

Summary
Sterile alpha motif domain containing 7 (SAMD7) is a component of the Polycomb repressive complex 1, which inhibits transcription of many genes, including those activated by the transcription factor Cone-Rod Homeobox (CRX). Here we report bi-allelic mutations in SAMD7 as a cause of autosomal-recessive macular dystrophy with or without cone dysfunction. Four of these mutations affect splicing, while another mutation is a missense variant that alters the repressive effect of SAMD7 on CRX-dependent promoter activity, as shown by in vitro assays. Immunostaining of human retinal sections revealed that SAMD7 is localized in the nuclei of both rods and cones, as well as in those of cells belonging to the inner nuclear layer. These results place SAMD7 as a gene crucial for human retinal function and demonstrate a significant difference in the role of SAMD7 between the human and the mouse retina.
Keywords: retina, macular dystrophy, SAMD7, mutation, inherited retinal diseases
Graphical abstract
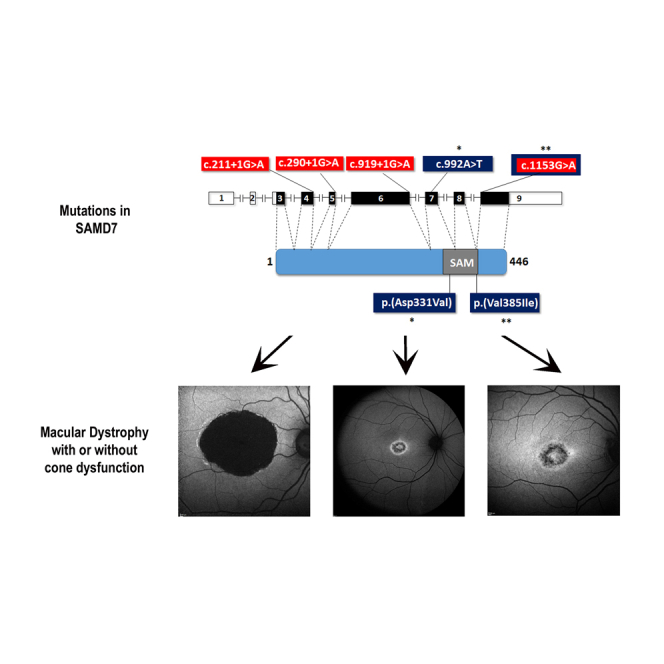
Macular dystrophies are a group of inherited eye diseases that affect the retina and significantly impair central visual functions. Here we report mutations in SAMD7 as a cause for autosomal-recessive macular dystrophy. This finding places SAMD7 as a gene crucial for human retinal function.
Main text
The retina of the eye includes two types of photosensitive neurons, or photoreceptors: rods, which are responsible for night vision, and cones, which are responsible for day vision and color vision. Rods are the predominant photoreceptors in the retinal periphery, while cones are the predominant photoreceptors in the macula (a defined region in the center of the retina) and the only type of photoreceptors in the fovea centralis (a depression in the center of the macula, which is responsible for high visual acuity). Inherited retinal diseases (IRDs) are a clinically and genetically heterogeneous group of diseases that cause visual loss due to improper development or premature death of photoreceptors and/or retinal pigment epithelium (RPE) cells.1 Macular dystrophies (MDs) are a subgroup of IRDs characterized by bilateral, relatively symmetrical macular abnormalities that significantly impair central visual functions.2 MD can appear as part of a general cone or cone-rod dystrophy (CD/CRD) or as an isolated degeneration of cones in the macular region only. To date, pathogenic variants in approximately 16 MD-associated genes have been identified (RetNet, Retinal Information Network). The most common form of MD is Stargardt disease type 1 (MIM: 248200), caused by bi-allelic variants in ABCA4 (MIM: 601691).3
Despite the large number of IRD genes that have been identified, approximately one-third of affected individuals remain genetically undiagnosed, indicating that a substantial number of mutations or disease genes are yet to be discovered.4,5,6,7 To identify previously unrecognized IRD-associated genes, we ascertained multiple IRD-affected individuals of various ethnicities who had undergone genomic investigations. The study was approved by the Institutional Review Boards of all participating institutions and informed consent was obtained from all participants and their relatives.
