

Abstract
Neoantigen (neoAg)-based cancer vaccines expand preexisting antitumor immunity and elicit novel cancer-specific T cells. However, at odds with prophylactic vaccines, therapeutic antitumor immunity must be induced when the tumor is present and has already established an immunosuppressive environment capable of rapidly impairing the function of anticancer neoAg T cells, thereby leading to lack of efficacy. To overcome tumor-induced immunosuppression, we first vaccinated mice bearing immune checkpoint inhibitor (CPI)-resistant tumors with an adenovirus vector encoding a set of potent cancer-exogenous CD8 and CD4 T cell epitopes (Ad-CAP1), and then “taught” cancer cells to express the same epitopes by using a tumor-retargeted herpesvirus vector (THV-CAP1). Potent CD8 effector T lymphocytes were elicited by Ad-CAP1, and subsequent THV-CAP1 delivery led to a significant delay in tumor growth and even cure.
Keywords: MT: Regular Issue, cancer vaccine, adenovirus vectors, herpesvirus vectors, immune checkpoint inhibitors, neoantigens
Graphical abstract
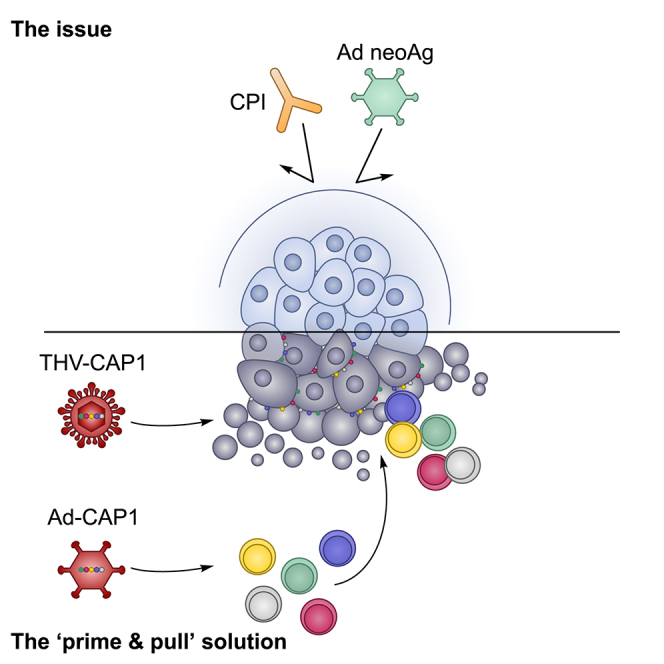
D’Alise and colleagues have developed a novel “prime and pull” approach with replication incompetent adenovirus and tumor retargeted oncolytic HSV vectors encoding potent cancer exogenous epitopes. Sequential administration of these vectors induced potent T cell immunity and antitumoral efficacy against checkpoint inhibitors and neoantigen vaccine-resistant cancer.
Introduction
Immune checkpoint inhibitor (CPI) therapy has proven to be an effective therapeutic approach in a variety of cancers. However, despite the clinical success of CPI, only a subset of patients achieves durable responses, and some tumor types are resistant to CPI treatment.1 The consensus of opinion is that a number of different immune pathways are involved in the effective killing of cancer cells, collectively called the cancer immunity cycle,2 with CPIs being active only on some of them. Therefore, novel immunotherapeutic approaches are needed to face CPI efficacy and even resistance.
Cancer vaccines are reemerging as a strategy to kill cancers cells through their ability to reexpand antitumor T cells and induce novel T cell reactivities. The renaissance of cancer vaccines rests on two recent scientific achievements. First, the development of DNA and RNA sequencing technologies (collectively called next-generation sequencing [NGS]) allowing the rapid and accurate identification of all of the somatic mutations (cancer mutanome), giving rise to amino acid changes only in cancer cells. These mutant protein fragments represent novel cancer-specific antigens (henceforth called neoantigens [neoAg]) with no established immune tolerance and good safety given their presence only in cancer cells. Evidence on the relevance of neoAgs for the success of cancer vaccines comes from the strong correlation observed between tumor mutational burden and the efficacy of checkpoint blockade (CPB).3 The second scientific milestone is the development of genetic vaccine technologies based on adenoviral vectors and modified mRNA formulated in lipid nanoparticles (LNPs), with the approval of coronavirus disease 2019 (COVID-19) vaccines and neoAg cancer vaccines showing promising clinical results.4,5 Genetic vaccines encode the antigen(s) leading to its intracellular production, and resulting in efficient presentation in the context of major histocompatibility complex class I (MHC class I) and consequent recognition by CD8 T killer cells, which are the most relevant T cell population to fight tumor growth.
We have previously shown that neoAg-based adenovirus vectored vaccines display antitumor activity when administered in a prophylactic or early therapeutic setting in mouse models.6 However, vaccination with neoAg-based adenovirus vectors was not effective when administered to mice with large established tumors, presumably due to the rapid exhaustion of T cells upon reaching the tumor microenvironment.6 Coadministration of neoAg vaccine and CPI partially reversed this phenotype and led to tumor clearance in up to 50% of treated animals.6 To improve vaccine efficacy in a therapeutic setting and to extend its use to CPI-resistant tumors, we vaccinated mice with an adenovirus encoding an array of potent CD8 and CD4 T cell epitopes (CAP1 string) derived from viruses and bacteria or from heterologous cancer mutanome (Ad-CAP1), and then selectively drove the expression in vivo of the same epitopes in mouse tumors by using a tumor-targeted herpes simplex virus type 1 (THV) where the receptor binding domain of the attachment and entry viral glycoprotein D (gD) was substituted by a single-chain antibody fragment (scFv) recognizing the tumor-associated surface antigen human epidermal growth factor receptor 2 (THV-HER2).7,8,9,10 A THV-HER2 encoding the CAP1 string (THV-CAP1) was used to selectively express the CAP1 epitopes only in tumor cells, and make them susceptible to T cell killing by the vaccine-induced cellular response. Using this “prime and pull” approach, we were able to achieve reduction of neoplastic cell growth and tumor clearance in HER2 transgenic mice with large established syngeneic CPI-resistant Lewis lung carcinoma (LLC) tumors expressing HER2.
Results
LLC is highly resistant to CPI and adenovirus vectored neoAg-based vaccine treatment
We have previously shown that adenovirus vectored neoAg vaccines prevent tumor development in a prophylactic setting and cure large established tumors when used in combination with CPI in a therapeutic setting in different mouse tumor models.4,6,11 Here, we confirmed our previous findings by generating a new vaccine targeting 31 neoAgs selected from the CT26 mouse colon cancer cells (Ad-CT26; Table 1; materials and methods) by using the VENUS algorithm, a recently described bioinformatic tool that uses DNA and RNA NGS data to prioritize mutated peptides with high potential to be neoAgs.11
Table 1.
