

Abstract
The detection of circular RNA molecules (circRNAs) is typically based on short-read RNA sequencing data processed using computational tools. Numerous such tools have been developed, but a systematic comparison with orthogonal validation is missing. Here, we set up a circRNA detection tool benchmarking study, in which 16 tools detected more than 315,000 unique circRNAs in three deeply sequenced human cell types. Next, 1,516 predicted circRNAs were validated using three orthogonal methods. Generally, tool-specific precision is high and similar (median of 98.8%, 96.3% and 95.5% for qPCR, RNase R and amplicon sequencing, respectively) whereas the sensitivity and number of predicted circRNAs (ranging from 1,372 to 58,032) are the most significant differentiators. Of note, precision values are lower when evaluating low-abundance circRNAs. We also show that the tools can be used complementarily to increase detection sensitivity. Finally, we offer recommendations for future circRNA detection and validation.
Introduction
Circular RNAs (circRNAs) are a class of non-coding RNA molecules ubiquitous in humans and other eukaryotic species. For a long time, circRNAs were regarded as unimportant byproducts of splicing. However, since the advancement of RNA sequencing technologies and the development of circRNA detection bioinformatics pipelines there has been a significant increase in circRNA research, with a compound annual growth rate of scientific publications of 58% over the last 5 years (Fig. 1a)1.
Fig. 1 |. CircRNA scientific relevance, structure and detection.

a, Over the last decade, circRNA research has increased rapidly, as illustrated by the proportional growth of publications mentioning circRNA in Europe PubMed Central. b, CircRNAs are formed through back-splicing, which results in a circular molecule with a back-spliced junction (BSJ). Black boxes highlight the BSJ in the circRNA isoforms. c, CircRNAs can be detected with RT–qPCR using a BSJ-specific primer pair. The primer pair can bind only in a divergent manner (facing away from each other) to linear RNA, where no amplification will be possible, yet binds the circRNA in a convergent manner (facing towards each other), amplifying the BSJ sequence. d, Large-scale circRNA detection is typically performed using total RNA sequencing datasets and specialized computational tools. These tools identify BSJ-spanning reads, which map divergently (in reverse order) on the linear reference genome.
Although an in vivo function for most circRNAs remains unknown and functional analyses are typically restricted to in vitro experiments, some circRNAs have been linked to specific diseases, including cancer. CircRNAs have also been reported to be more stable than linear transcripts due to the absence of a free 5′ or 3′ end that can be recognized by exonucleases1. In line with this, a higher fraction of circRNA relative to linear RNA has been observed in a wide range of human biofluids, which makes them interesting biomarker candidates, with the potential to be used for minimally invasive tests for diagnosis or response monitoring2. Wang et al. reviewed 112 differentially expressed circRNAs in various biofluids from patients with different cancer types3. Furthermore, 15 clinical trials incorporating circRNAs as disease biomarkers have been initiated (ClinicalTrials.gov, accessed on 20 October 2022).
Eukaryotic circRNAs are formed through a process called back-splicing, in which the 5′ end of an RNA molecule forms a covalent bond with its own 3′ end, forming a circular molecule with a characteristic back-spliced junction (BSJ) sequence (Fig. 1b)1. CircRNAs consist of one or multiple exons and, analogous to linear RNA, they also have alternative splicing, in which circRNAs with the same BSJ sequence may have a different exon (and/or intron) composition1.
In a targeted manner, circRNAs can be quantified with reverse transcription–quantitative polymerase chain reaction (RT–qPCR) using BSJ-spanning primer pairs to amplify the region flanking the BSJ (Fig. 1c). These primer pairs are divergent (facing away from each other) when hybridizing to the linear host transcripts and can therefore amplify only the circRNA4. However, false positives resulting from alignment ambiguity, repeat sequences, trans-splicing or reverse transcription template-switching artifacts have been described5,6. In all of these cases, a linear RNA molecule is formed with the same exon orientation and, therefore, the same sequence as the circRNA BSJ. To prevent false-positive circRNA identification, linear RNA is often digested with the exonuclease ribonuclease R (RNase R) followed by RT–qPCR. RNase R typically degrades linear RNA, whereas circRNAs are generally not affected. Of note, it has been suggested that long circRNAs may be somewhat sensitive to RNase R degradation, and various challenges in the validation of circRNAs have been recognized7,8.
In general, high-throughput or exploratory circRNA detection is performed using bioinformatics approaches that analyze total RNA sequencing data. For this, the RNA sequencing reads are first mapped against a reference genome. The unmapped reads are subsequently used to identify BSJ-spanning reads that map divergently (in reverse order) on the linear genome (Fig. 1d).
Over the last decade, numerous computational circRNA detection tools have been developed and tested. Whereas multiple sets of circRNA detection tools using a bioinformatics approach have been compared (often when a novel tool is published)9–16, a systematic and comprehensive evaluation of many circRNA detection tools using an orthogonal validation method is still missing. In our benchmarking study the aim was to evaluate all currently available circRNA detection tools with an orthogonal approach using RT–qPCR, RNase R and amplicon sequencing (Fig. 2a). Our study highlights that although the precision of the tools is generally excellent, their sensitivities are highly variable.
Fig. 2 |. CircRNA detection tools predict a wide variety of circRNAs.
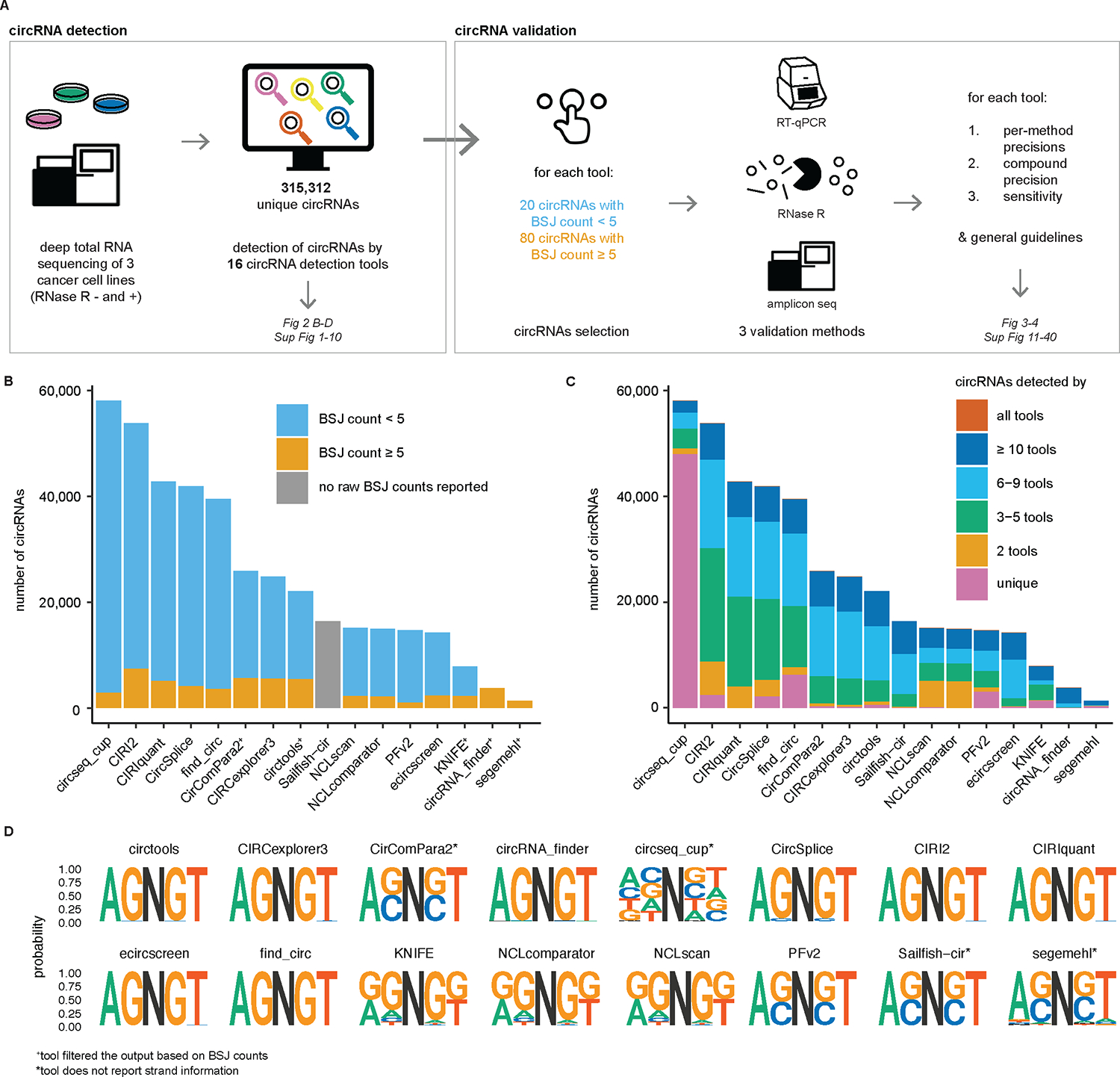
a, This study consists of a circRNA detection phase and a circRNA validation phase. For the former, 16 circRNA detection tools were used to predict circRNAs in three deeply sequenced cancer cell lines. For the latter, a set of circRNAs was selected per tool and validated using three orthogonal methods, generating tool-specific precision values for each method. This was also used to compute compound precision and both types of sensitivity values for each circRNA detection tool. b, The number of reported circRNAs differs greatly between tools (shown for HLF cells; similar results for the other cell lines are shown in Supplementary Fig. 1). The tools are ordered according to the total number of predicted circRNAs. The vast majority of circRNAs are predicted with a BSJ count below 5 (in blue). Two tools, circRNA_finder, and segemehl, filtered their results to report only circRNAs with a BSJ count of at least 5 (in orange). †Tool filtered the output based on BSJ count. c, The majority of circRNAs (49.9%) are detected by only one tool. Circseq_cup reports the largest set of unique circRNAs (shown for HLF cells; similar results for the other cell lines are shown in Supplementary Fig. 4). A small set of 55 circRNAs is detected by all tools (column n_db in Supplementary Table 2). d, CircRNA splice sites differ between circRNA detection tools. Most commonly, the canonical AGNGT pattern is observed, with AG being the splice acceptor, N the circRNA, and GT the splice donor. ‡Circseq_cup, CirComPara2, Sailfish-cir and segemehl do not report strand information. To be able to retrieve a splicing sequence for the circRNAs from these tools, it was assumed that the circRNA originated from the positive strand. This led to the ACNCT pattern (reverse complement of AGNGT), most probably from circRNAs that were assigned to the positive strand incorrectly. Last, there are some tools that also report a substantial number of circRNA BSJ sequences with a GGNGG splicing pattern.
