in UL24 and gH rescue efficient infection of an HSV vector retargeted to TrkA. Mol. Ther. Methods Clin. Dev. 2023;30:208–220. doi: 10.1016/j.omtm.2023.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ling A.L., Solomon I.H., Landivar A.M., Nakashima H., Woods J.K., Santos A., Masud N., Fell G., Mo X., Yilmaz A.S., et al. Clinical trial links oncolytic immunoactivation to survival in glioblastoma. Nature. 2023;623:157–166. doi: 10.1038/s41586-023-06623-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leoni V., Vannini A., Gatta V., Rambaldi J., Sanapo M., Barboni C., Zaghini A., Nanni P., Lollini P.-L., Casiraghi C., Campadelli-Fiume G. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakano K., Asano R., Tsumoto K., Kwon H., Goins W.F., Kumagai I., Cohen J.B., Glorioso J.C. Herpes simplex virus targeting to the EGF receptor by a gD-specific soluble bridging molecule. Mol. Ther. 2005;11:617–626. doi: 10.1016/j.ymthe.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Okano F., Storkus W.J., Chambers W.H., Pollack I.F., Okada H. Identification of a novel HLA-A∗0201-restricted, cytotoxic T lymphocyte epitope in a human glioma-associated antigen, interleukin 13 receptor alpha2 chain. Clin. Cancer Res. 2002;8:2851–2855. [PubMed] [Google Scholar]
- 58.Du X., Tabeta K., Hoebe K., Liu H., Mann N., Mudd S., Crozat K., Sovath S., Gong X., Beutler B. Velvet, a dominant Egfr mutation that causes wavy hair and defective eyelid development in mice. Genetics. 2004;166:331–340. doi: 10.1534/genetics.166.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyagawa Y., Marino P., Verlengia G., Uchida H., Goins W.F., Yokota S., Geller D.A., Yoshida O., Mester J., Cohen J.B., Glorioso J.C. Herpes simplex viral-vector design for efficient transduction of nonneuronal cells without cytotoxicity. Proc. Natl. Acad. Sci. USA. 2015;112:E1632–E1641. doi: 10.1073/pnas.1423556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolfe D., Craft A.M., Cohen J.B., Glorioso J.C. A herpes simplex virus vector system for expression of complex cellular cDNA libraries. J. Virol. 2010;84:7360–7368. doi: 10.1128/JVI.02388-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals . 8th ed. National Academies Press (US); 2011. Guide for the Care and Use of Laboratory Animals. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and materials described in this article will be available upon reasonable request to the corresponding author.