

Abstract
Metastatic and localized mismatch repair deficient (dMMR) tumors are exquisitely sensitive to immune checkpoint blockade (ICB). The ability of ICB to prevent dMMR malignant or pre-malignant neoplasia development in patients with Lynch syndrome(LS) is unknown. Of 172 cancer-affected patients with LS who had received ≥1 ICB cycles, 21 (12%) developed subsequent malignancies following ICB exposure, 91% (29/32) of which were dMMR, with median time to development of 21 months (IQR range 6–38). 39% (24/61) of the ICB-treated patients who subsequently underwent surveillance colonoscopy had premalignant polyps. Within matched pre- and post-ICB follow up periods, the overall rate of tumor development was unchanged; however, on subgroup analysis, a decreased incidence of post-ICB visceral tumors was observed. These data suggest ICB treatment of LS-associated tumors does not eliminate risk of new neoplasia development and LS-specific surveillance strategies should continue. These data have implications for immune-preventative strategies and provide insight into the immunobiology of dMMR tumors.
Lynch syndrome (LS), a pan-cancer predisposition syndrome,1 is defined by inactivating pathogenic germline alterations in the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2). Importantly, patients with LS are at elevated risk for both synchronous and metachronous cancers. Inactivation of the second wild-type allele causes impaired genome maintenance and renders cells unable to repair certain classes of mutations occurring during DNA replication. Over time this results in high tumor mutation burden (TMB) and enrichment of mutations in microsatellites, termed microsatellite instability (MSI).2 This high TMB, enriched for mutations encoding immunogenic proteins, sensitizes mismatch repair deficient (dMMR) tumors to immune checkpoint blockade (ICB)3. Efficacy of ICB in dMMR tumors, independent of tumor type, led to the first tumor agnostic approval of ICB in patients with metastatic disease.3
More recently, early-stage LS-associated dMMR cancers were found even more responsive to ICB than metastatic disease.4,5 This success has fuelled interest in ICB utility for cancer prevention in the LS population. The preventative role of ICB was recently suggested in a report of >46,000 solid tumor patients uninformed by germline analysis, where ICB-containing treatment of the first cancer was associated with decreased incidence of second primary cancers6.
Herein, we quantify the enduring risk of pre-malignant and malignant neoplasia in patients with LS post-ICB. Among 837 cancer-affected patients with LS at our institution, we identified 172 who received ICB between 03/2014–03/2023 (Extended Data Fig. 1) with median follow-up time of 3.12 years (37 months, IQR 11.25–43). Over 60% harbored MSH2 or MLH1 pathogenic germline variants; colorectal cancer was the first primary cancer diagnosis in 42% and endometrial cancer in 16%. Median age at first primary malignancy was 47 years (range, 10–81, IQR 39–56); 51% had multiple malignancies. (Extended Data Table 1).
In 63% of patients, ICB was given for the patient’s first cancer. ICB with an anti-PD-1 antibody was used in 80% and an anti-PD-L1 antibody in 10% (Extended Data Table 1). ICB was predominantly administered in the metastatic disease setting (66%). 82% of patients received ICB for a dMMR tumor. Median duration of treatment was 9 months (IQR 3.75–22).
A subsequent primary malignancy (SPM) developed in 12% (21/172) of LS cancer patients on or following ICB exposure (Supplementary Data Table 1/Extended Data Tabe 2, Fig. 1a/b). Median TMB in the initial tumors targeted with ICB was 42.3 mt/MB (range, 1– 93.9). 62% (13/21) of patients had regression in the initial tumor targeted by ICB; 33% (7/21) had tumor stability. Median time to development of SPM post-ICB exposure was 21 months (IQR range, 6–38). Eight patients developed multiple malignancies, for a total of 40 pathologically confirmed post-ICB tumors. All post-ICB malignancies were histologically distinct from the tumors for which patients were receiving ICB. Using a next-generation sequencing panel 7, paired pre- and post-ICB tumors were found to share 1 or fewer somatic mutations (Fig. 1a, Extended Data Fig. 2, patient case demonstration). Of the new cancers, immunohistochemical staining (IHC) for MMR was available on 32; 91% (29/32) were dMMR. Eight patients were diagnosed with SPM ≤8 months after first ICB exposure, these tumors were not present clinically nor radiologically evident prior to ICB initiation.
Figure 1. Patient case example, clinical timeline, and tumor characteristics of Lynch syndrome patients with subsequent primary malignancies (SPM) after immune checkpoint blockade (ICB).
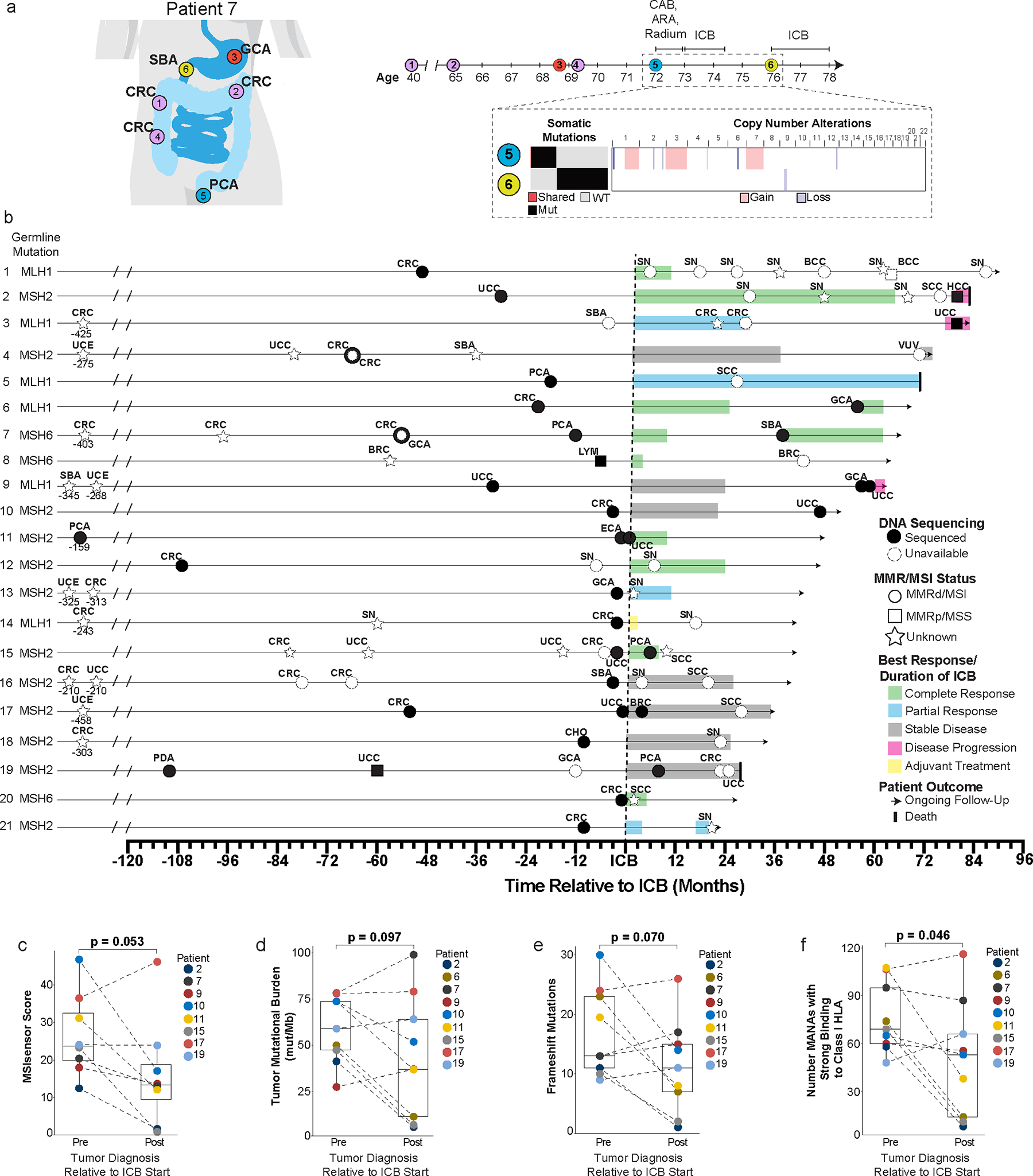
a, Case example of patient with multiple primary malignancies pre-ICB exposure. (Patient 7 from Figure 1b; Extended Data Table 1) Timeline of cancer diagnoses and sequential local and systemic interventions including combined androgen blockade (CAB), androgen receptor antagonist (ARA) and ICB. Targeted next-generation sequencing (MSK-IMPACT) comparing pre-ICB prostate cancer (PCA) and post-ICB small bowel cancer (SBA) by tumor mutational burden, somatic mutational profile, and copy-number alterations. Single shared somatic mutation, PTPRT at chromosome 20.
b, Timeline for SPM development post-ICB exposure (n=21 patients) inclusive of: SBA: Small bowel, CRC: Colorectal, GCA: Gastric, PCA: Prostate, SN: Sebaceous neoplasm, HCC: Hepatocellular, UCC: Urothelial, SCC: Squamous cell, CHO: Cholangiocarcinoma, ECA: Esophageal, BRC: Breast, LYM: Lymphoma, UCE: Uterine, PDA: Pancreatic, VUV: Vulvar.
c-f, MSISensor score (c), tumor mutational burden (d), frameshift mutations (e), and comparative analysis of strong binders to HLA Class 1 MHC (f) between pre- and post-ICB tumors. P-values are derived from pairwise t-testing for the paired pre and post tumor from each patient without replicates for each separate analysis.
Analyses included nine patients with 9 pre and 9 post tumors except analysis c (MSISensor score) which included 8 patients with 8 pre and 8 post tumors due to an MSI score not available for patient 6.
*MSI status on 1 sample with inadequate tumor purity was confirmed via MiMSI
We next evaluated colonic pre-neoplasia in patients with LS post-ICB exposure. Post-ICB completion, 61 patients underwent surveillance colonoscopy with 39% (24/61) diagnosed with ≥1 pre-malignant polyp(s) (tubular, tubulovillous or serrated adenomas); 54% (33/61) had a history of prior CRC. (Extended Data Table 3). Median time to polyp development was 14 months (IQR, 10.5–22) from last colonoscopy and 11.5 months (IQR, 6.5–22.5) from last ICB dose. In 14 patients with colonic polyps, the tumor treated by ICB was CRC; 5 of these patients also developed post-ICB tumors (Extended Data Table 3, patients 1, 7, 11, 12 and 14). MMR IHC staining in 19/24 patients’ polyps, identified 6 (32%) patients with dMMR adenomas, including three ≤ 3mm in size, with protein loss pattern consistent with the underlying LS-variant.
We previously reported adenoma rate in a LS prospective endoscopic surveillance cohort, without ICB exposure8. Baseline colonoscopy identified adenoma(s) in 33% (33/100) of patients and 29% (28/89) had interval adenomas at 1–2 years of follow-up with half of this cohort having had a prior CRC. In a recently published LS cohort (n=163)9, inclusive of 60% with CRC history, baseline adenoma detection rate was 21%; the mean number of adenomas per colonoscopies performed in this cohort was 0.54 with a mean duration of follow up of 8.7 years. The mean number of post-ICB adenomas per colonoscopies performed in our cohort (n=61) was 0.57 with a mean duration of follow up of 2.14 years (range, 1–79 months) post-ICB completion.
