

Abstract
In the past decade, hydrogels have attracted growing interest for emerging applications in flexible electronic devices, human–machine interactions, energy supply, or energy storage. Developing a multifunctional gel architecture with superior ionic conductivity and good mechanical flexibility is a bottleneck to overcome. Herein, poly(vinyl alcohol)/sulfuric acid (PVA–H2SO4) hydrogels were prepared via a freeze–thaw method. With the aim of tuning the formulation in view of a possible application in energy storage, the effects of different combinations in terms of the molecular weight (MW) of PVA and PVA–H2SO4 weight ratio were investigated. Moreover, exploiting the self-healing properties of these hydrogels and the easy possibility of functionalizing them, i.e., introducing a conducting polymer such as poly(2-acrylamido-2-methyl-1-propane) sulfonic acid doped polyaniline (PANI_PAMPSA), a sandwiched all-in-one double-layer hydrogel (electrode/electrolyte configuration) was prepared (PVA–H2SO4–PANI_PAMPSA/PVA–H2SO4). Results showed that the water content is independent of the PVA amount and MW; the polymer concentration has a significant effect on the formation of crystalline domains and therefore on swelling degree, whereas the cross-linking degree depends on the MW. The PVA MW has the maximum effect on the swelling percentage normalized with respect to the polymer fraction and the tensile properties of the hydrogel. The assembled all-in-one electrode/electrolyte shows promising ionic conductivity (439.7 mS cm–1) and specific capacitance performance (0.297 mF cm–2 at a current density of 0.025 mA cm–2), as well as excellent flexibility and considerable self-healing properties. These results will promote the development of self-healing symmetrical supercapacitors for storage devices in wearable electronics.
1. Introduction
The increasing energy demand produced by technology progressing is a problem we are facing as a society.1 In this perspective, energy storage devices have made great progress in this field because of their fast charging and discharging process, high power density, and good life cycle.2−4 Moreover, with the rapid development of modern wearable and portable electronics as well as intelligent clothes, which often can be subjected to various mechanical stresses (e.g., stretching and/or bending), energy storage devices require having self-healing abilities, good flexibility, good ionic conductivity, and good mechanical properties.2,5 All of the device elements should be developed to satisfy these specifications. Focusing on electrolytes, common supercapacitors generally provide energy through charge collection at electrodes in an aqueous or organic medium. Ions can move freely in a solution, facilitating a fast charge/discharge process even at a high achievable voltage and with a long life cycle.6,7 The risk of leaking and release of toxic compounds for humans or the environment hinders their use, especially in wearable devices. Moreover, the encapsulation of the aqueous interlayer represents a further step in the overall preparation of the supercapacitor, leading to an additional cost. To face this issue, solid-like or quasisolid-like phases represent a possible solution, using dry polymers, inorganic compounds, and gel polymers as electrolytes. Hydrogels, characterized by a three-dimensional cross-linked porous microstructure, have emerged as one of the most promising candidates due to their similarities to natural tissues and excellent flexibility.8−10
Hydrogels are defined as flexible three-dimensional cross-linked networks that can swell, retaining significant fractions of water, due to the presence of hydrophilic functional groups on the macromolecular structure.11,12 Thanks to their ability to maintain a colloidal-like structure and the resulting flexibility, hydrogels have garnered particular interest for energy storage and biomedical applications.12 For these reasons, the use of hydrogel-based electrolytes, as well as hydrogel-based electrodes for energy storage applications, has rapidly increased in recent years.13 The number of papers based on conductive hydrogels has also grown due to the rising attention on these systems.14 Conductive hydrogels can exhibit two conductivity mechanisms: ionic and electronic. To observe ionic conduction, free ions have to be incorporated via (i) directly doping electrolytes (i.e., by acids such as H2SO4, alkalis, salts, etc.), (ii) treating preformed hydrogels with salt solutions, or (iii) introducing polyelectrolytes with counterions.14 On the other side, electronic conductivity requires the presence of electronic conductive fillers in the hydrogel structure (i.e., nanofillers as metal nanoparticles, graphene and its derivatives, carbon nanotubes, or conductive polymers).15−17 The design and tuning of these devices have highlighted several critical points due to the lack of all-around high-performing materials, and therefore, optimization of the critical parameters, previously discussed, is necessary.
Hydrogels can be prepared from natural compounds (e.g., polysaccharides, polynucleotides) or can be derived from synthetic polymers (e.g., poly(acrylic acid) (PAA), poly(methacrylic acid) (PMAA), poly(ethylene glycol) or poly(ethylene oxide) (PEG or PEO), poly(vinyl alcohol) (PVA)).11,12,18 Hydrogel produced from natural sources are usually biocompatible and sustainable, but the starting components are not always pure, and tuning the chemical composition and consequent chemical–physical properties can be challenging. On the other hand, when hydrogels are prepared from synthetic compounds, their chemical structure can be varied to adapt the material to specific applications.
In particular, PVA-based hydrogels have been intensively studied for their physical properties such as high hydrophilicity, processability, and biocompatibility.14 Since they present hydroxyl groups on the main chain, PVA can form physically cross-linked systems through hydrogen bonds at low temperatures. The simplicity of this PVA hydrogel preparation lies in the freeze–thaw technique, which induces the formation of the structure though a freezing treatment that aligns macromolecules and forms water microcrystalline regions.19,20 Additionally, hydrogen bonds are weak interactions that can be used as an advantage for a potential self-healing ability in the preparation of electronic devices, allowing the field of wearable electronics to flourish. In this context, PVA hydrogels are already considered a valid tool for electronics and energy storage.2,21 However, a proper study of the influence of PVA molecular weight on mechanical and electrical performances is lacking in the literature.
In this work, our initial focus was on exploring the potential use of PVA-based ionic conductive hydrogels as a potential candidate for electrolytes and/or electrodes in the preparation of all-in-one flexible energy storage devices. To obtain the final materials called PVA–H2SO4, we have employed an acid direct doping electrolyte method and a physical freezing–thawing cross-linking strategy. The uncoiling and reorganization of the polymer chain in the PVA hydrogel contribute to the isotropic tensile properties of the final material.22 The freeze–thaw method avoids component leaching associated with traditional chemical methods and ensures the preparation of hydrogels with a nontoxic nature, enhanced mechanical properties, and high flexibility.23 A fixed H2SO4 concentration of 1.0 M has been chosen based on literature data24 as the optimal concentration for providing free ions capable of acting as charge carriers in the hydrogel. The effect of different combinations in terms of the molecular weight (MW) of PVA and the PVA–H2SO4 weight ratio on the mechanical properties and ionic conductivity has been investigated, with the aim of fine-tuning the formulation for possible application in the preparation of energy storage devices.
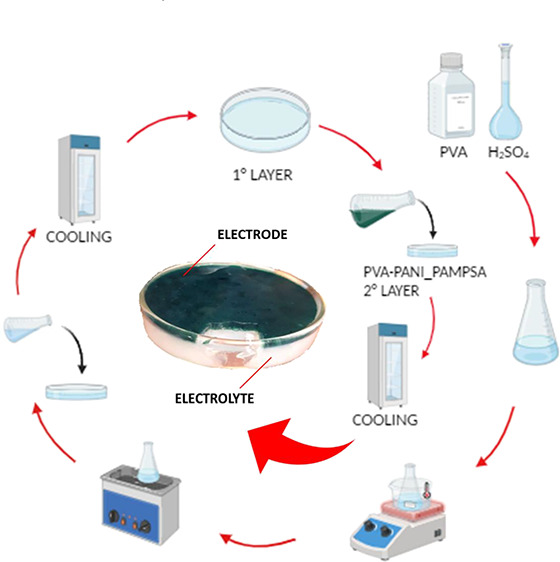
Furthermore, considering that these hydrogels could be easily functionalized by introducing other purposeful components (i.e., conducting polymers, nanoparticles, fibers, etc.), the PVA–H2SO4 system has been blended with a conducting polymer. Poly(2-acrylamido-2-methyl-1-propane) sulfonic acid doped polyaniline (PANI_PAMPSA) has been chosen for a number of reasons: its versatility, redox chemistry, charge-switching ability, ease of preparation, good chemical stability, and good conductivity.25,26 The hydrogel termed PVA–H2SO4–PANI_PAMPSA has been prepared with the addition of a 50% PANI_PAMPSA suspension (w/w). Finally, exploiting the self-healing ability of these hydrogels and the freeze–thaw physical method, a sandwiched all-in-one electrode/electrolyte hydrogel configuration has been prepared employing PVA–H2SO4 and PVA–H2SO4–PANI_PAMPSA layers with the aim of investigating the possibility of forming an all-in-one integral structure for flexible energy storage devices.
