

Abstract
Co‐administration of clesacostat (acetyl‐CoA carboxylase inhibitor, PF‐05221304) and ervogastat (diacylglycerol O‐acyltransferase inhibitor, PF‐06865571) in laboratory models improved non‐alcoholic fatty liver disease (NAFLD)/non‐alcoholic steatohepatitis (NASH) end points and mitigated clesacostat‐induced elevations in circulating triglycerides. Clesacostat is cleared via organic anion‐transporting polypeptide‐mediated hepatic uptake and cytochrome P450 family 3A (CYP3A); in vitro clesacostat is identified as a potential CYP3A time‐dependent inactivator. In vitro ervogastat is identified as a substrate and potential inducer of CYP3A. Prior to longer‐term efficacy trials in participants with NAFLD, safety and pharmacokinetics (PK) were evaluated in a phase I, non‐randomized, open‐label, fixed‐sequence trial in healthy participants. In Cohort 1, participants (n = 7) received clesacostat 15 mg twice daily (b.i.d.) alone (Days 1–7) and co‐administered with ervogastat 300 mg b.i.d. (Days 8–14). Mean systemic clesacostat exposures, when co‐administered with ervogastat, decreased by 12% and 19%, based on maximum plasma drug concentration and area under the plasma drug concentration–time curve during the dosing interval, respectively. In Cohort 2, participants (n = 9) received ervogastat 300 mg b.i.d. alone (Days 1–7) and co‐administered with clesacostat 15 mg b.i.d. (Days 8–14). There were no meaningful differences in systemic ervogastat exposures when administered alone or with clesacostat. Clesacostat 15 mg b.i.d. and ervogastat 300 mg b.i.d. co‐administration was overall safe and well tolerated in healthy participants. Cumulative safety and no clinically meaningful PK drug interactions observed in this study supported co‐administration of these two novel agents in additional studies exploring efficacy and safety in the management of NAFLD.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Combinations of novel agents with pre‐approved drugs, either as add‐on therapy or fixed‐dose combinations, is becoming routine. Similarly, clinical development of novel–novel drug combinations is emerging.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study investigated the safety and pharmacokinetic (PK) drug interaction risks of a novel–novel combination, acetyl‐CoA carboxylase inhibitor clesacostat (PF‐05221304) with diacylglycerol O‐acyltransferase inhibitor ervogastat (PF‐06865571) in early clinical development.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Although in vitro ervogastat was predominantly cleared by CYP3A and was also a potential inducer of CYP3A and clesacostat was a potential time‐dependent inactivator of CYP3A, co‐administration of ervogastat 300 mg b.i.d. with clesacostat 15 mg b.i.d. in this study established the safety and lack of clinically meaningful PK drug interaction. These results supported the progression of this novel combination to a subsequent phase II study in patients with non‐alcoholic fatty liver disease (NAFLD).
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This article describes the investigation of clinical drug interaction of a novel–novel combination in early clinical development.
INTRODUCTION
Non‐alcoholic steatohepatitis (NASH) is a progressive liver disease characterized by accumulation of liver fat, inflammation, and ballooning appearance of hepatocytes, and may lead to cirrhosis, hepatocellular carcinoma, and death. 1 Disordered metabolism, including elevated hepatic de novo lipogenesis (DNL), contributes to excess accumulation of fat in the liver in non‐alcoholic fatty liver disease (NAFLD)/NASH. Reducing fat accumulation in the liver is hypothesized to reduce lipotoxicity, leading to improvements in hepatic inflammation, hepatocellular injury, and possibly fibrosis. Acetyl‐CoA carboxylase (ACC) and diacylglycerol O‐acyltransferase 2 (DGAT2) are key enzymes in the lipogenic pathway, and inhibition of these enzymes may lead to reductions in steatosis and possibly downstream benefits for the treatment of NASH. 2 In mice, hepatic DGAT2 deficiency reduced diet‐induced hepatic steatosis, suggesting the reversal of NAFLD severity by reducing triglycerides (TGs), 3 which has been shown to cause reductions in markers of liver function in healthy participants. 4 In addition, a systemically available DGAT2 inhibitor, ervogastat (PF‐06865571), also showed reduction in TGs and liver fat in participants with NAFLD. 5 The liver‐directed ACC inhibitor clesacostat (PF‐05221304) inhibited hepatic DNL and reduced liver fat accumulation in primary hepatocytes and Western‐diet fed rats and improved markers of inflammation and fibrosis in liver injury and liver fibrosis models. 6 Similarly, dose‐dependent inhibition of DNL was reported in humans following repeated administration of the ACC inhibitor clesacostat (PF‐05221304). 7 , 8 In a 16‐week phase IIa trial in patients with presumed NAFLD or NASH, clesacostat markedly lowered liver fat content, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) levels, and hemoglobin A1c, as well as cytokeratin 18, a marker of hepatocyte apoptosis. 5 However, marked dose‐dependent elevations in serum TG concentrations, a known consequence of ACC inhibition, 9 were also observed. 5 As DGAT2 inhibition was hypothesized to directly reverse the mechanism of ACC inhibitor‐induced TG elevations, non‐clinical studies were undertaken to assess the effects of clesacostat and ervogastat co‐administration on circulating TGs, liver fat, hepatic inflammation, and fibrosis. These studies demonstrated that co‐administration of clesacostat and ervogastat mitigated clesacostat‐mediated increases in circulating TGs and produced greater liver fat, inflammation, and fibrosis efficacy than either monotherapy. 5
In vitro data (unpublished) suggest that the primary clearance mechanism of clesacostat is hepatic uptake mediated by organic anion‐transporting polypeptides (OATPs) specifically OATP1B1, OATP1B3, OATP2B1, and sodium‐taurocholate cotransporting polypeptide, and CYP3A metabolism. Ervogastat is primarily metabolized by CYP3A. The terminal elimination half‐life of clesacostat is 14–18 h following single oral doses of 1–240 mg, 7 whereas the terminal elimination half‐life of ervogastat is 1.45–5.22 h following single oral doses of 5–1500 mg. 10 In vitro, both compounds were substrates for efflux transporters P‐glycoprotein (Pgp) and breast cancer resistance protein (BCRP); ervogastat may inhibit Pgp and BCRP. In addition, in vitro studies demonstrated that clesacostat is a time‐dependent inhibitor of CYP3A and ervogastat is a CYP3A inducer (data on file). The data represent the first instance of co‐administering clesacostat and ervogastat in humans. Here, we assessed the safety and overall pharmacokinetic (PK) drug interactions following co‐administration of clesacostat and ervogastat in healthy participants (NCT03534648) prior to evaluation of these two novel agents co‐administered in participants with NASH and liver fibrosis.