Results
CircRNA detection tools predict a wide variety of circRNAs
CircRNA detection tools differ in detection strategies and filtering.
For this study, 16 different circRNA detection tools were assessed: CIRCexplorer3 (ref. 17), CirComPara2 (ref. 11), circRNA_finder18, circseq_cup19, CircSplice20, circtools21, CIRI2 (ref. 22), CIRIquant23, ecircscreen (unpublished tool), find_circ24, KNIFE15, NCLscan25, NCLcomparator26, PFv2 (ref. 27), Sailfish-cir28 and segemehl29 (Table 1 and Supplementary Table 1). CircRNA detection tools differ in their circRNA detection approach (including strand assignment), reliance on linear annotation, and filtering methods. CircRNAs can be detected from RNA sequencing data using the pseudo-reference-based approach (also called the candidate-based approach) or the fragmented-based approach (also called the segmented read-based approach)12,14. The former approach uses a reference list of potential BSJ sequences, often based on all possible combinations of known annotated exons in a gene. This approach is therefore limited to species with annotated genomes and to previously annotated genes and will detect only circRNAs that use the same splicing sites as the linear RNAs. The latter approach splits unmapped sequencing reads into shorter sequences and remaps these against the reference genome. Last, integrative tools, such as CirComPara2 and ecircscreen, combine the results of multiple tools.
Table 1 |.
CircRNA detection tools with their circRNA detection approach, strand assignment approach, reliance on linear annotation and filtering approach
| Tool | Approach | circRNAs detected in… | Strand assignmenta | Splicing | BSJ count filterb | Minimum circRNA length (in nucleotides)b | Maximum circRNA length (in nucleotides)b |
|---|---|---|---|---|---|---|---|
| CIRCexplorer3 | Segmented read-based | Entire genome | Based on linear annotation | AGNGT | None | None | None |
| CirComPara2 | Integrativec | Entire genome | No strand reported | AGNGT, ACNCT | ≥2 | 299 | 2,304,996 |
| circRNA_finder | Segmented read-based | Entire genome | Based on mapping to genome | AGNGT | ≥5 | 200 | 100,000 |
| circseq_cup | Based on segemehl, with full-length circRNA assembly | Entire genome | No strand reported | Non-canonical | None | None | 5,000 |
| CircSplice | Segmented read-based | Known splice sites | Based on linear annotation | AGNGT, ACNCT | None | 78 | None |
| circtools | Segmented read-based | Entire genome | Based on mapping to genome | AGNGT | ≥2 in ≥2 samples | 31 | 1,000,000 |
| CIRI2 | Segmented read-based | Entire genome | Based on GT–AG splice sites | AGNGT | None | 135 | 200,000 |
| CIRIquant | Based on CIRI2, with improved quantification | Entire genome | Based on GT–AG splice sites | AGNGT | None | 135 | 200,000 |
| ecircscreen | Integrativec | Entire genome | Based on consensus from tools | AGNGT | None | None | None |
| find_circ | Segmented read-based | Entire genomed | Based on mapping to genomed | AGNGT | None | None | 100,000 |
| KNIFE | Candidate-based | Entire genome | Based on linear annotation | Non-canonical | ≥2 | None | 1,000,000 |
| NCLscan | Candidate-based | Known splice sites | Based on linear annotation | Non-canonical | None | 100 | None |
| NCLcomparator | Filtered results of NCLscan | Known splice sites | Based on linear annotation | Non-canonical | None | 100 | None |
| PFv2 | Segmented read-based | Entire genome | Based on mapping to genome | AGNGT, ACNCT | None | 50 | 1,000,000 |
| Sailfish-cir | Based on CIRI2 v2.0.6 | Entire genome | No strand reported | AGNGT, ACNCT | No BSJ counts reported | 135 | 200,000 |
| segemehl | Segmented read-based | Entire genome | No strand reported | Non-canonical | ≥5 | None | 200,000 |
Some tools did not report strand information for this study, but the (updated) circRNA tool might report circRNA strand information.
The BSJ count and the minimum and maximum circRNA length filters are the filters used for this specific study. The user can choose these parameters freely. Of note, the minimum and maximum length filters are based on the estimated circRNA length with introns, calculated by subtracting the start position from the end position of the BSJ.
Integrative tools combine the results of multiple circRNA detection tools. This includes CirComPara2 (combining CIRCexplorer2 (v2.3.8), Segemehl (v0.3.4), CIRI2 (v2.0.6), DCC (v0.4.8) and find_circ (v1.2), and then filtering all circRNAs detected by at least two methods) and ecircscreen (combining CIRI2 (v2.0.6), circRNA_finder (v1.2), PFv2 (v2.0.0), find_circ (v1.2) and CIRCexplorer (v1.1.10), and then filtering all circRNA detected by at least three methods).
Inferred based on publication and available code.
The number of detected circRNAs differs greatly between tools.
A total of 315,312 unique circRNA predictions (corresponding to 1,137,099 unique circRNA–strand–tool–sample tuples) were detected using 16 different tools based on deeply sequenced total RNA from three human cancer cell lines (Supplementary Table 2, because of large file size, available only at https://github.com/OncoRNALab/circRNA_benchmarking). The circRNA detection tools were run by their developers (details in Methods and Supplementary Notes). There is a striking almost 40-fold difference between the tool with the highest number of predicted circRNAs (circseq_cup with 58,032 circRNAs) and the tool with the lowest number of predicted circRNAs (segemehl with 1,372 circRNAs) for one of the cell lines (Fig. 2b shows results for HLF (human lung fibroblast) cells; similar results for the other cell lines are shown in Supplementary Fig. 1).
Most circRNAs are characterized by low BSJ counts.