Utilizing targeted NGS analysis on paired pre-ICB and post-ICB tumors from 9 patients (Fig. 2 c–f), we noted post-ICB tumors exhibited fewer frameshift mutations although this did not reach statistical significance (p=0.07). TMB in 7/8 post-ICB dMMR tumors was >10 mt/MB (mean TMB=41.5, range 7–98.9). MSISensor score, a measure of MSI level10, was significantly lower in post-ICB tumors (p=0.05). Post-ICB tumor MSISensor score was also lower than the reference average for MSI tumors defined for each cancer type from the broader MSK clinical sequencing cohort (Supplementary Data Table 1).
Figure 2. Incidence of subsequent primary malignancies (SPM) and site of origin in the study cohort and comparator Lynch syndrome (LS) cohorts.
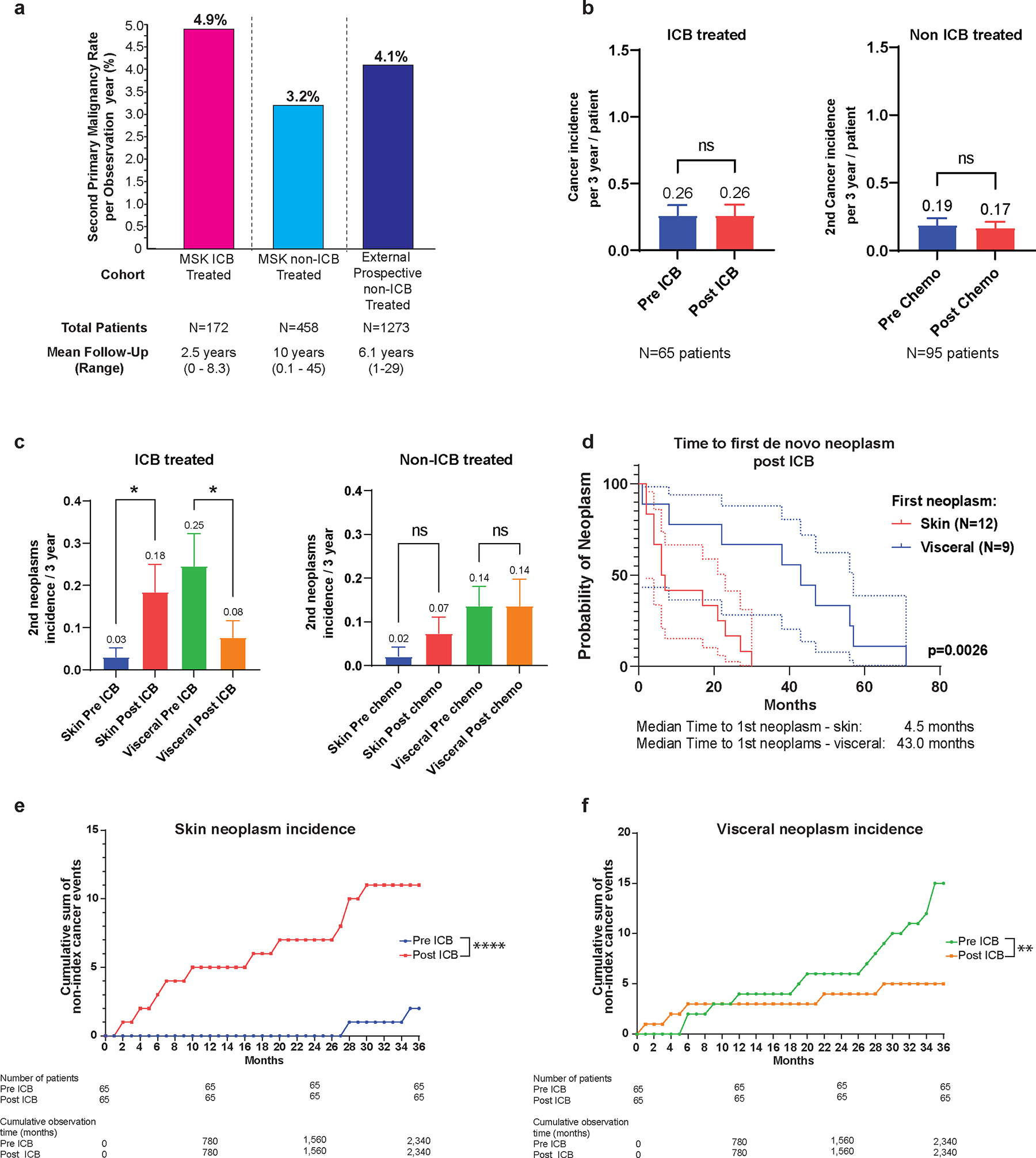
a, Rate of SPM per observation year in the ICB treated cohort (pink) and comparator cohorts inclusive of internal non-immune checkpoint blockade (non-ICB) exposed LS patients (light blue) and external multi-center LS patients undergoing prospective surveillance after the first cancer diagnosis (dark blue)11.
b, Overall SPM incidence at 3-years matched follow-up in the ICB treated (pre- and post-ICB) and non-ICB treated cohort (pre- and post-chemotherapy). The number of matched patient pre/post ICB or pre/post chemotherapy is reported below the figure. The 3 year incidence of 2nd neoplasm for each clinical condition is reported above the box plot representing the mean and standard deviation (SEM). Pre and post 3 year incidence were compared using paired t test. ns: non significant, P>0.05.
c, Incidence of cutaneous and visceral neoplasms at 3-years matched follow-up in patients pre- and post-ICB, and pre- and post-chemotherapy. The numbers of matched patient pre/post ICB or pre/post chemotherapy are respectively 65 and 95 patients. The 3 year incidence of 2nd neoplasm category (skin or visceral) for each clinical condition is reported above the box plot representing the mean and standard deviation (SEM). Pre and post 3 year incidence were compared using repeated measure one way ANOVA test corrected for multiple comparisons comparing 3 year incidences of de novo cutaneous or visceral neoplasms. *: P≤0.05 and P>0.01, ns: non significant, P>0.05.
d, Time to first cutaneous or visceral neoplasm post-ICB exposure. The number of patients is reported on the figure, each patient being accounted only once based on first 2nd neoplasm type (skin or visceral). The median time to first 2nd neoplasm post ICB is reported. Survival curves were compared using log rank test and the p-value is reported on the figure. The dashed lines represent the upper and lower limit of the confidence interval at 95% for each conditions with matched colors.
e, f, Cumulative incidence of cutaneous (e) versus visceral (f) post-ICB SPM. Patients with a matched follow up pre- and post-ICB of 36 months were selected and all non-index cancers occurring during the window of observation were accounted for. Cumulative incidence is reported as the cumulative sum of non-index cancers occurring during the period of observation. Number of patients and cumulative observation time in months is reported. Fisher’s exact test comparing the cumulative observation time without a neoplasm event to the number of months with an event was used for statistical analysis. ****: P<0.0001 ; **: P≤0.01 and >0.001.
We further assessed predicted 9’mer mutation associated neoantigens (MANA) in paired tumors pre-ICB and post-ICB(Fig. 1e/f, Extended Data Fig. 3). Post-ICB tumors exhibited significantly fewer (53, IQR 14–66) unique MANA predicted to strongly bind (threshold <=1%) to patient-specific Class I MHC compared to pre-ICB tumors (69, IQR 60–95). These trends were preserved in sensitivity analyses when assessing only dMMR gastrointestinal or only urothelial cancers (Extended Data Fig. 4). MANA peptides were compared across the 9 patients with sequenced pre-ICB and post-ICB tumors (Extended Data Table 4). Nearly all (98.8%) assessed MANA peptide sequences identified were unique to the individual tumor sample without overlap with other tumor(s) from the same patient (Extended Data Table 4). We assessed for HLA loss of heterozygosity (LOH) as a potential mechanism of immune escape. No HLA LOH in any of the sequenced tumors that developed either on ICB or post-ICB, was observed.
We also evaluated expression of CD8, PD1 and PD-L1 IHC in pre-ICB and post-ICB exposed tumors (15 tumors) with a statistically significant reduction in CD8+TILs in tumors developing on ICB that was not seen in tumors that developed post-ICB completion. (Extended Data Fig. 5a/b/c).
To contextualise our finding of enduring neoplasia risk amongst patients with LS who had received ICB, we evaluated the rate of SPM both in our internal cohort of patients with LS without ICB exposure and in a prospective LS multicentre study11 (Fig. 2a). The rate of SPM per mean observation years was comparable at 3.2%, 4.1%, and 4.9% in the non-ICB exposed internal cohort, the external cohort, and our ICB exposed cohort, respectively.
Given the limitations of historical cohort comparisons, we assessed intra-patient tumor development rates in patients with matched duration of follow-up pre-ICB and post-ICB. Rate of tumor development in the pre-ICB and post-ICB period was equivalent with 3-years of follow-up (Fig. 2b). We replicated this analysis in non-ICB exposed patients with LS (n=95) from our institution and demonstrated unchanged rates of tumor development pre- and post-chemotherapy exposure at 3-years of matched follow-up (Fig. 2b). When extended to 5-years of follow-up, the results were consistent in both ICB and chemotherapy groups (Extended Data Fig. 6a). Separating SPM into cutaneous versus visceral malignancies, we identified a reduction in post-ICB visceral tumor rate whilst conversely, the rate of cutaneous neoplasm increased significantly (Fig. 2 c–f). In our historical comparator cohort, the overall rate of cutaneous malignancies was 12%; in the post-ICB cohort with SPM, 48%, (10/21), of patients developed ≥1 cutaneous malignancy. On exploratory analysis, when restricted to high-risk patients with a history of 2 or more malignancies pre-ICB exposure, the rate of SPM was significantly lower at 3-years and 5-years of follow-up (Extended Data Fig. 6b–d).
Immunoprevention with ICB in patients with LS is an attractive hypothesis given the success of ICB in locally advanced and metastatic dMMR tumors3–5. Our findings that 12% of ICB-treated patients with LS developed a new cancer < 2 years after first ICB exposure and 39% harbored a pre-malignant colonic polyp provides initial evidence that ICB does not durably eliminate the risk of pre-malignant neoplasia or subsequent malignant neoplasia. This is even more surprising since the majority (91%) of new cancers and a subset of pre-malignant neoplasia identified post-ICB treatment were dMMR.
Subsequent primary tumors following exposure to ICB, in addition to being predominately dMMR, still harbored high TMB, but were found to be genetically ‘younger’ than the dMMR tumors targeted by ICB as evidenced by the lower MSI level and a trend towards lower number of frameshift mutations, which are known to increase in dMMR tumors over time and contribute proportionally to tumor immunogenicity12. Notably, despite the lower genomic measures of immunogenicity, including lower unique MANA, some SPM still responded to ICB, including three complete radiographic responses.
Our data suggest that there is a window of time between malignant precursor lesion formation and development of an invasive carcinoma where tumor precursors may not be susceptible to ICB. There are several potential interpretations, but dMMR alone may not be sufficient and stage of tumor development appears to be a factor. This has been modeled in prior studies12, 13 and may explain recent data wherein genetic ablation of MMR alone was insufficient in inducing tumor immunogenicity14. Mandal and Germano12,13 both show that conversion to a immunogenic phenotype incell lines from the point of MMR ablation by CRISPR required months of passaging in tissue culture and that immunogenicity was proportional to time and to the accumulation of single nucleotide variants, frameshift mutations and elevated levels of MSI3,12. It may be that during the earliest period of tumor development, dMMR tumors are not immunogenic and therefore resistant to the effect of ICB.
