

Abstract
The relative positioning of cells is a key feature of the microenvironment that organizes cell-cell interactions. To study the interactions between cells of the same or different type, micropatterning techniques have proved useful. DNA Programmed Assembly of Cells (DPAC) is a micropatterning technique that targets the adhesion of cells to a substrate or other cells using DNA hybridization. The most basic operations in DPAC begin with decorating cell membranes with lipid-modified oligonucleotides, then flowing them over a substrate that has been patterned with complementary DNA sequences. Cells adhere selectively to the substrate only where they find a complementary DNA sequence. Non-adherent cells are washed away, revealing a pattern of adherent cells. Additional operations include further rounds of cell-substrate or cell-cell adhesion, as well as transferring the patterns formed by DPAC to an embedding hydrogel for long-term culture. Previously, methods for patterning oligonucleotides on surfaces and decorating cells with DNA sequences required specialized equipment and custom DNA synthesis, respectively. We report an updated version of the protocol, utilizing an inexpensive benchtop photolithography setup and commercially available cholesterol modified oligonucleotides (CMOs) deployed using a modular format. CMO-labeled cells adhere with high efficiency to DNA-patterned substrates. This approach can be used to pattern multiple cell types at once with high precision and to create arrays of microtissues embedded within an extracellular matrix. Advantages of this method include its high resolution, ability to embed cells into a three-dimensional microenvironment without disrupting the micropattern, and flexibility in patterning any cell type.
Introduction
The positioning of cells with respect to one another in a tissue is an important feature of the microenvironment1 , 2 , 3 , 4 . Techniques used to pattern live cells into spatially controlled arrangements are valuable experimental tools for studying differentiation4 , 5 , 6 , 7 , 8, cell motility9, morphogenesis10 , 11 , 12, metabolism13, and cell-cell interactions7 , 14. A variety of methods exist for patterning cells, each with their own advantages and drawbacks3 , 4. Methods that create adhesive islands of extracellular matrix (ECM) proteins, such as microcontact printing and laser-cut stencils, are simple and scalable. However, it is difficult to pattern more than one or two cell types at a time because the adhesive properties of different cell types to different ECM molecules are often similar15 , 16 , 17. More complex micropatterns can be created with light-induced molecular adsorption (LIMAP), a technique that uses UV light to ablate PEG-coated regions and allow for subsequent protein adsorption18 , 19. This process can be repeated to create high-resolution micropatterns with multiple cell types. However, cross-binding of cells to the different protein patches can occur, resulting in poor pattern specificity19. Physical methods such as seeding cells onto micromechanical reconfigurable culture devices can create structured co-cultures with dynamic control, but without the flexibility in pattern design of microcontact printing or LIMAP14 , 8. Unlike the other techniques, bioprinting can create three-dimensional arrangements of cells within hydrogels20 , 21. However, bioprinted constructs have much lower resolution than other micropatterning techniques, with an average feature size on the order of hundreds of microns22. An ideal cell patterning method would have high resolution, pattern multiple cell types, use equipment and reagents that are easily accessible, and have the ability to embed successful patterns into a hydrogel for three-dimensional (3D) cell culture. In this article, we present CMO-DPAC, a cell micropatterning technique that uses the flexibility and speed of DNA hybridization to target cell adhesion to a substrate. This method has been adapted from our previous protocols23 , 24 to make it more affordable, modular, and accessible. Using the current protocol, any lab should be able to set up a fully functional system without any specialized equipment or expertise.