CircRNA abundance is reflected by the BSJ count, which is the number of reads uniquely assigned to a given circRNA. The majority of circRNAs (86.6%) that are detected have a BSJ count below 5 (Fig. 2b), with only 46.1% of the detected circRNAs having a BSJ count of at least 2 (detailed distribution in Supplementary Fig. 2). To increase confidence, circRNA_finder and segemehl filtered their results to report only circRNAs with a BSJ count of at least 5, and CirComPara2 and KNIFE filtered for circRNAs with a BSJ count of at least 2. Circtools filtered circRNAs with a BSJ count of at least 2 in at least two samples. Of note, Sailfish-cir does not report raw BSJ counts, but transcripts per million (TPM) instead. The similarity of circRNA BSJ counts between tool pairs is reasonable, according to regression analysis (linear models with median r2 = 0.86, median slope = 0.70, all P < 0.001, Supplementary Fig. 3).
CircRNA detection tools predict different sets of circRNAs.
Half of all circRNAs in this study (49.9%) are reported only by one tool, which is largely due to circseq_cup’s high number of uniquely predicted circRNAs (Fig. 2c for HLF cells, similar results for the other cell lines are shown in Supplementary Fig. 4). The overlap of circRNA predictions among different tools is visualized in a heatmap for each cell line in Supplementary Fig. 5. Of 16 circRNA detection tools, eight exclusively report circRNAs flanked by canonical splice sites (with an AGNGT pattern, where AG is the splice acceptor, N represents the circRNA sequence and GT is the splice donor) (Fig. 2d). CirComPara2, circseq_cup, Sailfish-cir and segemehl do not report circRNA strand orientation, which explains most of the ACNCT patterns (reverse complement of AGNGT) given that all of their predicted circRNAs were automatically assigned to the positive strand to retrieve the surrounding splicing sequence. Two-thirds of all predicted circRNAs in this study (68.5%) are novel compared with a set of previously reported circRNAs extracted from 13 published circRNA databases (Circ2Disease, circad, CircAtlas, circbank, circBase, CIRCpediav2, CircR2disease, CircRiC, circRNADb, CSCD, exoRBase, MiOncoCirc and TSCD) (Supplementary Fig. 6)30. Of note, approximately half of these novel circRNA candidates originate solely from circseq_cup. Looking at the tools individually, circseq_cup, KNIFE, NCLscan and NCLcomparator report a higher number of novel circRNAs (87.8%, 53.9%, 53.4%, 53.3%, respectively) compared with the other tools (median, 19.7%; interquartile range (IQR): 4.9–34.8%). Tools were further compared based on the predicted circRNA length, strand information, correspondence to linear annotation and predicted exon composition (Supplementary Data 1–4 and Supplementary Figs. 7–10). No notable differences were observed between tools, except for CIRI2 and PFv2, which have a higher number of circRNAs for which no canonical linear annotation match was found, compared with the other circRNA detection tools (that is, the BSJ position of the circRNA did not match any known intron–exon splicing position based on the canonical transcripts from Ensembl GRCh38.103). Across all tools, 53.7% of circRNAs uniquely match one canonical linear transcript, 10.3% match more than one canonical transcript, and 35.9% do not match any canonical transcript. CircRNAs were found for 17,461 different canonical human transcripts, demonstrating the pervasive nature of back-splicing (28.9% of canonical transcripts from Ensembl GRCh38.103). Of note, this is an underestimation, given that for 46.3% of circRNAs no (unique) annotation match could be found.
CircRNA validation with empirical methods
CircRNA primer design inherently introduces a selection bias.
Based on previous experiments (Supplementary Data 5 and Supplementary Fig. 11), for each tool we aimed to select 80 random high-abundance circRNAs with a BSJ count of at least 5 and 20 random low-abundance circRNAs with a BSJ count below 5. Importantly, the precision values for both abundance groups (described in the following paragraphs) cannot be directly compared due to differences in the sample size. Of note, circRNA primer design inherently introduces a bias caused by the discarding of primer pairs (and therefore circRNAs) with predicted off-target amplification (Supplementary Data 6 and Supplementary Fig. 12). A selection of 1,560 circRNAs was obtained (Supplementary Table 3, BSJ count distribution in Supplementary Fig. 13). Given that some circRNAs were selected more than once (by chance, for different tools or in different cell lines), the total number of unique circRNA–sample pairs was 1,516, from here on termed ‘selected circRNAs’ (detailed description in Supplementary Fig. 14). Furthermore, a second bias is introduced within the group of low-abundant circRNAs, given that three tools (CirComPara2, circtools and KNIFE) filter circRNAs with a BSJ count of at least 2, whereas all other tools in this category also allow circRNAs with a BSJ count of 1.
High BSJ detection precision using RT–qPCR validation.
Of the 1,516 selected circRNAs, 1,479 (97.6%) could be validated with RT–qPCR, that is, the primer pair flanking the BSJ site resulted in a detectable amplicon. For the low-abundance circRNAs there is some variation in the tool-specific precision values (median, 95.0%; range, 80.0–100%), which is expected. High-abundance circRNAs have high RT–qPCR precision for most tools (median, 98.8%; range 90.0–100%) (Fig. 3a; the cumulative plot of the RT–qPCR precision as a function of the BSJ count is shown in Supplementary Fig. 15). It is important to note that RT–qPCR-based validation is the net result of a successful primer pair and the actual presence of a sufficiently abundant circRNA in the amount of RNA tested.
Fig. 3 |. The precision of circRNA detection tools is generally high and similar, whereas tools largely differ with respect to the number of predicted circRNAs.

a–c, The plots are separated based on circRNA BSJ count below 5 (low-abundance, in blue, 20 circRNAs selected per tool) or a BSJ count of at least 5 (high-abundance, in orange, 80 circRNAs selected per tool). Sailfish-cir reports TPM (transcripts per million) instead of BSJ count, and is therefore shown separately. Given that circRNA_finder and segemehl do not report any circRNAs with a BSJ count < 5, these tools are not included in the blue bar plots. a, CircRNAs were validated using three different techniques: RT–qPCR detection, resistance to degradation by RNase R, and amplicon sequencing (seq). Low-abundance circRNAs are in general more difficult to validate. Of note, the precision for low-abundance circRNAs is based on a limited set of circRNAs. High-abundance circRNAs have good precision for most tools and most validation methods. The error bars represent the 95% confidence intervals (CI). †A set of circRNAs was excluded because their abundance was too low to enable assessment of their resistance to RNase R, resulting in a variable number of circRNAs per tool instead of 20 or 80 for low-abundance and high-abundance circRNAs, respectively (range, 10–18 or 71–80 circRNAs per tool, respectively, details in Supplementary Table 6). A random subset of circRNAs was included in the amplicon sequencing experiment, resulting in a variable number of circRNAs per tool for amplicon sequencing validation as well (range, 11–20 or 54–74 circRNAs per tool, respectively, details in Supplementary Table 6). b, The vast majority of circRNAs produce the same results based on the three different validation methods. However, some circRNAs have conflicting results. For example, there are 13 circRNAs that are detectable by RT–qPCR but also are degraded upon RNase R (RR) treatment and for which the primers seem to amplify the wrong product. c, The compound precision is used to calculate the theoretical number of true-positive circRNAs by multiplying it with the original number of circRNAs detected by that tool (that is, the extrapolated sensitivity) (shown for HLF; similar results for the other cell lines are shown in Supplementary Fig. 26).
RNase R treatment degrades 1 in 16 predicted circRNAs.
RNase R was used as a second, more stringent validation approach. RNase R selectively degrades linear transcripts, ensuring that the RT–qPCR primers amplify a circular molecule. For 112 out of 1,516 selected circRNAs (7.4%), RNase R treatment could not be evaluated because their abundance in the untreated sample was too low, leaving no room to confirm RNase R degradation in the event of a false-positive circRNA (hence labeled as NAs). In the remaining set of 1,404 predicted circRNAs, 1,319 circRNAs (93.9%) could be successfully validated using RT–qPCR on RNase R-treated RNA. For most tools, high RNase R precision was observed for high-abundance circRNAs (median, 96.3%; range, 74.0–100%). PFv2 has the lowest precision (74.0%). For low-abundance circRNAs, lower precision was observed (median, 86.7%; range, 50.0–100%) (Fig. 3a; the cumulative plot of the RNase R precision as a function of the BSJ count is shown in Supplementary Fig. 15). Of note, the number of circRNAs per tool in this bin is lower than the original 20 that were selected, given that more circRNAs were excluded due to abundancy being too low (resulting in only 10–18 circRNAs per tool, with a median of 14 circRNAs). A comparison with matched RNase R-treated and -untreated sequencing data is given in Supplementary Data 7 and shows that the RNase R precision calculated from sequencing results is mostly high and similar between tools, with PFv2 having the lowest precision (Supplementary Figs. 16–19 and Supplementary Tables 4 and 5, because of large file size, available only at https://github.com/OncoRNALab/circRNA_benchmarking).