Individual 1-1 is a female from an Israeli family of Yemenite Jewish ancestry (Figure 1A). Whole-exome sequencing (WES) was performed, resulting in 64,035 high-quality variants, of which 246 were rare variants (allele frequency [AF] ≤1% in the Genome Aggregation Database [gnomAD], Exome Sequencing Project 6500 [ESP6500], 1000 Genomes database, and over 1,000 exomes of Israeli individuals with IRD), leading to stop gain (nonsense), stop loss, start gain, start loss, deletions/insertions, missense (with deleterious or uncertain predictions), and candidate splicing mutations. We first focused on variants located in known IRD-associated genes, but found only three heterozygous variants, which were ruled out (Table S1). We then went back to the other 243 variants and sorted them based on zygosity, pathogenicity predictions, associated phenotypes, and expression in the retina (Table S1). The most promising was a homozygous putatively pathogenic variant in SAMD7 (GenBank: NM_001304366.2). This variant is a G>A transition at position +1 of intron 5, affecting the conserved donor splice site (GRCh38: chr3:169,925,137G>A [c.290+1G>A]) (Figure S1). This rare variant (gnomAD aggregated AF = 0.0004%) is predicted to alter splicing by several in silico prediction tools (Table 1).
Figure 1.

Genetic analysis in families segregating SAMD7 mutations
(A) Pedigrees segregating SAMD7 mutations. Black-filled symbols represent affected individuals, whereas clear symbols represent unaffected individuals. Gray-filled symbols represent individuals reporting visual difficulties, with no specific clinical data. A double line represents a consanguineous marriage. Genotypes of tested individuals are indicated below them.
(B) A schematic representation of the human SAMD7 gene and protein. In the human gene illustration, filled boxes represent coding exons, while open boxes represent non-coding exons. In the protein illustration the SAM domain is indicated. Also shown are the locations of mutations identified in affected individuals. The locations of splice-altering mutations (highlighted in red) are indicated on the gene illustration, while the locations of missense mutations (highlighted in blue and marked by asterisks) are indicated on both the gene and the protein illustrations. c.1153G>A (p.Val385Ile) is both a missense and a splice-altering mutation.
(C) Evolutionary conservation of SAMD7 amino acids affected by two of the mutations. The analysis was performed with The ConSurf Server based on 149 SAMD7 orthologs. e, exposed amino acid; b, buried amino acid; f, functionally important amino acid (conserved and exposed); s, structurally important amino acid (conserved and buried).
Table 1.
Demographic and genetic data of families segregating SAMD7 mutations
| Family | Ethnicity | Consanguinity | Mutationa | Location | gnomAD aggregated MAF | Mutation Tasterb |
Splice-site predictionsb |
In vitro splice assay outcome |
Missense predictions |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SpliceAI | dbscSNV Ada | dbscSNV RF | MutScoreb | Varityb | REVELb | SIFTc | ||||||||
| 1 | Yemenite Jewish | unknown | c.290+1G>A | intron 5 | 0.000004 | D (1) | splice-altering (0.99) | D (1) | D (0.93) | exon 5 skipping | N/A | N/A | N/A | N/A |
| 2 | Berber/Morocccan | yes | c.919+1G>A | intron 6 | – | D (1) | splice-altering (1) | D (1) | D (0.9) | intron 6 retention, exon 6 skipping | N/A | N/A | N/A | N/A |
| 3 | Pakistani | yes | c.211+1G>A | intron 4 | – | D (1) | splice-altering (0.76) | D (1) | D (0.88) | exon 4 skipping | N/A | N/A | N/A | N/A |
| 4 | Pakistani | yes | ||||||||||||
| 5 | African | unknown | c.1153G>A (p.Val385Ile) |
exon 9 | – | D (1) | splice-altering (low) (0.42) | D (1) | D (0.86) | exon 9 inclusion (normal splicing), exon 9 skipping, alternative splicing of exon 9 | B (0.39) | B (0.11) | B (0.15) | B (0.11) |
| 6 | Pakistani | yes | c.992A>T (p.Asp331Val) | exon 7 | – | D (1) | B (0) | N/A | N/A | N/A | D (0.89) | D (0.89) | D (0.74) | D (0) |
B, benign; D, deleterious; MAF, minor allele frequency; N/A, not applicable.
Corresponding to GenBank accession number NM_001304366.2.
The score can range from 0 to 1, when higher values are more likely of being deleterious (D).
The score can range from 0 to 1, when a score below 0.001 corresponds with supporting pathogenic evidence.