DNA Programmed Assembly of Cells (DPAC) is a powerful tissue engineering technique that patterns cells at single-cell resolution with precise control over cell-cell spacing and tissue geometry. In DPAC, cell membranes are decorated with DNA oligonucleotides (oligos) using two lipid-modified oligos designed to hybridize on the cell membrane. Because the oligos are conjugated to hydrophobic lipids, they rapidly partition to the cell membrane25 where they hybridize, increasing the net hydrophobicity of the non-covalently bound molecules, and thereby enhancing their lifetime at the cell surface26. The oligos are presented on the cell surface in a manner where they can hybridize with complementary oligos on other cells or DNA-functionalized glass slides to create defined 2D or 3D cell patterns with prescribed composition, cell-cell spacing, and geometry23 , 24. The patterned microtissues can be cleaved off of the surface enzymatically and embedded into a hydrogel for prolonged 3D culture. When used in combination with primary cells or stem cells, the resulting collections of cells can undergo morphogenesis and form into organoids23 , 27 , 28 . DPAC has been applied to investigate the dynamics of adult neural stem cell fate in response to competing signals6 , 29, to study self-organization of mammary epithelial cells23 , 28, and to generate “tissue origami” through mesenchymal condensation27.
DPAC allows for the precise placement of multiple cell populations and has substantially better resolution than extrusion-based bioprinters (on the order of microns)22 , 23. In addition, unlike ECM-based patterning methods such as microcontact printing, DPAC does not require differential adhesion of the different cell types to an ECM-coated surface15 , 23. It is ideal for answering questions about how the composition of a tissue affects its behavior, how cells integrate multiple cellular and microenvironmental cues when making decisions6 , 29, and how pairs of cells interact with each other. An advantage of this method over other micropatterning methods is that it can be used for 3D cell culture in a single imaging plane, facilitating time-lapse studies of tissue self-organization and organoid morphogenesis23 , 27 , 30.
Despite these advantages, successful implementation of DPAC has required the synthesis of custom oligonucleotide reagents and access to specialized equipment for DNA patterning23 , 24, limiting widespread adoption. For example, the optimal lipid-modified oligos (LMOs) used in the original protocol must be custom synthesized, modified with lignoceric acid or palmitic acid, and purified26. This process requires the use of a DNA synthesizer and a high-performance liquid chromatography instrument, as well as the purchasing of the associated reagents such as methylamine, a controlled substance that is subject to both institutional and federal regulations. As an alternative, LMOs can be custom purchased in bulk, but this requires a significant up-front investment in the technology.
To overcome these limitations, we have developed a revised version of DPAC that uses commercially available cholesterol-modified oligos (CMOs) in place of the custom-synthesized LMOs. To further reduce costs and to increase the flexibility of the platform, we have changed to a modular, three-oligo system. Instead of ordering a new cholesterol-modified oligo for each unique cell population, a user of this protocol can instead use the same cholesterol-modified oligos (“Universal Anchor” and “Universal Co-Anchor”) for every cell population and then employ an inexpensive, unmodified oligo (“Adapter Strand”) that hybridizes with both the Universal Anchor and either the amine-functionalized DNA on the surface or the Adapter Strand of another cell type.
Another limitation of the original DPAC protocol was that it created the DNA-patterned slides by using a high-resolution liquid printer (e.g., Nano eNabler, BioForce Nanosciences)23 , 24. While this instrument boasts extraordinary resolution and low reagent requirements, it is not available to most institutions and has a relatively low printing rate (approximately 1 feature patterned per second). Recently, two photolithographic methods have been developed to pattern DNA features onto surfaces. Viola and colleagues used a polyacrylamide and benzophenone coating that covalently bound single-stranded DNA oligos upon exposure to UV light30. Using this method, they were able to create tissue scaffolds that underwent large-scale, programmed shape changes as a result of cell contractility and self-organization. Scheideler et al. developed a method that uses UV exposure of a positive photoresist to selectively expose amine-modified DNA oligos to an aldehyde-functionalized slide29. After baking and reductive amination, the amine-modified DNA is covalently bound to the surface. This method was used to investigate the response of adult neural stem cells to spatially presented self-renewal and differentiation cues. This article adapts Scheideler et al.’s protocol to create the DNA patterns that will capture CMO-labeled cells. This photopatterning protocol can be performed without using a clean room. It uses inexpensive and commercially available equipment that is easily deployed on a benchtop or fume hood. The use of inexpensive or DIY (do-it-yourself) photolithography equipment increases accessibility to researchers without access to clean room facilities and allows researchers to try the technique without a large investment of time or resources31 , 32. However, better resolution and the alignment of multiple DNA features can be achieved by using the commercial spin coater and mask aligner commonly found in cleanroom facilities.