Amplicon sequencing is the most stringent validation method.
The RT–qPCR amplicons of the untreated RNA were sequenced for further validation of the circRNAs. A random subset of circRNAs (1,179 of 1,516, 77.8%) was included in the amplicon sequencing experiment, resulting in a variable number of circRNAs per tool instead of 20 or 80 for low-abundance and high-abundance circRNAs, respectively (range, 11–20 or 54–74 circRNAs per tool, respectively). For this subset of 1,179 circRNAs, 1,014 circRNAs (86.0%) could be readily validated with amplicon sequencing, that is, the majority of reads aligned to the expected BSJ sequence. Most tools have similar amplicon sequencing precision for high-abundance circRNAs (median, 95.5%; range, 30.0–100%), with PFv2 having a very low (30.0%) amplicon sequencing precision. Of note, given that PFv2 was developed to retain repeat sequences, it is expected to result in more false positives. The most obvious are caused by linear read-through between exons in neighboring tandemly repeated gene clusters and interspersed repeats, and these tend to be abundant. For low-abundance circRNAs, performance is more diverse, with generally lower amplicon sequencing precision (median, 73.3%; range, 17.6–94.1%) (Fig. 3a; the cumulative plot of the amplicon sequencing on-target amplification rate and the cumulative plot of the amplicon sequencing precision as a function of the BSJ count are shown in Supplementary Figs. 20 and 15, respectively).
Different validation methods should be used in concert.
Although the three validation strategies were used independently, it is interesting to evaluate to what extent they support each other (Fig. 3b and Supplementary Figs. 21 and 22). Considering 1,103 circRNAs for which all three validation results are available, 957 circRNAs (86.8%) pass all validation methods, 128 circRNAs (11.6%) fail one or two of the validation methods, and 18 circRNAs (1.6%) fail all three validation methods. This shows that orthogonal validation with different empirical approaches is important to compensate for their inherent limitations. It is beyond the scope of this study to investigate why there are some discrepancies among the validation results (some hypotheses are considered in the Discussion). First, they are rare (for most circRNAs, the different methods completely agree); and second, the same methods are used to compare the tools, whereby no tool should be favored over the other.
The three orthogonal validation methods were combined to label each circRNA as a true or false positive and the compound precision was calculated for each tool. Similar to the separate precision values, the compound precision is high and similar for most tools when looking at high-abundance circRNAs (median, 93.1%; range, 27.1–98.3%; IQR: 90.5–95.3%), and lower and more variable for low-abundance circRNAs (median, 63.6%; range, 5.9–88.2%; IQR: 53.8–76.5%) (Supplementary Figs. 15 and 23).
CircRNA detection tools differ greatly in sensitivity.
Tool sensitivity was evaluated using two different methods. In the first method, sensitivity was calculated based on the total number of true-positive circRNAs (n = 957) (Supplementary Figs. 24 and 25). Of note, this sensitivity metric should be used with caution because it is based on a biased set of circRNAs collected from the 100 circRNAs selected per tool, which overlap and are not a representative random sample of all circRNAs (see Methods). In the second method, the theoretical number of true-positive circRNAs for each tool was computed by multiplying the total number of detected circRNAs by the compound precision (that is, the extrapolated sensitivity) (Fig. 3c for HLF cells; similar results for the other cell lines are shown in Supplementary Fig. 26). There is a significant positive correlation between the sensitivity values for the two methods (Spearman rank correlation of 0.84 with P < 0.001, S = 58, for low-abundance circRNAs, and a Spearman rank correlation of 0.80 with P < 0.001, S = 113, for high-abundance circRNAs). Both methods show great variability in tool sensitivity, with a median sensitivity of 75.1% (range, 29.7–87.1%) for low-abundance circRNAs and 65.7% (range, 18.7–87.4%) for high-abundance circRNAs. To visualize the relationship between sensitivity and compound precision, a precision–recall (sensitivity) dot plot for all tools is shown in Supplementary Fig. 27.
All metrics described above ((compound) precisions and sensitivity) and the tool ranking for each metric are available in Supplementary Table 6. The user can easily filter and order the circRNA detection tools based on their preferences. Reproducibility evaluations were performed and are described in Supplementary Data 8–10 (Supplementary Figs. 28–32).
Evaluation of precision as a function of circRNA annotation.
To evaluate the precision as a function of circRNA annotation, we restrict the analyses to high-abundance circRNAs with information for all validation techniques. Furthermore, a strict validation definition was used, whereby all circRNAs failing for at least one technique were classified as unvalidated. CircRNAs previously described in databases have a higher likelihood of being validated (χ2 = 181.0, d.f. = 1, P < 0.001, odds ratio (OR) = 13.1). Nevertheless, false-positive circRNAs according to our data are still present in multiple published databases (Supplementary Figs. 33–35). For example, the false-positive circRNA chr6: 47526627–47554766 (hg38, 0-based) is present in CircAtlas (as hsa-CD2AP_0048) and in exoRBase (as exo_circ_65199). A difference in circRNA validation was observed depending on the splicing pattern, with better validation of circRNAs surrounded by canonical splice sites (χ2 = 45.4, d.f. = 1, P < 0.001, OR = 5.0). Similarly, circRNAs that originate from a region with an annotated linear transcript have higher validation rates (χ2 = 185.8, d.f. = 1, P < 0.001, OR = 17.1). Surprisingly, single-exon circRNAs had significantly lower validation rates than multi-exon circRNAs (χ2 = 20.0, d.f. = 1, P < 0.001, OR = 3.8). Last, while tools with a candidate-based approach seem more precise than tools using the segmented read-based approach (χ2 = 9.4, d.f. = 1, P = 0.0022, OR = 2.6), we cannot be sure that these results are not confounded by other algorithmic differences.
Evaluation of sensitivity in circRNA annotation.
There is a significantly higher sensitivity for tools reporting circRNAs surrounded by canonical splice sites, resulting in a median difference in sensitivity of 38.5% (two-sided Mann–Whitney U = 55, P = 0.0022, large effect size of 0.78, 95% CI: 0.56–0.85, n1 = 11 (canonical), n2 = 5 (non-canonical), only high-abundance circRNAs). However, no link could be found between sensitivity and tool approach, use of linear annotation, strand annotation method or BSJ count filtering.
Evaluation of tool combinations to improve performance
For the combination of two or more tools, both the intersection and the union have been proposed11 (Supplementary Tables 7 and 8). Although not evaluated here, the increased time and resource consumption should also be taken into account when considering the use of multiple tools. A list of the top-performing combinations is available in Supplementary Table 9 and can be used as a reference.
A circRNA predicted by two tools can be a false result.
Figure 4a shows that circRNAs uniquely detected by a single tool generally have lower precision. In line with this, circRNAs detected by at least two tools have a higher chance of being validated (χ2 = 333.1, d.f. = 1, P < 0.001, OR = 53.8). By contrast, out of 1,380 unique circRNAs detected by at least two tools, 7 circRNAs (0.5%) failed all three validation methods and 137 (9.9%) failed at least one of the validation methods (Supplementary Fig. 36), showing that the practice of using the intersection is not a guarantee to avoid false-positive results.
Fig. 4 |. The intersection or union of two circRNA detection tools decreases the number of false positives, or increases the overall number of detected circRNAs, respectively.

a, CircRNAs detected by multiple tools generally have higher precision. However, the often-used practice of using the intersection of two tools is not necessarily a guarantee of avoiding false-positive results. b, By considering the union of two circRNA detection tools, the number of circRNAs can be significantly increased while keeping the number of false-positive predictions low (shown for the HLF cell line; similar results for the other two cell lines are shown in Supplementary Fig. 37). For the y-axis, the percentage of detected circRNAs is calculated by dividing the number of circRNA detected by that tool combination by the total number of predicted circRNAs for that sample taking the union of all tools (13,087 circRNAs for the HLF sample). For this analysis, the compound precision of high-abundance circRNAs was used. Some circRNA detection tools are integrative and combine the results of multiple other tools. It is therefore assumed that an integrative tool would have large similarities with its underlying tools. However, a difference in tool version and filtering can still produce a different set of circRNAs. For example, CirComPara2 is an integrative tool that combines CIRCexplorer2, CIRI2, DCC and find_circ, but nevertheless, the combination of CirComPara2 and CIRCexplorer3 still produces a significant increase in detected circRNAs (corresponding to 10% of all circRNA predictions for that cell line).