SAMD7 is a sterile alpha motif (SAM) domain containing protein. It has two main transcripts in RefSeq (GenBank: NM_001304366.2 and NM_182610.4) that encode identical protein products and differ only by a part of exon 2, which is entirely non-coding. The human orthologue is 446 amino acids long (Figure 1B). SAMD7 is a component of the Polycomb repressive complex 1 (PRC1), an essential complex for maintaining a transcriptionally poised structural state of chromatin.8 SAMD7 expression is regulated by Cone-Rod Homeobox (CRX), a master transcription factor in photoreceptors, and it also acts as a transcriptional repressor involved in fine tuning of CRX-regulated gene expression.9 In the mouse retina, immunostaining revealed SAMD7 present mainly in rods.9 Correspondingly, Samd7-null mice showed rod dysfunction as well as ectopic expression of non-rod genes in rod photoreceptors, indicating that in the mouse retina SAMD7 is involved in defining rod photoreceptor cell identity by silencing non-rod gene expression.10 In humans, it was suggested that specific non-coding cis-regulatory variants in CRX-binding regions of SAMD7 acted as modifiers of the phenotype elicited by a homozygous rhodopsin mutation.11 However, a retinal phenotype exclusively linked to SAMD7 deficiency has not been reported to date. Based on these data, we considered the SAMD7 variant observed in individual 1-1 as a strong candidate for causing her retinal phenotype.
To test the effect of c.290+1G>A on splicing, we used an in vitro splicing assay. In brief, genomic segments spanning wild-type (WT) or mutant exon 5 of SAMD7 were subcloned into a pSPL3_2096 exon-trapping vector. HEK293T cells were transfected with constructs, followed by total RNA extraction and reverse transcription. The cDNA was PCR amplified with pSPL3 exon primers and sequenced (see details in supplemental information). This assay revealed that the c.290+1G>A variant leads to complete exon 5 skipping and absence of correctly spliced cDNA (Figure 2). Skipping of exon 5 is expected to cause a frameshift which would yield a nonsense-mediated decay (NMD)-sensitive RNA12; transcripts that survive NMD are expected to yield an aberrant truncated protein, p.Trp72Glnfs∗20.
Figure 2.

Analysis of the effect of SAMD7 variants on splicing by in vitro splicing assays
pSPL3_2096 plasmids containing exons 4, 5, 6, and 9 were transfected into HEK293T cells to investigate the splice effect of c.211G>A, c.290+1G>A, c.919+1G>A, and c.1153G>A, respectively.
(A) An agarose gel image is shown, representing the reverse transcription-polymerase chain reaction (RT-PCR) products derived from WT (G) and mutant (A) constructs for each depicted variant. Two replicates are shown for each construct: R1 and R2. Complete aberrant splicing with no formation of WT product was noted for c.211G>A, c.290+1G>A, and c.919+1G>A. For c.211G>A and c.290+1G>A, exon skipping was observed. For c.919+1G>A, however, the most abundant aberrant splice product is formed by (partial) intron 6 retention. A more complex splice pattern was noted for c.1153G>A, including both normal and aberrant splicing products, consistent with its position in the last exon of the gene and the more complex composition of the splice construct. +E, including exon; ΔE, excluding exon; +I, including intron. A cartoon of the used minigene vector pSPL3_2096, containing two HIV-TAT exons (TAT1, TAT2) and the inserted regions, is shown on the right. Arrows depict the position of the RT-PCR primers.
(B) Resulting sequences from RT-PCR products for each depicted variant are shown. WT, wild-type; MUT, mutant; TAT1/2, cDNA of pSPL3 HIV-TAT exons, which are co-amplified using the RT-PCR primers.
To identify other IRD-affected individuals harboring SAMD7 mutations, we screened whole-exome and whole-genome sequencing data from additional cohorts, which resulted in identification of four additional homozygous SAMD7 mutations in seven individuals from five families (Figures 1A and 1B; Table 1).
Individual 2-1 is a male who belongs to a consanguineous Berber/Morocccan family (Figure 1A). He is homozygous for a G>A transition at position +1 of intron 6, affecting the conserved donor splice site (chr3:169,927,182G>A [c.919+1G>A]) (Figures 1B and S1). This rare variant (not previously reported) is predicted to alter splicing by several in silico prediction tools (Table 1). Indeed, an in vitro splicing assay revealed that c.919+1G>A does not produce any correctly spliced cDNA, but instead leads to aberrant splicing, with predominantly intron 6 retention and some exon 6 skipping being observed (Figure 2). Either complete retention of intron 6 or skipping of exon 6 would lead to frameshifts and premature stop codons early on. Therefore, these transcripts would be subjected to NMD12; transcripts that survive NMD are expected to yield aberrant truncated proteins (p.Gly307Aspfs∗27 and p.Thr97Asnfs∗13, respectively). Segregation analysis revealed that the patient’s unaffected mother is heterozygous for this variant, while his father (individual 2-2) is homozygous. Thorough clinical examination of individual 2-2 revealed that he was also affected (see supplemental note).