METHODS
ACC inhibitor clesacostat (PF‐05221304) and DGAT2 inhibitor ervogastat (PF‐06865571) drug products were manufactured at Pfizer. The study was conducted at the Pfizer Clinical Research Unit (CRU), located in Brussels, Belgium, and registered on clinicaltrials.gov (NCT03534648). This study was performed in compliance with ethical principles originating in or deriving from the Declaration of Helsinki and in compliance with all International Conference on Harmonisation Good Clinical Practice Guidelines and the International Ethical Guidelines for Biomedical Research Involving Human Subjects (Council for International Organizations of Medical Sciences 2002). Approval from the local institutional review board (Comité d'Ethique Hospitalo‐Facultaire Erasme‐ULB) was obtained prior to the start of the study, in the interest of the greater protection and safety of the study participants.
Participant eligibility criteria
All relevant medical and non‐medical information was taken into consideration and participants were expected to meet all inclusion criteria to be eligible for enrollment. Healthy male and non‐child‐bearing female participants aged 18 to 55 years inclusive, with a body mass index of 17.5 to 30.5 kg/m2 and total body weight of >50 kg (110 lb), who provided a signed and dated informed consent form and who were willing and able to comply with the study plan, scheduled visits, treatment plan, laboratory tests, and all other study procedures, were eligible for inclusion in this study.
Study design
NCT03534648 was a phase I, open‐label, non‐randomized, fixed‐sequence, multiple‐dose study designed to investigate the potential of a PK drug–drug interaction (DDI) between clesacostat and ervogastat. The study was conducted in two different cohorts (Figure 1). Participants were screened within 28 days prior to their first dose and admitted to the CRU the day before dosing Day 1. By design, as a non‐randomized, fixed‐sequence study, the first seven enrolled participants were dosed in Cohort 1, followed by the next nine enrolled participants in Cohort 2.
FIGURE 1.
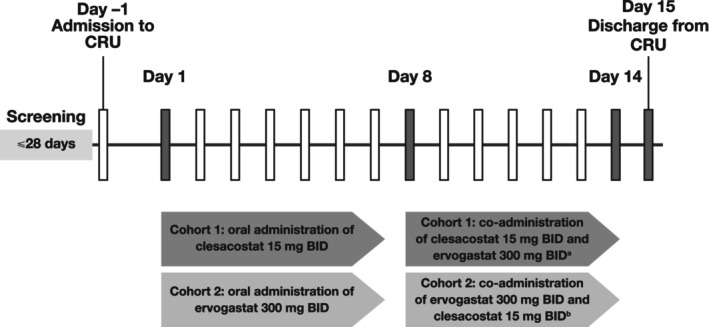
Study design. aOn Day 14, clesacostat 15 mg was co‐administered with ervogastat 300 mg in the morning only; no afternoon dose was administered. bOn Day 14, ervogastat 300 mg was co‐administered with clesacostat 15 mg in the morning only; no afternoon dose was administered. BID, twice daily; CRU, Clinical Research Unit.
Dose selection
A dose of clesacostat 15 mg b.i.d. was selected in this study based on observed cumulative safety, PK, and pharmacodynamics in humans. 7 At this dose, no change in platelet count was anticipated with ≥80% inhibition of DNL. Ervogastat 300 mg b.i.d. was selected based on all available cumulative safety, PK, and pharmacodynamic data generated in a previous clinical study (NCT03092232) and because this dose and regimen were under investigation as a pharmacologically relevant dose in participants with NAFLD. 11 The observed clinical PK of clesacostat supported a once‐daily dosing regimen. 7 However, a b.i.d. dosing regimen was selected to match that of ervogastat. This dose and dosing regimen (clesacostat 15 mg b.i.d. plus ervogastat 300 mg b.i.d.) was expected to be administered in a subsequent phase IIa study in participants with NAFLD (NCT03776175) 5 ; therefore, the same dose and dosing regimens of clesacostat and ervogastat were co‐administered in the current DDI study. All doses were administered as immediate‐release tablet formulations following standard meals, as food has been shown to significantly increase ervogastat exposure.