The union of tools increases the number of true circRNAs.
To maximize detection sensitivity and maintain precision, we evaluated the union of pairs or triples of circRNA detection tools. Generating all possible combinations of the better tools with individual compound precision ≥ 90% for high-abundance circRNAs (n = 12 tools) consistently results in higher detection sensitivity while maintaining a high weighted precision. The median increase in the number of detected circRNAs. for combinations of two or three tools, is 37.0% (IQR: 16.5–129.7%) and 79.6% (IQR: 33.1–215.7%), respectively. In other words, when combining very precise tools, the number of false positives does not counteract the gain in additional true positives. A subset of tool combinations with an increase of at least 1,000 circRNAs is shown in Fig. 4b (shown for HLF cells; similar results for the other cell lines are shown in Supplementary Fig. 37; the combination of three tools is shown in Supplementary Fig. 38). One obvious consideration when selecting two different tools is their circRNA detection approach, their reliance on linear annotation and their filtering methods. For example, when combining two tools with a different detection approach (the pseudo-reference-based or fragmented-based approach), the median increase in the number of detected circRNAs is 61.1%, compared with 35.4% for two tools with the same detection approach (two-sided Mann–Whitney U = 58,554.5, P < 0.001, small effect size of 0.16, 95% CI: 0.08–0.23, n1 = 336, n2 = 294). Similarly, when combining two tools with the same splice site settings (both canonical or both non-canonical), the median increase in the number of detected circRNAs (32.6%) is significantly smaller than that for the combination of two tools with different splice site settings (one canonical and one non-canonical) (76.2%) (two-sided Mann–Whitney U = 65,356.5, P < 0.001, small effect size of 0.28, 95% CI: 0.21–0.35, n1 = 300, n2 = 330). A similar analysis for the combination of tools that rely or do not rely on linear annotation was not significant.
Discussion
Multimodal orthogonal validation of bioinformatics tools that predict circular RNAs from total RNA sequencing data is currently lacking. Hence, their precision and sensitivities are unknown and scientific data are confounded with false-positive and false-negative predictions. To accommodate this lacuna, we set up a large-scale international collaborative circRNA detection tool benchmarking study (Fig. 2a). First, a deeply sequenced total RNA sequencing dataset was processed by the developers of 16 different circRNA detection tools. Next, three empirical validation strategies were used to evaluate a random selection of 1,560 circRNAs representing each tool: first, RT–qPCR to determine whether the candidate circRNA BSJ sequence was detectable; second, RNase R treatment to confirm that the detected RNA was most likely to be circular and not linear; and third, amplicon sequencing to confirm the circRNA BSJ sequence. Of note, both circRNA RT–qPCR and RNAse R validation protocols were extensively validated4,31.
The precision is similarly high among tools (Fig. 3a), especially when considering the subset of high-abundance circRNAs (with BSJ count ≥5). In contrast, the number of predicted circRNAs and the sensitivity varies greatly between tools, in line with previous studies based on simulated data11,13,14 (Supplementary Data 11 and Supplementary Figs. 39 and 40). The striking differences in sensitivity are in part dependent on the operator applying BSJ count filters.
The three validation methods each have their own strengths and biases, with conflicting results for several circRNAs (Fig. 3b, discussed in detail in Supplementary Discussion 1). In total, 957 (86.8%) circRNAs are validated by all three methods. However, 22 circRNAs are validated with qPCR and amplicon sequencing but are degraded by RNase R (with a decrease of concentration of at least 87.5%, that is, a difference of at least 3 cycles). A possible explanation could be that some bona fide circRNAs are susceptible to RNase R degradation8 or that the primers amplify a mixture of circular and linear RNA. Another subset of 92 circRNAs pass RT–qPCR validation and RNase R validation but fail amplicon sequencing. These could be (repetitive) RNAs resistant to RNAse R due to a secondary structure, either internal or through base pairing with orthologs8. These examples underscore the importance of using different validation methods to compensate for their intrinsic limitations and to increase the validation status confidence (as previously suggested in ref. 8).
Although long-read sequencing has been implemented to study full-length circRNAs32–35, the bulk of currently available data is still short-read sequencing. Therefore, this benchmarking study evaluated circRNA detection tools for short-read sequencing data, which typically report circRNAs by their BSJ position (chr, start, end, strand). However, it remains unknown whether the detected BSJ corresponds to one circRNA, or multiple alternatively spliced circRNAs with different exon and intron compositions. Hence, the prediction precision reported here might be influenced by more than one circRNA with the same BSJ. Given that this study is focused on circRNA detection in short-read sequencing data, the internal circRNA composition was not evaluated. Furthermore, no distinction can be made between circRNAs on the positive strand or negative strand using RT–qPCR and amplicon sequencing (9.4% of circRNAs were reported to originate from different strands according to different tools).
Based on a pilot study (Supplementary Data 5 and Supplementary Fig. 11), a cut-off was set at a BSJ count of 5, given that circRNAs under this cut-off approached the qPCR limit to reliably detect RNase R-based degradation of falsely predicted circRNAs. Although very deep sequencing of a large RNA input amount was performed, it is beyond the scope of this study to evaluate whether the BSJ count should be reconsidered with regard to the sequencing depth. However, given that the majority of predicted circRNAs have a BSJ count below 5, we decided to include at least a subset of these low-abundance circRNAs to calculate the corresponding prediction precision. It is no surprise that the precision for low-abundance circRNAs is significantly lower than that for high-abundance circRNAs (χ2 = 76.7, d.f. = 1, P < 0.001, OR = 3.8). This difference is likely to be due to the detection limits of the applied validation strategies in conjunction with the sampling bias of low-abundance analytes, and not due to inherently more false-positive predictions for circRNAs with a lower count. Of note, it can be presumed that weakly expressed circRNAs are less relevant for both functional studies and biomarker research.
Focusing on high-abundance circRNAs, interesting associations were found between circRNA annotation and validation rates. As such, circRNAs had higher validation rates when they were detected by multiple tools, when they were previously reported in a circRNA database, when they were surrounded by canonical splice sites, and when they originate from a region with an annotated linear transcript. CircRNA detection tools with a candidate-based approach are more precise than tools using the segmented read-based approach, which is in line with the higher validation likelihood of circRNAs originating from known linear genes and surrounded by canonical splice sites.
Based on our study, we compiled a list of recommendations for circRNA detection and validation, and for the future development of circRNA detection tools and their performance evaluation (Table 2). Ideally, publicly available (spike-in) reference material (consisting of known synthetic circRNAs) should be used to benchmark existing and novel circRNA detection tools. However, such reference material is currently not available. Given that the main goal of this study was to perform a neutral assessment of circRNA detection tool sensitivity and precision, the developers of the tools were asked to run the tools themselves. Therefore, execution time, memory usage and ease of use could not be compared and were not assessed here.
Table 2 |.
CircRNA research recommendations
| circRNA detection | 1. An orthogonal validation method must be used to validate a predicted circRNA; qPCR validation on its own is not sufficient, at least qPCR + RNase R treatment or preferably qPCR + amplicon sequencing should be used. 2. Filtering based on a minimum BSJ count is recommended to increase the likelihood of successful empirical validation. |
| circRNA validation | 3. For a precision-focused approach, the intersection of two tools with a high individual precision (for example, ≥90%) should be used. 4. For a sensitivity-focused approach, the union of two tools with a high individual precision (for example, ≥90%) should be used. 5. The choice of tools to be combined may be informed based on the tools’ underlying principles (circRNA detection approach, reliance on linear annotation and canonical splicing, and filtering). |
| circRNA tool development | 6. Tools should report the originating strand information, the BSJ count evidence, and the chromosomal start and end position of the BSJ. |
| circRNA tool validation | 7. For evaluation of sensitivity, novel and updated tools are encouraged to use the empirically validated set of 957 true-positive circRNAs. 8. For evaluation of precision, a random set of 100 predicted circRNAs should be validated with empirical methods. |
Furthermore, this study resulted in a circRNA resource containing > 315,000 circRNAs detected by different tools in three human cancer cell lines from different tissue origins and provides validation results for 1,500 circRNAs that can be used as a reference for the development of new or improved circRNA detection tools. Finally, our study can also serve as an example framework for empirical validation of benchmarking results from other bioinformatics tools in the future.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-023-01944-6.