2. Experiments
2.1. Materials
Commercial PVA powders of three different MWs (Table 1) were purchased from Merck KGaA, Darmstadt, Germany; a 1.0 M H2SO4 solution was prepared from dilution of 95–98% H2SO4 (Sigma-Aldrich).
Table 1. Properties of PVA Used in This Work.
| PVA | PVA MW | hydrolysis degree | hydrogel obtained |
|---|---|---|---|
| A-PVA | 13,000–23,000 | 98.0–98.8% | HyA |
| B-PVA | 31,000–50,000 | 98.0–98.8% | HyB |
| C-PVA | 146,000–186,000 | 99+% | HyC |
2.2. Preparation of the PVA–H2SO4 Hydrogel
A typical hydrogel preparation via the freezing–thawing method is reported.2,10 2.6 g of PVA powder were added to 15 mL of aqueous 1.0 M H2SO4 solution (PVA/H2SO4 weight ratio equal to 1/5.8) and heated in a water bath at 70–90 °C under vigorous stirring to obtain a transparent solution. The dissolution time and temperature strictly depended on the polymer MW. The solution was then ultrasonically treated to remove bubbles and poured into a Petri dish (8 cm in diameter). The Petri dish was placed in a refrigerator at −18 °C for 3 h and then was kept at room temperature to thaw the hydrogel for 1 h. After three freeze–thaw cycles, the PVA–H2SO4 hydrogel membrane was obtained (Figure S1). This procedure was changed appropriately (PVA/H2SO4 weight ratio) depending on the PVA MW used. All samples were wrapped with cling film for food and stored in sealed food bags until use.
2.3. Preparation of the PVA–H2SO4–PANI_PAMPSA Hydrogel and Self-Healing Double-Layer Hydrogel
The PVA–H2SO4–PANI_PAMPSA hydrogel (HyPVA–PANI_PAMPSA) was prepared starting from a just-synthesized PANI_PAMPSA polymer obtained via an oxidative polymerization process27 and from B-PVA, keeping the (PANI_PAMPSA + PVA)/H2SO4 weight ratio equal to 1/5.8. Briefly, 1.3 g of B-PVA was dissolved in 7.6 mL of 1.0 M H2SO4 solution at 80 °C under stirring at 350 rpm (solution A). Then, 1.3 g of the PANI_PAMPSA suspension (3.5% w/w PANI_PAMPSA) was added to solution A under vigorous stirring, and then the mixture was sonicated at 70 °C for 6 min. To obtain the self-healing double-layer hydrogel (HyPVA–PANI_PAMPSA/HyB#1), a layer of HyB#1 was first deposited into a Petri dish of 8 cm diameter and subjected to one freeze–thaw cycle of 30 min. Afterward, a HyPVA–PANI_PAMPSA layer was deposited on top of HyB#1. Finally, the double-layer hydrogel was subjected to six freeze–thaw cycles.
2.4. Material Characterizations
Before analyzing PVA–H2SO4 hydrogel samples by means of total reflectance (ATR–FTIR) spectroscopy and scanning electron microscopy (SEM), it is necessary to dry them through a process of lyophilization to ensure the complete removal of water. To avoid acid contamination of the freeze dryer during the process, the hydrogels obtained from low, medium, and high PVA MWs (HyA, HyB, and HyC, respectively) were washed many times with distilled water to fully remove H2SO4, frozen at −19 °C overnight, and then placed into a Labconco lyophilizer working at −50 °C and 0.850 mbar for 24 h.
For thermal analysis (thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)), the PVA–H2SO4 hydrogels were kept in an oven for 48 h at 50 °C to remove the excess water from the polymeric matrix.
ATR–FTIR analyses were performed using a PerkinElmer Spectrum Two spectrophotometer, equipped with a Universal ATR accessory, with a resolution of 0.5 cm–1 in the range 4000–400 cm–1 using 40 scans.
Morphological observations were carried out on Au-coated specimens using a Renishaw field-emission scanning electron microscope, equipped with an InLens detector, operating at 10 kV and a current of 80 pA.
TGA was carried out under inert atmosphere (N2) heating from 25 to 600 °C, at a scan rate of 20 °C/min using a Netzsch TG 209F1 Libra instrument.
The presence of crystalline domains derived from the freeze–thaw method has been mentioned by different authors employing several techniques, for instance, DSC and wide-angle X-ray diffraction (WAXD).28
DSC analyses were performed with a TA Instruments Q2000, using unsealed aluminum pans, and loading 3 to 5 mg of the crude sample with the treatment carried out in an oven. The DSC analyses were carried out by applying two subsequent heating ramps from 0 to 250 °C (heating/cooling ramps at 10 °C/min). The DSC degree of crystallinity was determined as the ratio between the heat of fusion (ΔHm) of the PVA hydrogel sample (normalized for the mass of the polymer in the hydrogel) and the enthalpy of melting of 100% crystalline PVA (ΔH°m):28
| 1 |
with ΔH°m = 150 J/g.
Moreover, DSC thermograms were employed even to investigate the glass transition temperature (Tg) of the hydrogels after each freeze–thaw cycle.
Moreover, WAXD was used to determine the crystallinity of the freeze–thawed PVA–H2SO4 hydrogels.29,28 WAXD profiles were collected by using a Philips X’Celerator diffractometer equipped with a graphite monochromator. The 2θ range was from 10° to 60°/2 theta with a step size of 0.300° and a time step of 10 s. Cu Kα radiation (40 mA, 40 kV, 1.54 Å) was used.28 WAXD crystallinity (or relative crystallinity) was calculated using eq 2 as reported by Ricciardi et al.:28
| 2 |
where the PVA crystalline peak area is the area of the weak diffraction reflection centered at about 19.4°/2 theta, while the total area is the entire area within the 2θ range of 10–60°. The X’Pert HighScore Plus software was used to evaluate peak areas.
The obtained crystallinity represents a relative value and can be compared to those of other samples if the same testing method is used; however, it does not represent an exact measurement of the crystallinity percentage as the equation is not normalized using known crystallinity values.28
2.5. Mechanical Test
The mechanical tensile properties of PVA–H2SO4 hydrogel membranes were evaluated by an LBG UDI24Pro instrument with a 1 kN load cell at room temperature. The hydrogel membranes were cut into strips with dimensions of 40 mm × 10 mm. Tensile measurements were carried out by uniaxially stretching the hydrogel strip at a rate of 10 mm min–1. The stress σ and the strain ε were obtained by the following formulas, respectively:2
| 3 |
where F (kN) is the force and A is the cross-section area (mm2), and
| 4 |
where L0 is the initial length and L is the length of the specimen in tension. Young’s modulus was calculated from the average slope of the linear portion (1–5% strain) of stress versus strain curves. A compression test was performed on the same instrument with a 10 kN load cell at room temperature.29 The samples were approximately 15 mm in diameter and 3 or 5 mm in height. Three independent samples were used for each set of hydrogels.
2.6. Water Content and Swelling Percentage
To obtain the water content (Wc%) of the different PVA–H2SO4 hydrogels, the weight of the hydrogel samples was measured before (Wi) and after (Wd) drying in an oven at 40 °C for 27 h. The water content was calculated as follows:30,31
| 5 |
To estimate the degree of swelling, the dried samples were immersed in distilled water at 25 °C for 72 h to reach swelling equilibrium. The samples were removed from the bath; the excess of unabsorbed water was quickly removed with filter paper before being weighed on an analytical balance. The swelling percentage (Sw%) was calculated according to the following formula:30,31
| 6 |
where Ws is the weight of the swollen gel and Wd is the weight of the dry gel.
To focus on the PVA amount (SPVA%), the swelling data were normalized with respect to the polymer fraction (f) in the hydrogel according to the following formula:
| 7 |
The porosity of the dried samples was determined from eq 8 by measuring the adsorbed volume of cyclohexane versus the total volume of each sample using a pycnometer:32
| 8 |
where md is the mass of the dry gel, mw is the mass of the wet hydrogel in cyclohexane, m1 is the mass of the apparatus filled with cyclohexane, and m2 is the mass of the apparatus with cyclohexane and hydrogel.
2.7. Measurement of Moisture Content
The moisture content (Mn%) of the hydrogels was measured gravimetrically using eq 9:33
| 9 |
in which Wi is the initial wet weight of the sample and Wd is weight of the sample after drying.