Study treatments
In Cohort 1, participants received clesacostat 15 mg b.i.d. alone on Days 1–7 and clesacostat 15 mg b.i.d. plus ervogastat 300 mg b.i.d. on Days 8–13. On Day 14, participants received a single oral dose of clesacostat 15 mg plus ervogastat 300 mg in the morning only. In Cohort 2, participants received ervogastat 300 mg b.i.d. alone on Days 1–7 and ervogastat 300 mg b.i.d. plus clesacostat 15 mg b.i.d. on Days 8–13. On Day 14, participants received a single oral dose of clesacostat 15 mg plus ervogastat 300 mg in the morning only. Participants were scheduled to be discharged on Day 15. A follow‐up visit was planned for Day 22 ± 2 and a follow‐up telephone contact was planned for Day 44 ± 2.
Safety assessments
The safety and tolerability of clesacostat and ervogastat (alone and in combination) were evaluated by continuous monitoring of adverse events (AEs) during inpatient and follow‐up parts of the study. In addition, brief physical examinations at admission, intermittent sampling of safety‐related laboratory parameters, 12‐lead electrocardiograms (ECGs), and vital signs were assessed during the study.
PK assessments
PK sample collection and bioanalyses of clesacostat and ervogastat in plasma using liquid chromatography–tandem mass spectrometry (LC–MS/MS)
Blood samples (3 mL each) were collected to provide approximately 1 mL plasma. Samples were collected on Day 7 and Day 14 at pre‐dose, 0.5, 1, 1.5, 2, 4, 6, 8, 10, and 12 h post‐morning dose. Plasma samples were stored at −20°C until analysis for drug levels of clesacostat only (Cohort 1) or ervogastat only (Cohort 2). All plasma samples were assayed at Covance Bioanalytical Services, LLC using a validated, sensitive, and specific high‐performance liquid chromatography–tandem mass spectrometry (HPLC‐MS/MS) bioanalytical method in compliance with Pfizer standard operating procedures (SOPs). The mass spectrometer (API 4000; Sciex) settings and acquisition parameters are listed in Table S1.
Plasma samples were analyzed for clesacostat at Covance. Briefly, clesacostat and its stable labeled internal standard, PF‐06894391, were extracted from human plasma using a protein precipitation extraction procedure. Following centrifugation, the supernatant was diluted with 200 μL of acetonitrile:water:formic acid (20:80:0.1, v:v:v). The mixture was vortexed and analyzed using liquid chromatography coupled with MS/MS using electrospray ionization (ESI) in positive mode. The chromatographic separation was achieved using a Xbridge C8/Waters/3.5 μm column (Waters Corporation) and gradient elution, using 0.1% formic acid in 5 mM ammonium formate in water as mobile phase A and 50:50:0.1 acetonitrile:methanol:formic acid as mobile phase B. Calibration standard responses were linear over the range from 2.00 to 1000 ng/mL, using weighted (L/concentration2) linear least squares regression. The quality control (QC) concentrations were 6.00, 50, 450, 800, and 16,000 ng/mL. The performance of the method during validation is documented internally (Report No. C1179005). During sample analysis, the interday assay accuracy ranged from −0.7% to 4.6% and the between‐day precision was ≤10.4%. Clinical specimens with plasma clesacostat concentrations below the lower limit of quantification (LLOQ) were reported as below the LLOQ (<2.00 ng/mL).
Plasma samples were assayed for ervogastat at Syneos Health (301D College Road East Princeton, New Jersey, NY, USA) using a validated bioanalytical method in compliance with Pfizer SOPs. Briefly, ervogastat and its stable labeled internal standard PF‐06865571‐[13C2, 15N2] were extracted from human plasma by a liquid–liquid extraction procedure. After mixing, the supernatant was evaporated to dryness, reconstituted, vortexed, and analyzed by HPLC‐MS/MS using ESI in positive ionization mode. Chromatographic separation was achieved using a Restek Raptor Biphenyl 2.1 × 50 mm, 5 μm column and isocratic elution (Restek) using 0.01% trifluoroacetic acid in 60/40 (v/v) methanol/water as mobile phase A. Calibration standard responses were linear over the range from 0.250 to 250 ng/mL using weighted (L/concentration2) linear least squares regression. The QC concentrations were 0.750, 125, 200, and 4000 ng/mL. The performance of the method during validation is documented internally (Report No. C2549001). During sample analysis, the interday assay accuracy ranged from −3.00% to 0.933% and the between‐day precision was ≤8.16%. Clinical specimens with plasma ervogastat concentrations below the LLOQ were reported as below the LLOQ (<0.250 ng/mL).
PK parameter evaluations of clesacostat and ervogastat alone and after co‐administration
Plasma PK parameters following oral administration of clesacostat and ervogastat were determined from the observed plasma concentration–time profile, using non‐compartmental analyses, as per Pfizer internal SOPs and as data permitted. The PK parameters of the maximum plasma drug concentration (C max), the area under the plasma drug concentration–time curve during the dosing interval (AUCtau), the apparent clearance (CL/F), the apparent steady‐state volume of distribution, and the plasma elimination half‐life were determined using non‐compartmental analysis, as data permitted. Concentrations below the LLOQ were recorded as 0 ng/mL.