Here, we describe a method to pattern cells at single-cell resolution using DNA-based adhesion. First, photopatterning with a positive photoresist is used to create high-resolution patterns of amine-modified DNA onto an aldehyde-modified glass substrate. Next, the slide is treated to reduce non-specific cell attachment and PDMS flow cells are created to confine cells over patterned regions. Cells are then labeled with short DNA oligonucleotides that are functionalized with cholesterol and as a result insert into the cell membrane. The cells are then flowed over the DNA micropatterns. Hybridization between the cell-surface DNA and the DNA on the glass surface results in specific adhesion of the cells to the DNA pattern. Non-adherent cells are washed away, revealing the adherent cell pattern. This process can be repeated to pattern multiple cell types or to create multi-layered structures. If desired, the cells can be fully embedded into an ECM for 3D cell culture.
Protocol
1. Design experiment
Plan out the desired experiment, considering feature size, feature spacing, number of cell types involved, and the arrangement of cells with respect to one another. Refer to Supplemental File 1, a guide for experimental design, and Supplemental File 2, which contains example oligo sequences.
-
Design photomask using computer-aided design software. An example photomask is provided in Supplemental File 3.
Draw a rectangle of the dimensions of a standard microscope slide (25 mm x 75 mm).
Draw four rectangular regions 10 mm wide and 10 mm long, distributed evenly across the slide.
Within each region, draw features that are the desired size, shape, and spacing for the experiment. Cells will adhere only to these features in the experiment.
To create aligned photomasks for multiple cell types, create a master drawing with all sets of features, then save versions that correspond to each cell type.
Order a high-resolution (at least 20,000 dots per inch) transparency photomask from this CAD drawing with the features drawn in 1.2.3 transparent and the larger regions black.
2. Photopattern DNA onto aldehyde-functionalized slides (protocol adapted from Scheideler et al.29)
-
If patterning multiple cell types, fabricate fiducial markers on the aldehyde-functionalized slide before any DNA patterning to facilitate alignment of features. Alternative methods for creating fiducial markers are suggested in Supplemental File 1.
To create metal fiducial markers, apply S1813 positive photoresist as described in steps 2.3 – 2.11. Use a photomask that contains large features that will be easy to align later. Incorporate these features into the design of the photomasks that will be used for DNA patterning.
Deposit a thin film (100 Angstroms) of titanium onto the slide using electron-gun evaporation29. Remove excess metal and photoresist using acetone, and then proceed to the DNA photopatterning.
-
Prepare a 20 µM solution of a 5’-amine-modified oligo in DNA buffer (50 mM of sodium phosphate in water, pH = 8.5). See Supplemental File 2 for suggested oligo sequences.
NOTE: It is possible to use as little as 5 µM of amine-modified oligo for some patterns and applications, so surface DNA concentration may need to be optimized.
Pre-heat a hot plate to 100 °C.
-
Use double-sided tape or a vacuum to attach an aldehyde-functionalized glass slide to the rotor of a spin coater.
CAUTION: Slide detachment during spin-coating is a safety risk. Always use the spin coater in an enclosed container with a lid, such as an acrylic box.
NOTE: Label a corner of the slide by using a diamond scribe or similar implement to scratch the glass. This helps with slide identification and orientation after the photoresist has been washed away.
Use a disposable pipette to drop the positive photoresist onto the aldehyde slide. For even coatings, add small drops of the photoresist across the slide, instead of one large drop in the middle (Supplemental Figure 1A).
Using the spin coater, spin the slide at 3000 rpm for 30 s.
Place the slide on 100 °C hotplate for 1.5 min (soft bake) to crosslink photoresist.