In line with these studies, the assumption of later acquisition of dMMR status in LS-associated polyps has been questioned by evidence of colonoscopically ‘invisible’ precursor lesions or alternative routes to LS-associated CRC development15. dMMR crypt foci have been identified in the normal mucosa of LS patients16, and an alternative 2-in-1 model of shortcut carcinogenesis has been proposed in MLH1-associated LS-CRC17. Molecular analyses of LS-associated CRC suggests that dMMR can be an early event preceding adenoma formation15 and furthermore an immune profile independent of MMR or neoantigen formation has been demonstrated in LS-associated polyps18. Indeed in our study 6 post-ICB colonic polyps were dMMR, 3 of which were >8 mm concordant with prior studies19.
Of particular note are two significant observations with potential clinical implications for surveillance and prevention. The first is higher rates of cutaneous SPM and lower rates of visceral SPM post-ICB, suggesting that visceral SPMs may be decreased by ICB exposure or that there is a shift in the type of SPM that occurs. This may impact survival since visceral tumors can be lethal if not successfully treated. Also, dermatological screening, even in patients with LS without prior diagnosis of the Muir-Torre variant, should be considered given high number of cutaneous SPM. Second, high-risk individuals with multiple malignancies prior to ICB exposure demonstrated significantly reduced rates of SPM. Whether this was attributable to improved surveillance or the impact of ICB requires future validation studies.
Additionally, we demonstrate a lack of shared MANA and lower number of MANA with strong binding to Class 1 HLA between pre- and post-ICB tumors. Exploiting shared frameshift MANA among dMMR tumors underpins frameshift pepride-based vaccination approaches in LS20; it remains to be elucidated whether such strategies will be as effective in patients previously subject to ICB exposure.
Our study has limitations. This is a single center retrospective study with mixed distribution of stage and tumor types requiring longer follow-up to validate preliminary findings. However, it highlights the unique immunobiology of dMMR tumors and demonstrates persistent risks of both new SPM and pre-neoplastic lesions after ICB exposure in patients with LS. This has immediate clinical implications in that high-risk surveillance of patients with LS should be continued, if clinically appropriate, both throughout and after ICB treatment.
Online Methods
Study Population
The Memorial Sloan Kettering LS database was queried for all cancer affected patients with pathogenic or likely pathogenic germline variants in MLH1, MSH2 (including EPCAM terminal deletions with epigenetic silencing of MSH2), MSH6, or PMS2 diagnostic of LS (n=837). LS patients who received ≥1 cycle of ICB between 03/2014, and 03/2023 (n=172) were then identified with annotation from the electronic medical record for baseline demographics, tumor stage and pathology, cancer treatment and response, and endoscopic surveillance. Excluding patients with congenital mismatch repair deficiency, all patients with Lynch syndrome (LS) were eligible for inclusion in this study regardless of sex as LS predisposes to the development of multiple dMMR (deficient microsatellite repair) malignancies in both sexes; the pivotal studies of ICB have included both sexes and furthermore there is no data available in the published literature to suggest that the immuno-preventative effect of ICB exposure should differ between the sexes. ICB included the CTLA4 inhibitor ipilimumab, PD-1 inhibitors: pembrolizumab, nivolumab, dostarlimab and PD-L1 inhibitors: avelumab, atezolizumab, durvalumab. All patients with further tumor profiling provided informed written consent for an institutional review board–approved prospective protocol (NCT01775072) for tumor and matched normal DNA sequencing via MSK-IMPACT (MSK–Integrated Mutation Profiling of Actionable Cancer Targets), a clinical NGS platform FDA authorized to identify genetic variants in up to 505 cancer-related genes as well as MSI status21, 22.
We additionally assessed the non-ICB treated cohort (n=665). Patients whose last date of follow-up was 2019 or earlier or without documentation of death were excluded from the analysis (n=207) as we could not reliably exclude the possibility of additional tumors. The remaining comparator cohort of non-ICB exposed patients (n=458) was annotated for baseline demographics, all tumors inclusive of cutaneous malignancies and cancer treatment. For further comparisons with ICB exposure, we considered a subgroup of patients who received chemotherapy (N=136).
Post-ICB pre-neoplasia rate:
We previously reported the adenoma rate in a LS prospective endoscopic surveillance cohort, without ICB exposure8. In a recently published LS cohort (n=163)9, inclusive of 60% with CRC history, Del Carmen et al calculated the mean number of adenomas per procedure (MAP) over the number of all colonoscopies performed reporting a MAP of 0.54 with a mean duration of follow up of 8.7 years. We replicated this analysis in our internal cohort of patients who underwent colonoscopies post-ICB exposure (n=61) and we report the mean number of adenomas per procedure over all post-ICB colonoscopies performed.
Tumor Assessment for dMMR and MSI
Tumors were designated as dMMR or MSI utilizing one or more methods of analysis. Immunohistochemical (IHC) staining for the MMR proteins was performed as previously described23 with a tumor being designated as dMMR if IHC staining demonstrated loss of protein expression in one or more of the 4 MMR proteins corresponding to the underlying germline alteration known to be present in the patient. When sufficient tissue was available, IHC was also performed on colonic adenomas from colonoscopy surveillance post-ICB exposure. For MMR ICH we used the following antibodies: MLH1 (clone G168–728, diluted 1:250; BD PharMingen or clone M1, ready-to-use, Ventana), MSH2 (clone G219.1129, ready-to-use, Cell Marque), MSH6 (clone 44, ready-to-use, Ventana), and PMS2 (clone A16.4, diluted 1:100, BD Biosciences).
For MSI, the MSIsensor algorithm was applied as previously described10. MSIsensor scores ≥ 10 defined MSI status, scores ≥ 3 to < 10 an indeterminate (MSI-I) status, and scores < 3 microsatellite stable (MSS) status. If discordance existed between IHC and MSIsensor, miMSI, an alternative classifier-based bioinformatics tool which has a better sensitivity on low tumor samples, was used to further assess MSI24. Tumor mutation burden (TMB) was calculated by dividing the total number of reported non-synonymous mutations by the genomic size where mutations were reported (Mut/MB) for each sample.
Neoantigen Prediction and HLA binding affinity analysis
Computational predictions of MHC class I-binding affinity as a surrogate for immunogenicity are routinely used in epitope identification and vaccine discovery studies.
Patients in the cohort with at least one sample before and after ICB treatment (N=9) were evaluated for similarity in tumor molecular features. Nonsynonymous somatic mutations from the MSK IMPACT variant call format (VCF) output from each sequenced patient tumor were input into the SnpEff program v 5.1d (https://pcingola.github.io/SnpEff/) to predict translated amino acid sequences for each respective mutation. For missense mutations, 9’mer peptides were predicted with a sliding reading frame that was centered around the mutation site (to account for the different possibilities of the mutated amino acid in the 1–9 position). For frameshift mutations, 9’mers were defined starting 8 amino acids upstream of the insertion or deletion event and possible 9’mers were consecutively defined continuing along the mutated peptide sequence. The number of unique MANAs was determined by taking the defined peptide sequence and evaluating all potential contiguous 9’mer variations using a sliding reading frame. Patient-specific Human Leukocyte Antigen (HLA) Class I (HLA-A, HLA-B, HLA-C) subtypes was determined from MSK-IMPACT. The patient-specific HLA class-I binding affinity of all possible unique 9’mer sequences was determined using NetMHCpan version 4.0 (DTU Health); a widely-used artificial neural network-based method25,26 trained on quantitative binding data and mass-spectrometry derived MHC ligands to predict MHC peptide binding affinity. Strong HLA-binding NAs for each patient were identified if the NA sequence was predicted to have a binding threshold of less than or equal to 1%. Patient pre and post samples were paired and assessed with paired t-testing. If a patient had more than one tumor either before or after ICB, the median value of the respective dependent variable was determined for that patient to produce a single data point pre and post ICB for each patient. Patient 19’s pre-ICB urothelial carcinoma was not included due to inadequate sample coverage of MSK IMPACT (57X). Sensitivity analyses were performed in mismatch-repair deficient gastrointestinal cancers only (Extended Data Fig 4a) and mismatch-repair deficient urothelial cancers only (Extended Data Fig 4b). For these analyses, Student’s t-tests were used without pairing.
Pre- and post-ICB SPM incidence calculation
To assess the 3-year or 5-year incidence of de novo neoplasms pre- and post-ICB, we selected patients with at least 36 months or 60 months of post-ICB follow up, respectively. After exclusion of the index cancer for which the patient received ICB and concomitant cancers (< 1 months between diagnoses), we assessed the total number of non-index cancers occurring during the defined observation time of 3-year or 5-year incidence pre- and post-ICB. Comparison of the incidence of pre-ICB and post-ICB on matched follow up was performed using a paired t test. (Fig 2b, Extended data Fig 6). The same methodology was used for patients not exposed to ICB, using initiation of chemotherapy as a reference for matched pre- and post-chemotherapy 3-year and 5-year incidence calculation.
Post-ICB SPM were further subdivided by visceral versus cutaneous site of origin; we compared incidence of cutaneous tumors, and incidence of visceral tumors, pre- and post-ICB exposure versus pre- and post- chemotherapy exposure (Fig. 2c). We subsequently compared time to first neoplasm post ICB exposure (Fig. 2d) and incidence of cutaneous versus visceral tumors pre- and post-ICB (Fig. 2 e/f). The sebaceous neoplasms in our study have been labelled as such as a consequence of the inherent pathological challenges of categorizing sebaceous adenomas and carcinomas due to the considerable overlap of histologic criteria for sebaceous adenoma, sebaceoma and sebaceous carcinoma. This is particularly true when tumor content is small as in a biopsy or when a carcinoma is low grade or well differentiated.
High risk patients were defined as all cumulative patients presenting with multiple non concomitant cancers (>=2) in a pre-ICB delay matched with post-ICB follow up. To allow enough post-ICB follow-up, patients with a post-ICB follow up of less than 18 months, corresponding to the median ICB duration +6 months, were excluded from the analysis. In case of other cancers occurring outside of the matched pre-ICB post -CB follow-up window, these extra cancers were not considered for the definition of high risk. As for the whole population, the 3-year and 5-year incidence calculation was performed excluding the index cancer and concomitant cancers (<1 month between diagnoses of cancers). Comparison of the incidence of pre-ICB and post-ICB on matched follow up in high risk patients was performed using a paired t test.(Extended Data Fig. 6) The same methodology was used for patients not exposed to ICB, using initiation of chemotherapy as a reference for matched pre- and post-chemotherapy 3-year and 5-year incidence calculation.
For cumulative incidence calculation in high risk patients of de novo cancers, (Extended Data Fig. 6), we selected patients with a matched follow up pre- and post-ICB of 36 months and accounted for all non-index cancers occurring during the window of observation. Cumulative incidence is reported as the cumulative sum of non-index cancers occurring during the period of observation. Number of patients and cumulative observation time in months is reported. Fisher’s exact test comparing the cumulative observation time without a metastatic event to the number of months with an event was used for statistical analysis.
CD8, PD-1, PDL1 Immunohistochemistry and Analysis
IHC analysis of 15 tumors (18 samples overall, 5 matched pre and post ICB) for CD8, PD1 and PDL1 expression was possible. Antibodies used include: CD8 (clone C8/144B, diluted 1:1000, Dako), PD-1 (clone NAT105, ready to use, Cell Marque) and PDL1 (clone E1L3N, diluted 1:100, Cell signalling). The regions with best representation of tumor, away from edges or other artifacts were chosen for analysis. The relevant cells were counted in three 40x High Power Fields (HPF); the final result is expressed as count/HPF. For CD8 and PD1, the counts include TIL (tumor-infiltrating cells), Stromal (within tumor area), and the Sum. For PDL1, the counts include the number of tumor cells staining, and immune cell staining. Additionally, for PDL1, an overall CPS score is provided.