The plasma PK parameters were summarized descriptively by treatment. To assess any effect of ervogastat on clesacostat exposures or vice versa, natural log‐transformed C max and AUCtau were analyzed using a mixed‐effects model with treatment as a fixed effect and participant as a random effect. The adjusted mean differences and 90% confidence intervals (CIs) for the differences obtained from the model were exponentiated to provide estimates of the ratio of adjusted geometric means (Test/Reference) and 90% CIs for the ratios. The comparisons of interest were:
Cohort 1: Effect of ervogastat on clesacostat exposures with Day 7 PK as the Reference treatment and clesacostat Day 14 PK as the Test treatment.
Cohort 2: Effect of clesacostat on ervogastat exposures with Day 7 PK as the Reference treatment and ervogastat Day 14 PK as the Test treatment.
Statistical analyses
Sample size determination
A sample size of approximately 16 participants was planned for the study, with the goal of attaining approximately 14 completers (6 for Cohort 1 and 8 for Cohort 2). The sample size was based on a precision approach.
Sample size determination in Cohort 1: Effect of ervogastat on the PK of clesacostat
A sample size of six participants was selected to provide sufficient precision to detect a 50% increase in exposure, as measured by 90% CIs for the difference between the Day 14 (Test) and Day 7 (Reference) measurements of C max and AUCtau of clesacostat on the natural log scale. For the estimated effect of 1.5 (Test/Reference), the expected 90% CI with 80% coverage probability was (1.23, 1.83) and (1.36, 1.65) for C max and AUCtau, respectively. The calculations assumed intra‐participant standard deviations (SDs) of 0.14 and 0.07 for log dose‐normalized C max and AUCtau of clesacostat, respectively, based on single‐dose PK data from a previous study (NCT02871037).
Sample size determination in Cohort 2: Effect of clesacostat on the PK of ervogastat
A sample size of eight participants was selected to provide sufficient precision to detect a 50% increase in exposure, as measured by 90% CIs for the difference between Day 14 (Test) and Day 7 (Reference) measurements of C max and AUCtau of ervogastat on the natural log scale. For the estimated effect of 1.5 (Test/Reference), the expected 90% CI with 80% coverage probability was (1.23, 1.84) and (1.31, 1.72) for C max and AUCtau, respectively. Intra‐participant SDs for log dose‐normalized C max and AUCtau of ervogastat were 0.18 and 0.12, respectively, as obtained from the PK data in a previous study (NCT03092232).
Analysis populations
The safety analysis set was defined separately for each cohort as all participants who received at least one dose of study medication in the respective cohort. Safety evaluations were summarized in accordance with the sponsor reporting standards. The PK concentration populations were defined as all participants who received at least one dose of clesacostat (Cohort 1) or ervogastat (Cohort 2) and in whom at least one plasma concentration value was reported. The PK parameter analysis population was defined as all participants who received at least one dose of clesacostat (Cohort 1) or ervogastat (Cohort 2) and had at least one of the PK parameters of interest calculated.
RESULTS
Participant demographics and disposition
This study enrolled 16 healthy adult male participants (n = 7 in Cohort 1 and n = 9 in Cohort 2). The demographics of all participants enrolled are summarized in Table 1. In Cohort 1, one participant was discontinued on Day 9 (i.e., 7 days of clesacostat alone followed by 2 days of co‐administration of clesacostat + ervogastat) due to elevations in alanine transaminase (ALT), aspartate transaminase (AST), and gamma‐glutamyl transferase (GGT), determined by the investigator to be of moderate intensity. There were no discontinuations in Cohort 2.
TABLE 1.
Demographics of study participants.
| Parameter | Cohort 1 | Cohort 2 |
|---|---|---|
| Participants (n) | 7 | 9 |
| Age, years; mean (range) | 38.0 (24, 54) | 33.8 (22, 49) |
| Weight, kg; mean (range) | 77.1 (61.7, 94.5) | 72.5 (58.5, 86.2) |
| Body mass index, kg/m2; mean (range) | 24.4 (19.7, 29.7) | 23.2 (18.6, 28.1) |
| Sex, male (n) | 7 | 9 |
| Race, White (n) | 7 | 9 |
Safety of clesacostat (PF‐05221304) and ervogastat (PF‐06865571) alone or in combination
The safety of repeated dosing with clesacostat 15 mg b.i.d., ervogastat 300 mg b.i.d., and co‐administration was evaluated in healthy adult participants. The AEs reported are presented in Table S2. All AEs were mild to moderate in severity. There were no serious AEs, no severe AEs, and no deaths in this study and no other clinically significant findings observed in laboratory parameters, vital signs, or ECGs. One participant in Cohort 1 was noted to have elevations in ALT (2.8× upper limit of normal [ULN] of 49 U/L), AST (3.5× ULN of 40 U/L), and GGT (2.8× ULN of 73 U/L) following 7 days of dosing of clesacostat, with these elevations continuing to Day 9 (ALT = 3.8× ULN, AST = 2.9× ULN, and GGT = 3.3× ULN) at which time a decision was made to stop dosing, with monitoring continued until these liver function tests returned to baseline (Day 15 for GGT, Day 20 for AST, and Day 22 for ALT).