Individual 3-1 is a female from a consanguineous Pakistani family (Figure 1A). She is homozygous for a G>A transition at position +1 of intron 4, affecting the conserved donor splice site (chr3:169921339G>A; c.211+1G>A) (Figures 1B and S1). This rare variant (not previously reported) is predicted to alter splicing by several in silico prediction tools (Table 1), as confirmed by an in vitro splicing assay, demonstrating complete exon 4 skipping (Figure 2). Skipping of exon 4 is expected to cause a frameshift which would yield an NMD-sensitive RNA12; transcripts that survive NMD are expected to yield an aberrant truncated protein (p.Asp30Leufs∗5).
Individuals 4-1 and 4-2 are siblings from another consanguineous Pakistani family (Figure 1A). Both are homozygous for the same c.211+1G>A mutation identified in individual 3-1.
Individual 5-1 is a female of African ancestry. Her father was also reported to have visual difficulties, but his clinical data and DNA were not available (Figure 1A). She is homozygous for the rare missense variant (not previously reported) chr3:169938318G>A (c.1153G>A; p.Val385Ile) (Figure S1). Despite the fact that valine at position 385 is located within the conserved SAM domain of the protein (Figure 1B) and is predicted to be structurally important by the ConSurf Server13 (Figure 1C), this valine to isoleucine missense change was not estimated to be pathogenic by several in silico prediction tools. However, the G>A transition at position c.1153 was estimated to affect pre-mRNA splicing of SAMD7 by several prediction tools, as it is located at the first base and within the acceptor site of the last exon (exon 9) (Figure S1; Table 1). Of note, exon 9 includes the end of SAMD7 open reading frame and the 3′ untranslated region (3′ UTR) (Figure 1B). The effect of c.1153G>A on splicing was tested in vitro, revealing both normal and aberrantly spliced products, in line with its lower predicted splice-altering score, compared to the other splice-altering mutations. Normally spliced cDNA with exon 9 was still produced by the mutant construct (62.7% of produced transcripts), albeit 60% less abundant compared to the WT construct. Nevertheless, this transcript contains the c.1153G>A change (p.Val385Ile). Aberrantly spliced products either lacking exon 9 (24.4%) or with alternative splicing of exon 9 (12.9%) were also observed (Figure 2). Skipping of exon 9 (including the 3′ UTR) is expected to result in the formation of a truncated protein product (p.Ser386Trp∗). Since skipping involves the last exon in the transcript, NMD is not to be expected, although exceptions to this rule have been observed.14
Individual 6-1 originates from a consanguineous Pakistani family (Figure 1A). She is homozygous for a rare missense variant (not previously reported), chr3:169,928,529A>T (c.992A>T; p.Asp331Val) (Figure S1). The variant was located in a large region of homozygosity of 28.1 Mb as detected by AutoMap.15 Aspartic acid at position 331 is located within the conserved SAM domain (Figure 1B). Based on a bioinformatic analysis performed with the ConSurf Server, this amino acid is predicted to be functionally important (highly conserved and exposed) (Figure 1C). Indeed, this variant is predicted to be pathogenic by several in silico missense prediction tools (Table 1). We therefore reasoned that it may affect the core properties of SAMD7 as a transcriptional repressor, and we tested this hypothesis by using a previously described in vitro system ascertaining this very function.9 In brief, we used four plasmids, bearing (respectively) a CRX-responsive promoter fused to the luciferase cDNA (reporter plasmid), a constitutively expressed human CRX cDNA (CRX-expressing plasmid), a constitutively expressed murine Samd7 WT cDNA (Samd7-expressing plasmid), and a constitutively expressed murine Samd7 cDNA that bore the equivalent of the human p.Asp331Val variant, i.e., p.Asp328Val (Figure S2). HEK293FT cells were co-transfected with the reporter plasmid, the CRX expression plasmid, various amounts of murine Samd7 WT or mutant expression plasmids, and various amounts of a β-galactosidase plasmid. Twenty-four hours after transfection, cell lysates were subjected to luciferase activity assay (see details in supplemental information). As shown in Figure 3, the expression of the luciferase gene following co-transfection with the CRX-bearing plasmid was progressively reduced by increasing amounts of plasmids carrying the WT Samd7 cDNA, in line with the repressive nature of SAMD7. However, this inhibition was significantly reduced when the Asp>Val variant was present, indicating a clear functional effect for this mutation.
Figure 3.