CT26 neoantigens encoded by Ad-CT26 vaccine
| Position | CHR coord (mm10) | Mapping | NeoAg sequence (reference/mutation) | Best IC50 mutation (nM) | MHC class I |
|---|---|---|---|---|---|
| 1 | 16:57133128 | Tomm70a | AQAAKNKGNKYF[K/Q]AGKYEQAIQCYT | 459.45 | H-2-Kd |
| 2 | 1:33777642 | Zfp451 | VLLYCHISEGSR[P/L]PCDLHLFSQPKI | 883.69 | H-2-Kd |
| 3 | 5:48219745 | Slit2 | QDLHNLNLLSLY[D/A]NKLQTVAKGTFS | 206.17 | H-2-Kd |
| 4 | 1:66706508 | Rpe | GHPVVESLRKQL[G/D]QDPFFDMHMMVS | 503.57 | H-2-Ld |
| 5 | 14:34343682 | Glud1 | LRTAAYVNAIEK[V/I]FKVYNEAGVTFT | 141.15 | H-2-Kd |
| 6 | 2:129208197 | Slc20a1 | KPLRRNNSYTSY[T/I]MAICGMPLDSFR | 118.03 | H-2-Kd |
| 7 | 1:30804436 | Phf3 | PGPQNFPPQNMF[G/E]FPPHLSPPLLPP | 41.5 | H-2-Ld |
| 8 | 12:51365554 | G2e3 | FEGSLAKNLSLN[S/F]QAVKENLYYEVG | 467.97 | H-2-Kd |
| 9 | 7:48871007 | E2f8 | ILPQAPSGPSYA[I/T]YLQPAQAQMLTP | 320.56 | H-2-Kd |
| 10 | 7:55873449 | Cyfip1 | YIEQATVHSSMN[E/K]MLEEGQEYAVML | 929.32 | H-2-Kd |
| 11 | 7:65663891 | Tarsl2 | AIKIFKNSSTYW[E/K]GNPEMETLQRIY | 585.18 | H-2-Kd |
| 12 | 4:86583172 | Haus6 | DSNLQARLTSYE[A/T]LKKSLSKIREES | 18.22 | H-2-Kd |
| 13 | 2:72970297 | Sp3 | ITLTPVQTLTLG[Q/H]VAAGGALTSTPV | 659.55 | H-2-Kd |
| 14 | 1:143699294 | Cdc73 | VWGTGKEGQPRE[Y/H]YTLDSILFLLNN | 799.16 | H-2-Dd |
| 15 | 18:5767633 | Zeb1 | GAQEEPQVEPLD[L/F]SLPKQQGELLER | 121.04 | H-2-Ld |
| 16 | 17:34205757 | Tap2 | LSRPDLPFLIAA[F/V]FFLVVAVWGETL | 86.9 | H-2-Ld |
| 17 | 2:180713221 | Gid8 | MSYAEK[P/S]DEITKDEWMEKL | 67.01 | H-2-Kd |
| 18 | 1:173926892 | Ifi203 | INPTMIISNTLS[E/K]SAIATPKISYLL | 445.82 | H-2-Kd |
| 19 | 10:36993650 | Hdac2 | GAGKGKYYAVNF[P/S]MRDGIDDESYGQ | 39.48 | H-2-Kd |
| 20 | 11:58188928 | Gm12250 | TGLYFRKSYYMQ[N/K]YFLDTVTEDAKV | 103.63 | H-2-Kd |
| 21 | new12:91825363 | Sel1l | LIAHMILGYRYW[A/T]GIGVLQSCESAL | 118.72 | H-2-Kd |
| 22 | 7:35547980 | Nudt19 | IHSPYVYEIYMT[L/F]PSENKHVYPRNY | 852.11 | H-2-Kd |
| 23 | 10:121615052 | Xpot | PRGVDLYLRILM[A/P]IDSELVDRDVVH | 41.56 | H-2-Kd |
| 24 | 7:28814084 | Hnrnpl | VLLFTILNPIYS[I/R]TTDVLYTICNPC | 613.79 | H-2-Kd |
| 25 | 19:38812346 | Noc3l | ALASAILSDPES[H/Y]IKKLKELRSMLM | 101.21 | H-2-Kd |
| 26 | 17:35117867 | Csnk2b | QPMLPIGLSDIP[G/D]EAMVKLYCPKCM | 202.23 | H-2-Ld |
| 27 | 7:45881542 | Kdelr1 | WTFSIYLESVAI[L/M]PQLFMVSKTGEA | 836.76 | H-2-Kd |
| 28 | 17:29332778 | Mtch1 | SWIHCWKYLSVQ[G/S]QLFRGSSLLFRR | 13.46 | H-2-Kd |
| 29 | 4:153960954 | A430005L14Rik | SIFLDDDSNQPM[P/A]VSRFFGNVELMQ | 78.13 | H-2-Ld |
| 30 | 12:59160182 | Ctage5 | ALKKLIYAAKLN[A/T]SLKALEGERNQV | 971.41 | H-2-Kd |
| 31 | 19:53635752 | Smc3 | IETQQRKFKASR[D/A]SILSEMKMLKEK | 250.7 | H-2-Kd |
CHR coord (mm10), chromosome coordinates from mouse genome mm10.
The 31 neoAgs were selected by analyzing the mutanome of CT26. MHC class I binding predictions were performed with IEDB 2.17 software.
Vaccination with Ad-CT26 induced potent T cell immunity against five neoAgs reaching over 1,500 antigen-specific interferon-γ (IFN-γ)-secreting lymphocytes/million splenocytes (Figure 1A), and protected mice from tumor development when used in a prophylactic mode (Figure 1B). In a therapeutic setting, monotherapy vaccine was not effective and induced tumor regression in approximately 50% of the treated animals only when combined with anti-programmed cell death protein 1 (PD1) treatment (Figure 1C).
Figure 1.

In vivo immunogenicity and efficacy of Ad neoAgs-based vaccines in CT26 and LLC murine models
(A and D) Predicted neoAgs from CT26 or LLC tumors were cloned in tandem in an Ad vector. Immunogenicity of Ad-CT26 vaccine (A) and Ad-LLC (D) vaccine was tested in vivo. T cell responses were measured by IFN-γ ELISpot on splenocytes of naive mice 3 weeks postimmunization with 5 × 108 vp of Ad-CT26 (green) or Ad-LLC (light blue). Responses against individual neoAgs found to be immunogenic are shown (n = 5); neoAgs ID matches the position in the neoAgs string. Peptide diluent DMSO and concanavalin A were used as negative and positive controls, respectively. Data are representative of 2 independent experiments. (B and E) Activity of Ad-CT26 vaccine (B) and Ad-LLC (E) in prophylactic efficacy setting. Mice (n = 6–8/group) were vaccinated with Ad-CT26 or Ad-LLC; 2 weeks after immunization, tumor cells were injected subcutaneously (CT26 or LLC) and tumor growth was monitored over time. Percentage of tumor-bearing mice at day 30 in Ad vaccinated or not vaccinated (gray bars, mock) groups is shown (2-tailed Mann-Whitney U test; ∗∗∗∗p < 0.0001). n.s., not significant by 2-tailed Mann-Whitney U test. (C and F) Schematic of therapeutic efficacy setting. Seven days after tumor cell injection (CT26 or LLC), mice bearing large established tumors (tumor volume average: 60–90 mm3) were treated with Ad vaccine alone, or αPD1 or the combination Ad vaccine/αPD1. Untreated tumors were used as negative control. Ad vaccination was performed once, at day 0. αPD1 was injected twice per week for 3 weeks. Tumor growth was monitored over time. Each line represents an individual tumor (gray lines for nonresponders, black or green lines for responders to αPD1 or combo, respectively); n = 8–10. Percentages on the graphs indicate the rate of complete response (cured tumors). Data are representative of 2 independent experiments (Fisher exact test; ∗p < 0.05; ∗∗p < 0.01; ns, no significant difference).