Analyses of immune populations was performed by comparing quantification per high power field of CD8+, PD-1+ or PD-L1+ cells in 15 different MMRd tumors from 10 patients. Tumors were classified according to the timing of diagnosis compared to ICB exposure: pre-ICB, on ICB or post-ICB discontinuation and means compared using ordinary one way ANOVA test corrected for multiple comparisons. A p-value <0.05 was considered as significant.
HLA Loss of Heterozygosity Analysis
HLA genotyping was performed by Polysolver27. The copy number profile for each tumor was found using FACETS (Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing) v0.5.1428 compared to its matched normal in a two-pass manner: an initial run for purity and ploidy estimation followed by a second run for focal event detection. Each fit was manually reviewed to evaluate the quality of the fit. Loss of heterozygosity over HLA-A, HLA-B, and HLA-C was identified using LOHHLA (Loss Of Heterozygosity in Human Leukocyte Antigen)29 with the purity and ploidy estimates from FACETS. A loss of heterozygosity event was defined as copy number of an alternate allele <0.5 and the p-value related to this allelic imbalance <0.001.
Statistical Analyses
According to parameters, categorical variables are expressed in the form of frequencies or/and percentages and are compared where appropriate using two-sided Fisher’s exact testing. Continuous variables are calculated as mean and/or median with variation expressed as the mean and standard deviation (SEM) and interquartile range, respectively. For continuous variables minimum and maximum values are reported when appropriate. Continuous variables are compared with two sided student t testing or two-sided paired t test when matched samples from the same patients where available. For multiple comparisons, we used two sided one-way ordinary ANOVA tests, corrected for multiplicity using the Dunnet test. In all cases, a p-value less than or equal to 0.05 was considered significant. Survival curve estimates calculated with the Kaplan Meier method and compared with log-rank test. Statistics for incidence and cumulative incidence calculation are reported in designated method section. Data originating from the whole cohort was used repeatedly in subgroup analyses, including for the high-risk patients analyses as defined in the related method section.
Data has been analyzed with GraphPad Prism V9 software (Dotmatics, MA, USA) and R (v4.0.0; https://www.R-project.org/) software package. The level of significance displayed in all the figures across the article is the following: ns: non significant, P>0.05; *: P≤0.05 and P>0.01; **: P≤0.01 and >0.001; ***: P≤0.001 and P>0.0001; ****: P≤0.0001.
Extended Data
Extended Data Fig. 1. Study Cohort of patients with Lynch Syndrome from MSKCC.
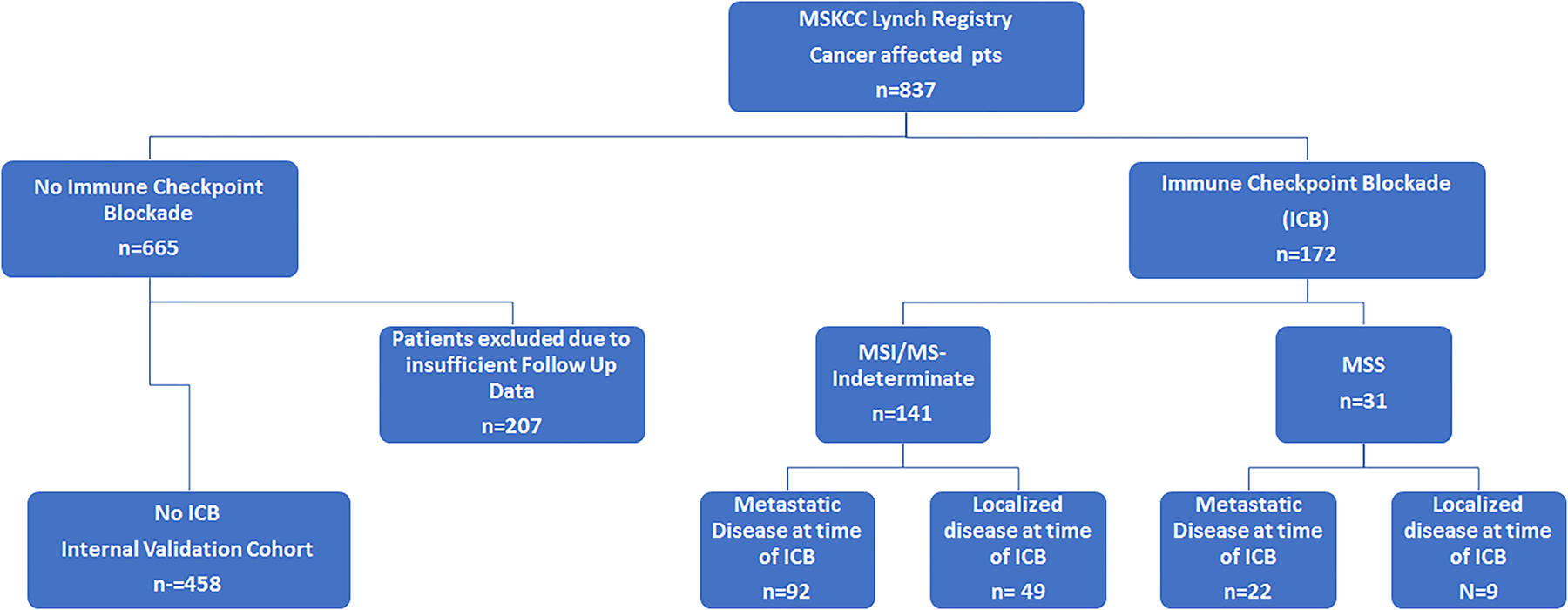
Consort diagram of study cohort. Segregated by microsatellite status as designated by MSIsensor14. MSKCC: Memorial Sloan Kettering Cancer Center; MSI: Microsatellite unstable; MSS: Microsatellite stable.
Extended Data Fig. 2. Histological evaluation of patient 7′s sequential tumors demonstrating distinct histological and immunohistochemical features.
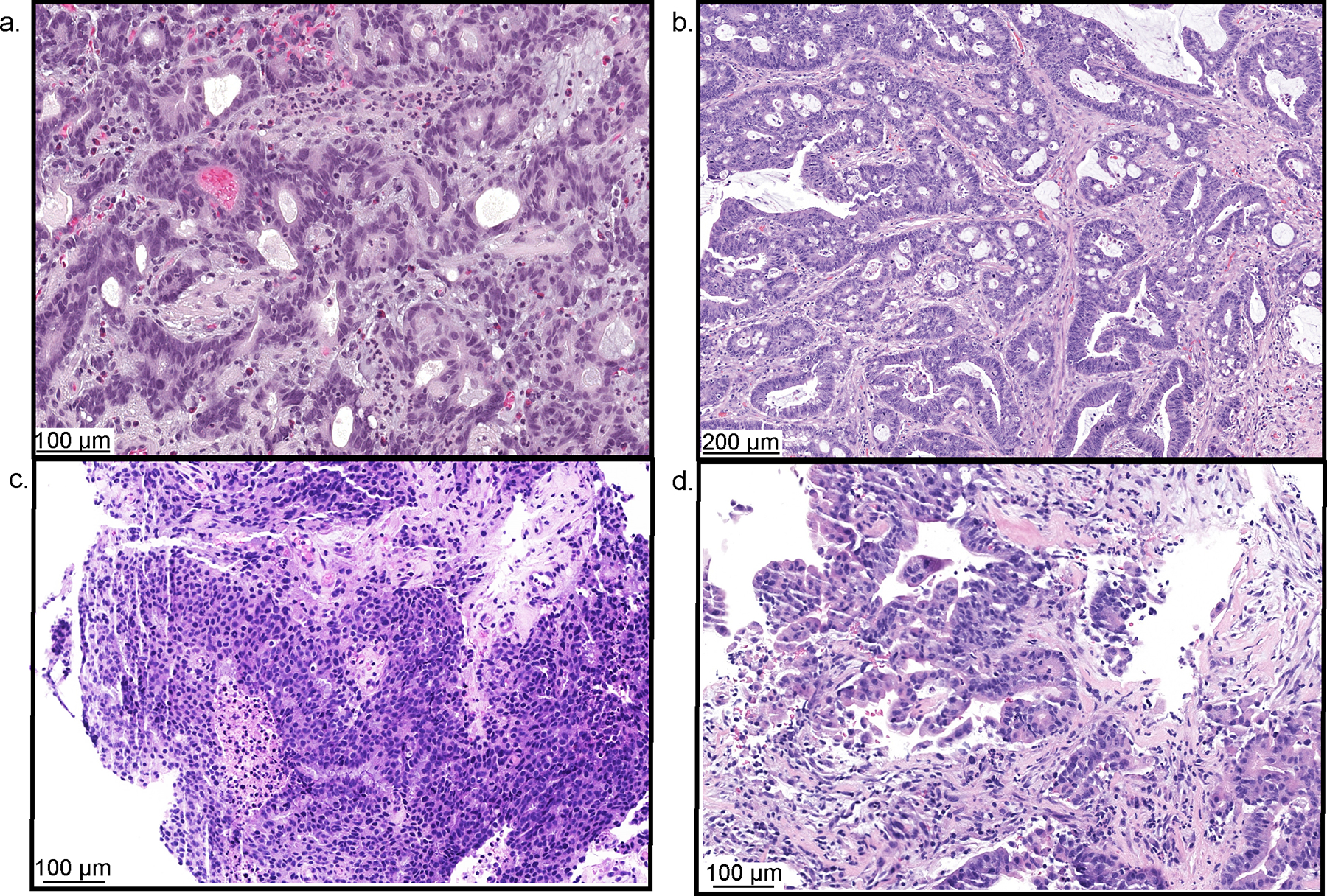
(Patient example Fig. 1a, patient 7 Fig. 1b, Extended Data Tables 3/4). a, Tumor 3: Gastric adenocarcinoma. b, Tumor 4: Colorectal adenocarcinoma. c, Tumor 5: Prostate adenocarcinoma. d, Tumor 6: Small bowel adenocarcinoma. The relevant stain, hematoxylin and eosin, was run on each chosen study tissue block once. All antibodies and staining protocols are validated and optimized to current standards.
Extended Data Fig. 3. Paired analysis of pre- and post-immune checkpoint blockade (ICB) exposed tumors using MSK-IMPACT next-generation sequencing derived data22.