2.8. Rheological Characterization
The rheological properties of PVA–H2SO4 hydrogels were determined using an MCR 102 parallel-plate rheometer (Anton Paar, Graz, Austria) in plate–plate geometry with a diameter of 25 mm (PP-25 plate) and a gap of 1.2 mm. Disks of the hydrogel with a diameter of 25 mm and a thickness of 1.2 mm were deposited onto the plate of the rheometer. Subsequently, the upper plate was lowered until it made contact with the surface of the sample. The excess material was removed with a spatula, and the trap was filled with distilled water to avoid evaporation phenomena. An oscillatory frequency sweep test was carried out to assess the self-healing property of hydrogels at 25 °C in a frequency range from 500 to 0.1 rad s–1, with a fixed strain value equal to 1%. Oscillatory amplitude sweep analysis was performed at 25 °C to assess the stress value at which chains flow and the interactions are broken. The frequency value was kept constant at 1 rad s–1, while the amplitude of the shear strain varied from 0.01 to 1000%. Finally, a three-interval thixotropic test (3ITT) was carried out in controlled shear rate (CSR) mode to assess the hydrogel’s mechanical properties after the application of high shear rates: the rest condition of the hydrogel was simulated by applying a low shear rate (0.1 s–1) at 25 °C; then, a high shear rate (100 s–1) was applied to simulate the breaking of chain interactions, and finally, the resting conditions were restored to evaluate the % recovery of material properties. The value of the high shear rate applied (100 s–1) was chosen on the basis of the amplitude sweep test results.
2.9. Ionic Conductive Properties and Galvanostatic Charge/Discharge Curves
The ionic conductive properties of hydrogel membranes were analyzed by electrochemical impedance spectroscopy (EIS) using an Autolab GSTAT128 N potentiostat/galvanostat (Metrohm-Autolab) controlled by the NOVA 2.10 software. The electrochemical measurements were carried out in a two-electrode Swagelok-type cell, and the samples were sandwiched between the two 316 stainless steel electrodes with a testing diameter of 1.0 cm (area = 0.785 cm2, Figure S2). The electrochemical impedance study was conducted at room temperature, at an AC voltage amplitude of 10 mV, and within the frequency range of 0.01–105 Hz. The impedance was obtained from the Nyquist plot of the hydrogels. The ionic conductivity (σc, S cm–1) was calculated with the following equation:2
| 10 |
where s is the thickness of the sample (cm), R is the ionic resistance (Ω), and A (cm2) is the surface of the analyzed sample that is in contact with the electrodes.
A galvanostatic charge/discharge (GCD) test was carried out on the self-healing double-layer hydrogel at a current density of 0.025 mA cm–2 in a voltage range from 0 to 0.9 V. The specific capacitance (Cp) of HyPVA–PANI_PAMPSA/HyB#1 was evaluated from GCD curves according to the equation
| 11 |
where I (A) is the discharge current, A (cm2) is the surface of the hydrogel of the semicell, Δt is the difference between the end-of-discharge time and the end-of-charge time, and ΔV is the difference between the end-of-charge potential and the end-of-discharge potential.34
3. Results and Discussion
3.1. PVA–H2SO4 Hydrogel Preparation and Characterization
The PVA–H2SO4 hydrogel electrolytes were easily synthesized by physical cross-linking through hydrogen bond formation using a freeze–thaw method.20 The PVA MW and the percentage amount of polymer employed in the hydrogel formation (PVA/H2SO4 weight ratio) are the two key parameters whose effects are analyzed in this paper in terms of the water content and mechanical and conductive properties of the obtained hydrogels. On the opposite, the PVA hydrolysis degree % and the number of freeze–thaw cycles are always kept constant. The descriptions of the different samples are reported in Table 2.
Table 2. PVA–H2SO4 Samples (Three Freeze–Thaw Cycles).
| hydrogel sample | PVA MW | PVA/H2SO4 weight ratio (w/w) | PVA% | hydrogel formation |
|---|---|---|---|---|
| HyA#1 | 13,000–23,000 | 1/5.8 | 17 | no |
| HyB#1 | 31,000–50,000 | 1/5.8 | 17 | yes |
| HyC#1 | 146,000–186,000 | 1/5.8 | 17 | no |
| HyA#2 | 13,000–23,000 | 1/3 | 33 | yes |
| HyB#2 | 31,000–50,000 | 1/3 | 33 | yes |
| HyC#2 | 146,000–186,000 | 1/10 | 9 | yes |
Samples HyA#1, HyB#1, and HyC#1 were prepared starting from PVA with different MWs, with a fixed PVA/H2SO4 weight ratio of 1/5.820 and three freeze–thaw cycles, as reported by Ma et al.2 and described in the Experiments section. During dissolution, A-PVA and B-PVA give, in the acidic medium, a transparent solution in a few minutes (10–15 min). C-PVA tends to agglomerate and creates a gelatinous sticky suspension mixed with residual solid. After three freeze–thaw cycles, a whitish and opaque PVA–H2SO4 hydrogel can be obtained only with B-PVA (HyB#1). HyA#1 appears partially jellified, and HyC#1 is completely solidified. Several attempts have led to the optimized formation of hydrogels with C-PVA only by decreasing the PVA/H2SO4 weight ratio up to 1/10 (HyC#2), whereas a ratio of 1/3 was enough to obtain a hydrogel starting from low- and medium-MW PVA (HyA#2, HyB#2). Finally, samples HyB#1 and HyB#2 have the same MW but different PVA/H2SO4 weight ratios, whereas samples HyB#2 and HyA#2 have the same PVA/H2SO4 weight ratio but different MWs.
The morphology of the obtained hydrogels was observed by SEM of the lyophilized samples;29 the images are reported in Figure 1.
Figure 1.

Scanning electron micrographs of lyophilized hydrogels; the scale bar indicates 2 μm.
Pores, cracks, and droplets are evident in HyA#2, HyB#1, and HyB#2. As water freezes during the freeze–thaw cycles, the microstructure of the PVA phase undergoes remodeling, leading to the formation of regions with a concentrated polymer. Additionally, during each cycle, the water freezes and expands, pushing the PVA chains into close contact with each other. This mechanism decreases the distance between the PVA chains, facilitating physical cross-links among the PVA chains via hydrogen bonding and formation of crystalline regions.29 The addition of a higher amount of H2SO4 (HyC#2) promotes an improvement of the structural integrity with no phase separation and formation of a more compact structure.
The ATR–FTIR spectra of lyophilized samples and pristine PVA are shown in Figure 2. The presence of the typical bands of PVA at 3400 cm–1 (−OH stretching), 2950 cm–1 (−CH2 stretching), and 1090 cm–1 (C–O stretching) confirms the hydrogel structure. Moreover, it is possible to observe a band at 1142 cm–1, associated with the crystalline domains of PVA.35
Figure 2.

ATR–FTIR spectra of HyA#2, HyB#1, HyB#2, and HyC#2. Inset: spectrum of PVA.
The DSC curves related to the first heating and cooling ramp of PVA–H2SO4 hydrogels are reported in Figure 3A in the range 150–250 °C. All thermograms highlighted a peak in the range 210–240 °C in the heating cycle deriving from the melting of crystalline domains formed during the freeze–thaw process and a subsequent recrystallization process in the cooling ramp. As expected, a slight shift was observed increasing the MW of the PVA while in the case of HyB no significant changes were revealed modifying the polymer amount in the samples. The crystallinity degree was calculated from the melting enthalpy followingeq 1;23 the data are reported in Figure 3B. Analysis of the DSC crystallinity data results showed that the final hydrogels exhibited a low crystallinity degree (7–9%).
Figure 3.

(A) DSC thermograms in the range 150–250 °C for each prepared hydrogel; (B) melting temperature (°C), melting enthalpy (J/g), and crystallinity (%) of different gels obtained by DSC and WAXD.