PK of clesacostat (PF‐05221304) 15 mg b.i.d. alone on Day 7 (Reference) and in combination with ervogastat (PF‐06865571) 300 mg b.i.d. on Day 14 (Test): Cohort 1
The median plasma concentration–time profile of clesacostat in participants in Cohort 1 on Day 7 (Reference) and Day 14 (Test) is summarized in Figure 2a. Following multiple oral doses of clesacostat 15 mg b.i.d. administered alone (Day 7), median (range) time to maximum plasma drug concentration (T max) was 4.0 (2.0 to 6.0) h post‐dose followed by a decline in plasma concentrations. Following co‐administration of clesacostat 15 mg b.i.d. with ervogastat 300 mg b.i.d., the median (range) T max of clesacostat was 2.0 (1.5 to 4.0) h post‐dose. Co‐administration with ervogastat resulted in a slight increase in CL/F and peak‐to‐trough ratio within one dosing interval (PTR) of clesacostat (Table 2). Interparticipant variability for clesacostat exposure, based on geometric percent coefficient of variability (%CV), was similar for both treatments ranging from 20%–25% for C max and 23%–26% for AUCtau. The Day 14 (Test)/Day 7 (Reference) ratio of the adjusted geometric mean (90% CI) for clesacostat C max was 88.46% (79.77%, 98.10%) and for AUCtau was 80.51% (73.59%, 88.09%). Statistical analysis of clesacostat exposure parameters (with and without ervogastat) is summarized in Table 2.
FIGURE 2.
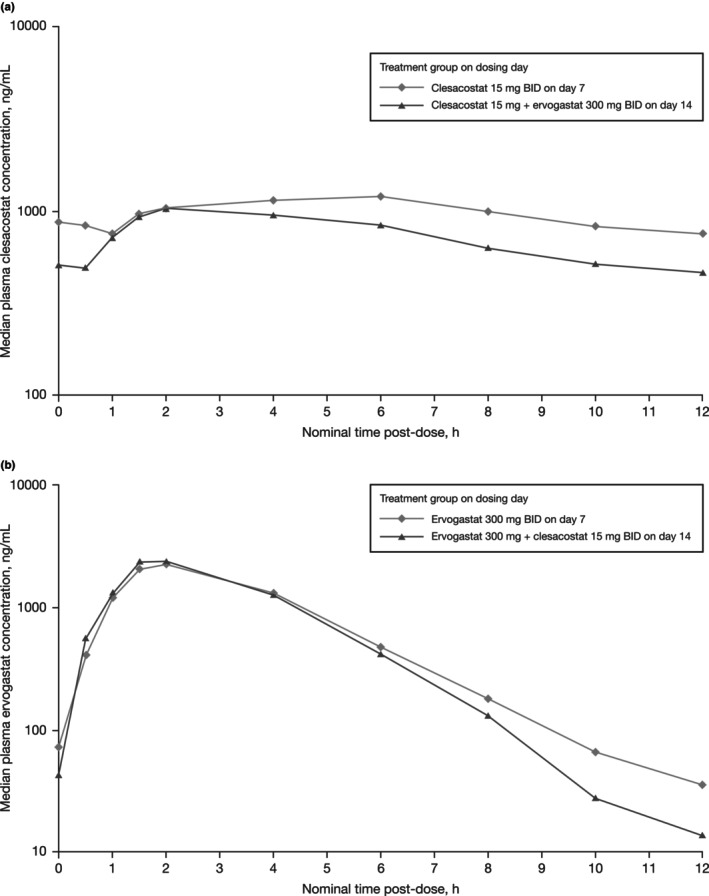
Median total plasma concentration–time profile of (a) clesacostat (PF‐05221304) 15 mg BID alone on Day 7 (Reference) and following co‐administration with ervogastat (PF‐06865571) 300 mg BID on Day 14 (Test) and (b) ervogastat (PF‐06865571) 300 mg BID alone on Day 7 (Reference) and following co‐administration with clesacostat (PF‐05221304) 15 mg BID on Day 14 (Test). BID, twice daily; h, hour.
TABLE 2.
Descriptive and statistical summary of plasma pharmacokinetics of clesacostat (PF‐05221304, Cohort 1) or ervogastat (PF‐06865571, Cohort 2) alone on Day 7 (Reference) or co‐administered on Day 14 (Test).
| Parameter | Cohort 1 | Cohort 2 | ||||
|---|---|---|---|---|---|---|
| Clesacostat 15 mg b.i.d., Day 7 | Clesacostat 15 mg + ervogastat 300 mg b.i.d., Day 14 | Ratio (%) (90% CI) a | Ervogastat 300 mg b.i.d., Day 7 | Ervogastat 300 mg + clesacostat 15 mg b.i.d., Day 14 | Ratio (%) (90% CI) a | |
| Participants (n) | 7 | 6 | NA | 9 | 9 | NA |
| T max, h | 4.00 (2.00–6.00) | 2.00 (1.50–4.00) | NA | 2.00 (1.00–4.00) | 2.00 (1.00–2.00) | NA |
| CL/F, L/h | 1.24 (26) | 1.58 (23) | NA | 34.41 (19) | 31.76 (34) | NA |
| PTR, h | 1.92 | 2.41 | NA | 94.35 | 149.7 | NA |
| AUCtau, ng*h/mL | 12,140 (26) | 9477 (23) | 80.51 (73.59, 88.09) | 8715 (19) | 9450 (34) | 108.43 (97.08, 121.10) |
| C max, ng/mL | 1337 (25) | 1128 (20) | 88.46 (79.77, 98.10) | 2386 (10) | 2580 (21) | 108.14 (97.91, 119.43) |
Note: Data are geometric mean (%CV) for all parameters except median (range) for T max.