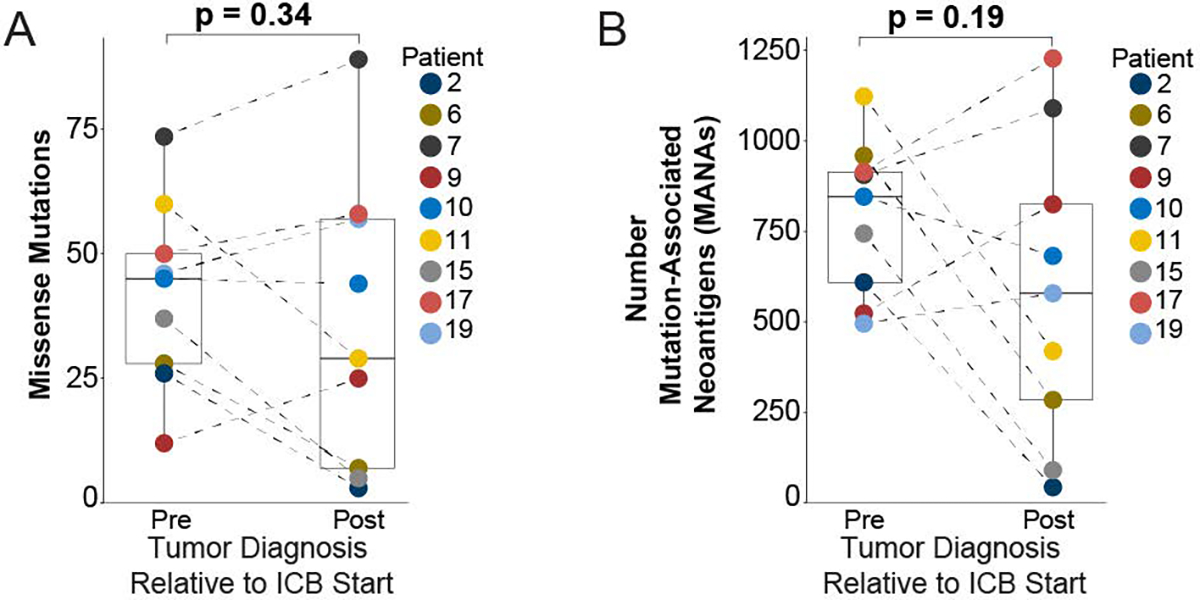
Nonsynonymous somatic mutations from the MSK IMPACT variant call format (VCF) output from each sequenced patient tumor were input into the SnpEff program v5.1d (https://pcingola.github.io/SnpEff/) to predict translated amino acid sequences for each respective mutation. The number of unique mutation associated neoantigens (MANA) was determined by taking the defined peptide sequence and evaluating all potential contiguous 9′mer variations using a sliding reading frame. a, Comparison of total missense mutations in pre- and post-ICB tumors. b, Comparison of mutation associated neoantigens between pre- and post-ICB tumors. All boxplots are composed of median (central line), 25th–75th percentile (box edges), and minimum and maximum values (whiskers). All analyses included 9 distinct pre and 9 distinct post tumors paired for 9 unique patients. p-values are derived from pairwise t-testing based on patient.
Extended Data Fig. 4. Sensitivity analyses of pre- and post-immune checkpoint blockade (ICB) exposed tumors using MSK-IMPACT next generation sequencing derived data22.
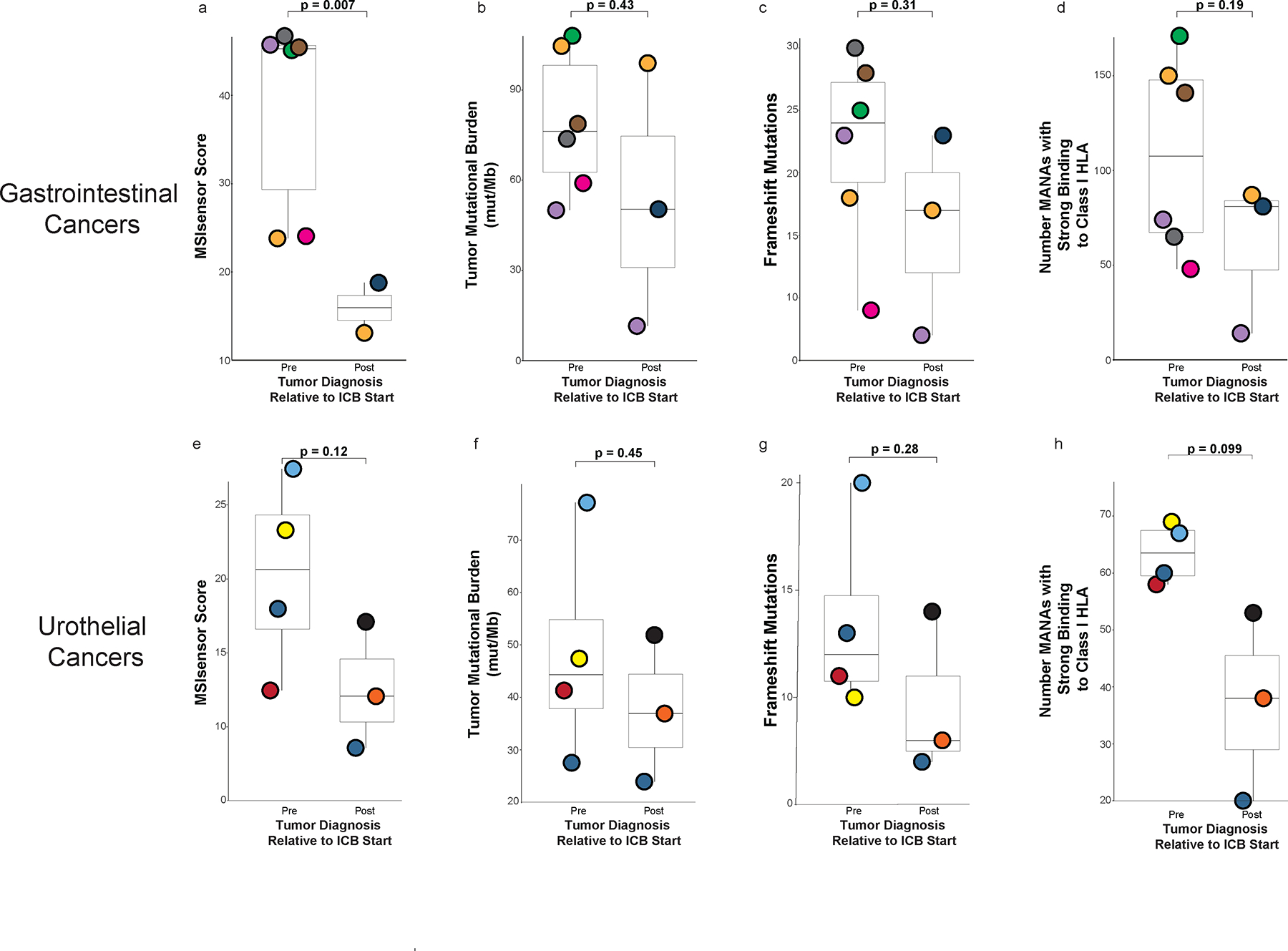
a, d, MSIsensor score, tumor mutational burden (TMB), frameshift mutation rates and number of mutation associated neoantigens (MANA) between pre- and post-ICB exposed tumors when grouped by gastrointestinal cancer only. e-h, MSIsensor score, tumor mutational burden (TMB), frameshift mutation rates and number of mutation associated neoantigens (MANA) between pre- and post-ICB exposed tumors when grouped by urothelial cancers only. All boxplots are composed of median (central line), 25th–75th percentile (box edges), and minimum and maximum values (whiskers). P-values are derived from student’s t-test (unpaired) between pre and post tumors agnostic of patient identity. Analysis a (MSIsensor score) includes 8 unique tumors (6 pre and 2 post) from 7 unique patients, and the other analyses, b-h, include 9 unique tumors (6 pre and 3 post) from 7 unique patients.
Extended Data Fig. 5. Immunohistochemical analysis of immune cell infiltration of pre-immune checkpoint blockade (ICB) and post-ICB tumors.
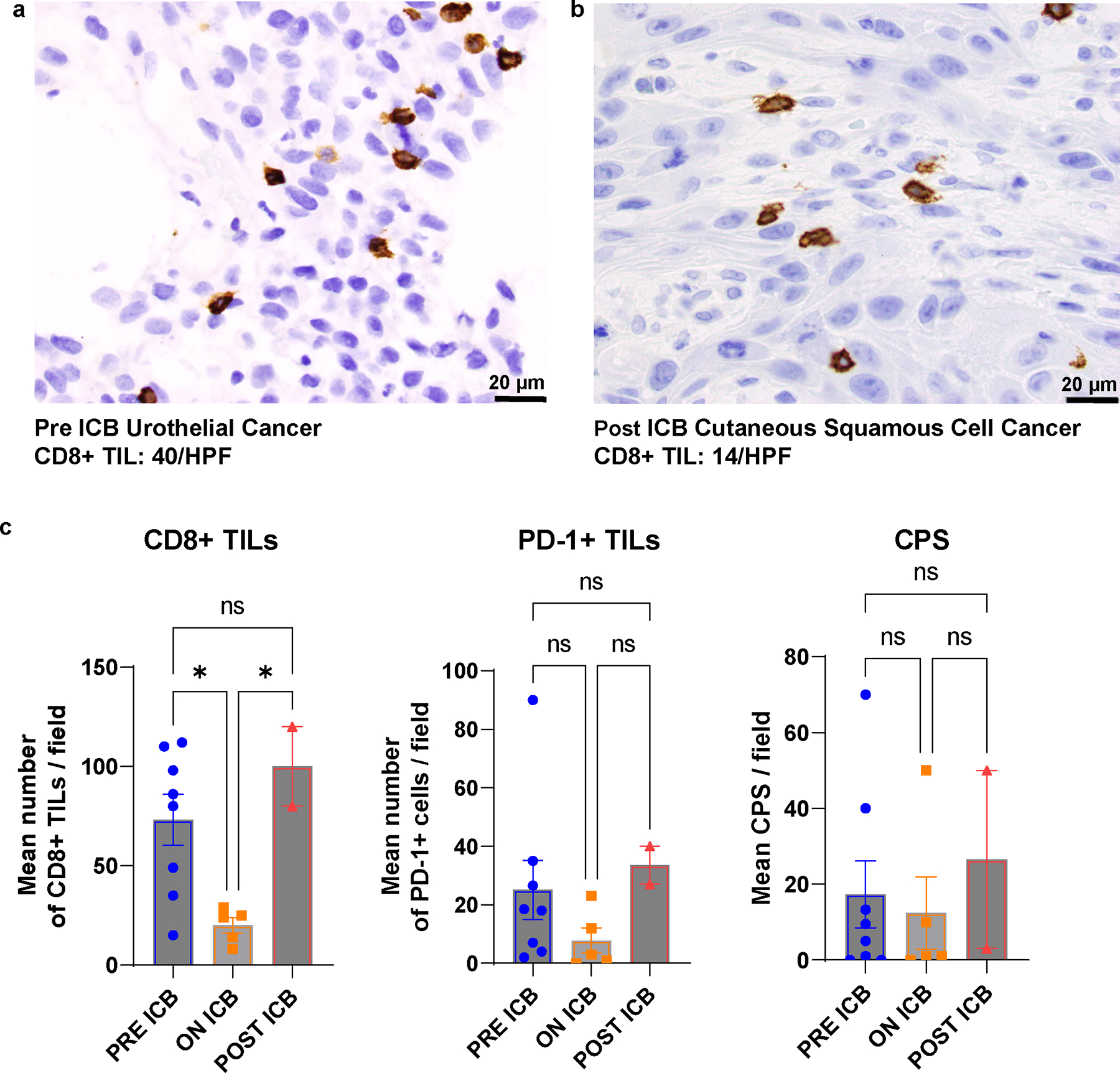
a, and b, Intra-patient comparison of paired pre- (a) and post-ICB tumors (b). CD8 tumor infiltrating lymphocytes (CD8 TILS) in the non-ICB exposed urothelial cancer and the post-ICB exposure cutaneous squamous cell carcinoma. HPF: High Power Field. c, Interpatient comparisons of CD8 TILS, programmed cell death 1 protein (PD1) positive TILS and programmed cell death ligand 1 (PDL1) combined positive score (CPS) in tumors arising prior to ICB exposure (N = 8), tumors arising whilst patients were on ICB (N = 5) and tumors arising post completion of ICB (N = 2). Mean and standard deviation (SEM) are represented and compared using ordinary one way ANOVA test corrected for multiple comparisons.*: P ≤ 0.05 and P > 0.01, ns: non significant. Stains with the relevant antibodies, CD8 antibody (clone C8/144B, Catalog # sc-53212, diluted 1:100, Dako), PD-1 (clone NAT105, Catalog # 760–4895, ready to use, Cell Marque), and PD-L1 (cloneE1L3N, Catalog # 13684 S, diluted 1:100, Cell Signaling) were run on each chosen study tissue block once. All antibodies and staining protocols are validated and optimized to current standards.