The relative crystallinity was also measured using WAXD, as reported in the literature.28 In fact, the gelation process results in the formation of a porous network in which polymer crystallites act as junction points; however, the microcrystalline domains are very small, and their extent increased on increasing the number of cycles, the aging of the hydrogels, and the solvent content. The collected WAXD spectra are shown in Figure S3: all of the diffractograms exhibit two halos centered at about 25 and 42°/2 theta, even if the diffraction profiles are slightly different due to different PVA MWs. The weak peak occurring at 19.5 ± 0.1°/2 theta, in agreement with the literature, demonstrates the presence of a low amount of crystalline PVA aggregates, and it is clearly observable only for HyA#2 and HyB#2 samples, while sample HyB#1 shows only a barely visible reflection, demonstrating a strong correlation between crystallinity, PVA concentration, and MW. In fact, as reported by Ricciardi et al., the characteristic pattern leading to the determination of the extent of crystallinity can be clearly observed by means of XRD only for hydrogels with a PVA content over the range 10–15% (w/w) when the MW is about 115,000.36 The absence of the PVA crystalline peak in the pattern of HyC#2 and the broad reflection obtained from HyB#1 confirmed this statement, as the hydrogels are obtained respectively from a 9% (w/w) polymer solution and from one with a higher concentration but a lower MW (see Table 2). The relative WAXD crystallinity was calculated according to eq 2; the values reported in Figure 3B are in agreement with the range reported in the literature29 (from 0.03 to 5.3, depending on the polymer concentration and number of cycles performed); no relative crystallinity was calculated for the HyC#2 sample. Moreover, the increase in the amorphous polymer content in HyB#1 and HyC#2 can be due to the increase in the amount of sulfuric acid present in the hydrogel (PVA/H2SO4 weight ratio = 1/5.8 and 1/10).34
Gel porosity and pore size are strictly related to the number of freeze–thaw cycles, PVA concentration, and PVA MW.29 As the number of cycles was kept constant, the differences observed in the samples investigated can be attributed to the PVA/H2SO4 weight ratio or to the PVA MW used. Data reported in Figure 4A show a decrease in the percentage porosity (calculated from eq 8) when changing the PVA/H2SO4 weight ratio from 1/5.8 to 1/3 at the same MW (compare samples HyB#1 and HyB#2), whereas an increase of the MW, keeping the PVA/H2SO4 weight ratio constant, leads to a decrease in the porosity (compare samples HyA#2 and HyB#2). A similar porosity was obtained for samples HyB#2 and HyC#2.
Figure 4.

(A) Porosity, (B) water content (Wc), and (C) swelling ratio (Sw) results.
The water content (Wc%) and the swelling percentage (SW%) calculated using eqs 5 and 6(8,37) are reported in Figure 4B,C. The results of the swelling percentage normalized with respect to the polymer fraction (SPVA%), calculated using eq 7,37 are 10, 11, 3, and 22 for HyA#2, HyB#1, HyB#2, and HyC#2, respectively. During the swelling phase, the water molecules penetrate the polymer network and coordinate themselves with the oxidrilic moieties of the macromolecular chains, with a consequent increase of the hydrogel volume. Therefore, the whole increase in the hydrogel volume after swelling is due to the increase in volume of the non-crystalline (amorphous) regions of PVA.37 From the data in Figure 4 and Figure S4, it appears evident that SPVA decreases with the increase of the crystallinity of the material, and a higher value is obtained for HyC#2, the hydrogel with a PVA/H2SO4 weight ratio of 1/10 and a higher PVA MW.
The thermal stability of freeze–thaw hydrogels was evaluated by TGA (Figure 5). Water is present in different states inside the PVA hydrogels, i.e., absorbed water molecules (weakly bound), which prefer to be on the external or internal surface without interacting with the matrix, and water molecules strongly bound to the hydroxyl groups. The TGA curves of PVA–H2SO4 hydrogels in a nitrogen atmosphere exhibit three weight loss regions, which are similar to those reported in the literature.38,37 The first region between 50 and 200 °C can be attributed to the loss of the absorbed water molecules, while the second region between 200 and 340 °C is related to the loss of water bound to the polymer matrix. The third region between 340 and 450 °C is associated with the decomposition of the polymer.39
Figure 5.
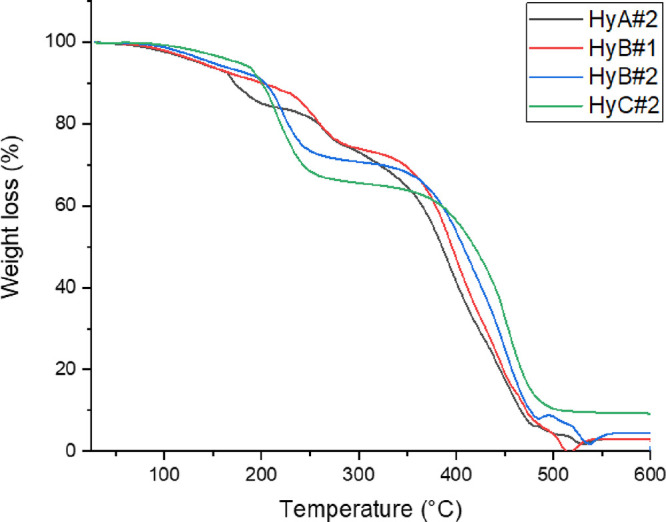
TGA curves of the prepared hydrogels.
The increase in the degradation temperature observed in the third step for HyB#1 and HyC#2 with respect to HyA#2 can be attributed to the increase of the PVA MW; the breaking of the polymeric chains occurs at a higher temperature than the breaking of shorter macromolecules. Comparison of HyB samples showed that the increase of PVA% in the hydrogel led to a slight shift of the final degradation step toward higher temperatures.
Moreover, to investigate the glass transition temperature (Tg) of PVA–H2SO4 hydrogels during the three freeze–thaw cycles, DSC thermograms were used.37,40,41 The DSC measurements were carried out after each freeze–thaw cycle (three in total) on all of the hydrogels previously treated at 50 °C for 48 h to remove the excess water. The midpoint of the step change was used to evaluate the glass transition temperature (Tg). Since the heating/cooling DSC cycles can change the thermal properties of physically cross-linked materials due to the breaking of weak bonds, only the Tg values related to the first DSC heating scan are reported in Table S1 for each freezing cycle investigated.
Due to the different consistence of HyA#2, the first freeze–thaw cycle cannot be used to determine the Tg of this sample, and only in this case do the measurements start from the second cycle. The increase in Tg observed as a function of the number of freeze–thaw cycles can be ascribed to the physical cross-links provided upon hydrogen bond formation between PVA chains and consequent decrease in the free volume throughout amorphous regions.41 For HyA#2 and HyB#1, the Tg values slightly increase with the number of freezing cycles, confirming the rising of the cross-linking degree during the process, whereas for HyC#2, no significant changes in the Tg can be observed and the value is similar to that obtained with HyB#1 after three cycles. This can be explained from the high number of conformations that a polymer can assume; with increasing MW, the number of polymer conformations also becomes higher, and therefore, statistically, the favorable conformations to form efficient cross-linking interactions decrease. This explanation is in keeping with the observed trend, where the shift of the Tg after each cycle (i.e., from 24 to 29 °C for HyA#2) is more prominent for lower-MW hydrogels. Moreover, the samples HyB#1 and HyB#2 prepared with different PVA weight percentages (w/w%) highlighted no significant change, demonstrating that this parameter does not influence the thermal properties of the materials; the result is in agreement with the TGA and DSC data described previously. Furthermore, as expected, the different Tg values observed for the three samples derived from the different MWs of the PVA used.
3.2. Mechanical Properties
A uniaxial tensile test was conducted to investigate the hydrogel’s mechanical properties. To have a stable sample, each hydrogel was freshly prepared 3 days prior to the test as hydrogels tend to dehydrate after a few hours from their preparation and then reach an equilibrium, which stabilizes them for about 3 weeks.42 The tensile tests were conducted on rectangular samples at room temperature and 70.5% RH. As shown in Figure S5A, HyC#2 results in a hydrogel with a higher tensile stress at break. The data are reported in Table 3. Due to their fragility, samples HyA#2 and HyB#2 did not provide reliable results.
Table 3. Mechanical Characterization of Investigated Hydrogel Samples (Average of Three Samples).
| sample | strain (%) | tensile strength (kPa) | toughness (kPa/m3) | Young’s modulus (kPa) | compressive strength (kPa) |
|---|---|---|---|---|---|
| HyB#1 | 24 ± 4 | 14 ± 2 | 2.39 ± 0.03 | 60 | 78 |
| HyC#2 | (1.6 ± 0.5) × 102 | (1.4 ± 0.5) × 102 | (1.1 ± 0.7) × 102 | 41 | 53 |
As observable from Table 3, sample HyB#1 shows a lower tensile strength due to the presence of shorter chains, which can easily slip when exposed to stress. In other words, the free ends of chains act as defects in the system and reduce the mechanical strength.34 Moreover, it is known from the literature that the presence of an acidic electrolyte in the hydrogel increases its elongation at break with respect to that of the pure PVA hydrogel.34 The bonds between polymer chains are weakened by the acid at the molecular level, and at the same time, the free volume or interchain spaces are increased, making the polymer chains more flexible. The increase of the cross-linking degree due to the increase of the −OH groups in the same macromolecule as in HyC#2 allows an alignment and stretching of the linear macromolecules of the hydrogel and improves the overall mechanical strength.43
The compression test was conducted with circular samples, each about 1.4–1.5 cm in diameter. The compression strength was calculated from the strain/stress curve at a 60% compressive strain ratio. From the curves reported in Figure S5B, it is possible to observe the decrease of the compressive strain with the decrease of the MWs of PVA used.