We then performed the same experiments in mice inoculated subcutaneously with the LLC cells, previously described as nonresponsive to immune CPB.12,13,14 Vaccination with an adenovirus vectored vaccine encoding 31 neoAgs derived from LLC cells (Ad-LLC; Table 2) induced potent T cell immunity in naive mice, with 5 immunogenic neoAgs out of 31, as measured by ex vivo IFN-γ ELISpot (Figure 1D). The antitumor activity of Ad-LLC was evaluated in the two settings of prophylactic and therapeutic vaccination. Interestingly, differently from the CT26 model, in both settings of prophylactic and therapeutic vaccination, no efficacy was observed upon immunization in either group receiving vaccine alone and in combination with anti-PD1, highlighting the high resistance of this model (Figures 1E and 1F).
Table 2.
LLC neoantigens encoded by Ad-LLC vaccine
| Position | CHR coord (mm10) | Mapping | NeoAg sequence (reference/mutation) | Best IC50 mutation (nM) |
MHC class I | |
|---|---|---|---|---|---|---|
| 1 | 6:30812772 | Copg2 | ATFYLNVLQQRQ[M/I]ALNATYIFNGLT | 370.7 | H-2-Db | |
| 2 | 11:45906430 | Clint1 | SSQSMNFSLMST[N/S]TVGLGLPMSRSQ | 56.34 | H-2-Db | |
| 3 | 4:114987061 | Cmpk1 | LVPSCPPLTR[T/R]LRLFPLHLMKPLVV | 820.97 | H-2-Kb | |
| 4 | 9:7451277 | Mmp3 | PEFYLISSFWPS[L/F]PSNMDAAYEVTN | 224.13 | H-2-Kb | |
| 5 | 15:64094446 | Asap1 | HHLSLDRTNIPP[E/K]TFQKSSQLTELP | 227.03 | H-2-Kb | |
| 6 | 4:135966395 | Gale | QLLEIMRAHGVK[N/S]LVFSSSATVYGN | 392.97 | H-2-Kb | |
| 7 | 11:4922008 | Thoc5 | KVVSRLVKWVII[T/A]HEDYMELHFTKD | 309.79 | H-2-Kb | |
| 8 | 14:57598032 | Xpo4 | SRTDSVIRLLSA[V/L]LRVSEVESRAIR | 71.16 | H-2-Kb | |
| 9 | 11:119542649 | Nptx1 | GGGFDATQAFVG[E/Q]LAHFNIWDRKLT | 177.06 | H-2-Kb | |
| 10 | 13:73328436 | Ndufs6 | MAA[V/A]LTFRRLLTLPRA | 36.53 | H-2-Kb | |
| 11 | 4:127048903 | Zmym1 | ICGQAYDSATNF[T/R]VKLNEVVAEFKK | 117.03 | H-2-Kb | |
| 12 | 6:17540629 | Met | VFEPFEKPVMIS[I/M]GNENVVEIKGNN | 173.03 | H-2-Db | |
| 13 | 13:38050902 | Riok1 | LQVIQYMRKMYQ[D/Y]ARLVHADLSEFN | 45.2 | H-2-Kb | |
| 14 | 11:5104143 | Rhbdd3 | WQECHLGTVRFL[H/Y]NSTVLALATGLL | 239.21 | H-2-Kb | |
| 15 | 4:88397026 | Focad | YLLVSMPLWAKH[V/M]SDEQIQGFVENL | 327.19 | H-2-Kb | |
| 16 | 11:49095454 | Ifi47 | FISAFLKGASEN[S/N]FQQLAKEFLPQY | 743.41 | H-2-Kb | |
| 17 | 9:123436316 | Lars2 | GPVPVPLQDLPV[I/T]LPSIASLTGRGG | 213.2 | H-2-Kb | |
| 18 | 6:33971925 | Exoc4 | READLDFARQYY[E/V]MLYNTADELLNL | 44.9 | H-2-Kb | |
| 19 | 15:98813679 | Prkag1 | NLDVSVTKALQH[R/L]SHYFEGVLKCYL | 211 | H-2-Kb | |
| 20 | 4:132371088 | Phactr4 | IAELSQAMNSGT[V/L]LSKPSPPLPPKR | 100.55 | H-2-Db | |
| 21 | 4:88297060 | Focad | EESVDLSIPGAC[F/Y]LRLLTITAPSVL | 684.99 | H-2-Kb | |
| 22 | 4:132672942 | Eya3 | HILSVPVSETTY[P/S]GQTQYQTLQQSQ | 322.43 | H-2-Kb | |
| 23 | 6:48025275 | Zfp777 | SPERGSAFNPKH[S/L]LKPRPKSPSSGS | 387.52 | H-2-Kb | |
| 24 | 4:86661976 | Plin2 | NSGVDNAITKSE[L/M]LVDQYFPLTQEE | 608.55 | H-2-Kb | |
| 25 | 4:133586018 | Gpn2 | SLATMLHVELPH[I/V]NLLSKMDLIEHY | 563.28 | H-2-Kb | |
| 26 | 1:131129591 | Dyrk3 | VIHMLESFTFRN[H/N]VCMAFELLSIDL | 87.37 | H-2-Kb | |
| 27 | 10:80689958 | Mob3a | RVFVHVYIHHFD[R/P]IAQMGSEAHVNT | 193.14 | H-2-Kb | |
| 28 | 4:136014429 | Eloa | PKKLLKYLKKLS[I/V]LPITVDILVETG | 992.64 | H-2-Kb | |
| 29 | 6:49055683 | Gpnmb | NGVLISIGCLAV[L/F]VTMVTILLYKKH | 508.89 | H-2-Kb | |
| 30 | X:134238207 | Trmt2b | HYRVVRAIRNCR[T/A]IHTLVFVSCKPH | 42.99 | H-2-Db | |
| 31 | 5:115348675 | Cox6a1 | SARMWKALTYFV[A/P]LPGVGVSMLNVF | 17.09 | H-2-Kb | |
The 31 neoAgs were selected by analyzing the mutanome of Lewis lung tumor. MHC class I binding predictions were performed with IEDB 2.17 software.