The murine SAMD7 p.Asp328Val variant (equivalent of human p.Asp331Val) impairs repression of CRX activity
A reporter plasmid, carrying the luciferase gene under the control of a universal CRX-dependent promoter, was used to transfect HEK293FT cells. Alone, it could drive the expression of the luciferase gene only to minimal levels (Control bar), as assessed by measuring the luminescence of transfected cells (RLU, or relative light units). Upon co-transfection with a plasmid bearing a CRX sequence, the promoter upstream of the luciferase gene could be activated, resulting in high RLU levels (CRX bar). Additional co-transfection with increasing amounts of plasmids (in ng, per transfection) carrying the wild-type murine Samd7 sequence (WT) resulted in a dose-dependent inhibition of CRX activity. However, such inhibition was significantly lower when plasmid with mutated Samd7 (bearing p.Asp328Val, equivalent of human SAMD7 p.Asp331Val, MT) were used. Main bars indicate average values of n = 14 to 16 technical replicates; error bars show standard errors. Statistical comparison between pairs of conditions was performed by the two-tailed unpaired t test, following the ascertainment that all primary values were normally distributed. Three stars indicate p values ≤ 0.001; four stars indicate p values ≤ 0.0001.
Clinically, all individuals with bi-allelic SAMD7 mutations presented with MD. Mean age at first presentation was 34.8 years and ranged from 14 to 51. In addition to MD, six individuals had CD and two had normal global retinal function. Four individuals (1-1, 3-1, 5-1, and 6-1) presented hyperautofluorescent speckled changes, and individual 1-1 was initially diagnosed with suspected Stargardt MD. Individuals 4-1 and 4-2 are siblings who presented with an identical phenotype, showing MD with a central ring of mottled hypoautofluorescence, surrounded by a hyperautofluorescent ring. Individual 2-1 showed a fundus phenotype identical to that of 4-1 and 4-2, while his asymptomatic father, individual 2-2, showed a pattern dystrophy-type maculopathy limited to the fovea and parafovea (Table 2; Figures 4 and S3 and supplemental note).
Table 2.
Clinical data of study participants
| Individual (Sex) | Lab ID | Age at first diagnosis (y) | Age at most recent diagnostic exam (y) |
Visual acuity |
Fundus |
FF-ERGa |
FAF | OCT | Diagnosis | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | Scotopic | Photopic | ||||||||
| 1-1 (F) | R1124 | 25 | 50 | 6/60 | 6/60 | macular atrophy | a-wave WNR, b-wave MiR | MiR | complete hypoautofluorescence extending to the arcades, surrounded by hyperautofluorescent speckled changes | complete RPE and outer retinal atrophy, surrounded by focal RPE irregularities | MD with CD |
| 2-1 (M) | D1804026 | 14 | 23 | 6/40 | 6/30 | macular atrophy | a- and b-wave MiR | MiR | foveolar hyperautofluorescence surrounded by mottled hypo-autofluorescence with hyperautofluorescent border | foveal thinning due to outer retinal atrophy; temporal peripapillary retinal nerve fiber layer thinning | MD with CD |
| 2-2 (M) | D2309613 | 51 | 51 | 6/10 | 6/6 | maculopathy | a- and b-wave MR | MR | foveolar hyper- and hypo-autofluorescent rings and strongly hyperautofluorescent linear lesions of the parafovea | foveal thinning due to outer nuclear layer thinning; irregular ellipsoid zone and RPE thickenings; normal peripapillary nerve fiber layer thickness | MD with CD |
| 3-1 (F) | GC22085 | 49 | 57 | 6/24 | 6/6 | maculopathy | WNR | WNR | central speckled hyperautofluorescence (pattern-like) | central outer retinal disruption with intraretinal hyperreflective deposits (OD), perifoveal outer retinal disruption (OS), thickening of the RPE and ellipsoid zone (OU) | MD without CD |
| 4-1 (F) | GC28331.1 | 35 | 38 | 6/75 | 6/60 | macular atrophy | WNR | a-wave WNR, b-wave MiR | central ring of mottled hypoautofluorescence, surrounded by a hyperautofluorescent ring | central outer retinal loss (OU) | MD with CD |
| 4-2 (M) | GC28331.2 | 26 | 29 | 6/12 | 6/18 | macular atrophy | WNR | a-wave WNR, b-wave MiR | central ring of mottled hypoautofluorescence, surrounded by a hyperautofluorescent ring | central outer retinal loss (OU) | MD with CD |
| 5-1 (F) | D2112274 | 42 | 50 | 6/15 | 6/15 | maculopathy | WNR | WNR | central mottled hypoautofluorescence surrounded by a hyperautofluorescent rim in both eyes, few hyperautofluorescent speckles in the right eye | irregularity of the ellipsoid zone and focal thickening of the RPE, with small hyperreflective linear lesions migrating above the external limiting membrane | MD without CD |
| 6-1 (F) | CHlaus0429 | 37 | 39 | 6/6 | 6/7.5 (SE -0.5 diopters) | macular atrophy | WNR | MiR | central mottled hypoautofluorescence with few hyperautofluorescent speckles, surrounded by a hyperautofluorescent rim in both eyes | irregularity of the ellipsoid zone and focal thickening of the RPE, with small hyperreflective linear lesions migrating above the external limiting membrane | MD with CD |
F, female; M, male; FAF, fundus autofluorescence; MD, macular dystrophy; CD, cone dysfunction; FAF, fundus autofluorescence; ND, not done; OCT, optical coherence tomography; OD, right eye; OS, left eye; OU, both eyes; RPE, retinal pigmented epithelium; y, years.