3.3. Electrochemical Properties
The ionic conductivity of the hydrogel is a very important parameter for possible use in all-in-one supercapacitors. The ionic conductivity of the PVA–H2SO4 hydrogels was calculated at room temperature, and the resistance was obtained from the Nyquist plots.38 From Figure 6A, the bulk resistance R, which is the intercept on the real part Z′ axis in the high-frequency region, can be obtained. The ionic conductivity through the plane is calculated for the samples using the respective R values (eq 8). The conductive and resistance data as well as the porosity %, calculated with eq 7, are reported in Figure 6B,C. The ionic conductivity reported in the literature2 for similar samples (PVA/H2SO4 1 M) is 580 mS cm–1.
Figure 6.

(A) Nyquist plots; inset: enlargement of the high-frequency region. (B) Ionic conductivity (σc) and (C) resistance values of HyA#2, HyB#1, HyB#2, and HyC#2.
With the rise in PVA MW at a constant PVA/H2SO4 weight ratio (samples HyA#2 and HyB#2), the physical interaction between polymer chains and bulk resistance of the hydrogel are increased. Although the high MW relates to high viscosity, the amount of free end of chains and free volume of the polymer are decreased, resulting in low ionic motion, which leads to a decrease in ionic conductivity.34,44 Moreover, at a low polymer concentration at the same MW (samples HyB#1 and HyB#2), the enhancement of the ionic conductivity can be explained by the “breathing polymeric chain model”.34 According to this model, a gel electrolyte is composed of polymer chains, solvent, and dissociate or aggregate ions.
At a low PVA concentration, the gel electrolyte behaves as a liquid electrolyte, and the polymer chains participate in the charge-transfer process, contributing to the higher ionic conductivity.34 At the same time, the change in the PVA/H2SO4 weight ratio affects the ion’s mobility or the increase or decrease of channels for ion transfer by changing the viscosity.34 In a dilute polymer solution, the polymeric chains do not attract one another, while in a concentrated polymer solution, the polymeric coils are overlapped. PVA with a higher MW has more chain entanglements, holding more water molecules; the higher amount of water molecules leads to a more considerable amount of dissociated free ions, which results in a higher ionic conductivity.34 The higher conductivity observed for HyC#2 (PVA/H2SO4 = 1/10) despite its lower porosity can be justified by the two possible mechanisms of proton transport:38 (i) the hopping (Grotthuss) mechanism and (ii) the vehicle mechanism. According to the former, a proton jumps between the hydrolyzed ionic sides through the hydrogel and the porosity of the sample becomes less influential. In contrast, the free volume between the polymer chains is accountable for the latter mechanism; in this case, the ions are transported by upwardly moving water molecules forming H3O+, which diffuses in the aqueous medium through the free volumes driven by the electrochemical potential gradient.
3.4. Hydrogel Conservation and Aging
The need to keep hydrogels’ properties (mechanical, hydration, conductivity) unaltered over time is crucial to ensure their employment as part of devices. As previously described, all of the hydrogel samples were wrapped with cling film for food and stored at room temperature in sealed food bags until used. It can be seen from Figure 7 that under such conditions, all the hydrogels lose around 70% of moisture up to the first week of study and became almost dry (25%) after 4 weeks. The physical appearance of the hydrogel after 3 months of storage is shown in Figure S6 (HyA#2 and HyC#2), and the stability of hydrogels is closely related with both the %PVA and MW used.
Figure 7.

Moisture % loss profiles of hydrogels with respect to aging time.
The ionic conductivity after 4 weeks results in almost 57% of the initial one (302.2, 219.3, and 458.2 mS cm–1 for HyA#2, HyB#1, and HyC#2, respectively). For checking the possibility of regeneration of the hydrogels, sample HyB#1 (used as a test sample) was dipped in 1.0 M H2SO4 for 24 h, with an increase of the ionic conductivity from 219.3 to 306.6 mS cm–1, corresponding to 79% of the initial value (385.2 mS cm–1).
3.5. Self-Healing Double-Layer Hydrogel
A sandwiched all-in-one electrode/electrolyte hydrogel configuration was prepared by exploiting the self-healing ability of these hydrogels and the freeze–thaw physical method. PVA-based self-healing hydrogels, obtained using the freezing–thawing method, are widely reported in the literature.45−47 For both the hydrogel layers used, the HyB#1 conditions (Table 1) were used. The first layer contains only PVA (HyB#1), whereas the second one contains PVA and the conductive PANI_PAMPSA polymers with a 50% w/w ratio (HyPVA–PANI/PAMPSA) characterized by a deep green color due to the presence of the conductive emeraldine form of PANI.48 The all-in-one double-layer hydrogel HyPVA–PANI_PAMPSA/HyB#1 is easily obtained after six freeze–thaw cycles. The double-layer hydrogel has a thickness of 9 mm with two layers of 3 mm for PVA/PANI_PAMPSA, easily recognizable from the characteristic green color, and 6 mm for PVA (see Figure 8A).
Figure 8.
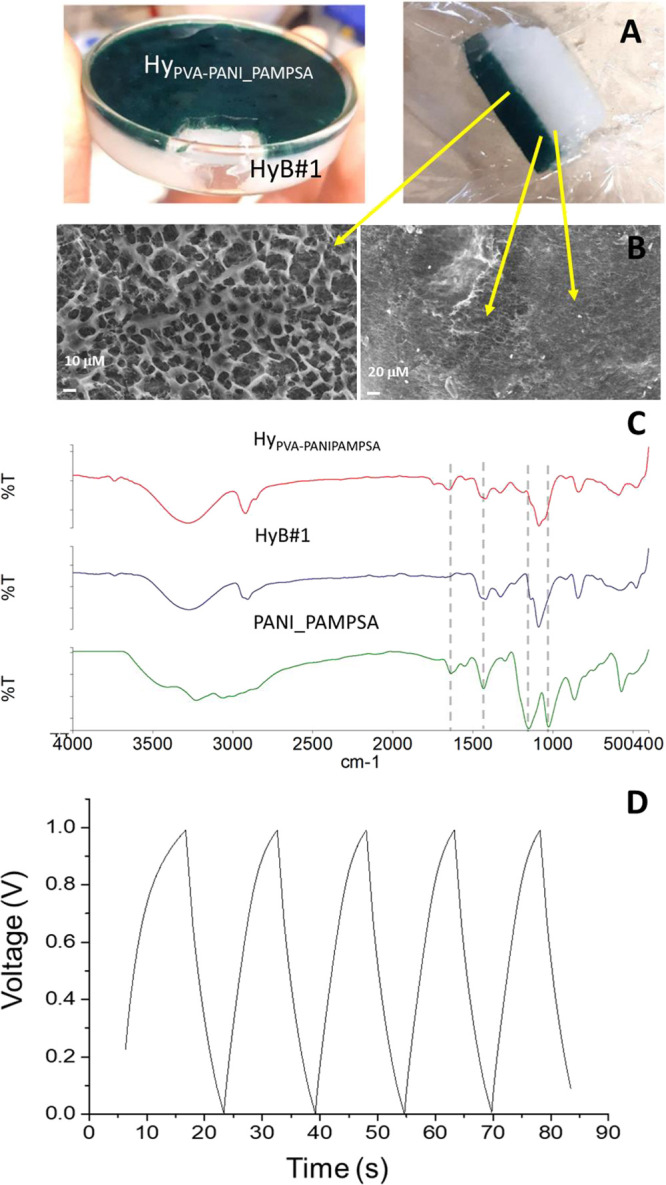
(A) Self-healing double-layer hydrogel HyPVA–PANI_PAMPSA/HyB#1. (B) Micrographic images of lyophilized hydrogel recorded using SEM (magnification of 1000×). (C) ATR–FTIR spectra of HyPVA–PANI_PAMPSA, HyB#1, and PANI_PAMPSA. (D) GCD curves.