Extended Data Fig. 6. Incidence of subsequent primary malignancies (SPM) in the immune checkpoint blockade (ICB) treated and non-ICB (chemotherapy) treated patients with Lynch syndrome.
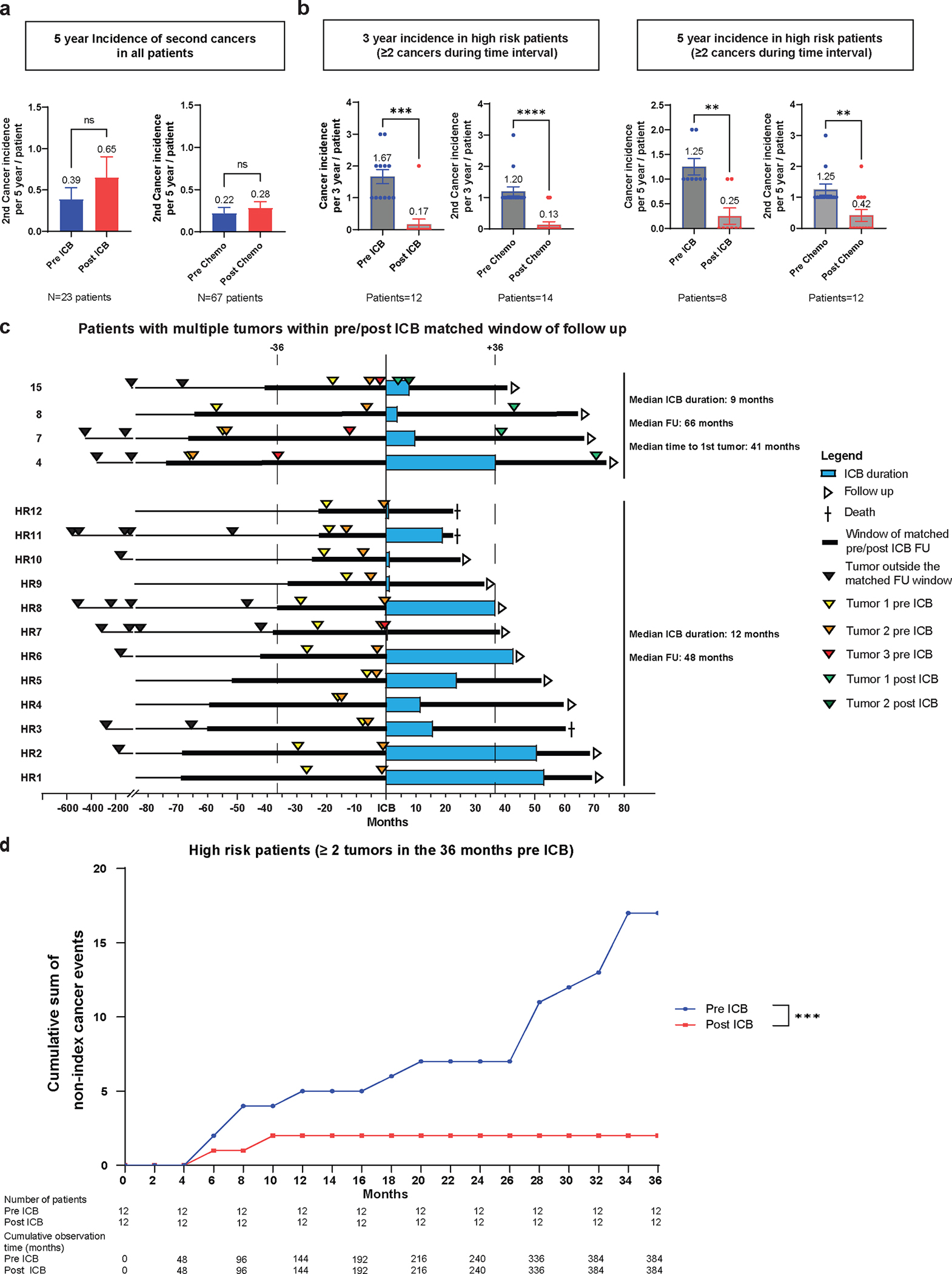
a, SPM incidence at 5-years matched follow-up in the ICB treated (pre- and post-ICB) and non-ICB treated cohort (pre- and post-chemotherapy). The number of matched patient pre/post ICB or pre/post chemotherapy is reported below the figure. The 5 year incidence of 2nd neoplasm for each clinical condition is reported above the box plot representing the mean and standard deviation (SEM). Pre and post 5 year incidence were compared using paired t test. ns: non significant, P > 0.05. b, SPM incidence in patients defined as high risk (HR). HR-patients were defined as patients presenting with multiple non concomitant cancers (> = 2) in a pre-ICB delay matched with post-ICB follow up. 3-year and 5-year incidence calculation was performed excluding the index cancer and concomitant cancers ( < 1 month between diagnoses of cancers). The number of matched patient pre/post ICB or pre/post chemotherapy for 3-year and 5-year incidence of 2nd neoplasm is reported below the figure. The 3-year and 5-year incidence of 2nd neoplasm for each clinical condition is reported above the box plot representing the mean and standard deviation (SEM). Pre and post 3-year or 5-year incidence were compared using paired t test. ns: non significant, P > 0.05. c, Swimmers plot demonstration cancer occurrence pre- and post-ICB exposure in the HR patient cohort over time in months for patients with match pre and post ICB follow up. The patient numbers for the ones developing post ICB neoplasms correspond to those reported in Fig. 1 (patients 15, 8, 7 and 4). Other high-risk patients (HR) are numerated from 1 to 12. Median ICB duration and follow up (FU) are reported for patients developing post ICB neoplasms and for the patients not developing post ICB neoplasm. The order of neoplasm occurring during the matched pre and post ICB follow up is reported. No statistical comparison was performed. d, Cumulative post-ICB cancer incidence in the ICB-exposed HR-patient cohort. High risk Patients with a matched follow up pre- and post-ICB of 36 months were selected and all non-index cancers occurring during the window of observation were accounted for. Cumulative incidence is reported as the cumulative sum of non-index cancers occurring during the period of observation. Number of patients and cumulative observation time in months are reported. Fisher’s exact test comparing the cumulative observation time without a neoplasm event to the number of months with an event was used for statistical analysis. ***: P < 0.001.
Extended Data Tabe 1.
Clinicodemographic characteristics of Lynch syndrome patients treated with immune checkpoint blockade.
| Entire Cohort (N=172) | MSI/MS-Indeterminate (N=141) | MSS (N=31) | |
|---|---|---|---|
|
| |||
| Characteristic: | %/(n/N) | %/(n/N) | %/(n/N) |
|
| |||
| Sex: | |||
| Male | 56 (97/172) | 57 (81/141) | 52 (16/31) |
| Female | 44 (75/172) | 43 (60/141) | 48 (15/31) |
|
| |||
| Pathogenic Germline Variant: | |||
| MSH6 | 17.4 (3 0/172) | 16 (23/141) | 23(7/31) |
| PMS2 | 17 (29/172) | 1 3 (18/141) | 35 (11/31) |
| MSH2 | 38 (66/172) | 44 (62/141) | 13 (4/31) |
| MLH1 | 23 (39/172) | 2 4 (34/141) | 16 (5/31) |
| EPCAM | 3.4 (6/172) | 1.4 (2/141) | 13 (4/31) |
| MSH2+EPCAM | 0.6 (1/17 2) | 0.7 (1/141) | - |
| MLH1+MSH2 | 0.6 (1/172) | 0.7 (1/141) | - |
|
| |||
| FPM: | |||
| Colorectal | 42 (72/172) | 48 (67/141) | 16 (5/31) |
| Endometrial | 16 (27/172) | 16 (23/141) | 13 (4/31) |
| Gastric/ esophageal | 6.4 (11/172) | 6.4 (9/141) | 6.5 (2/31) |
| Small bowel | 2.3 (4/172) | 3 (4/141) | - |
| Pancreas/ biliary | 5.8 (10/172) | 6.4 (9/141) | 3 (1/31) |
| Urothelial | 4.6 (8/172) | 5 (7/141) | 3 (1/31) |
| Ovary | 3 (5/172) | 2 (3/141) | 6.5 (2/31) |
| Melanoma | 1.2 (2/172) | 1.4 (2/141) | - |
| Brain/CNS | 4 (7/172) | 2 (3/141) | 13 (4/31) |
| Breast | 3.4 (6/172) | 1.4 (2/141) | 13 (4/31) |
| Sarcoma | 2.3 (4/172) | 0.7 (1/141) | 10 (3/31) |
| Prostate | 1.7 (3/172) | 2 (3/141) | - |
| Skin, non-melanoma | 0.6 (1/172) | 0.7 (1/141) | - |
| Testicular/germ cell tumor | 0.6 (1/172) | 0.7 (1/141) | - |
| Other | 6.4 (11/172) | 4.3 (6/141) | 16 (5/31) |
|
| |||
| Multiple Primary Malignancies: | |||
| Yes | 51 (88/172 ) | 50.4 (7 1/14 1) | 55(17/31) |
| No | 49 (84/172 | 49.6 (70/141) | 4 (14/31) |
|
| |||
| Median Age at FPM: (Interquartile range) | 47 (39–56) | 46 (38–55) | 50 (45.5–58) |
|
| |||
| Extent of Disease at Time of ICB Exposure: | |||
| Local | 34 (58/172) | 3 5 (49/141) | 29 (9/31) |
| Metastatic | 66(114/172) | 65 (92/141) | 71 (22/31) |
|
| |||
| Immune checkpoint Blockade: | |||
| Single agent PD1i | 8 0 (138/172) | 80.1 (1 13 /141) | 81 (25/31) |
| Single Agent PDLli | 1 0 (1 7/1 72) | 9.2 (13 /141) | 13 (4/31) |
| Combination (CTLA4i +PD1i) | 6.4 (11/172) | 6.4 (9/141) | 6 (2/31) |
| Sequential PDli, Combination (CTLAi+PDli) | 3 (5/172) | 3.5 (5/141) | - |
| Sequential | 0.6 (1/172) | 0.7 (1/141) | - |
| PDL1i,PD1i,Combination(CTLA4i+PD1i) | |||
FPM: First Primary Malignancy, MSI: Microsatellite Instability, MSS: Microsatellite Stable, MS-I: Microsatellite indeterminate, PD-1i: Programmed Cell Death Protein 1 Inhibitor, PD-L1i: Programmed Cell Death Ligand1 Inhibitor, CTLA4i: Cytotoxic T Lymphocyte protein inhibitor
Extended Data Table 2.