Methods
Study set-up
Given that this study involves the execution and evaluation of circRNA detection tools, the co-authors can be divided into two groups: an independent group (with no circRNA detection tool of their own) that initiated and designed the study and performed all of the wet-lab work and data analysis (the validation co-author group), and a group of tool developer co-authors who detected circRNAs using their own circRNA detection tools according to their expertise (the circRNA prediction co-author group) (details in Author Contribution section). During the study, meetings and emails were used to share the results (initially in a blinded manner) and discuss the final manuscript with the circRNA prediction co-authors.
Cell culture
Three cancer cell lines of different cellular origin were randomly chosen as biological replicates. Ethics approval was obtained for this study (EC014–202, Ghent University Hospital) and the cell lines were purchased from the JCRB (Japanese Collection of Research Bioresources) Cell Bank (HLF and NCI-H23) or ECACC (European Collection of Authenticated Cell Cultures) (SW480). SW480 cells were cultured at 37 °C, 0% CO2 in Leibovitz’s L-15 medium (31415–029, ThermoFisher). HLF cells and NCI-H23 cells were cultured at 37 °C, 5% CO2 in DMEM, low glucose, GlutaMAX Supplement, pyruvate (21885025, ThermoFisher) and RPMI 1640 Medium, HEPES (52400041, ThermoFisher), respectively. Also, 10% FBS (F7524, Sigma) and 1% penicillin–streptomycin (10,000 U ml−1) (15140122, ThermoFisher) were added to all three media.
RNA isolation
RNA was isolated from the cells using the miRNeasy Mini kit (217004, Qiagen) according to the manufacturer’s instructions, including the optional on-column DNase treatment (79254, Qiagen). For each cell line, a sufficient number of cells was cultured to be able to collect a minimum of 330 μg RNA. The RNA concentration was measured spectrophotometrically using a NanoDrop instrument, and the RNA integrity was evaluated using the Fragment Analyzer system. For each cell line, the RNA was pooled and aliquoted (1,000 ng RNA in 100 μl nuclease-free water per aliquot) and stored at −80 °C, making a uniform RNA collection to use for all downstream experiments.
RNase R treatment, library preparation and sequencing
For each cell line, two aliquots of 1,000 ng input RNA (in 10 μl nuclease-free water) were used. First, ribosomal RNA was removed with the NEBNext rRNA Depletion Kit (E6350X, New England Biolabs), following the manufacturer’s instructions. Next, RNAse R treatment was performed according to our previously described protocol4. In summary, one aliquot of each cell line was treated with RNase R (RNR07250 (250 U), Lucigen), and one aliquot of each cell line was treated as a buffer control. This was followed by a clean-up step using Vivacon 500, 10,000 MWCO (molecular weight cut-off) Hydrosart columns (VN01H02, Sartorius). Subsequently, the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760L, New England Biolabs) was used in combination with the NEBNext Multiplex Oligos for Illumina (E7600S, New England Biolabs) to index and prepare the samples for sequencing. The library preparation protocol was adjusted to obtain relatively long insert sizes (average size of 636 nucleotides measured using the Fragment Analyzer system): RNA fragmentation of 7.5 min; and a first-strand complementary DNA synthesis elongation step of 50 min instead of 15 min. The last bead clean-up step was performed twice to completely remove all indexes from the samples. Finally, the samples were pooled equimolarly and sequenced on a NovaSeq 6000 instrument using a NovaSeq 6000 S1 Reagent Kit v1.5 (300 cycles) (20028317, Illumina), resulting in approximately 300 million paired-end 150-nucleotide reads per sample. Raw FASTQ files are stored in the Sequence Read Archive (PRJNA789110: SRX13414572 (untreated HLF), SRX13414573 (untreated NCI-H23), SRX13414574 (untreated SW480), SRX13414575 (RNase R-treated HLF), SRX13414576 (RNase R-treated NCI-H23), SRX13414577 (RNase R-treated SW480)).
CircRNA detection
In November 2020 a comprehensive list of all published circRNA detection tools was compiled, and all developers were invited to collaborate. Upon consent, they were asked to detect circRNAs using their own circRNA detection tool as appropriate for the data that were provided. The circRNA detection steps for each tool are detailed in the Supplementary Notes. Often, the default parameters were used given that most of the methods included in our benchmarking underwent continuous development during the last several years and their parameters have been optimized for standard RNA sequencing data (as is the case in this study). We were unable to contact the authors of find_circ24 and decided to run this tool ourselves, given that it is one of the most frequently cited and broadly used circRNA detection tools. Unfortunately, other well-performing tools (according to refs. 10,14), such as MapSplice36, could not be included. More recent tools, such as Circall13 and CYCLeR37, have been published after the validation experiments were performed, and are therefore not included.
After collecting all circRNA detection results, a uniform list of circRNAs defined by their BSJ position (chr, start, end, strand) and the BSJ count for each tool was compiled (Hg38, 0-based).
CircRNA selection and primer design
Guided by a pilot experiment assessing circRNA RT–qPCR detectability depending on circRNA abundance and RNA input amount (Supplementary Data 5 and Supplementary Fig. 11), for each tool, 80 high-abundance circRNAs (with a BSJ count of at least 5), and 20 low-abundance circRNAs (with a BSJ count below 5) were selected (as two separate count bins). Primer pairs were designed using our primer design tool CIRCprimerXL31. All primer sequences are available in Supplementary Table 3. If no primer pair could be designed for a given circRNA, a substitution was randomly selected from the complete dataset, considering the BSJ count bin. In total, 1,560 circRNA–tool–cell line tuples were selected. Given that some circRNAs were selected more than once (for different tools) the total number of unique circRNA–cell line pairs is 1,516, and the number of unique circRNAs (not taking into account the strand) is 1,457 (Supplementary Fig. 14). Additionally, most of the selected circRNAs are detected by multiple tools (for which they were not selected). For the precision calculations, only the 20 low-abundance and 80 high-abundance circRNAs selected for a specific tool were used to evaluate that tool to maintain an equal number of observations for each tool, even though more of its predicted circRNAs might have been validated. However, for the sensitivity calculations, the complete set of circRNAs had to be used (see below).
RNase R and RT–qPCR
The RNA aliquots derived from the three cell lines were used for the circRNA RT–qPCR validation. A total of 1,080 μl, 900 μl and 780 μl RNA (100 ng μl−1) was required to validate 579, 500 and 437 circRNAs in HLF, NCI-H23 and SW480 cells, respectively. RNase R treatment was performed according to our previously reported protocol4, adapted for this large-scale experiment. In summary, one RNA aliquot of a given cell line was treated with RNase R (RNR07250 (250 U), Lucigen) and another was treated as a buffer control, for a total of 92 RNase R-treated replicates and 92 buffer control replicates (2 × 36 for HLF, 2 × 30 for NCI-H23 and 2 × 26 for SW480 RNA). All volumes were doubled during the buffer and RNase R reaction (total reaction volume of 20 μl). This was followed by a clean-up step using Vivacon 500, 10,000 MWCO Hydrosart columns (VN01H02, Sartorius). Next, reverse transcription was carried out on the 184 separate replicates using the iScript Advanced cDNA Synthesis Kit (172–5038, Bio-Rad), according to the manufacturer’s instructions. After reverse transcription, the cDNA was diluted 1:2 and an aliquot (2.5 μl) was further diluted 1:4 to evaluate the success of the RNase R reaction for each individual replicate. For this, ACTB and a known circRNA (chr1: 117402185–117420649) previously described4 (primer sequences available in Supplementary Table 10) were measured with qPCR using 2.5 μl 2x SsoAdvanced Universal SYBR Green Supermix (172–5274, Bio-Rad), 0.5 μl forward and reverse primer (5 nM), and 2 μl cDNA per well, with qPCR duplicates. Once the RNase R treatment was successfully validated, all cDNA replicates were pooled per cell line and treatment condition. The cDNA was diluted 1:5 in 2× SsoAdvanced Universal SYBR Green Supermix (172–5274, Bio-Rad). All 1,560 circRNA primer pairs were ordered from IDT in 96-well plates at a concentration of 100 μM in nuclease-free water. All primers were diluted 1:160 to obtain a 0.625 μM concentration. In each well of a qPCR plate, 2 μl diluted primers and 3 μl cDNA–master mix combination were added, resulting in an equivalent of 25 ng input RNA per qPCR reaction. Each assay (circRNA) was measured four times to include qPCR duplicates and to measure the abundance in both an RNase R-untreated and -treated sample, resulting in a total of more than 6,000 qPCR reactions. A pipetting robot (EVO100, TECAN L) was used to dilute the primers and fill the qPCR plates. The qPCR reactions were run on a CFX384 instrument (Bio-Rad). Cq (quantification cycle) calling was done using the Bio-Rad CFX Manager (v3.1), with the ‘regression’ settings. The plates were stored at −20 °C prior to amplicon sequencing.