The self-healing property was investigated with different tests.45−47 The self-healing hydrogel assembled using the two differently colored hydrogels (green for HyPVA–PANI_PAMPSA and white for HyB#1) was examined visually and with optical and SEM microscopy. Optical (not shown) and SEM images (Figure 8B) show a perfect connection between HyPVA–PANI_PAMPSA and HyB#1 layers. Moreover, the self-healing double-layer hydrogel was stretched to check for resistance against stretching and splitting. The tensile measurements were carried out under the conditions previously reported, and a maximum tensile strain (%) of 53 ± 4 was obtained without splitting.
Moreover, according to a previous study,49 a self-healing material should satisfy three requirements from a rheological point of view: (1) exhibit a terminal flow pseudoplastic behavior, (2) be characterized by a chain flow relaxation time (τf) on a reasonable time scale, and (3) behave as a viscous fluid at low frequencies. To verify the self-healing nature of the developed PVA–H2SO4 hydrogel, a rheological frequency sweep was conducted at 25 °C, and the dynamic viscosity (η′) curve (Figure S7A) showed that η′ tends to a plateau value at low frequencies. The absolute value of the slope of the initial η′ calculated for this sample is equal to 0.07, confirming the Newtonian character of the hydrogel at a low frequency, typical of self-healing materials.49 Indeed, self-healing hydrogels behave as pseudoplastic fluids, with the η′ trend increasing and then showing a plateau in the lower Newtonian flow region (LNFR) at a low frequency. Furthermore, frequency sweep tests were conducted, and the storage modulus (G′) and loss modulus (G″) were recorded as they provide important information about the chain relaxation behavior of self-healing materials (Figure S7B). Generally, a polymeric hydrogel is characterized by G′ > G″, exhibiting a plateau across the entire range of measured frequencies. For self-healing materials, a crossover between the two moduli (G′ = G″) is observed, and G″ becomes higher than G′ approaching low frequency values, indicating a behavior more akin to a polymeric solution rather than a gel-like network. The results reported in Figure S7B suggest that the dynamic bond-based gel network forms a pseudostructure with minimal steric hindrance for the chain flow, owing to the short lifetime of the dynamic bond. Indeed, it is possible to calculate the value of τf, which is given by the reciprocal value of the crossover frequency. No self-healing materials exhibit extremely low chain relaxation times, making it impossible to observe and calculate τf. A τf value of 0.03 s was calculated for the PVA–H2SO4 hydrogel, confirming that the chain flow relaxation strongly contributes to self-healing on a reasonable time scale. To deeply characterize the PVA–H2SO4 hydrogel, the trend of the loss factor (tan δ) was reported as a function of the angular frequency, providing further insights into the nature of the polymer network. If tan δ exhibits a downward (or upward) trend, the polymeric system becomes more solid-like (or liquid-like). This trend is particularly important at low frequencies since self-healing occurs primarily in the static state. Specifically, when the tan δ value exceeds 1, the liquid component of the material is predominant over the solid one. Figure S7C clearly shows an upward trend of tan δ, confirming the hydrogel liquid-like behavior, as a consequence of the chain mobility and/or bond exchange, linked to the self-healing property. Taken together, the obtained results indicate that the PVA-based hydrogel exhibits a terminal flow pseudoplastic behavior, is characterized by a finite τf, and behaves as a viscous fluid at low frequencies; therefore, it can be classified as a self-healing material.
Finally, an oscillatory amplitude sweep test was performed to determine the breaking point of the PVA–H2SO4 hydrogel. The crossover between G′ and G″ occurred at 4.7% shear strain and at 8.2 Pa of shear stress (Figure S7D). This information was utilized to perform the 3ITT analysis (Figure S7E). The first and third intervals simulate static conditions (shear rate = 0.1 s–1), while the second interval simulates the breaking of bond interactions. The shear rate applied (100 s–1) corresponds to a shear stress value equal to 17 Pa, which falls in the region where G″ > G′ in the amplitude sweep test, corresponding to the liquid-like range where chains are able to flow and the interactions are broken. From the resulting 3ITT curve, it was possible to calculate the % recovery of the viscosity of the hydrogel after the high shear rate applied, which amounted to 64% after 2 min.
Figure 8C reports the ATR–FTIR spectrum of lyophilized HyPVA–PANI_PAMPSA. By comparison with HB#1 and PANI_PAMPSA spectra, the presence of the typical bands of PVA and the bands at 1641 and 1032 cm–1 attributed to the C=O stretching and to the symmetric O=S=O stretching of the carbonyl and sulfonic groups of PAMPSA, and at 1546 and 1440 cm–1 for quinoid and benzenoid groups, is evident.50
The self-healing double-layer hydrogel was investigated as a semicell for flexible energy storage. GCD curves were recorded (Figure 8D) and showed symmetric triangular shapes, with a relatively short charge/discharge time, indicating a capacitance close to that of a common capacitor. The specific capacitance (Cp) of HyPVA–PANI_PAMPSA/HyB#1, evaluated from GCD curves according to eq 11, results in 0.297 ± 0.002 mF cm–2, at a current density of 0.025 mA cm–2. The conductivities obtained by EIS measurements are reported in Table 4.
Table 4. Ionic Conductivity Values of HyPVA–PANI_PAMPSA and HyPVA–PANI_PAMPSA/HyB#1, Obtained with Six Freeze–Thaw Cycles.
| sample | porosity (%) | ionic conductivity, σc (mS cm–1) |
|---|---|---|
| HyPVA–PANI_PAMPSA | 17 | 676.0 ± 0.1 |
| HyB#1 | 17 | 261.2 ± 0.2 |
| HyPVA–PANI_PAMPSA/HyB#1 | 439.7 ± 0.1 |
The values of Cp reported in the literature for supercapacitors based on hydrogel and PANI5,51,52 (Table S2) are orders of magnitude higher than that of our device. This is because the current stage of development of the fabrication technique allows only the preparation of asymmetric devices. The performance of the entire device is compromised as one electrode is metallic and cannot accommodate a significant charge, resulting in capacity loss. To achieve better performance, we are studying the freeze–thaw procedure in order to produce a symmetric supercapacitor with a more balanced capacitance between the two electrodes.
As expected, the HyPVA–PANI_PAMPSA layer shows a higher conductivity than HyB#1, due to the presence of both the ionic conductor (H2SO4) and the conductive polymer. The decrease of the conductivity observed in HyB#1 with increasing freeze–thaw cycles during the preparation (i.e., from 385.2 to 261.2 increasing from three to six cycles) can be ascribed to an increase of cross-linking between the polymeric chains that reduces the porosity of the material. However, the presence of PANI_PAMPSA in the hydrogel does not change its porosity.
Finally, the HyPVA–PANI_PAMPSA/HyB#1 double-layer hydrogel remains flexible.
4. Conclusions
A simple one-pot physical cross-linking by freezing–thawing methods was employed to prepare PVA–H2SO4 hydrogels. The effects of the MW as well as the PVA amount (inversely correlated with the H2SO4 amount) on the mechanical properties and ionic conductivity were investigated. From the experimental data obtained, it was found that the increase of H2SO4 amount (when a lower quantity of PVA is used) in the hydrogels results in a decrease in the elastic modulus but an increase in the elongation at break and the specimen’s toughness or resistivity under crack propagation. The increase of PVA MW had a significant effect on the mechanical properties of the hydrogel due to the increase of the cross-linking degree with the increasing amount of the OH– groups in PVA, which allows alignment and stretching of the linear macromolecules of the hydrogel. HyC#2 presents a higher ionic conductivity (802.9 ± 0.1 mS cm–1).
Our data clearly demonstrate that ionic conductivity and mechanical properties can be easily tuned by the PVA/H2SO4 ratio and PVA MW.
Additionally, the self-healing properties of the hydrogel, its easy functionalization, and the freeze–thaw method employed here allow for a simple preparation of a physically cross-linked dual-network integrated structure. The good conductivity and self-healing ability exhibited by the PVA–PVA/PANI_PAMPSA double-layer hydrogel make it a good candidate for the formulation of an all-in-one flexible and wearable energy storage device.
Acknowledgments
The authors wish to thank Dr. Fabrizio Tarterini and Dr. Emanuele Maccaferri, Department of Industrial Chemistry “Toso Montanari”, for SEM microscopy images and for providing mechanical measurements, respectively; Dr. Danilo Arcangeli for the 3D-printed Swagelok-type cell; and Prof. Maria Letizia Focarete for rheological experiment discussions. This research was supported by MIUR, Next Generation EU, and Italia Domani Piano Nazionale di Ripresa e Resilienza.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05392.