Abbreviations: %CV, coefficient of variability; AUCtau, area under the plasma drug concentration–time curve during the dosing interval; b.i.d., twice daily; CI, confidence interval; CL/F, apparent clearance; C max, maximum plasma drug concentration; h, hour; NA, not applicable; PTR, peak‐to‐trough ratio; T max, time to maximum plasma drug concentration.
Geometric mean ratio (Day 14 [Test]/Day 7 [Reference]); ratios and 90% CIs are expressed as percentages.
The concentration–time profiles by participant are shown in Figure S1.
PK of ervogastat (PF‐06865571) 300 mg b.i.d. alone on Day 7 (Reference) and in combination with clesacostat (PF‐05221304) 15 mg b.i.d. on Day 14 (Test): Cohort 2
The median plasma concentration–time profile of ervogastat in participants in Cohort 2 on Day 7 (Reference) and Day 14 (Test) is summarized in Figure 2b. Following multiple oral doses of ervogastat 300 mg b.i.d., when administered alone (Day 7) or in combination with clesacostat, median (range) T max was approximately 2.0 (1.0 to 4.0) h post‐dose followed by a decline in plasma concentrations. Co‐administration of ervogastat 300 mg b.i.d. with clesacostat 15 mg BID resulted in similar CL/F and PTR (Table 2). Interparticipant variability of ervogastat exposures, based on geometric %CV, was higher when co‐administered with clesacostat and increased from 10% to 21% for C max and from 19% to 34% for AUCtau. The Day 14 (Test)/Dday 7 (Reference) ratio of the adjusted geometric mean (90% CI) for ervogastat C max was 108.14% (97.91%, 119.43%) and for AUCtau was 108.43% (97.08%, 121.10%). Statistical analysis of ervogastat exposure parameters (with and without clesacostat) is summarized in Table 2.
DISCUSSION
Several successful combinations have been developed in oncology and cardiometabolic disorders and as antiretroviral therapies. 12 , 13 , 14 , 15 , 16 Safety, pharmacology, PK, and formulation uncertainties have to be assessed to quantify the overall benefit–risk for such combinations. In addition, regulatory recommendations should be considered during the development of novel combinations. 17 Clesacostat was identified as a potent, selective, and orally bioavailable ACC1/2 dual inhibitor, designed for preferential hepatic uptake and inhibition of hepatic DNL, while minimizing systemic ACC inhibition that has been shown to cause dose‐dependent reductions in platelet counts. 8 Ervogastat was designed as a potent, selective, and orally bioavailable DGAT2 inhibitor. The current work describes the investigation of the PK and safety of co‐administration of two novel molecules, clesacostat and ervogastat, in healthy adult participants. This investigation supported the progression of this combination in subsequent studies in patients with NAFLD (NCT03776175). 5
This phase I study was designed as a fixed‐sequence, non‐randomized trial in healthy volunteers. It should be considered that demographics such as race, age, and sex may impact the PK of clesacostat and ervogastat, given that CYP3A4 and OATP genes may show pharmacogenomic variations across global populations, leading to differences in hepatic clearance and resulting drug metabolism rates. 18 Demographic variations in CYP3A4 genes have the potential to impact on both clesacostat clearance and ervogastat‐mediated CYP3A induction. In vitro, clesacostat was identified as a potential CYP3A time‐dependent inhibitor (composite slopes for midazolam and testosterone based on the observed inactivation rate constant [k obs] 0–300 μM were 0.0271 and 0.0239 mL min−1 μmol−1, respectively); the inhibition constant (K I) and maximal rate of enzyme inactivation (k inact) could not be calculated, as the inactivation reaction rate when the enzyme is fully saturated by substrate (k inact) was not reached within the concentration range tested. As per US Food and Drug Administration (FDA) guidance, 19 the CYP3A time‐dependent inhibition potential for clesacostat was assessed using the static model 20 to assess the potential need for a clinical DDI study. At the planned clinical dose of clesacostat of 15 mg b.i.d., the risk for CYP3A TDI is predicted to be low (area under the plasma concentration–time curve ratio [AUCR] of a CYP3A probe substrate in the presence and absence of clesacostat ≤1.25). In vitro, ervogastat was identified as an inducer of CYP3A based on mRNA expression and enzyme activity in human hepatocytes (mRNA EC50 values ranging from 21.4 to 37.2 μM and E max values ranging from 12.5 to 58.1‐fold; data on file). As per FDA guidance, 19 at the planned clinical dose of ervogastat 300 mg b.i.d., the static model 20 predicted potential induction of CYP3A4, based on an AUCR of <0.8 for sensitive CYP3A substrates. Therefore, this phase I study was designed as a fixed‐sequence, non‐randomized trial to investigate the drug interactions following co‐administration of clesacostat 15 mg b.i.d. and ervogastat 300 mg b.i.d.