-
Remove the slide from hotplate. Place a photomask with the features desired for this experiment on top of the slide and weigh the photomask down with a piece of glass (Supplemental Figure 1B,C). Cover the entire setup in an opaque box (Supplemental Figure 1D). Expose with a UV lamp (365 nm wavelength, 360 mW, 5 inches from slide, total radiant energy density 100 mJ/cm2) for 2 min.
NOTE: UV light will break the polymer bonds in the photoresist underneath transparent regions of the photomask, creating regions where DNA will later be able to adhere.
Develop the slide by immersing in developer solution for 3–5 min (Supplemental Figure 1E).
Rinse away excess developer solution with water. Dry under a stream of air or nitrogen. (Supplemental Figure 1F).
-
Confirm that the photolithography was successful by looking at the slide under the microscope. Because the photoresist is UV-light sensitive, do this step quickly and then store the slide in the dark while preparing other slides (if applicable).
NOTE: A successfully patterned slide should have sharply defined edges for each feature, no cracking, and no feature distortion at the edges. Examples of correct and incorrect photolithography are provided in Supplemental Figure 2A. See Table 1 for troubleshooting suggestions if photolithography does not provide the desired feature quality.
Add a droplet of the 20 µM amine-modified oligo solution (Step 2.1) onto each photopatterned region of the slide. Use a pipette tip to gently spread the droplet across the entire region, being careful not to scratch the slide. (Supplemental Figure 1G).
Bake the slide in a 65–70 °C oven until the DNA solution has fully dried onto the slide surface (about 1 h).
-
Perform reductive amination by placing the patterned, baked slides in a 15 cm cell-culture dish and place in a fume hood on top of a shaker. Weigh out 100 mg of sodium borohydride. In a fume hood, add 40 mL of phosphate-buffered saline (PBS), gently mix, and add to the dish containing the patterned slides. Let the reaction proceed for 15 min with gentle shaking.
NOTE: The amine on the oligo first forms a Schiff base with the aldehydes on the slide surface. This is a reversible covalent bond that must be converted to an irreversible bond prior to use in DPAC. Addition of a reducing agent (sodium borohydride) converts the Schiff base to a secondary amine by reductive amination.
CAUTION: The reaction of sodium borohydride with water creates hydrogen gas and will continue to do so for hours or days after the reaction begins. Perform the reductive amination step in a fume hood and keep all sodium borohydride solution waste in an open or loosely capped container in the fume hood for at least 24 h.
Remove unreacted DNA by washing twice with 0.1% sodium dodecyl sulfate (SDS) in water, then three times with distilled water. Dry the slide under a stream of nitrogen or air.
-
Rinse the slide with acetone to remove the remaining photoresist.
NOTE: At this point, the DNA has been irreversibly and covalently attached to the slide and all unreacted aldehyde functional groups have been converted to alcohols. The photoresist is no longer needed.
-
If multiple oligos will be patterned, return to step 2.4, align the photomask with fiducial marks, and repeat.
NOTE: The experiment can be paused here. Store slides in a vacuum desiccator. Under dry conditions, the slides can be stored for up to 3 months without a loss of quality.
Table 1: A troubleshooting guide to identify and resolve potential failures that can arise from this protocol.
In particular, poor adhesion of cells to the pattern can have many root causes and this guide should help with the identification and resolution of those issues.