Generation and immunogenicity testing of an adenovirus vector encoding LLC exogenous epitopes
A possible explanation for the lack of protective activity against LLC tumor development by prophylactic administration of adenovirus vectored neoAg vaccines could be the potent and rapidly established T cell exhausted phenotype induced by this tumor cell line.14 Transcriptome-based cell-type quantification and flow cytometry analysis of LLC tumors showed that these tumors are less infiltrated of CD8 and CD4 T cells, significantly enriched of immunosuppressive macrophages M2, and with reduced proinflammatory M1 macrophages (Figures S1A and S1B), as compared to the anti-PD1 sensitive model CT26.
We used the LLC cells as a neoAg vaccine and CPI-resistant model to test our prime and pull approach. Fifteen previously characterized C57BL/6 CD8 and CD4 epitopes from common murine pathogens (murine cytomegalovirus [MCMV]; lymphocytic choriomeningitis virus [LCMV]), model antigens (ovalbumin [OVA]; β-galactosidase [β-gal]), or tumor neoAg unrelated to the LLC cell line (MC38 neoAg6,15) were chosen to generate a synthetic polyepitope string (Figures 2A and 2B). Among the 15 selected epitopes, 12 were CD8 and 3 were CD4 epitopes. Epitopes shorter than 25 amino acids were mapped on the original protein sequence and extended to 25–31 amino acid–long sequences by adding 8 to 9 amino acids present upstream and downstream in the native antigen sequence. The resulting peptide sequences were ordered according to a procedure that minimizes the presence of junctional epitopes (9-mer MHC class I epitopes with predicted half-maximal inhibitory concentration [IC50] ≥1,500 nM) and reduces the number of predicted proteasome cleavage sites within each epitope. The nucleotide sequences encoding the 15 epitopes were joined in a head-to-tail configuration to generate a single artificial open reading frame (ORF) of 428 amino acids called CAP1 (Figure 2A), and the corresponding optimized gene was cloned in a replication incompetent human Ad5 vector (Ad-CAP1).
Figure 2.

Design of Ad5-CAP1 vector
(A) Representation of the CAP1 cassette encoded by adenovirus and THV vector (Ad-CAP1 and THV-CAP1, respectively). Fifteen well-known C57BL/6 CD8/CD4 epitopes from common murine pathogens (MCMV; LCMV), potent model antigens (OVA; β-gal), or tumor-specific antigens unrelated to the LLC cell line (MC38 tumor neoantigens) were selected for inclusion in the CAP1 string. (B) List of antigens encoded by the CAP1 cassette and relative sequences; bold sequences denote known T cell epitopes.
CB6F1 mice, a cross between BALB/c and C57BL/6 mice, were used for the characterization of immune response. Vaccination of CB6F1 mice with Ad-CAP1 elicited potent T cell responses against the majority of CAP1 epitopes (Figure 3A), with the strongest reactivities observed on OVA SIINFEKL and MCMV M45 and M139 epitopes (Figures 3A and 3B). The phenotype of Ad-CAP1 vaccine-induced T cells was evaluated by blood tetramer staining at days 21 and 100 for the highest CAP1-specific CD8 T cell reactivities (OVA SIINFEKL; MCMV M45 and M139, >200 IFN-γ spot-forming cells [SFCs]/million splenocytes). This analysis indicated that most of the vaccine-induced CD8+ T cells had an effector (CD62L− CD127) or effector memory (CD162L− CD127+) phenotype (Figure 3C).
Figure 3.

CAP1 immunogenicity and antigen-specific CD8 frequency and phenotype in CB6 mice
(A) In vivo immunogenicity of Ad5-CAP1 vector in CB6 mice. Mice were injected intramuscularly with 5 × 108 vp of Ad5-CAP1 at day 0. At 28 and 100 days after, distinct groups of vaccinated mice were sacrificed for spleen removal. T cell responses were measured by IFN-γ ELISpot on splenocytes. Responses against individual CAP1 antigens, a peptide pool covering the major Ad5 capsid protein (Hexon) and HA tag are shown; peptide diluent DMSO and concanavalin A were used as negative and positive controls, respectively. Bars show mean ± SEM, n = 12. (B) Kinetics of the frequency of the major CAP1-specific CD8 T cells (OVA SIINFEKL; MCMV M45 and M139, >200 IFNγ SFCs/million splenocytes) in blood, as detected by fluorescent pentamers. Blood was collected at days 21, 50, and 100 postimmunization, pooled, and processed for staining and FACS analysis. Blood from naive, unvaccinated age-matched control mice was collected as control for pentamer background. (C) Phenotype of Ad5-CAP1 vaccine-induced T cells analyzed by flow cytometry. Antigen-specific CD8+ T cells in the blood were identified by pentamer staining and analyzed for effector (E), central memory (CM), and effector memory (EM) phenotype, based on the expression of memory markers CD62L and CD127.
Prime and pull with Ad-CAP1 vaccine and THV-CAP1 leads to tumor regression of LLC-resistant tumors and establishes long-lasting T cell memory response
To test the prime and pull approach, we took advantage of the THV-HER2 vector system. In THV-HER2, the receptor-binding domain of the gD surface glycoprotein was substituted with a scFv against the human (h)HER2 protein.9 THV-HER2 has lost its original tropism and has been redirected to an hHER2-displaying cell type.9 By infecting no other cells than the specifically targeted tumor cells, THV-HER2 displays a high safety profile, as demonstrated in our previous studies.16 We have shown that THV-HER2 selectively infects and expresses encoded exogenous proteins in hHER2 cells both in vitro and in vivo.17 We generated a THV-HER2 expressing the CAP1 polyepitope (THV-CAP1) and used it to selectively deliver the CAP1 string into hHER2-LLC tumors implanted in hHER2 C57BL/6 transgenic mice. Prime and pull treatment with Ad-CAP1 and THV-CAP1 was evaluated in a therapeutic setting of large established LLC tumors (Figure 4A). Transgenic hHER2 mice were first vaccinated with Ad-CAP1, showing a similar pattern of immune responses to the CB6F1 mice (Figure S2). At the peak of immune response elicited by the Ad-CAP1 vaccine (i.e., 14 days after immunization), mice were inoculated subcutaneously with the hHER2 LLC tumor cells until the development of established tumors. Tumor-bearing mice were then treated by intratumoral administration of THV-CAP1, according to the scheme of Figure 4A. Mice receiving Ad-CAP1 or THV-CAP1 as stand-alone treatments did not achieve antitumoral response. On the contrary, the sequential administration of Ad-CAP1 and THV-CAP1 resulted in complete tumor eradication in 30% of treated mice (Figure 4B). Interestingly, complete responder mice in the prime and pull group showed higher CAP1-specific T cell response than the nonresponder ones, further supporting the conclusion that the T cells elicited by the Ad-CAP1 vaccine against the LLC exogenous epitopes exerted a therapeutic effect in restricting and eventually clearing LLC tumors transduced by THV-CAP1 (Figure 4C). The combination of Ad-CAP1 and THV-CAP1 led to a significant increase in the overall survival compared to the monotherapies (Figure 4D). Mice that became tumor free upon combined prime and pull treatment were protected from a second tumor challenge, demonstrating effective induction of memory T cells against LLC tumor cell line antigens (Figure 4E). No antitumor efficacy was observed when using a THV encoding an unrelated protein (Figure S3).