One participant in Cohort 1 experienced elevated liver enzymes on Day 7, following treatment with clesacostat. These remained elevated for several days, at which point the participant discontinued treatment. Although the increases in liver enzymes were deemed related to study treatment by the investigator, this isolated finding in one participant has not been observed by others following clesacostat administration in a presumed/diagnosed NASH population. 5 In this population, clesacostat was associated with transient increases in liver enzymes; however, longer‐term treatment was associated with reductions from baseline in liver enzymes, 5 consistent with the anti‐steatotic effect of the mechanism.
To evaluate the net effect of CYP3A induction (due to ervogastat) and time‐dependent inactivation (due to clesacostat), the NCT03534648 study was conducted in two separate cohorts and the compounds were co‐administered over 7 days after administration of each of the monotherapies to steady state, as shown in Figure 1. The dose and dosing regimens of clesacostat and ervogastat were chosen to match those planned for the subsequent 6‐week phase II study (NCT03776175). 5 In the current study, administration of clesacostat, ervogastat, and co‐administration were found to be generally safe and well tolerated. When clesacostat 15 mg b.i.d. was co‐administered with ervogastat 300 mg b.i.d., a decrease in mean systemic exposures of 12% (C max) and 19% (AUCtau) was observed (Table 2), suggesting no clinically meaningful drug interaction. The decrease in AUCtau (19%) was consistent with the slightly higher CL/F and PTR of clesacostat co‐administered with ervogastat. While in vitro ervogastat is identified as a CYP3A inducer, the small magnitude of change in the systemic exposures of clesacostat when co‐administered with ervogastat may be because the predominant clearance mechanisms of clesacostat are via hepatic uptake by OATPs and CYP3A and potentially because clesacostat is not a CYP3A‐sensitive substrate (CYP3A fm was ~0.25; data on file). Furthermore, in healthy adults, a total daily dose of ervogastat ≤720 mg indicated no changes in 4β‐hydroxycholesterol, a biomarker of CYP3A activity, 21 , 22 , 23 suggesting that ervogastat may not be a clinically significant inducer at 300 mg b.i.d. Overall, while a decrease in the AUCtau of clesacostat in the presence of ervogastat was observed, dose adjustment of ervogastat or clesacostat for co‐administration was not deemed necessary due to the small magnitude of interaction. This approach is further supported by previous co‐administration study findings, wherein the combination of these agents was demonstrated. 5
In Cohort 2, the exposures of ervogastat remained unchanged in the presence of clesacostat. While based on preliminary reaction phenotyping in human liver hepatocytes, ervogastat is cleared predominantly by CYP3A (f m ~68%) and clesacostat is identified as a potential time‐dependent inactivator of CYP3A and no significant drug interaction was observed when ervogastat 300 mg b.i.d. was co‐administered with clesacostat 15 mg b.i.d.
Based on cumulative clinical DDI study outcomes, this co‐administration of clesacostat and ervogastat was safe and, overall, well tolerated in healthy participants. As the observed drug interaction was not clinically meaningful, dose adjustment of either of the two monotherapies was not deemed necessary. The overall observed safety and PK of this combination enabled its progression in a subsequent early sign‐of‐efficacy study. 5
AUTHOR CONTRIBUTIONS
A.S.‐B., A.J.B., R.A.C., S.T., J.R.G., W.P.E., and J.M. wrote the manuscript. A.S.‐B., A.J.B., R.A.C., W.P.E., and J.M. designed the research. A.S.‐B., A.J.B., R.A.C., and J.M. performed the research and analyzed the data. L.MdC. performed the research. S.T. reviewed bioanalytical data and managed the PK samples bioanalysis and reporting. W.P.E. was responsible for leadership of the overall program strategy and contributed to the introduction section of the manuscript. All authors critically reviewed the manuscript.
FUNDING INFORMATION
This study was sponsored by Pfizer Inc.
CONFLICT OF INTEREST STATEMENT
Authors are present or past employees and shareholders of Pfizer Inc. The authors declared no competing interests for this work.
Supporting information
FIGURE S1
TABLE S1
TABLE S2
ACKNOWLEDGMENTS
The authors would like to thank the healthy adults, the investigators, and the site staff who participated in this study. The authors would like to acknowledge Zhiwu Lin for performing the in vitro induction assay and Heather Eng for performing the CYP3A time‐dependent inactivation assay and providing the data included in this article. The authors would like to acknowledge Neeta B. Amin for the constructive input offered on the presented content. Editorial support was provided by Claire Cairney, PhD, CMC Connect, a division of IPG Health Medical Communications, and was funded by Pfizer Inc, New York, NY, USA, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022;175(9):1298‐1304. doi:10.7326/M22‐1460).
Sawant‐Basak A, Bergman AJ, Mancuso J, et al. Investigation of pharmacokinetic drug interaction between clesacostat and DGAT2 inhibitor ervogastat in healthy adult participants. Clin Transl Sci. 2024;17:e13687. doi: 10.1111/cts.13687
Clinical trial number NCT03534648.