| Result | Possible Cause(s) | Suggested Fixes |
|---|---|---|
| Photolithography – features are cracked | Inconsistent or inadequate soft-bake | Increase time of soft-bake up to 3 minutes; verify actual temperature of hotplate and increase temperature as necessary |
| Photolithography – features are not sharp or have photoresist remaining within them | Under-development | Increase time that slide spends in developer solution; incorporate gentle agitation |
| Photolithography – features inconsistent across slide | UV light may not be centered or not focused properly | Adjust UV light setup to ensure collimated light of uniform intensity |
| Cells don’t adhere to patterned spots with high efficiency | Not enough DNA on surface | Confirm that DNA is present on surface by hybridizing the slide with fluorescent complementary oligos and then imaging under microscope |
| Cells are inadequately labeled with CMO | Add fluorescent complementary oligos to cell suspension and confirm fluorescence via flow cytometry | |
| Not enough cells over pattern | Collect cells by washing out from PDMS flow cell, centrifuge, and re-suspend in lower volume to concentrate the cells | |
| Too much remaining CMO in cell suspension, hybridizing with DNA on slide | Add another wash step. Be sure to remove as much supernatant as possible with each wash. | |
| Too much internalization of CMO due to time and temperature | Work quickly after labeling the cells with CMO; keep cells and slide on ice and use ice-cold reagents | |
| Cells clump | Cells were not adequately separated during trypsinization | Use PBS + 0.04% EDTA during cell washes; pass cell suspension through 35 µm filter before the final wash |
| Cells adhere non-specifically | If in one specific area – could be due to scratches on slide, misalignment of PDMS flow cells, or spillage of DNA outside the pattern region | Avoid scratches, be careful to align the PDMS flow cells to the pattern region |
| If cells are adhering everywhere – inadequate blocking or washing | Add in more washes after patterning the cells; pipet more vigorously during washes; block with 1% BSA for longer before starting cell patterning; silanize slide (optional step 3) or confirm silanization was successful by measuring contact angle of water droplet | |
| Bubbles form within flow cell | Pipetting errors, uneven hydrophilic surface created during plasma oxidation | If bubbles are small, add PBS to the inlet of the flow cell and they may be washed out. If bubbles are larger, apply gentle pressure to the PDMS flow cell, nudging the bubbles towards the inlet or outlet. |
| Cells initially adhere to pattern but are removed during washes, patterning of other cell types, or adding the hydrogel precursor | The shear forces from pipetting too vigorously can cause the cells to detach from the surface | Pipet more gently during subsequent washes, rounds of cell patterning, or adding hydrogel precursors. Because the hydrogel precursors are viscous, they are more likely to cause the pattern to dislodge, so take extra caution. Multilayered structures tend to be top-heavy and are more susceptible to being dislodged. |
| Tissue deforms during 3D transfer | Hydrogel sticks to slide | Confirm hydrophobicity of slide using contact angle measurements |
| Use razor blade to lift PDMS fully on both edges, allowing PBS to float under the tissue | ||
| This can happen with pure collagen hydrogels – consider adjusting the protein concentration or composition of hydrogel | ||
| Cells don’t transfer with the hydrogel and remain on the slide | Increase Turbo DNAse concentration or increase incubation time | |
| Hydrogel is not solid enough | Increase incubation time and/or the gelation mechanism for the hydrogel in question (e.g. for collagen, make sure pH is correct) | |
| Hydrogel tears upon removing PDMS | Make PDMS flow cells hydrophilic using plasma oxidation before beginning experiment so that they detach easily upon adding media. Use forceps very gently to detach the PDMS. |
3. Make slide hydrophobic (optional) (protocol adapted from Todhunter et al. 24)
NOTE: It is advantageous, but not required, to modify the slide’s surface chemistry to render it more inert and hydrophobic. Non-specific cell attachment is reduced on these surfaces33, thereby alleviating non-specific binding of cells to un-patterned areas of the slide. Additionally, if the patterned cells will ultimately be embedded within a hydrogel and transferred off the slide, the surface treatment is essential for reliable movement of the cell-laden hydrogel across the slide without distortion or tearing. Silanizing with (tridecafluoro-1,1,2,2-tetrahydrooctyl) dimethylchlorosilane results in the presence of hydrophobic fluoroalkyl groups on the slide surface.
CAUTION: Perform all steps from 3.1 onward in a chemical fume hood to prevent exposure to acetic acid and methylene chloride fumes.
Rinse slide with 10% acetic acid and then dry under an air stream.
-
In a glass Coplin jar, prepare a solution of 60 mL methylene chloride (dichloromethane), 0.6 mL of triethylamine, and 0.6 mL of (tridecafluoro-1,1,2,2-tetrahydrooctyl) dimethylchlorosilane. Stir with a metal spatula to mix.