Figure 4.

Efficacy in therapeutic setting of Ad-CAP1 and/or THV-CAP1 vector
(A) Schematic of therapeutic efficacy setting. The therapeutic effect of Ad5-CAP1 or THV-CAP1 as stand-alone therapies or the combination therapy was evaluated in HER2-LLC-derived large established tumors. Ad5-CAP1-treated transgenic hHER2 mice were intramuscularly injected with 5 × 108 vp, 13 days before HER2-LLC cells inoculum (subcutaneous, day 0). THV-CAP1-treated mice were intratumorally injected at days 7 and 9 with 2.5 × 108 PFU upon mice randomization according to tumor volume (tumor volume average: 60–90 mm3). The combination treated mice received Ad5-CAP1 and THV-CAP1 as described for the single treatments. Not-treated tumors were used as negative control. (B) Tumor growth monitoring over time until day 28. Each line represents an individual tumor (gray lines for nonresponders, dark red lines for responders to Ad5-CAP1/THV-CAP1 combination; n = 10–12). Percentage on the graphs indicates the rate of response. Data are representative of 2 independent experiments (Fisher exact test; ∗p < 0.05). (C) In vivo immunogenicity in responder and nonresponder mice to Ad5-CAP1/THV-CAP1 combination. At day 20 post tumor challenge, responder (n = 4) and nonresponder (n = 4) mice to combination were sacrificed. T cell responses were measured by IFN-γ ELISpot on splenocytes. Responses against the pooled antigens of CAP1 string are shown; peptide diluent DMSO and concanavalin A were used as negative and positive controls, respectively. Bars show mean ± SEM; dark red bar for responders, gray bar for nonresponders. Statistics were generated using an unpaired Mann-Whitney nonparametric test. Data are representative of 2 independent experiments. (D) Overall survival is shown for mice treated with Ad-CAP1 (blue line) or THV-CAP1 (dark green line) as monotherapies and with the combination (dark red line). Untreated mice represent the negative control (black line). (E) A total of 60 days after HER2-LLC first subcutaneous injection, tumor-free mice treated with Ad-CAP1/THV-CAP1 combination were challenged with a second HER2-LLC tumor inoculum. Animals’ survival after the second challenge was monitored until day 120 postfirst challenge (log rank test; ∗∗∗p < 0.001).
Discussion
CPB is now an established anticancer treatment in different indications resulting in a long-term cure, but its efficacy is limited to only a fraction of the patients treated.18 Cancer vaccines may provide a valuable option to improve CPI efficacy. Therapeutic efficacy of cancer vaccination depends on (1) the selection of the most appropriate antigens, (2) the ability to induce a robust T killer lymphocyte response, and (3) the concomitant treatment with CPI to counteract the immunosuppressive tumor microenvironment. The advent of NGS techniques for the accurate and rapid sequencing of the DNA exome and mRNA transcriptome of cancer cells from patients’ biopsies has allowed the identification of cancer-specific somatic mutations giving rise to neoAgs, which constitute a source of highly immunogenic and safe antigens for cancer vaccine generation.19,20 Furthermore, genetic vaccine technologies based on mRNA formulated in LNPs and viral vectors represent a potent platform to address the second requirement, as witnessed by preliminary results in cancer vaccine clinical trials.4,5 However, the combination of CPI and neoAg-based vaccine cannot be used in tumors that are resistant to CPI treatment and/or do not present a sufficient number of druggable mutations via neoAg-based vaccines.
We sought to address the issue of CPI-resistant (and/or neoAg lacking) tumors by devising a vaccination strategy based on potent cancer exogenous antigens and a prime and pull modality of administration. To this end, we chose the mouse LLC model, which was previously shown to be insensitive to CPI treatment alone or to a combination of CPI and a peptide vaccine reproducing an LLC neoAg despite the induction of neoAg-specific CD8+ tumor-infiltrating lymphocytes.14 We confirmed and expanded these observations by using an adenovirus vectored neoAg vaccine containing 31 LLC neoAgs. This immune hostile phenotype was shown to be due to a blunted IFN-γ response as a result of high basal expression of suppressor of cytokine signaling 1 (SOCS1), an inhibitor of IFN-γ.12 Furthermore, although several studies performed in different mouse tumor models showed that neoAg-based vaccine monotherapy is efficacious in a prophylactic setting, we found that this paradigm is not confirmed in the LLC model.14 We hypothesized that given their resistance to immunotherapy even when given prophylactically, LLC tumor cells would lead to an exceptionally potent and rapid establishment of T lymphocyte immunosuppression. Therefore, one possible way to increase the efficacy of a vaccine would be to use it in a pseudo-prophylactic setting, namely by inducing a functional antigen-specific T cell immunity against a preset number of epitopes, and then having only the tumor cells to express the same vaccine-targeted epitopes. As carrier for vaccine antigens, we chose a replication incompetent adenovirus vector for its ability to encode large artificial antigens generated by joining several fragments from different proteins (i.e., over 2,000 amino acids) and to induce potent and functional CD8 T cell responses in humans.21
As a vaccine antigen, we selected 15 known T cell epitopes derived from viruses, bacterial proteins, or neoAgs from tumor cell lines different from the LLC with demonstrated immunogenicity in the mouse strain used for our experiments. We used an unbalanced ratio between CD8 and CD4 epitopes (12 out of 15) to ensure the induction of a potent antitumor cytolytic response.22 The nucleotide sequences encoding the 15 epitopes were joined in a head-to-tail configuration to generate an ORF of 428 amino acids (CAP1). The CAP1 epitopes are not present in the mouse proteome and are not homologous to LLC protein sequences, thus representing safe and potent exogenous antigens not subjected to tolerance or immune editing. In view of a human application, this approach is superior to the neoAg approach because it relies on previously characterized epitopes of known immunogenicity and allows one to generate an off the shelf vaccine with a potential pan-cancer indication. Consistently, Ad-CAP1 was highly immunogenic and elicited large populations of antigen-specific CD8+ T cells with a phenotype of T effector and T effector memory cells with lytic capability and effector functions, which have been associated with effective immunity against infectious diseases and cancer.23
To make tumors susceptible to the vaccine-induced adaptive immunity, we used a tumor-targeted herpesvirus17 as delivery vector to express the exogenous CAP1 antigen only in the tumor. The combined Ad-CAP1 and THV-CAP1 treatment led to tumor delay and clearance in 30% of the animals. These results are due to the prime and pull of CAP1-specific T cells since neither Ad-CAP1 or THV-CAP1 alone nor a combination of Ad-CAP1 and a THV encoding an unrelated protein did achieve antitumor efficacy. Although efficacy was observed only in less than half of the treated animals, these results must be put in the context of the difficult-to-treat LLC tumor model, which is resistant to different immunotherapies, including anti-PD1, anti-CTLA4, and neoAg vaccines. Relevant to this point, it was previously shown that a triple therapy combining radiotherapy, anti-PD-L1, and L19–IL2 cured only 38% of LLC tumors.24
We could show that there is a significant difference (almost 3-fold) in the extent of T cell response measured in responding versus nonresponding mice. Administration of anti-PD1 did not improve the rate of cure (Figure S4).