Clinical and treatment characteristics of immune checkpoint blockade (ICB) exposed patients with and without development of a subsequent primary malignancy (SPM).
| ICB exposed who developed SPM N=21 | ICB exposed no SPM N=151 | |
|---|---|---|
|
| ||
| Characteristic: | %/N | %/N |
|
| ||
| Pathogenic germline variant: | ||
| MSH6 | 14 (3/21) | 18 (27/151) |
| PMS2 | 0 | 19 (29/151) |
| MSH2 | 57 (12/21) | 36 (54/151) |
| MLH1 | 29 (6/21) | 22 (33/151) |
| EPCAM | 0 | 4 (6/151) |
| MSH2+EPCAM | 0 | 0.7 (1/151) |
| MLH1+MSH2 | 0 | 0.7 (1/151) |
|
| ||
| Rank order of the target tumor: | ||
| 1 | 43 (9/21) | 66 (99/151) |
| 2 | 14 (3/21) | 20 (30/151) |
| 3 | 14 (3/21) | 7 (11/151) |
| 4 | 10 (2/21) | 2 (3/151) |
| 5 | 19(4/21) | 3 (4/151) |
| 7 | - | 1 (2/151) |
| 9 | - | 0.7 (1/151) |
| 10 | - | 0.7(1/151) |
|
| ||
| Extent of disease at the time of ICB exposure | ||
| Local | 33.3 (7/21) | 34 (51/151) |
| Metastatic | 66.7(14/21) | 66 (100/151) |
|
| ||
| Immune checkpoint blockade: | ||
| Single agent PD1i | 81 (17/21) | 80 (121/151) |
| Single agent PDL1i | 5 (1/21) | 11 (16/151) |
| Combination (CTLA4i +PD1i) | 0 | 7 (11/151) |
| Sequential PD1i, Combination (CTLA4i+PD1i) | 9(2/21 | 2 (3/151) |
| Sequential PDL1i, PD1i, Combination (CTLA4i+PD1i) | 5 (1/21) | 0 |
|
| ||
| Patients who received less than 6 months of therapy: | 24 (5/21) | 48 (72/151) |
|
| ||
| Median duration of ICB: | ||
| Time(months) (IQR) | 24 (10–27) | 7 (3–17) |
Given risk of multiple primary cancers in Lynch syndrome, the tumor targeted by the ICB was ranked as to the order in which it had occurred.
IQR: Interquartile range
Extended Data Table 3.
Malignant precursor lesions post-immune checkpoint blockade (ICB) exposure.
| Total Unique 9’mer Neoantigens | Number Unshared Neoantigens | Tumor Pairs (Number Neoantigens Shared) | Shared Mutation Per Tumor Pair | |
|---|---|---|---|---|
| Patient 2 | 652 | 652 (100%) | 0 | 0 |
| Patient 6 | 1243 | 1243 (100%) | 0 | 0 |
| Patient 7 | 2902 | 2898 (99.9%) | Prostate and Small Bowel (7) | PTPRT R260W |
| Patient 9 | 2063 | 2038 (98.9%) | Gastric and Urothelial (25) | KMT2C K2797Rfs*26 |
| Patient 10 | 1528 | 1497 (98.0%) | Urothelial and Colorectal (31) | MSH3 K383Rfs*32 |
| Patient 11 | 2665 | 2590 (97.2%) | Esophagus and Prostate (50) Esophagus and Urothelial (25) |
ZFHX3 ( R329Vfs*56, L3434Sfs*51) AR Q58L |
| Patient 15 | 834 | 834 (100%) | 0 | 0 |
| Patient 17 | 3106 | 3059 (98.5%) | Breast and Colorectal (15) Colorectal and Urothelial (29) Breast and Urothelial (3) |
JAK1 K860Nfs*16 KMT2D P2354Lfs*30 INPP4B R818Efs*4 |
| Patient 19 | 1080 | 1073 (99.4%) | Pancreas and Prostate (7) | KMT2D R3536H |
| Total | 16073 | 15884 (98.8%) |
TT: target tumor; TTP: time to polyp; TA: tubular adenoma; TVA: tubulo-villous adenoma; SA: serrated adenoma; IHC: immunohistochemistry; dMMR: deficient DNA mismatch repair; pMMR: proficient DNA mismatch repair.
Incomplete baseline colonoscopy pre-ICB exposure
NA: Not available
Extended Data Table 4.
Analysis of shared mutations and mutation associated noeantigens (MANA) between pre- an post-immune checkpoint blockade exposed tumors.
| Tumor Targeted by IC B (TT) | Germline Mutation | Patient Age (years) | Microsatellite Status TT | Polyps prior to ICB | Time from Last colonoscopy to ICB start | Scope on ICB | TTP from last colonoscopy (months) | TTP from last dose of ICB (months) | Post ICB Malignancy | Pathological Description | Polyp size (mm) | IHC Protein Loss | MMR Status | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Colorectal | MLH1 | 43 | dMMR | Yes | 24 | No | 6 | 69 | Multiple cutaneous neoplasms | TA | 5 | Not possible to assess | NA |
| Patient 7 | Prostate | MSH6 | 75 | dMMR | Yes | 10 | Yes | 11 | 13 | dMMR SB Ca | TA | 10 | MSH6 | dMMR |
| Patient 11 | Esophageal | MSH2 | 69 | dMMR | No | 4 | No | 15 | 27 | dMMR Upper tract urothelial | TA | 2 | NA | NA |
| Patient 12 | Colorectal | MSH6 | 78 | dMMR | No | 1 | No | 18 | 10 | SCC MMR status UNK | TA X2 | 2, 3 | - | pMMR |
| Patient 14 | Colorectal | MLH1 | 53 | dMMR | No | 2 | Yes | 27 | 25 | dMMR Sebaceous adenoma | TVA with HGD | 25 | MLH1/ PMS2 | dMMR |
| Patient 22 | Colorectal | MSH2 | 38 | dMMR | No | <1 | Yes | 10 | 9 | - | TA | 3 | MSH2/MSH6 | dMMR |
| Patient 23 | Urothelial | MSH2 | 65 | dMMR | No | 91 | Yes | 14 | 11 | - | TA x3 | 3 x3 | Loss of MSH2 in 1 | dMMRx1, pMMRx2 |
| Patient 24 | Endometrial | MLH1 | 51 | dMMR | No | 55 | No | 6 | 11 | - | TA | 2 | PMS2 | dMMR |
| Patient 25 | Colorectal | MSH2 | 52 | dMMR | Yes | <1 | No | 22 | 20 | - | TA | 10 | MSH2/MSH6 | dMMR |
| Patient 26 | Endometrial | PMS2 | 63 | dMMR | Nil prior | Nil prior | No | NA | 7 | - | TA | 5 | - | pMMR |
| Patient 27 | Colorectal | MLH1 | 42 | dMMR | Yes | 9 | No | 31 | 3 | - | TA | 4 | - | pMMR |
| Patient 28 | Colorectal | EPCAM | 40 | dMMR | No | 4 | Yes | 39 | 22 | - | TA | 3 | - | pMMR |
| Patient 29 | Colorectal | MSH2 | 32 | dMMR | No | 1 | No | 12 | 20 | - | TA | 2 | - | pMMR |
| Patient 30 | Endometrial | MSH2 | 61 | dMMR | Nil prior | Nil prior | Yes | 5 | 4 | - | TA | 2 | - | pMMR |
| Patient 31 | Colorectal* | PMS2 | 60 | dMMR | Incomplete, obstructing primary | 2 | No | 9 | 1 | - | TVA × 1, TA ×12 | 35, 7–10×7, 7–12×5 | - | pMMR all blocks |
| Patient 32 | Urothelial | MSH6 | 62 | pMMR | Yes | 4 | Yes | 13 | 9 | - | SSP | 5 | NA | NA |
| Patient 33 | Colorectal | MSH6 | 38 | dMMR | No | 4 | Yes | 12 | 35 | - | TA | 5 | NA | NA |
| Patient 34 | Colorectal | MSH2 | 47 | dMMR | No | 1 | No | 13 | 2 | - | TA | 5 | - | pMMR |
| Patient 35 | Colorectal | MSH6 | 74 | dMMR | Yes | 0.23 | No | 16 | 0.4 | - | TA ×2 | 4, 6 | - | pMMR |
| Patient 36 | Colorectal | MLH1 | 33 | dMMR | No | 7 | Yes | 22 | 20 | - | TA | 10 | - | pMMR |
| Patient 37 | Colorectal | MSH6 | 75 | dMMR | Yes | <1 | No | 10 | 5 | - | TA x3, SA | 10,5 4,4 | NA | NA |
| Patient 38 | Gastric | MSH2, EPCAM | 77 | dMMR | Nil prior scop | Nil prior scope | Yes | 23 | 24 | - | TA x3 | 8, 2,4 | - | pMMR all blocks |
| Patient 39 | Urothelial | MSH2 | 37 | dMMR | No | 49 | No | 35 | 39 | - | TA | 5 | pMMR | |
| Patient 40 | Gastric | MSH2 | 73 | dMMR | Yes | 25 | Yes | 20 | 12 | - | TA ×3 | 5 | - | pMMR all blocks |
Tumor pairs with shared neoantigen number are aligned with the corresponding shared mutation(s) for that specific pair.
Supplementary Material
Supplementary Data Table 1. (excel format) Characteristics of tumors targeted (TT) by immune checkpoint blockade (ICB) and the subsequent primary malignancies (SPM) post-ICB.
SPM: Second primary malignancy; SNV: Short nucleotide variant; ICB: Immune checkpoint blockade; ADT: Androgen deprivation therapy; CR: Complete response; SD: Stable disease; POD: Progressions of disease; PSA: Prostate Specific Antigen; TVA: Tubulovillous adenoma; SCC: Squamous cell carcinoma of skin; HCC: Hepatocellular carcinoma; DCIS: Ductal carcinoma in situ; *Not present radiologically on baseline imagine
Acknowledgments:
This work was supported in part through the Marie-Josée and Henry R. Kravis Center for Molecular Oncology at Memorial Sloan Kettering (DBS, MFB, ZKS) ; the Precision, Interception and Prevention Program at Memorial Sloan Kettering (ZKS, LD); the Robert and Kate Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering (ZKS, KO); the Romeo Milio Lynch Syndrome Foundation (ZKS), the Fieldstone Fund (ZKS), Swim Across America (LD), the National Insitutes of Health National Cancer Institute Exploratory/ Developmental Grant R21 CA252519 (LD, AC), National Institutes of Health 1K08CA279922 (MBF), the Chris4Life Colorectal Cancer Alliance Early Investigator Award (MBF) and the National Institutes of Health National Cancer Institute Cancer Center Support Grants P30 CA008748 (all authors), P50-CA221745(DBS, JER) and P50-CA092629 (DBS, DR).
The authors would also like to acknowledge the contributions of Dennis Stephens who assisted with the use of SnpEff program v 5.1d (https://pcingola.github.io/SnpEff/) to predict translated amino acid sequences for each respective mutation derived from the MSKCC IMPACT NGS panel.
Footnotes
Competing interests:
Emily Harrold (EH) has received funding from the Conquer Cancer ASCO Foundation. She also served as a consultant to Pfizer Ireland on one occasion in 2021. She reports education grants from Merck and Amgen to attend GI ASCO in 2020.
Benoit Rousseau (BR) serves as a consultant for Neophore and has a patent: Methods and composition for cancer immunotherapy.
Fergus Keane (FR) has received funding from the Conquer Cancer ASCO Foundation.
Andrea Cercek (AC) serves as a consultant for Bayer, GlaxoSmithKline, Janssen Biotech, Merck, Seagen Inc, and receives research funding from GlaxoSmithKline, Inspirna, Seagen Inc.
Rona Yaeger (RY) serves as a consultant for Array BioPharma/ Pfizer, Amgen, Mirati Therapeutics, and Natera, and receives research funding to her institution from Pfizer, Boehringer Ingelheim, Mirati Therapeutics and Daiichi Sankyo
Dana Rathkopf (DR) is an uncompensated Advisor/Steering Committee member and Research Support (PI): Janssen, Astra Zeneca, Bayer, Myovant, Genentech, Promontory, BMS/Celgene
Neil Segal (NHS) serves as a consultant for ABL Bio, Amgen, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Immunocore, Novartis, Psioxus, Puretech, Revitope, Roche/ Genentech and Numab and receives grant/contracts from: AstraZeneca, Bristol Myers Squibb Company, Immunocore, Merck, Pfizer, Puretech, Regeneron Pharmaceuticals Inc., Roche/ Genentech and Agenus.