NOTE: These reagents are sensitive to water. They should be stored under dry conditions and used as fresh as possible.
Add the slide to the Coplin jar containing the silane solution. Place Coplin jar on an orbital shaker (set to 60–80 rpm) and allow the reaction of the silane and the slide to progress for 15 min.
Use metal forceps to remove the slide from the silane solution. Immerse slide in a Coplin jar containing methylene chloride for 1 minute to remove excess silane from the slide.
-
Immerse the slide in a 50 mL conical tube containing ethanol. Agitate. Immerse the slide in a 50 mL conical tube containing distilled water. Agitate.
NOTE: Methylene chloride and water are not miscible, so an ethanol rinse is needed to remove excess methylene chloride before the final water rinse.
-
Remove the slide from the water and inspect it. The slide should be fairly dry, with any water droplets having a contact angle of greater than 90°. Allow slides to dry fully and store in vacuum desiccator until use.
NOTE: The experiment can be paused here. Store the slide under dry conditions.
4. Prepare PDMS flow cells and slide for experiment
NOTE: Rectangular PDMS flow cells are used to concentrate the cells over the patterned regions of the slide. For experiments cultured in 3D, the flow cells form a mold for the hydrogel.
-
Make SU-8 master to use as mold for PDMS flow cells.
Pre-heat hotplate to 95 °C.
Add 5 mL of SU-8 2075 to a silicon wafer.
Spin coat the SU-8 on the wafer at 500 rpm for 10s, followed by 1,000 rpm for 30s. This should create features up to 240 µm in height34.
Soft bake the wafer on the hotplate for at least 45 min.
Remove the wafer from the hotplate. Put the photomask (see Supplemental File 4) (emulsion side down) on top of the wafer and weigh it down with a glass disc to ensure contact between the photomask and the slide.
Expose with UV light (365 nm) for a radiant energy density of 350 mJ/cm2.
Bake wafer on the hotplate for 12–15 min.
Place wafer in wide glass container. Cover wafer with SU-8 developer solution. Place on a shaker and develop while agitating for at least 15 min.
Use forceps to remove the wafer from the developer solution. Rinse for 5 s by spraying more developer solution from a squirt bottle. Spray with isopropyl alcohol to rinse. If a white precipitate appears, return the wafer to the developer solution and develop for longer.
Dry wafer under a stream of air or nitrogen.
-
Bake slide for 5 min.
NOTE: Once the master wafer has been created, it can be reused indefinitely as long as the features remain intact.
-
Prepare PDMS.
In a weigh boat, add polydimethylsiloxane elastomer and crosslinker in a 10:1 ratio (by mass). Stir vigorously to ensure even mixing.
De-gas the PDMS in a vacuum desiccator for 15–30 min until no more bubbles are visible.
Place the master wafer in a 15 cm tissue culture dish. Pour PDMS over the wafer. If bubbles appear, de-gas in a vacuum desiccator for a few minutes.
-
Bake in 60 °C oven for 3 h.
NOTE: After baking, PDMS flow cells can be stored on the benchtop indefinitely.
-
Prepare PDMS flow cells for the experiment.
Shortly before starting a CMO-DPAC experiment, cut out the required number of PDMS flow cells from the master wafer. Plasma oxidize with 10 cc/min room air for 90 s to render the surface hydrophilic.
Cut out each individual flow cell so that there is 1–2 mm of PDMS remaining on each side, then cut open the top and bottom of the flow cell to create an inlet and outlet.
Retrieve patterned slide created in Steps 2 and 3. Align on top of photomask.
Using the photomask as reference, place the PDMS flow cells on the slide in the location of each patterned region.
-
Add 50 µL of phosphate buffered saline (PBS) + 1% bovine serum albumin (BSA) to the inlet of each flow cell, as shown in Supplemental Figure 1H. Confirm that the flow cell is completely filled by the PBS + 1% BSA and that there are no large bubbles. Proceed immediately to Steps 5 and 6.