Amplicon sequencing
After RT–qPCR, ~80% of the circRNAs were randomly included for amplicon sequencing. To construct the sequencing library, the amplicons were pooled by combining 2 μl of the PCR reaction from one of the untreated qPCR duplicates, per cell line. Next, the three samples were cleaned using Vivacon 500, 10,000 MWCO Hydrosart columns (VN01H02, Sartorius). The PCR product pools were analyzed using a TapeStation 4150 (Agilent) and the concentration was measured using a Qubit fluorometer (ThermoFisher). Next, the three pools were diluted in nuclease-free water to obtain 50 μl samples with a concentration of 20 ng μl−1. Finally, the samples were prepared for sequencing using the NEBNext Ultra II DNA Library Prep Kit for Illumina (E7645S, New England Biolabs) and NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) (E7600S, New England Biolabs). To retain all amplicons, no size selection was performed after adapter ligation, and 1.0x AMPure XP beads (A63881, Beckman Coulter) in a 1:1 sample : beads ratio was used instead. After library preparation, the samples were pooled equimolarly. The pool was sequenced on a NextSeq 500 instrument using a Mid Output Kit v2.5 (150 cycles) (20024904, Illumina), resulting in approximately 25–30 million paired-end 75-nucleotide reads per library.
Data analysis
Data analysis was mostly done using R38 (v4.2.1) in RStudio39 (v2022.07.1). The following R packages were used: tidyverse (v1.3.2), conflicted (v1.1.0), ggrepel (v0.9.1), ggseqlogo (v0.1), europepmc (v0.4.1), gplots (v3.1.3), ggpubr (v0.4.0), quantreg (v5.94), rstatix (v0.7.0) and UpSetR (v1.4.0). For sequencing data analyses, including circRNA detection and amplicon sequencing analysis, the Ghent University high-performance cluster was used. For this, Python3 (v3.6.8) (ref. 40), Bowtie2 (v2.3.4.1) (ref. 41), fastahack (v1.0.0), SAMtools (v1.11) (ref. 42) and BEDTools (v2.30.0) (ref. 43) were used. The human reference transcriptome was downloaded as a GTF file from Ensembl44. All data analysis scripts are available at https://github.com/OncoRNALab/circRNA_benchmarking.
Amplicon sequencing data analysis
For the amplicon sequencing data analysis, first, a custom Python script matches the primer sequences with the first 16-mer of each read (forward and reversed) and generates a separate FASTQ file per primer pair, containing all reads starting with that primer sequence. The FASTQ reads are then clipped to remove the primer sequences. Next, all FASTQ files are mapped against the reference genome (Ensembl GRCh38.101) supplemented with the theoretical BSJ amplicon sequences using Bowtie2 with default settings. Last, the Bowtie2 BAM files are converted to counts using another custom Python script and the percentage on-target amplification was calculated for each primer pair.
Determination of orthogonal precision values and sensitivity
Several strategies to filter the data prior to precision and sensitivity calculations were explored. For RT–qPCR, a circRNA was considered validated when at least one of the untreated RNA samples had Cq above 10. Multiple variations of this threshold and a potential upper Cq threshold were evaluated. For RNase R validation, a subset of circRNAs with at least one untreated replicate with Cq below 32 was selected to ensure that the enzymatic degradation of a false-positive circRNA could be measured. A circRNA was considered validated upon RNase R treatment if the difference in Cq between the untreated and treated RNA sample was equal to or less than 3 cycles, based on a previous study4. Given that there were two qPCR replicates available for each (un)treated sample, the best-case scenario was used to calculate the difference in Cq by subtracting the maximum untreated Cq replicate from the minimum treated Cq replicate. A circRNA with both untreated replicates having Cq above 32 was labeled as NA. For amplicon sequencing, a circRNA was considered validated if the primer pair was found in at least 1,000 reads and if at least 50% of these reads matched the expected amplicon upon mapping with Bowtie2. For a random subset of circRNAs, unintentionally no amplicon sequencing was performed; these were labeled as NA. A detailed description of the choice of performance metrics is available in Supplementary Data 12 and 13. To calculate precision per tool, BSJ count bin and validation method, the number of circRNAs that passed the validation was divided by the total number of circRNAs that were not NA for that validation method. We also determined the compound precision by considering qPCR, RNase R treatment and amplicon sequencing. For this, each circRNA was labeled as a true positive (that is, validated by all three methods), as a false positive (that is, not validated by at least one of the methods), or as NA (that is, not included in the amplicon sequencing run). Based on this summarizing label, compound precision was computed for each tool and BSJ count bin. The number of theoretically true-positive circRNAs was calculated by multiplying the total number of circRNAs predicted by that tool for that sample with the compound precision value (that is, the extrapolated sensitivity). The sensitivity was also calculated as the percentage of circRNAs that each tool detected from the validated set of true-positive circRNAs (that is, the circRNAs labeled as true positives over all three methods). This metric should be used with caution because it is based on a biased selection of circRNAs due to the overlap between tools (Supplementary Data 12). To calculate the sensitivity per BSJ count group, the median BSJ count of each circRNA was used (given that most circRNAs are detected by multiple tools and therefore have multiple BSJ count values).
Annotation of circRNAs
To obtain the circRNA splice site information, the BSJ-flanking nucleotides were extracted from the reference genome using fastahack (Ensembl GRCh38.104). To compare BSJ positions with known linear annotation, BEDtools intersect was used with a list of canonical transcripts from Ensembl with their positions based on the corresponding Ensembl GTF file (Ensembl GRCh38.103). When a circRNA mapped to multiple isoforms, the annotation was labeled as ‘ambiguous’ and the circRNA was not taken into account for further annotation-based calculations and figures. The annotation was used to compute the length of each circRNA excluding introns, and the number of exons per circRNA. CircRNAs smaller than their host gene exon were labeled ‘single-exon’ circRNAs. For the length of each circRNA including introns, the BSJ start position was simply subtracted from the BSJ end position. Furthermore, for each circRNA, annotation was added to indicate whether the BSJ start and end positions match known exon boundaries. When comparing predicted circRNAs to circRNAs previously described in databases, strand information was discarded.
Combination of tools
To compare the circRNA tools, the union and intersection of all circRNAs predicted by each tool pair and triple were calculated. The weighted precision was calculated for each combination of tools as follows: ((compound_precision_1 × total_n_1) + (compound_precision_2 × total_n_2))/(total_n_1 + total_n_2). For this, strand information was discarded, given that 4 out of 16 tools did not report circRNA strands and would therefore have been excluded. These calculations were performed for each cell line separately. To determine the correspondence between tools, the Jaccard distance was calculated and heatmap clusters were generated. The tools were compared based on the presence or absence of a circRNA. Also, for the calculation of how many tools detected a given circRNA, circRNA strand information was discarded.
Statistical analyses
To evaluate the effect of circRNA characteristics on circRNA validation, the chi-squared test was used (chisq.test) function in R). For every test, the set of used circRNAs was slightly different depending on the availability of annotation information. All of the tests had all expected values in the contingency table above 5, therefore no correction for small sample size was necessary. Seven different characteristics were tested, and no multiple testing correction was performed. To evaluate the effect of circRNA detection tool methods on sensitivity and to evaluate the effect of the combination of tools with different approaches, the two-sided Mann–Whitney U-test was used (rstatix::wilcox_test) function in R). For correlation analysis between the sensitivity and the extrapolated sensitivity, the Spearman rank correlation was used (cor.test(method = ‘spearman’) function in R). For correlation analysis between circRNA BSJ counts from different tools, or between circRNA BSJ counts and Cq values, or between Cq values in different cell lines, linear models were used (lm) function in R). To evaluate the contribution of the cell lines (in contrast to the tools) to precision and sensitivity, an ANOVA test was used (aov) function in R).