Eileen M. O’Reilly (EO) Receives research from Genentech/Roche, Celgene/BMS, BioNTech, AstraZeneca, Arcus, Elicio, Parker Institute, NIH/NCI, and serves as a consultant for: Cytomx Therapeutics (DSMB), Rafael Therapeutics (DSMB), Seagen, Boehringer Ingelheim, BioNTech, Ipsen, Merck, IDEAYA, Silenseed, Novartis, AstraZeneca, BioSapien, Astellas, Thetis, Autem, Novocure, Neogene, BMS, ZielBio, Merus, Tempus, Fibrogen. An immediate family member serves as a consultant for Agios, Genentech-Roche, Eisai and Servier.
Diane Reidy (DR) Receives research funds from Merck, Novartis, and Ipsen, and is on the Scientific Advisory Board for Chiasma, Novartis, and Advanced Accelerator Applications (AAA).
Yelena Y.Janjigian (YYJ) Receives research funding from Bayer, Bristol-Myers Squibb, Cycle for Survival, Department of Defense, Eli Lilly, Fred’s Team, Genentech/Roche, Merck, NCI, RGENIX, serves on the advisory boards/is a consultant for Amerisource Bergen, Arcus Biosciences, Astra Zeneca, Basilea Pharmaceutica, Bayer, Bristol, Myers Squibb, Daiichi-Sankyo, Eli Lilly, Geneos Therapeutics, GlaxoSmithKline, Imedex, Imugene, Lynx Health, Merck, Merck Serono, Mersana Therapeutics, Michael J. Hennessy Associates, Paradigm Medical Communications, PeerView Institute, Pfizer, Research to Practice, RGENIX, Seagen, Silverback Therapeutics, Zymeworks Inc., and has stock options in RGENIX.
Yonina R. Murciano-Goroff (YRMG) reports travel, accommodation, and expenses from AstraZeneca and LOXO Oncology/ Eli Lilly. She acknowledges honoraria from Virology Education and Projects in Knowledge (for a CME program funded by an educational grant from Amgen). She acknowledges associated research funding to the institution from Mirati Therapetuics, Loxo Oncology at Eli Lilly, Elucida Oncology, Taiho Oncology, Hengrui USA, Ltd/ Jiangsu Hengrui Pharmaceuticals, Luzsana Biotechnology, Endeavor Biomedicines, and AbbVie. She is an employee of Memorial Sloan Kettering Cancer Center, which has an institutional interest in Elucida. She acknowledges royalties from Rutgers University Press and Wolters Kluwer. She acknowledges food/beverages from Endeavor Biomedicines. Y.R. Murciano-Goroff acknowledges receipt of training through an institutional K30 grant from the NIH (CTSA UL1TR00457). She has received funding from a Kristina M. Day Young Investigator Award from Conquer Cancer, the ASCO Foundation, endowed by Dr. Charles M. Baum and Carol A. Baum. She is also funded by the Fiona and Stanley Druckenmiller Center for Lung Cancer Research, the Andrew Sabin Family Foundation, the Society for MSK, and a Paul Calabresi Career Development Award for Clinical Oncology (NIH/NCI K12 CA184746).
Ying L. Liu (YLL) reports research funding from AstraZeneca, GSK, and Repare Therapeutics outside this work.
Jonathan E. Rosenberg (JER) has received research support for clinical trials from Bayer, Seagen, Astellas, AstraZeneca, and Roche/Genentech. He has served as an advisor or consultant to Bayer, Seagen, Astellas, AstraZeneca, Roche/Genentech, BMS, Merck, Pfizer, Boehringer Ingelheim, GSK, Janssen, Mirati, EMD-Serono, Gilead, Alligator Biosciences, Eli Lilly, Tyra Biosciences, Infinity, IMVax, Aadi, Century Therapeutics, Emergence Therapeutics, Hengrui
Martin R Weiser (MRW) – Copyright: participate on online tumor board. Licensee (Precisa), Editor of Colorectal Section. Licensee (UpToDate)
Anthony M. Rossi (AMR) serves as Regeneron: Consultant; Evolve CME: Consultant; Almirall: Consultant; Mavig: Travel; Merz: Consultant; Dynamed: Consultant; Canfield Scientific: Consultant; AllerganInc: Advisory Board; Evolus: Consultant; Biofrontera: Consulatant; QuantiaMD: Consultant; Lam Therapeutics; Consultant; Cutera: Consultant; Skinfix, advisor; L’oreal, travel, DAR companies: Founder; Skinpass Board. AMR received research/study funding from ASLMS: A Ward Memorial Research Grant, Skin Cancer Foundation, Regen, LeoPharma, Biofrontera and serves as Editorial Board: Lasers in Surgery and Medicine; CUTIS, Editorial Board: Journal of the American Academy of Dermatology (JAAD); Dermatologic Surgery, Board Member: ASDS, Committee Member and / or Chair: AAD; ASDS; ASLM.
Kenneth Offit (KO) is a founder (uncompenstated; shares not alloted) of AnaNeo Therapeutics, Inc.
Patents, Royalties, Other Intellectual Property: Diagnosis and treatment of ERCC3-mutant cancer; inventors: Joseph Vijai, Sabine Topka, Kenneth Offit; US National Stage Patent Application No.: 16/493,214; filing date: September 11, 2019 (Inst)
Michael F. Berger (MFB) serves as a consultant for Eli Lilly and Astra Zeneca, and receives research support from Boundless Bio.
David B. Solit (DBS) has served as a consultant for/received honorarium from Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, Scorpion Therapeutics, FORE Therapeutics, Fog Pharma, Elsie Biotechnologies, and BridgeBio
Leonard Saltz (LS) serves as consultant and is a member of the Scientific Advisory Board for Genor Biopharma Ltd.
Jinru Shia (JS) serves as a consultant for Paige AI.
Luis Diaz (LD) is a member of the board of directors of Jounce Therapeutics and Epitope. He is a compensated consultant to PetDx, Innovatus CP, Se’er, Delfi, Blackstone, Kinnate and Neophore. LD is an inventor of multiple licensed patents related to technology for circulating tumor DNA analyses and mismatch repair deficiency for diagnosis and therapy. Some of these licenses and relationships are associated with equity or royalty payments to the inventors. He holds equity in Epitope, Jounce Therapeutics, PetDx, Se’er, Delfi, Kinnate and Neophore. He divested his equity in Personal Genome Diagnostics to LabCorp in February 2022 and divested his equity in Thrive Earlier Detection to Exact Biosciences in January 2021. His spouse holds equity in Amgen. The terms of all these arrangements are being managed by Memorial Sloan Kettering in accordance with their conflict-of-interest policy.
Zsofia K. Stadler’s (ZKS) immediate family member serves as a consultant in Ophthalmology for Adverum, Genentech, Neurogene, Novartis, Optos Plc, Outlook Therapeutics, and Regeneron outside the submitted work. ZKS serves as an Associate Editor for JCO Precision Oncology and as a Section Editor for UpToDate.
The remaining authors have no competing interests.
Prior Presentation: This work, in part, was an Oral Presentation at the American Society of Clinical Oncology Annual Meeting in 2022. DOI: 10.1200/JCO.2022.40.16_suppl.10505 Journal of Clinical Oncology 40, no. 16_suppl (June 01, 2022) 10505–10505.
Supplementary Information is available for this paper.
Data Availability Statement:
All information regarding the study cohort and pre and post ICB malignancies is available in the extended data and supplementary tables. Additional information can be provided by the corresponding author in accordance with institutional regulatory approval within 8 weeks of request. Genomic data derived from MSK-IMPACT is available in aggregated form via the cBioPortal for Cancer Genomics (http://cbioportal.org).
References:
- 1.Win Aung Ko, et al. Journal of clinical oncology 30.9 (2012): 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Latham Alicia, et al. Journal of clinical oncology 37.4 (2019): 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Dung T., et al. Science 357.6349 (2017): 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cercek Andrea, et al. New England Journal of Medicine (2022). [Google Scholar]
- 5.Chalabi Myriam, et al. Nature medicine 26.4 (2020): 566–576. [DOI] [PubMed] [Google Scholar]
- 6.Heudel Pierre, et al. ESMO open 6.1 (2021): 100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zehir Ahmet et al. Nature Medicine 23.6 (2017): 703–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan et al. Cell host and microbe 27.4 (2020): 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmen Del et al. Frontiers in Oncology 13:(2023): 1710 [Google Scholar]
- 10.Middha et al. JCO Precision Oncology 1(2017): 1–17 [Google Scholar]
- 11.Moller Pal et al. Gut 66.9 (2017): 1657–166427261338 [Google Scholar]
- 12.Mandal Rajarsi, et al. Science 364.6439 (2019): 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amodio Vito, et al. Cancer Cell (2022). [Google Scholar]
- 14.Westcott Peter MK, et al. bioRxiv (2021). [Google Scholar]
- 15.Ahadova Aysel, et al. International Journal of Cancer 148.4 (2021): 800–811. [DOI] [PubMed] [Google Scholar]
- 16.Kloor Matthias, et al. The lancet oncology 13.6 (2012): 598–606. [DOI] [PubMed] [Google Scholar]
- 17.Ahadova Aysel et al. Gastroenterology (2023) [Google Scholar]
- 18.Chang Kyle et al. JAMA Oncology 4.8 (2018): 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yurgelun Matthew B., et al. Cancer prevention research 5.4 (2012): 574–582.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roudko Vladimir, et al. Cell 183.6 (2020): 1634–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Only References:
- 21.Cheng DT, Prasad M, Chekaluk Y, et al. : Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics 10:33, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251–264, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shia Jinru. “The diversity of tumours with microsatellite instability: molecular mechanisms and impact upon microsatellite instability testing and mismatch repair protein immunohistochemistry.” Histopathology 78.4 (2021): 485–497. [DOI] [PubMed] [Google Scholar]
- 24.Ziegler John, et al. “MiMSI-a deep multiple instance learning framework improves microsatellite instability detection from tumor next-generation sequencing.” bioRxiv (2020). [Google Scholar]
- 25.Jurtz Vanessa, et al. “NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data.” The Journal of Immunology 199.9 (2017): 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul Sinu, et al. “Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system.” PLoS computational biology 16.5 (2020): e1007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla SA et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33, 1152–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen R & Seshan VE FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGranahan N et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data Table 1. (excel format) Characteristics of tumors targeted (TT) by immune checkpoint blockade (ICB) and the subsequent primary malignancies (SPM) post-ICB.
SPM: Second primary malignancy; SNV: Short nucleotide variant; ICB: Immune checkpoint blockade; ADT: Androgen deprivation therapy; CR: Complete response; SD: Stable disease; POD: Progressions of disease; PSA: Prostate Specific Antigen; TVA: Tubulovillous adenoma; SCC: Squamous cell carcinoma of skin; HCC: Hepatocellular carcinoma; DCIS: Ductal carcinoma in situ; *Not present radiologically on baseline imagine
Data Availability Statement
All information regarding the study cohort and pre and post ICB malignancies is available in the extended data and supplementary tables. Additional information can be provided by the corresponding author in accordance with institutional regulatory approval within 8 weeks of request. Genomic data derived from MSK-IMPACT is available in aggregated form via the cBioPortal for Cancer Genomics (http://cbioportal.org).