FF-ERG, full-field electroretinogram; WNR, within normal range; MiR, mildly reduced; MR, moderately reduced.
Figure 4.

Clinical findings in individuals with SAMD7 mutations
Optical coherence tomography (OCT) (a and b subpanels), fundus autofluorescence (FAF) (c and d subpanels), and fundus photographs (e and f subpanels) of affected individuals with MD due to SAMD7 mutations.
The SAMD7 mutations we identified are located throughout the gene (Figure 1B) and point to loss of SAMD7 function as a mechanism of disease (splice defects leading to predicted NMD or protein truncations being predominant). All individuals presented with MD, but the age of onset ranged between the second and the sixth decades of life. This variable age of onset was found even between individuals homozygous for the same mutation (2-1 and 2-2, both homozygous for c.919+1G>A; 3-1, 4-1 and 4-2, all homozygous for c.211+1G>A). Individual 5-1, homozygous for c.1153G>A, which leads to partial exon skipping, had one of the latest ages of onset in our cohort (42 years). These results indicate that additional genetic and/or environmental factors affect the final phenotypic outcome of SAMD7 deficiency.
Previous immunostaining of mouse retinal sections demonstrated that the murine SAMD7 was localized mainly in nuclei of rod photoreceptors,9,10 while single-cell RNA sequencing (scRNA-seq) of mouse retina showed that Samd7 is expressed in both rods and cones (Figure S4).16 Analysis of scRNA-seq datasets from human retina and human-derived retinal organoids revealed that SAMD7 is expressed in all these tissues in both rods and cones, as well as in other retinal cell types (Figure S4).17 To further determine the localization of SAMD7 in the human retina, we applied immunofluorescence staining with an anti-SAMD7 antibody on human retinal paraffin sections (see details in supplemental information). This staining revealed the presence of SAMD7 in the retinal inner layer, where nuclei of retinal neurons are located, and in the outer nuclear layer, where nuclei of rod and cone photoreceptors are placed (Figure 5).
Figure 5.

SAMD7 localizes to the inner and outer nuclear layers of the human retina and to the nuclei of both rods and cones
(A) Immunofluorescence staining of transverse sections of paraffin-embedded human retina, displaying localization of SAMD7 (green) in the inner and outer nuclear layers of this tissue. PNA (peanut agglutinin, magenta) identifies cones and DAPI (4′,6-diamidino-2-phenylindole, blue) stains nuclei. ONL, outer nuclear layer; INL, inner nuclear layer. Scale bars: 20 μm.
(B) Magnified image of a different region of the retina, focusing on photoreceptors, demonstrating the presence of SAMD7 in both rods and cones. Scale bars: 10 μm.
Notably, in a subset of our cohort (specifically individuals 1-1, 2-1, 2-2, 4-1, and 4-2), electroretinographic assessments revealed a mild attenuation of scotopic rod b-waves, indicative of inner retinal dysfunction. In individual 2-2 there were no peripheral signs of retinal degeneration at age 51, suggesting that rod involvement is unlikely in this older individual. Hence, the presence of a relatively reduced rod-specific response, which is b-wave only, is likely due to inner retinal dysfunction. Therefore, the reduced a-wave of the combined rod-cone responses to the high-intensity flashes (3 and 10 cd s/m2) is likely entirely due to the reduction of cone-specific phototransduction. In addition, the reduction of the b-to-a wave ratio of the combined rod-cone responses further suggests inner retinal dysfunction. These electroretinographic observations are congruent with our anatomical findings, thereby substantiating the pathophysiological implications of SAMD7 mutations.