Supplementary Material
Acknowledgements
The authors thank S. Lefever for his contribution to primer design in the early stage of this project. The computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, FWO and the Flemish Government – department EWI. This work was supported by the Foundation Against Cancer grant STK F/2018/1,267 (M.V., J.V., P.-J.V.), the Standup Against Cancer grant STIVLK2018000601 (J.V., P.-J.V.), two Concerted Research Action of Ghent University grants BOF16/GOA/023 (J.V.), BOF/24J/2021/244 (M.V., J.V., P.-J.V.), the Research Foundation – Flanders FWO grant 1253321N (P.-J.V.), the Fondazione AIRC per la Ricerca sul Cancro Investigator Grant 2017 20052 (S.B.), the Italian Ministry of Education, Universities, and Research PRIN 2017 grant 2017PPS2X4_003 (S.B.), the EU within the MUR PNRR ‘National Center for Gene Therapy and Drugs based on RNA Technology’, Project no. CN00000041 CN3 RNA (S.B.), the ‘HPC, big data and quantum computing’ CN1 HPC (S.B.), the Department of Molecular Medicine of the University of Padova (S.B.), the National Health Research Institutes Taiwan grant NHRI-EX110–11011B1 (T.-J.C.), the German Science Foundation grant DI 1501/13–1 (C.D.), the Wellcome Trust grant WT108749/Z/15/Z (P.F.), the Fondazione Umberto Veronesi Fellowship 2020 (E.G.), the German Federal Ministry of Education and Research grant BMBF 031L0106D (S.H.), the NIH grant R01-NS083833 (E.C.L.), the MSK Core Grant P30-CA008748 (E.C.L.), the German Federal Ministry of Education and Research grant BMBF 031L0164C (P.S.), the Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab (J.W.), the National Natural Science Foundation of China (NSFC) grant 31925011 (L.Y.), the Ministry of Science and Technology of China (MoST) grant 2021YFA1300503 (L.Y.), and the National Science Foundation of China (31871589) (C.-Y.Y.).
Footnotes
Competing interests
The authors declare no competing interests.
Code availability
All of the scripts used to compute the metrics described in the study and generate the figures are available at https://github.com/OncoRNALab/circRNA_benchmarking.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41592-023-01944-6.
Peer review information Nature Methods thanks Eduardo Andrés-León and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Rita Strack, in collaboration with the Nature Methods team. Peer reviewer reports are available.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
We anticipate that this study will serve as a future resource for the circRNA community. The information on all predicted circRNAs (n = 315,312), including the large extensively validated circRNA set (n = 1,516), along with the validation results are available in the GitHub repository (https://github.com/OncoRNALab/circRNA_benchmarking) and as Supplementary Tables. The set of circRNAs previously described in databases (Circ2Disease, circad, CircAtlas, circbank, circBase, CIRCpediav2, CircR2disease, CircRiC, circRNADb, CSCD, exoRBase, MiOncoCirc and TSCD) is also included in the GitHub repository. All databases were accessed in the context of a previous study30. Raw FASTQ files are stored in the Sequence Read Archive (PRJNA789110: SRX13414572 (untreated HLF), SRX13414573 (untreated NCI-H23), SRX13414574 (untreated SW480), SRX13414575 (RNase R-treated HLF), SRX13414576 (RNase R-treated NCI-H23), SRX13414577 (RNase R-treated SW480)). Source data are provided with this paper.
References
- 1.Kristensen LS et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Hulstaert E et al. Charting extracellular transcriptomes in the Human Biofluid RNA Atlas. Cell Rep. 33, 108552 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Wang S et al. Circular RNAs in body fluids as cancer biomarkers: the new frontier of liquid biopsies. Mol. Cancer 20, 13. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vromman M et al. Validation of circular RNAs using RT-qPCR after effective removal of linear RNAs by ribonuclease R. Curr. Protoc. 1, e181 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Yu CY, Liu HJ, Hung LY, Kuo HC & Chuang TJ Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro?. Nucleic Acids Res. 42, 9410–9423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szabo L & Salzman J Detecting circular RNAs: bioinformatic and experimental challenges. Nat. Rev. Genet. 17, 679–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen AF et al. Best practice standards for circular RNA research. Nat. Methods 19, 1208–1220 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dodbele S, Mutlu N & Wilusz JE Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep. 22, e52072 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakobi T & Dieterich C Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA 10, e1528 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Hansen TB, Venø MT, Damgaard CK & Kjems J Comparison of circular RNA prediction tools. Nucleic Acids Res. 44, e58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaffo E, Buratin A, Dal Molin A & Bortoluzzi S Sensitive, reliable and robust circRNA detection from RNA-seq with CirComPara2. Brief. Bioinform. 23, bbab418 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeck WR & Sharpless NE Detecting and characterizing circular RNAs. Nat. Biotechnol. 32, 453–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen DT et al. Circall: fast and accurate methodology for discovery of circular RNAs from paired-end RNA-sequencing data. BMC Bioinformatics 22, 495 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng X, Lin W, Guo M & Zou Q A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13, e1005420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo L et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 16, 126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song X et al. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 44, e87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma XK et al. CIRCexplorer3: a CLEAR pipeline for direct comparison of circular and linear RNA expression. Genomics Proteomics Bioinformatics 17, 511–521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westholm JO et al. Genome-wide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 9, 1966–1980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye C-Y et al. Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol. 14, 1055–1063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng J et al. Genome-wide identification of cancer-specific alternative splicing in circRNA. Mol. Cancer 18, 35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakobi T, Uvarovskii A & Dieterich C Circtools: a one-stop software solution for circular RNA research. Bioinformatics 35, 2326–2328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao Y, Zhang J & Zhao F Circular RNA identification based on multiple seed matching. Brief. Bioinform. 19, 803–810 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Chen S, Yang J & Zhao F Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat. Commun. 11, 90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Memczak S et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Chuang TJ et al. NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 44, e29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CY & Chuang TJ NCLcomparator: systematically post-screening non-co-linear transcripts (circular, trans-spliced, or fusion RNAs) identified from various detectors. BMC Bioinformatics 20, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izuogu OG et al. Analysis of human ES cell differentiation establishes that the dominant isoforms of the lncRNAs RMST and FIRRE are circular. BMC Genomics 19, 276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M et al. Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 33, 2131–2139 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann S et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 15, R34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vromman M, Vandesompele J & Volders P-J Closing the circle: current state and perspectives of circular RNA databases. Brief. Bioinform. 22, 288–297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vromman M, Anckaert J, Vandesompele J & Volders P-J CIRCprimerXL: convenient and high-throughput PCR primer design for circular RNA quantification. Front. Bioinform. 2, 834655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J et al. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat. Biotechnol. 39, 836–845 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Rahimi K, Venø MT, Dupont DM & Kjems J Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat. Commun. 12, 4825 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xin R et al. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat. Commun. 12, 266 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z et al. circFL-seq reveals full-length circular RNAs with rolling circular reverse transcription and nanopore sequencing. Elife 10, e69457 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stefanov SR & Meyer IM CYCLeR: a novel tool for the full isoform assembly and quantification of circRNAs. Nucleic Acids Res. 51, e10 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.R Core Team. R: A Language and Environment for Statistical Computing (2019); https://www.R-project.org/
- 39.RStudio Team. RStudio: Integrated Development for R (2020); http://www.rstudio.com/
- 40.van Rossum G and Drake FL Python 3 Reference Manual (Createspace, 2009). [Google Scholar]
- 41.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cunningham F et al. Ensembl 2022. Nucleic Acids Res. 50, D988–D995 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We anticipate that this study will serve as a future resource for the circRNA community. The information on all predicted circRNAs (n = 315,312), including the large extensively validated circRNA set (n = 1,516), along with the validation results are available in the GitHub repository (https://github.com/OncoRNALab/circRNA_benchmarking) and as Supplementary Tables. The set of circRNAs previously described in databases (Circ2Disease, circad, CircAtlas, circbank, circBase, CIRCpediav2, CircR2disease, CircRiC, circRNADb, CSCD, exoRBase, MiOncoCirc and TSCD) is also included in the GitHub repository. All databases were accessed in the context of a previous study30. Raw FASTQ files are stored in the Sequence Read Archive (PRJNA789110: SRX13414572 (untreated HLF), SRX13414573 (untreated NCI-H23), SRX13414574 (untreated SW480), SRX13414575 (RNase R-treated HLF), SRX13414576 (RNase R-treated NCI-H23), SRX13414577 (RNase R-treated SW480)). Source data are provided with this paper.