The THV-CAP1 was injected in mice 3 weeks after vaccination with Ad-CAP1 and 1 week after tumor cell inoculation to allow for both immune response and tumor development to reach high levels. However, we cannot exclude that a different administration interval between the two vectors could result in a higher efficacy. Therefore, a fine-tuning of the modality of administration of the two vectors would be needed to fully appreciate the power of our proposed approach. More important, none of the cured animals developed tumors after a second subcutaneous challenge with LLC cells, which is reflective of the development of an immunological memory against LLC tumor cells triggered by the Ad-CAP1 and THV-CAP1 combination, further supporting the role of the vaccine-induced adaptive immunity. This memory T cell response cannot be targeting the CAP1 string because in the rechallenge experiment, we did not re-administer the THV-CAP1 vector, but it is likely to be the result of an epitope spreading mechanism25 directed at other LLC antigens not included in the administered vectors.
Previous studies have used the prime and pull approach to redirect an immune response primed in the periphery against an infectious agent to a specific tissue (i.e., liver, mucosa) with the objective of eliciting tissue resident memory cells to prevent a subsequent infection.26,27 In our work, we aimed to establish a protective immune response in a cancer therapeutic setting when tumor size had reached a significant value, and against a difficult tumor model in which not even a prophylactic mode of cancer vaccine administration could achieve any efficacy.
The clinically validated adenovirus platform represents a benefit of our approach versus other vaccine strategies with substantial translational challenges. In particular, different from previous work performed using a single model epitope to prime and pull anticancer T cells,28 we have shown here that the adenovirus and HSV1 technologies allow the delivery of several different epitopes embedded in a 25-amino acid–long sequence. This high-capacity feature6 may represent a significant advantage regarding the immunosuppressive tumor environment and simplifies the biomanufacturing process. Similarly, the herpesvirus has been used for a marketed product against metastatic melanoma (T-VEC),29 which shows the clinical translatability of our proposed pulling approach to tilt the balance between the tumor immunosuppressive activity and the vaccine-induced immunity toward the latter one leading to antitumoral efficacy in CPI-resistant cancers.
Materials and Methods
Mice
hHER2-transgenic C57BL/6 mice (B6.Cg-Pds5bTg (Wap ERBB2) 229Wzw/J) were obtained from The Jackson Laboratory (Bar Harbor, ME). CB6F1 mice were purchased from Envigo (Indianapolis, IN). Mouse colony management and all day-to-day care were performed by trained mouse house staff at Plaisant (Castel Romano, Italy). Female mice (6–8 weeks of age) were used for in vivo experiments.
All of the experimental procedures were approved by the Italian Ministry of Health (Authorizations 213/2016 PR) and have been done in accordance with the applicable Italian laws (D.L.vo 26/14 and following amendments), the Institutional Animal Care and ethic Committee of CEINGE and Allevamenti Plaisant S.r.l.
Cell culture
The HER2-LLC cell line was maintained in DMEM (Thermo Fisher Scientific, Waltham, MA), supplemented with 10% fetal bovine serum [FBS], 1% (v/v) penicillin-streptomycin, 2 mM l-glutamine (Thermo Fisher Scientific), and 2 mM puromycin (Sigma-Aldrich/Merck, St. Louis, MO) for selection. The CT26 colon carcinoma cell line was purchased from American Type Culture Collection and cultured in complete RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% FBS, 2 mM l-glutamine, and 1% (v/v) penicillin-streptomycin. The SKOV3 (human ovarian adenocarcinoma) cell line was cultured in RPMI 1640 Medium GlutaMAX (Thermo Fisher Scientific) and supplemented as described above. All of the cell lines were maintained at 37°C in 5% CO2.
Adenovirus vaccines generation and production
The mutanome detected in each tumor model was ranked by using our VENUS algorithm to design vaccines targeting 31 tumor-specific neopeptides with the highest potential to act as neoAgs. VENUS assigned to each tumor-specific somatic mutation a weighted score that combines the mutation allelic frequency, the abundance of the transcript coding for the mutation, and the likelihood to bind the mouse MHC class I alleles. The 31 best potential neoAgs according to the VENUS ranking score were selected to design an artificial transgene to be encoded by the adenovirus vectors. The leader sequence (amino acids 1–29) of the human tissue plasminogen activator protein (NP_000921.1) and a hemagglutinin (HA) tag were added at the N and C terminus of each transgene. CAP1, CT26 neoAg, and LLC neoAg transgenes were synthetized by GENEART (Thermo Fisher Scientific) and subcloned via SwaI or EcoRI-Not1 restriction enzymes (New England Biolabs, Ipswich, MA) into the respective shuttle plasmids containing the CMV promoter with two Tet Operator repeats and a BGH polyA. The expression cassette of CAP-1 was then transferred into pAd5 plasmid E1/E3 deleted.
The CT26 and LLC expression cassettes were transferred into pGAd plasmid, containing the E1/E3/E4 deleted, in which the E4 is replaced with Ad5 E4 ORF6. For all of the plasmids, the transgene cassettes were introduced in the E1 deletion locus of related pAdeno by homologous recombination in BJ5183 cells (Agilent, Santa Clara, CA).
hAd5 and GAd vectors were produced by transfection of adherent M9 cells (293 cells derivative) with Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) and amplification in suspension M9 cells. Vectors were then purified from infected cells by Vivapure Adenopack 20 RT (Sartorius, Goettingen, Germany).
CAP1 polyepitope design
The murine CAP1 polyepitope is constituted by 15 previously characterized immunogenic epitopes derived from C57BL/6 common murine pathogens (MCMV; LCMV), model antigens (OVA; β-gal) and tumor neoAg unrelated to the LLC cell line (MC38). The optimized nucleotide sequences encoding the epitopes (12 CD8 and 3 CD4) were joined in a head-to-tail configuration to generate a single artificial ORF (428 amino acids) and the corresponding gene cloned in a replication incompetent human Ad5 vector (Ad-CAP1). An HA tag was added at the C terminus to detect the polyepitope.
THV-CAP1 generation and production
THV encoding CAP1 was generated by recombineering. Briefly, the CAP1 coding sequence was cloned into a shuttle plasmid under the control of the CMV promoter. The entire expression cassette (CMV promoter, CAP1 coding sequence, and polyA signal) was then excised by PCR using oligonucleotides to incorporate homology arms to the intergenic locus into the THV BAC genome. Infectious viral particles were generated by the transfection of SKOV3 cells. Following single plaque isolation, THV-CAP1 was grown into SKOV3 cells for a few passages. The identity of the CAP1 transgene was confirmed by Sanger sequencing from infectious viral particles purified by iodixanol gradient.