Given the expression of SAMD7 in both rods and cones, as well as in other retinal cell types, it is surprising that SAMD7 deficiency affects only cone photoreceptors, leading to MD. Nevertheless, similar cases have been described. For example, elongation of very-long-chain fatty acids-like 4 (ELOVL4) is expressed in both rods and cones,18 but the main retinal phenotype associated with ELOVL4 mutations is MD (Stargardt disease type 3 [MIM: 600110]).19 This is probably due to different composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors.20
Previous studies on WT and Samd7-null mice suggested that SAMD7 plays a role as a transcriptional repressor of CRX-regulated gene expression in the retina, to determine cell identity of rod photoreceptors.9,10 In a proposed model, SAMD7 would interact with NR2E3, a rod-specific transcription factor, to silence non-rod gene expression in rod photoreceptors, via histone modifications and interaction with proteins of the Polycomb complex.10 Indeed, the phenotype observed in Samd7-null mice is rod dominated, as reflected by electroretinographic (ERG) responses, in which both a- and b-waves were significantly reduced under scotopic conditions. Photopic ERG responses in these mice were completely normal, indicating normal cone function.10 This is in contrast to the outcome of bi-allelic SAMD7 mutations in humans, which mainly involves cones in the macular region. Based on full-field ERG, rod function in our patients was normal, or only minimally affected. Cone function in the peripheral retina was mildly to moderately reduced in six of eight individuals, and normal in two of them; these two individuals had isolated MD. It should be noted that the murine retina lacks a macula, and therefore an MD phenotype cannot be observed in mice. Nevertheless, these results demonstrate a significant difference in the role of SAMD7 between the human and the mouse retina. Further studies are needed to provide more insights on the role of SAMD7 in photoreceptors, especially in cones, and in other types of retinal neurons, using different models mirroring the human retina.
In conclusion, the genes regulated by SAMD7 in the human retina and the pathophysiological mechanism underlying the maculopathy reported here in humans with SAMD7 deficiency remain to be determined. Nevertheless, the data presented here place SAMD7 as a crucial gene for human retinal function, which should be evaluated for pathogenic variation in individuals with MD.
Data and code availability
All SAMD7 variants identified in this study were submitted to the Leiden Open Variation Database (LOVD) (http://www.lovd.nl).
Acknowledgments
We are grateful to the affected individuals for their participation in this study. We thank Marc Folcher for technical assistance. We are also indebted to Francesca Cancellieri and Karolina Kaminska for their contribution in obtaining genetic data. T.B.-Y. was supported by a research grant from Israel Ministry of Health (3-12583); G.A. is funded by a Fight For Sight UK Early Career Investigator Award (5045/46), National Institute of Health Research Biomedical Research Centre (NIHR-BRC) at Moorfields Eye Hospital and UCL Institute of Ophthalmology, and NIHR-BRC at Great Ormond Street Hospital Institute for Child Health; C.R. is funded by the Swiss National Science Foundation (grant # 204285). E.D.B. and M.B. were supported by the Ghent University Special Research Fund (BOF20/GOA/023), EJPRD19-234 Solve-RET, FWO research project G0A9718N, and Foundation John W. Mouton Pro Retina & Marie-Claire Liénaert. E.D.B. is a Senior Clinical Investigator (1802220N) of the Research Foundation Flanders (FWO).
Author contributions
M.B., M.Q., Q.M., L.R., A.R.M., M.V.H., and E.C. conducted the experiments and analyzed data. D.Z., S.L., F.V.D.B., B.P.L., H.V.T., A.P.M., A.R.W., A.M., and M.M. collected and interpreted clinical data. M.B., G.A., C.R., E.C., M.Q., E.D.B., and T.B.-Y. conceived and designed the experiments. T.B.-Y. wrote the manuscript, which was edited and/or approved by all co-authors.
Declaration of interests
The authors declare no competing interests.
Published: January 24, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2024.01.001.