Preparation of PANI-PAMPSA, preparation of hydrogel membranes, additional material characterization (XRD patterns; plots of SPVA as a function of WAXD crystallinity, PVA MW, and PVA/H2SO4 w/w ratio; Tg values; stress/strain curves), and additional electrical measurements (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Renewables 2021 Global Status report (https://www.reportlinker.com/report-summary/Renewable-Energy/56034/Global-Renewable-Energy-Industry.html).
- Ma W. B.; Zhu K. H.; Ye S. F.; Wang Y.; Guo L.; Tao X. Y.; Guo L. T.; Fan H. L.; Liu Z. S.; Zhu Y. B.; Wei X. Y. A Self-Healing Hydrogel Electrolyte towards All-in-One Flexible Supercapacitors. J. Mater. Sci. Mater. Electron. 2021, 32, 20445–20460. 10.1007/s10854-021-06555-5. [DOI] [Google Scholar]
- Libich J.; Máca J.; Vondrák J.; Čech O.; Sedlaříková M. Supercapacitors: Properties and Applications. J. Energy Storage 2018, 17, 224–227. 10.1016/j.est.2018.03.012. [DOI] [Google Scholar]
- Winter M.; Brodd R. J. What Are Batteries, Fuel Cells, and Supercapacitors?. Chem. Rev. 2004, 104, 4245–4269. 10.1021/cr020730k. [DOI] [PubMed] [Google Scholar]
- Zou Y.; Chen C.; Sun Y.; Gan S.; Dong L.; Zhao J.; Rong J. Flexible, All-Hydrogel Supercapacitor with Self-Healing Ability. Chem. Eng. J. 2021, 418, 128616 10.1016/j.cej.2021.128616. [DOI] [Google Scholar]
- González A.; Goikolea E.; Barrena J. A.; Mysyk R. Review on Supercapacitors: Technologies and Materials. Renewable Sustainable Energy Rev. 2016, 58, 1189–1206. 10.1016/j.rser.2015.12.249. [DOI] [Google Scholar]
- Kotz R.; Carlen M. Principles and Applications of Electrochemical Capacitors. Electrochim. Acta 2000, 45, 2483–2498. 10.1016/S0013-4686(00)00354-6. [DOI] [Google Scholar]
- Haraguchi K.; Takehisa T. Nanocomposite Hydrogels: A Unique Organic-Inorganic Network Structure with Extraordinary Mechanical, Optical, and Swelling/De-swelling Properties. Adv. Mater. 2002, 14 (16), 1120–1124. . [DOI] [Google Scholar]
- Kopeček J. Hydrogel Biomaterials: A Smart Future?. Biomaterials 2007, 28, 5185–5192. 10.1016/j.biomaterials.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T.; Liao W.; Yu L.; Zhu J.; Wu M.; Peng Q.; Han L.; Zeng H. Recent Progress in Conductive Self-Healing Hydrogels for Flexible Sensors. J. Polym. Sci. 2022, 60, 2607–2634. 10.1002/pol.20210899. [DOI] [Google Scholar]
- Ahmed E.; Hydrogel M. Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121. 10.1016/j.jare.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir S.; Hina M.; Iqbal J.; Rajpar A. H.; Mujtaba M. A.; Alghamdi N. A.; Wageh S.; Ramesh K.; Ramesh S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 1–60. 10.3390/polym12112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.; Jiang C.; Sun N.; Tan D.; Li Q.; Bi S.; Song J. Recent Progress in Multifunctional Hydrogel-Based Supercapacitors. J. Sci. Adv. Mater. Devices 2021, 6, 338–350. 10.1016/j.jsamd.2021.06.002. [DOI] [Google Scholar]
- Zhang J.; Zhang Q.; Liu X.; Xia S.; Gao Y.; Gao G. Flexible and Wearable Strain Sensors Based on Conductive Hydrogels. J. Polym. Sci. 2022, 60, 2663–2678. 10.1002/pol.20210935. [DOI] [Google Scholar]
- Green R. A.; Hassarati R. T.; Goding J. A.; Baek S.; Lovell N. H.; Martens P. J.; Poole-Warren L. A. Conductive Hydrogels: Mechanically Robust Hybrids for Use as Biomaterials. Macromol. Biosci. 2012, 12, 494–501. 10.1002/mabi.201100490. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Yu R.; Guo B. Stimuli-Responsive Conductive Hydrogels: Design, Properties, and Applications. Mater. Chem. Front. 2021, 5, 2092–2123. 10.1039/D0QM00868K. [DOI] [Google Scholar]
- Liu K.; Wei S.; Song L.; Liu H.; Wang T. Conductive Hydrogels - A Novel Material: Recent Advances and Future Perspectives. J. Agric. Food Chem. 2020, 68, 7269–7280. 10.1021/acs.jafc.0c00642. [DOI] [PubMed] [Google Scholar]
- Madduma-Bandarage U. S. K.; Madihally S. V. Synthetic Hydrogels: Synthesis, Novel Trends, and Applications. J. Appl. Polym. Sci. 2021, 138, 1–23. 10.1002/app.50376. [DOI] [Google Scholar]
- Peppas N. A. Turbidimetric Studies of Aqueous Poly(Vinyl Alcohol) Solutions. Die Makromol. Chemie 1975, 176, 3433–3440. 10.1002/macp.1975.021761125. [DOI] [Google Scholar]
- Adelnia H.; Ensandoost R.; Shebbrin Moonshi S.; Gavgani J. N.; Vasafi E. I.; Ta H. T. Freeze/Thawed Polyvinyl Alcohol Hydrogels: Present, Past and Future. Eur. Polym. J. 2022, 164, 110974 10.1016/j.eurpolymj.2021.110974. [DOI] [Google Scholar]
- Peng H.; Lv Y.; Wei G.; Zhou J.; Gao X.; Sun K.; Ma G.; Lei Z. A Flexible and Self-Healing Hydrogel Electrolyte for Smart Supercapacitor. J. Power Sources 2019, 431, 210–219. 10.1016/j.jpowsour.2019.05.058. [DOI] [Google Scholar]
- Wan W. K.; Campbell G.; Zhang Z. F.; Hui A. J.; Boughner D. R. Optimizing the Tensile Properties of Polyvinyl Alcohol Hydrogel for the Construction of a Bioprosthetic Heart Valve Stent. J. Biomed. Mater. Res. 2002, 63, 854–861. 10.1002/jbm.10333. [DOI] [PubMed] [Google Scholar]
- Hassan C. M.; Peppas N. A. Structure and Morphology of Freeze/Thawed PVA Hydrogels. Macromolecules 2000, 33, 2472–2479. 10.1021/ma9907587. [DOI] [Google Scholar]
- Hu M.; Li Z.; Li G.; Hu T.; Zhang C.; Wang X. All-Solid-State Flexible Fiber-Based MXene Supercapacitors. Adv. Mater. Technol. 2017, 2, 1–6. 10.1002/admt.201700143. [DOI] [Google Scholar]
- Sarihan A. Development of High-Permeable PSf/PANI-PAMPSA Composite Membranes with Superior Rejection Performance. Mater. Today Commun. 2020, 24, 101104 10.1016/j.mtcomm.2020.101104. [DOI] [Google Scholar]
- Puggioni G.; Abd-Razak N. H.; Amura I. F.; Bird M. R.; Emanuelsson E. A. C.; Shahid S. Preparation and Benchmarking of Highly Hydrophilic Polyaniline Poly(2-Acrylamido-2-Methyl-1-Propanesulfonic Acid) PANI PAMPSA Membranes in the Separation of Sterols and Proteins from Fruit Juice. Food Bioprod. Process. 2022, 134, 109–120. 10.1016/j.fbp.2022.05.008. [DOI] [Google Scholar]
- Ragazzini I.; Gualandi I.; D’Altri G.; Di Matteo V.; Yeasmin L.; Cassani M. C.; Scavetta E.; Bernardi E.; Ballarin B. Polyaniline/Poly (2-Acrylamido-2-Methyl-1-Propanesulfonic Acid) Modified Cellulose as Promising Material for Sensors Design. Carbohydr. Polym. 2023, 316, 121079 10.1016/j.carbpol.2023.121079. [DOI] [PubMed] [Google Scholar]