Ex vivo immune analysis
IFN-γ ELISpot assays were performed on single-cell suspensions of spleens. MSIP S4510 plates (Millipore, Billerica, MA) were coated with 10 μg/mL of anti-mouse IFN-γ antibody (U-CyTech, Utrecht, the Netherlands) and incubated overnight at 4°C. After washing and blocking, mouse splenocytes were plated in duplicate at two different cell densities and stimulated overnight with single 25-mer peptides at a final concentration of 1 μg/mL. PepTivator-AdV5 Hexon pool (Miltenyi Biotec, Cologne, Germany) was used at a 1-μg/mL final concentration. The peptide diluent dimethyl sulfoxide (Sigma-Aldrich/Merck) and concanavalin A (Sigma-Aldrich/Merck) were used, respectively, as negative and positive controls. Plates were developed by subsequent incubations with biotinylated anti-mouse IFN-γ antibody (dilution: 1/100; catalog no. CT317-D; U-CyTech), conjugated streptavidin-alkaline phosphatase (dilution: 1/2,500; catalog no. 554065; BD Biosciences, San Jose, CA) and finally with 5-bromo-4-chloro-3-indoyl-phosphate/nitro blue tetrazolium 1-step solution (Thermo Fisher Scientific). An automated ELISpot video analysis system automated plate reader was used to analyze plates. ELISpot data were expressed as IFN-γ SFCs per million splenocytes. ELISpot responses were considered positive if all of the following conditions occurred: (1) IFN-γ production present in concanavalin A-stimulated wells, (2) the number of spots seen in positive wells was three times the number detected in the mock control wells (dimethyl sulfoxide), and (3) at least 30 specific spots per million splenocytes.
In vivo treatments
Adenovirus vaccines were administered via intramuscular injections in both quadriceps by delivering a volume of 50 μL per side at 5 × 108 viral particles (vp). THV-CAP1 was administered intratumorally with 2.5 × 108 plaque-forming units (PFU). For efficacy studies, α-mPD1 (BioXcell, Lebanon, NH; clone RMP114, catalog no. BE0146) was administered systemically (intraperitoneally) at a dosage of 200 μg twice per week for 3 weeks. For combination treatments, Ad-CAP1 and THV-CAP1 were administered as described for the single treatments upon mice randomization according to tumor volume (tumor volume average: 60–90 mm3).
In vivo tumor growth
For prophylactic experiments, 5 × 105 HER2-LLC cells and CT26 cells were injected subcutaneously into the lower right flank 14 days after immunization. For the established tumor setting experiments, 5 × 105 HER2-LLC1 cells were injected subcutaneously. After 5–7 days, tumor masses were measured with a digital caliper, applying the formula 0.5 × length × width2, where the length is the longer dimension. Animals were then randomized based on their tumor size (tumor size average per group 60–90 mm3) and treatments began (day 0). Tumor growth was measured every 3–4 days and mice were euthanized as soon as signs of distress or a tumor volume above 1,500 mm3 was reached. For rechallenge experiments, 5 × 105 HER2-LLC1 cells were injected subcutaneously in cured tumor-free mice 60 days after the first tumor challenge.
Immune phenotyping
Blood was collected from the retro-orbital plexus of CAP1-vaccinated or naive CB6 mice in tubes containing lithium heparin anticoagulant (lithium heparin tubes, BD Biosciences). In a 5-mL fluorescence-activated cell sorting (FACS) tube, 200 μL of blood from each vaccinated mouse (n = 12) or from the naive mouse were pooled (for the naive control, blood was drawn and processed from a single unvaccinated animal), and red blood cells were lysed twice with 3 mL ACK lysis buffer (Gibco, Thermo Fisher Scientific) by incubating 5 min at room temperature. After spinning for 6 min at 500 × g, the ACK lysis was repeated (1 mL ACK, 3 min incubation, spin). Cell pellet was resuspended in FACS buffer (PBS 0.1% BSA) and a 200-μL cell suspension was transferred into wells of a 96-well plate round bottom for staining. Plate was spun (5 min 300 × g) and pellets resuspended in 40 μL of FACS buffer; 10 μL of a FACS buffer solution containing prespun specific pentamer at 8 μg/μL (MCMV m45 985–993 APC, MCMV M139 419–426 PE, OVA SIINFEKL OVA 257–264 PE, all from Proimmune, Oxford, UK) was added to each well and incubated 20 min at room temperature. Cells were then washed with PBS and stained for viability with near-infrared LIVE/DEAD fixable dead cell stain (Invitrogen, Thermo Fisher Scientific) in PBS; incubating 20 min at room temperature protected from light. Cells were then washed with PBS and surface stained 20 min at room temperature protected from light with FACS buffer containing Fc block (BD Biosciences) and the following conjugated antibodies (all from BioLegend, San Diego, CA): CD3e FITC (clone 145-2C11), CD8 Pacific Blue (clone 53-6-7), CD4 BV605 (clone GK1.5), CD19 BV650 (clone 6D5), CD127 PECy7 (clone A7R34), and CD62L BV785 (clone MEL14). After washing with FACS buffer, the cells were resuspended in 200 μL FACS buffer-EDTA 2 mM and immediately acquired at a Cytoflex analyzer (Beckman Coulter, Brea, CA). Data were analyzed with FlowJo (BD Biosciences) version 10.8.1.
Data and code availability
All of the data relevant to the study are included in the article or uploaded as supplemental information. Materials are available from the corresponding authors on reasonable request.
Acknowledgments
We acknowledge the animal facility of Plaisant in Castel Romano (Rome, Italy) for maintenance and care of the mice used in this study. We thank Laura Seclì for her valuable help in preparation of the illustrations. A.N. and E. Sasso were supported by POR Regione Campania Covid-19; PNRR Centri Nazionali - Progetto “National Center for Gene Therapy and Drugs based on RNA Technology CN3; PNRR PARTENARIATO ESTESO PNRR, LINEA 13 – MALATTIE INFETTIVE EMERGENTI.
Author contributions
Study design, A.M.D., A.N., S.C., and G.L. Methodology and investigation, F.T., E. Sasso, G.R., S.A., M.D.S., M.E., G.F., and G.C. Bioinformatic analysis, G.L. Writing – original draft, A.M.D. and A.N. Writing – review & editing, A.M.D., A.N., S.C., and E. Scarselli. Supervision, A.M.D., A.N., and A.F. Guarantor, A.N.
Declaration of interests
E. Scarselli, A.F., and A.N. are co-founders of Nouscom. F.T., G.L., G.R., S.A., G.C., E. Scareselli, and A.M.D. are employees of Nouscom. All of the other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200760.