Web resources
BLASTP, https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins
Exome Sequencing Project (ESP) 6500, https://evs.gs.washington.edu/EVS/
GenBank, http://www.ncbi.nih.gov/Genbank/
Retnet (Retinal Information Network), https://web.sph.uth.edu/RetNet/
Supplemental information
References
- 1.Duncan J.L., Pierce E.A., Laster A.M., Daiger S.P., Birch D.G., Ash J.D., Iannaccone A., Flannery J.G., Sahel J.A., Zack D.J., et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018;7:6. doi: 10.1167/tvst.7.4.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman N., Georgiou M., Khan K.N., Michaelides M. Macular dystrophies: clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020;104:451–460. doi: 10.1136/bjophthalmol-2019-315086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cremers F.P.M., Lee W., Collin R.W.J., Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020;79 doi: 10.1016/j.preteyeres.2020.100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Yosef T. Inherited Retinal Diseases. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232113467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carss K.J., Arno G., Erwood M., Stephens J., Sanchis-Juan A., Hull S., Megy K., Grozeva D., Dewhurst E., Malka S., et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017;100:75–90. doi: 10.1016/j.ajhg.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Pozo-Valero M., Riveiro-Alvarez R., Martin-Merida I., Blanco-Kelly F., Swafiri S., Lorda-Sanchez I., Trujillo-Tiebas M.J., Carreño E., Jimenez-Rolando B., Garcia-Sandoval B., et al. Impact of Next Generation Sequencing in Unraveling the Genetics of 1036 Spanish Families With Inherited Macular Dystrophies. Invest. Ophthalmol. Vis. Sci. 2022;63:11. doi: 10.1167/iovs.63.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stone E.M., Andorf J.L., Whitmore S.S., DeLuca A.P., Giacalone J.C., Streb L.M., Braun T.A., Mullins R.F., Scheetz T.E., Sheffield V.C., Tucker B.A. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology. 2017;124:1314–1331. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Illingworth R.S. Chromatin folding and nuclear architecture: PRC1 function in 3D. Curr. Opin. Genet. Dev. 2019;55:82–90. doi: 10.1016/j.gde.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hlawatsch J., Karlstetter M., Aslanidis A., Lückoff A., Walczak Y., Plank M., Böck J., Langmann T. Sterile alpha motif containing 7 (samd7) is a novel crx-regulated transcriptional repressor in the retina. PLoS One. 2013;8 doi: 10.1371/journal.pone.0060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omori Y., Kubo S., Kon T., Furuhashi M., Narita H., Kominami T., Ueno A., Tsutsumi R., Chaya T., Yamamoto H., et al. Samd7 is a cell type-specific PRC1 component essential for establishing retinal rod photoreceptor identity. Proc. Natl. Acad. Sci. USA. 2017;114:E8264–E8273. doi: 10.1073/pnas.1707021114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Schil K., Karlstetter M., Aslanidis A., Dannhausen K., Azam M., Qamar R., Leroy B.P., Depasse F., Langmann T., De Baere E. Autosomal recessive retinitis pigmentosa with homozygous rhodopsin mutation E150K and non-coding cis-regulatory variants in CRX-binding regions of SAMD7. Sci. Rep. 2016;6 doi: 10.1038/srep21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhuvanagiri M., Schlitter A.M., Hentze M.W., Kulozik A.E. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010;430:365–377. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu M.K., Lin H.Y., Chen F.C. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS One. 2017;12 doi: 10.1371/journal.pone.0174798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinodoz M., Peter V.G., Bedoni N., Royer Bertrand B., Cisarova K., Salmaninejad A., Sepahi N., Rodrigues R., Piran M., Mojarrad M., et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 2021;12:518. doi: 10.1038/s41467-020-20584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karademir D., Todorova V., Ebner L.J.A., Samardzija M., Grimm C. Single-cell RNA sequencing of the retina in a model of retinitis pigmentosa reveals early responses to degeneration in rods and cones. BMC Biol. 2022;20:86. doi: 10.1186/s12915-022-01280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowan C.S., Renner M., De Gennaro M., Gross-Scherf B., Goldblum D., Hou Y., Munz M., Rodrigues T.M., Krol J., Szikra T., et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell. 2020;182:1623–1640.e34. doi: 10.1016/j.cell.2020.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agbaga M.P., Brush R.S., Mandal M.N.A., Henry K., Elliott M.H., Anderson R.E. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc. Natl. Acad. Sci. USA. 2008;105:12843–12848. doi: 10.1073/pnas.0802607105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K., Kniazeva M., Han M., Li W., Yu Z., Yang Z., Li Y., Metzker M.L., Allikmets R., Zack D.J., et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 20.Agbaga M.P., Merriman D.K., Brush R.S., Lydic T.A., Conley S.M., Naash M.I., Jackson S., Woods A.S., Reid G.E., Busik J.V., Anderson R.E. Differential composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors. J. Lipid Res. 2018;59:1586–1596. doi: 10.1194/jlr.M082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All SAMD7 variants identified in this study were submitted to the Leiden Open Variation Database (LOVD) (http://www.lovd.nl).