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
REFERENCES
- 1. Tokushige K, Ikejima K, Ono M, et al. Evidence‐based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis 2020. Hepatol Res. 2021;51:1013‐1025. [DOI] [PubMed] [Google Scholar]
- 2. Nakamuta M, Kohjima M, Morizono S, et al. Evaluation of fatty acid metabolism‐related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2005;16:631‐635. [PubMed] [Google Scholar]
- 3. Gluchowski NL, Gabriel KR, Chitraju C, et al. Hepatocyte deletion of triglyceride‐synthesis enzyme acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology. 2019;70:1972‐1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amin NB, Carvajal‐Gonzalez S, Purkal J, et al. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci Transl Med. 2019;11:eaav9701. [DOI] [PubMed] [Google Scholar]
- 5. Calle RA, Amin NB, Carvajal‐Gonzalez S, et al. ACC inhibitor alone or co‐administered with a DGAT2 inhibitor in patients with non‐alcoholic fatty liver disease: two parallel, placebo‐controlled, randomized phase 2a trials. Nat Med. 2021;27:1836‐1848. [DOI] [PubMed] [Google Scholar]
- 6. Ross TT, Crowley C, Kelly KL, et al. Acetyl‐CoA carboxylase inhibition improves multiple dimensions of NASH pathogenesis in model systems. Cell Mol Gastroenterol Hepatol. 2020;10:829‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bergman A, Carvajal‐Gonzalez S, Tarabar S, Saxena AR, Esler WP, Amin NB. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a liver‐targeting acetyl‐CoA carboxylase inhibitor (PF‐05221304): a three‐part randomized phase 1 study. Clin Pharmacol Drug Dev. 2020;9:514‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huard K, Smith AC, Cappon G, et al. Optimizing the benefit/risk of acetyl‐CoA carboxylase inhibitors through liver targeting. J Med Chem. 2020;63:10879‐10896. [DOI] [PubMed] [Google Scholar]
- 9. Kim CW, Addy C, Kusunoki J, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26:394‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amin NB, Saxena AR, Somayaji V, Dullea R. Inhibition of diacylglycerol acyltransferase 2 versus diacylglycerol acyltransferase 1: potential therapeutic implications of pharmacology. Clin Ther. 2023;45:55‐70. [DOI] [PubMed] [Google Scholar]
- 11. Saxena AR, Chidsey K, Somayaji V, Ogden A, Duvvuri S. Diacylglycerol acyltransferase 2 (DGAT2) inhibitor PF‐06865571 reduces liver fat by MRI‐PDFF after 2 weeks in adults with NAFLD [Abstract]. Hepatology. 2019;70(Suppl. 1):1260A. [Google Scholar]
- 12. Dagogo‐Jack S, Liu J, Eldor R, et al. Efficacy and safety of the addition of ertugliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sitagliptin: the VERTIS SITA2 placebo‐controlled randomized study. Diabetes Obes Metab. 2018;20:530‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Webster RM. Combination therapies in oncology. Nat Rev Drug Discov. 2016;15:81‐82. [DOI] [PubMed] [Google Scholar]
- 14. Hofmann WP, Soriano V, Zeuzem S. Antiviral combination therapy for treatment of chronic hepatitis B, hepatitis C, and human immunodeficiency virus infection. Handb Exp Pharmacol. 2009;189:321‐346. [DOI] [PubMed] [Google Scholar]
- 15. Wang B, Choudhry NK, Gagne JJ, Landon J, Kesselheim AS. Availability and utilization of cardiovascular fixed‐dose combination drugs in the United States. Am Heart J. 2015;169:379‐386.e371. [DOI] [PubMed] [Google Scholar]
- 16. Menendez‐Arias L, Delgado R. Update and latest advances in antiretroviral therapy. Trends Pharmacol Sci. 2022;43:16‐29. [DOI] [PubMed] [Google Scholar]
- 17. Orloff DG. Fixed combination drugs for cardiovascular disease risk reduction: regulatory approach. Am J Cardiol. 2005;96:28K‐33K; discussion 34K–35K. [DOI] [PubMed] [Google Scholar]
- 18. Guttman Y, Nudel A, Kerem Z. Polymorphism in cytochrome P450 3A4 is ethnicity related. Front Genet. 2019;10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. U.S. Food and Drug Administration . In vitro drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions guidance for industry. 2020; https://www.fda.gov/media/134582/download. Accessed October 6, 2022
- 20. Fahmi OA, Maurer TS, Kish M, Cardenas E, Boldt S, Nettleton D. A combined model for predicting CYP3A4 clinical net drug–drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab Dispos. 2008;36:1698‐1708. [DOI] [PubMed] [Google Scholar]
- 21. Bodin K, Bretillon L, Aden Y, et al. Antiepileptic drugs increase plasma levels of 4beta‐hydroxycholesterol in humans: evidence for involvement of cytochrome P450 3A4. J Biol Chem. 2001;276:38685‐38689. [DOI] [PubMed] [Google Scholar]
- 22. Bodin K, Andersson U, Rystedt E, et al. Metabolism of 4β‐hydroxycholesterol in humans. J Biol Chem. 2002;277:31534‐31540. [DOI] [PubMed] [Google Scholar]
- 23. Josephson F, Bertilsson L, Bottiger Y, et al. CYP3A induction and inhibition by different antiretroviral regimens reflected by changes in plasma 4beta‐hydroxycholesterol levels. Eur J Clin Pharmacol. 2008;64:775‐781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1
TABLE S1
TABLE S2
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.