Supplemental information
References
- 1.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen D.S., Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S., et al. Cancer immunology Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Alise A.M., Brasu N., De Intinis C., Leoni G., Russo V., Langone F., Baev D., Micarelli E., Petiti L., Picelli S., et al. Adenoviral-based vaccine promotes neoantigen-specific CD8(+) T cell stemness and tumor rejection. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abo7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B., et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 6.D'Alise A.M., Leoni G., Cotugno G., Troise F., Langone F., Fichera I., De Lucia M., Avalle L., Vitale R., Leuzzi A., et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun. 2019;10:2688. doi: 10.1038/s41467-019-10594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campadelli-Fiume G., Petrovic B., Leoni V., Gianni T., Avitabile E., Casiraghi C., Gatta V. Retargeting Strategies for Oncolytic Herpes Simplex Viruses. Viruses. 2016;8:63. doi: 10.3390/v8030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasso E., Froechlich G., Cotugno G., D'Alise A.M., Gentile C., Bignone V., De Lucia M., Petrovic B., Campadelli-Fiume G., Scarselli E., et al. Replicative conditioning of Herpes simplex type 1 virus by Survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci. Rep. 2020;10:4307. doi: 10.1038/s41598-020-61275-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menotti L., Nicoletti G., Gatta V., Croci S., Landuzzi L., De Giovanni C., Nanni P., Lollini P.L., Campadelli-Fiume G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA. 2009;106:9039–9044. doi: 10.1073/pnas.0812268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Froechlich G., Gentile C., Infante L., Caiazza C., Pagano P., Scatigna S., Cotugno G., D'Alise A.M., Lahm A., Scarselli E., et al. Generation of a Novel Mesothelin-Targeted Oncolytic Herpes Virus and Implemented Strategies for Manufacturing. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22020477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leoni G., D'Alise A.M., Tucci F.G., Micarelli E., Garzia I., De Lucia M., Langone F., Nocchi L., Cotugno G., Bartolomeo R., et al. VENUS, a Novel Selection Approach to Improve the Accuracy of Neoantigens' Prediction. Vaccines. 2021;9 doi: 10.3390/vaccines9080880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bullock B.L., Kimball A.K., Poczobutt J.M., Neuwelt A.J., Li H.Y., Johnson A.M., Kwak J.W., Kleczko E.K., Kaspar R.E., Wagner E.K., et al. Tumor-intrinsic response to IFNgamma shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci. Alliance. 2019;2 doi: 10.26508/lsa.201900328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Froechlich G., Caiazza C., Gentile C., D'Alise A.M., De Lucia M., Langone F., Leoni G., Cotugno G., Scisciola V., Nicosia A., et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers. 2020;12 doi: 10.3390/cancers12113407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S., Simoni Y., Zhuang S., Gabel A., Ma S., Chee J., Islas L., Cessna A., Creaney J., Bradley R.K., et al. Characterization of neoantigen-specific T cells in cancer resistant to immune checkpoint therapies. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025570118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yadav M., Jhunjhunwala S., Phung Q.T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T.K., Fritsche J., Weinschenk T., et al. Immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 16.Leoni V., Vannini A., Gatta V., Rambaldi J., Sanapo M., Barboni C., Zaghini A., Nanni P., Lollini P.L., Casiraghi C., Campadelli-Fiume G. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Lucia M., Cotugno G., Bignone V., Garzia I., Nocchi L., Langone F., Petrovic B., Sasso E., Pepe S., Froechlich G., et al. Retargeted and Multi-cytokine-Armed Herpes Virus Is a Potent Cancer Endovaccine for Local and Systemic Anti-tumor Treatment. Mol. Ther. Oncolytics. 2020;19:253–264. doi: 10.1016/j.omto.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 19.Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 20.Davar D., Dzutsev A.K., McCulloch J.A., Rodrigues R.R., Chauvin J.M., Morrison R.M., Deblasio R.N., Menna C., Ding Q., Pagliano O., et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595–602. doi: 10.1126/science.abf3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasso E., D'Alise A.M., Zambrano N., Scarselli E., Folgori A., Nicosia A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020;50 doi: 10.1016/j.smim.2020.101430. [DOI] [PubMed] [Google Scholar]
- 22.Ostroumov D., Fekete-Drimusz N., Saborowski M., Kühnel F., Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018;75:689–713. doi: 10.1007/s00018-017-2686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Hara G.A., Duncan C.J.A., Ewer K.J., Collins K.A., Elias S.C., Halstead F.D., Goodman A.L., Edwards N.J., Reyes-Sandoval A., Bird P., et al. Clinical assessment of a recombinant simian adenovirus ChAd63: a potent new vaccine vector. J. Infect. Dis. 2012;205:772–781. doi: 10.1093/infdis/jir850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olivo Pimentel V., Marcus D., van der Wiel A.M., Lieuwes N.G., Biemans R., Lieverse R.I., Neri D., Theys J., Yaromina A., Dubois L.J., Lambin P. Releasing the brakes of tumor immunity with anti-PD-L1 and pushing its accelerator with L19-IL2 cures poorly immunogenic tumors when combined with radiotherapy. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-001764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulley J.L., Madan R.A., Pachynski R., Mulders P., Sheikh N.A., Trager J., Drake C.G. Role of Antigen Spread and Distinctive Characteristics of Immunotherapy in Cancer Treatment. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Çuburu N., Khan S., Thompson C.D., Kim R., Vellinga J., Zahn R., Lowy D.R., Scheper G., Schiller J.T. Adenovirus vector-based prime-boost vaccination via heterologous routes induces cervicovaginal CD8(+) T cell responses against HPV16 oncoproteins. Int. J. Cancer. 2018;142:1467–1479. doi: 10.1002/ijc.31166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao T., Israelow B., Peña-Hernández M.A., Suberi A., Zhou L., Luyten S., Reschke M., Dong H., Homer R.J., Saltzman W.M., Iwasaki A. Unadjuvanted intranasal spike vaccine elicits protective mucosal immunity against sarbecoviruses. Science. 2022;378:eabo2523. doi: 10.1126/science.abo2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tähtinen S., Feola S., Capasso C., Laustio N., Groeneveldt C., Ylösmäki E.O., Ylösmäki L., Martins B., Fusciello M., Medeot M., et al. Exploiting Preexisting Immunity to Enhance Oncolytic Cancer Immunotherapy. Cancer Res. 2020;80:2575–2585. doi: 10.1158/0008-5472.CAN-19-2062. [DOI] [PubMed] [Google Scholar]
- 29.van Akkooi A.C.J., Haferkamp S., Papa S., Franke V., Pinter A., Weishaupt C., Huber M.A., Loquai C., Richtig E., Gokani P., et al. Retrospective Chart Review Study of Real-World Use of Talimogene Laherparepvec in Unresectable Stage IIIB-IVM1a Melanoma in Four European Countries. Adv. Ther. 2021;38:1245–1262. doi: 10.1007/s12325-020-01590-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data relevant to the study are included in the article or uploaded as supplemental information. Materials are available from the corresponding authors on reasonable request.