- Ricciardi R.; Auriemma F.; Gaillet C.; De Rosa C.; Lauprêtre F. Investigation of the Crystallinity of Freeze/Thaw Poly(Vinyl Alcohol) Hydrogels by Different Techniques. Macromolecules 2004, 37, 9510–9516. 10.1021/ma048418v. [DOI] [Google Scholar]
- Holloway J. L.; Lowman A. M.; Palmese G. R. The Role of Crystallization and Phase Separation in the Formation of Physically Cross-Linked PVA Hydrogels. Soft Matter 2013, 9, 826–833. 10.1039/C2SM26763B. [DOI] [Google Scholar]
- Zhang L.; Wang Z.; Xu C.; Li Y.; Gao J.; Wang W.; Liu Y. High Strength Graphene Oxide/Polyvinyl Alcohol Composite Hydrogels. J. Mater. Chem. 2011, 21, 10399–10406. 10.1039/c0jm04043f. [DOI] [Google Scholar]
- Tanpichai S.; Oksman K. Cross-Linked Nanocomposite Hydrogels Based on Cellulose Nanocrystals and PVA: Mechanical Properties and Creep Recovery. Compos. Part A Appl. Sci. Manuf. 2016, 88, 226–233. 10.1016/j.compositesa.2016.06.002. [DOI] [Google Scholar]
- Mecca T.; Ussia M.; Caretti D.; Cunsolo F.; Dattilo S.; Scurti S.; Privitera V.; Carroccio S. C. N-Methyl-D-Glucamine Based Cryogels as Reusable Sponges to Enhance Heavy Metals Removal from Water. Chem. Eng. J. 2020, 399, 125753 10.1016/j.cej.2020.125753. [DOI] [Google Scholar]
- Roy N.; Saha N.; Saha P. Stability Study of Novel Medicated Hydrogel Wound Dressings. Int. J. Polym. Mater. Polym. Biomater. 2013, 62, 150–156. 10.1080/00914037.2011.641697. [DOI] [Google Scholar]
- Alipoori S.; Torkzadeh M. M.; Mazinani S.; Aboutalebi S. H.; Sharif F. Performance-Tuning of PVA-Based Gel Electrolytes by Acid/PVA Ratio and PVA Molecular Weight. SN Appl. Sci. 2021, 3, 1–13. 10.1007/s42452-021-04182-7. [DOI] [Google Scholar]
- Paranhos C. M.; Soares B. G.; Oliveira R. N.; Pessan L. A. Poly(Vinyl Alcohol)/Clay-Based Nanocomposite Hydrogels: Swelling Behavior and Characterization. Macromol. Mater. Eng. 2007, 292, 620–626. 10.1002/mame.200700004. [DOI] [Google Scholar]
- Ricciardi R.; Auriemma F.; De Rosa C.; Lauprêtre F. X-Ray Diffraction Analysis of Poly(Vinyl Alcohol) Hydrogels, Obtained by Freezing and Thawing Techniques. Macromolecules 2004, 37, 1921–1927. 10.1021/ma035663q. [DOI] [Google Scholar]
- Reguieg F.; Ricci L.; Bouyacoub N.; Belbachir M.; Bertoldo M. Thermal Characterization by DSC and TGA Analyses of PVA Hydrogels with Organic and Sodium MMT. Polym. Bull. 2020, 77, 929–948. 10.1007/s00289-019-02782-3. [DOI] [Google Scholar]
- Gomaa M. M.; Hugenschmidt C.; Dickmann M.; Abdel-Hady E. E.; Mohamed H. F. M.; Abdel-Hamed M. O. Crosslinked PVA/SSA Proton Exchange Membranes: Correlation between Physiochemical Properties and Free Volume Determined by Positron Annihilation Spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 28287–28299. 10.1039/C8CP05301D. [DOI] [PubMed] [Google Scholar]
- Scurti S.; Ortolani J.; Ghirri A.; Maccaferri E.; Caretti D.; Mazzocchetti L. Phosphorylated Poly(Vinyl Alcohol) Surface Coatings as Intumescent Flame Inhibitor for Polymer Matrix Composites. Prog. Org. Coatings 2023, 177, 107457 10.1016/j.porgcoat.2023.107457. [DOI] [Google Scholar]
- Xue R.; Xin X.; Wang L.; Shen J.; Ji F.; Li W.; Jia C.; Xu G. A Systematic Study of the Effect of Molecular Weights of Polyvinyl Alcohol on Polyvinyl Alcohol–Graphene Oxide Composite Hydrogels. Phys. Chem. Chem. Phys. 2015, 17, 5431–5440. 10.1039/C4CP05766J. [DOI] [PubMed] [Google Scholar]
- Fathi E.; Atyabi N.; Imani M.; Alinejad Z. Physically Crosslinked Polyvinyl Alcohol-Dextran Blend Xerogels: Morphology and Thermal Behavior. Carbohydr. Polym. 2011, 84, 145–152. 10.1016/j.carbpol.2010.11.018. [DOI] [Google Scholar]
- Stammen J. A.; Williams S.; Ku D. N.; Guldberg R. E. Mechanical Properties of a Novel PVA Hydrogel in Shear and Unconfined Compression. Biomaterials 2001, 22, 799–806. 10.1016/S0142-9612(00)00242-8. [DOI] [PubMed] [Google Scholar]
- Wu X.; Li W.; Chen K.; Zhang D.; Xu L.; Yang X. A Tough PVA/HA/COL Composite Hydrogel with Simple Process and Excellent Mechanical Properties. Mater. Today Commun. 2019, 21, 100702 10.1016/j.mtcomm.2019.100702. [DOI] [Google Scholar]
- Aziz S. B.; Abdullah R. M.; Kadir M. F. Z.; Ahmed H. M. Non Suitability of Silver Ion Conducting Polymer Electrolytes Based on Chitosan Mediated by Barium Titanate (BaTiO3) for Electrochemical Device Applications. Electrochim. Acta 2019, 296, 494–507. 10.1016/j.electacta.2018.11.081. [DOI] [Google Scholar]
- Liu Y.; Hsu S. H. Synthesis and Biomedical Applications of Self-Healing Hydrogels. Front. Chem. 2018, 6, 1–10. 10.3389/fchem.2018.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Xiang L.; Gong L.; Hu W.; Huang W.; Chen Y.; Asha A. B.; Srinivas S.; Chen L.; Narain R.; Zeng H. Injectable, Self-Healing Hydrogel with Tunable Optical, Mechanical, and Antimicrobial Properties. Chem. Mater. 2019, 31, 2366–2376. 10.1021/acs.chemmater.8b04803. [DOI] [Google Scholar]
- Anupama Devi V. K.; Shyam R.; Palaniappan A.; Jaiswal A. K.; Oh T. H.; Nathanael A. J. Self-Healing Hydrogels: Preparation, Mechanism and Advancement in Biomedical Applications. Polymers 2021, 13, 3782. 10.3390/polym13213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Li F.; Ma L.-P.; Cheng H.-M. Advanced Materials for Energy Storage. Adv. Mater. 2010, 22, E28–E62. 10.1002/adma.200903328. [DOI] [PubMed] [Google Scholar]
- Shin M.; Shin S. H.; Lee M.; Kim H. J.; Jeong J. H.; Choi Y. H.; Oh D. X.; Park J.; Jeon H.; Eom Y. Rheological Criteria for Distinguishing Self-Healing and Non-Self-Healing Hydrogels. Polymer. 2021, 229, 123969 10.1016/j.polymer.2021.123969. [DOI] [Google Scholar]
- Shen J.; Shahid S.; Amura I.; Sarihan A.; Tian M.; Emanuelsson E. A. Enhanced Adsorption of Cationic and Anionic Dyes from Aqueous Solutions by Polyacid Doped Polyaniline. Synth. Met. 2018, 245, 151–159. 10.1016/j.synthmet.2018.08.015. [DOI] [Google Scholar]
- Zhao J.; Cao L.; Lai F.; Wang X.; Huang S.; Du X.; Li W.; Lin Z.; Zhang P. Double-Cross-Linked Polyaniline Hydrogel and Its Application in Supercapacitors. Ionics. 2022, 28, 423–432. 10.1007/s11581-021-04251-2. [DOI] [Google Scholar]
- Lai F.; Fang Z.; Cao L.; Li W.; Lin Z.; Zhang P. Self-Healing Flexible and Strong Hydrogel Nanocomposites Based on Polyaniline for Supercapacitors. Ionics. 2020, 26, 3015–3025. 10.1007/s11581-020-03438-3. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.