NOTE: Blocking with BSA minimizes non-specific cell adhesion to the slide surface.
5. Lift and label cells with cholesterol-modified DNA
-
Prepare the cholesterol-modified DNA solutions.
For each set of cells in the experiment, mix together 3 µL of a 100 µM stock solution of the cholesterol-modified Universal Anchor Strand with 3 µL of a 100 µM stock solution of an Adapter Strand. Incubate for 1 minute. This will pre-hybridize the oligos. Add 69 µL of phosphate-buffered saline (PBS) to create a 4 µM Universal Anchor + Adapter solution.
For each set of cells in the experiment, add 3 µL of a 100 µM Universal cholesterol-modified Co-Anchor Strand stock solution to 12 µL of PBS, creating a 20 µM solution.
-
Prepare the single-cell suspension(s).
For adherent cells, use trypsin or other dissociation agent to remove the cells from the culture flask. Add culture media to neutralize the trypsin and centrifuge to pellet the cells. For non-adherent cells, collect the cell suspension and centrifuge to pellet the cells.
-
Resuspend the cell pellet in 1 mL of ice-cold PBS or serum-free media. Transfer 1–3 million cells to a 1.5 mL microcentrifuge tube. Centrifuge at 160 × g for 4 min.
NOTE: If the cell type being used is prone to clumping/aggregation, use PBS without calcium and magnesium ions for all wash steps to reduce unwanted cell aggregation. If viability is a particular concern for the cell type being used, use serum-free media instead of PBS. Media containing fetal bovine serum is not recommended for cell labeling as it can hinder incorporation of lipid-modified oligos.35
-
Label the cells with cholesterol-modified oligos.
-
Resuspend the cell pellet in 75 µL of ice-cold PBS or serum-free media. Keep the cells in an ice bucket throughout the labeling and washing process to maximize cell viability and minimize loss of the cholesterol-modified oligos from the cell surface.
NOTE: Resuspending the cells before adding the DNA ensures that the distribution of DNA is uniform across the cell population.
Add the 75 µL of the 4 µM Universal Anchor + Adapter solution created in Step 5.1.1 to the microcentrifuge tube containing the cell suspension. Mix thoroughly by pipetting. Incubate for 5 min on ice.
Add 15 µL of the Universal Co-Anchor Solution to the microcentrifuge tube. Mix thoroughly by pipetting. Incubate for 5 min on ice.
-
Remove excess oligos from the cell suspension. Add 1 mL of ice-cold PBS or serum-free media to the microcentrifuge tube. Mix with a P1000 pipette. Centrifuge at 160 × g for 4 min at 4 °C. Discard the supernatant. Repeat two more times.
NOTE: If cells are prone to clumping, pass the cell suspension through a 40 µm filter before the final wash. If cells are prone to adsorption onto the side of the microcentrifuge tube, consider pre-blocking the tube with casein.
-
6. Pattern the DNA-labeled cells
-
Resuspend the cells in ice-cold PBS or serum-free media to create a cell-dense solution of at least 25 million cells/mL.
NOTE: For one slide using four of the 10 mm x 15 mm x 200 µm PDMS flow cells described in Step 4, about 100 µL of this dense cell suspension is required. Although most of these cells will not adhere to the pattern and will ultimately be discarded, having an extremely concentrated solution of cells over the pattern dramatically improves the efficiency of cell patterning.
Pick up slide and tilt it slightly. Add 25 µL of cell suspension to the inlet of each flow cell on the patterned slide. Remove the PBS + 1% BSA solution from the outlet, allowing the cell suspension to fill the PDMS flow cell. Incubate on ice or at room temperature for 30 s. NOTE: At this point, looking at the flow cell under a microscope should show densely packed cells with little to no gaps visible between cells. See Supplemental Figure 2B.
-
Aspirate 5 µL of cell suspension from the outlet of the slide and add it back into the inlet. Repeat 10 times per flow cell.
NOTE: The adhesion of CMO-labeled cells to the DNA-patterned slide is nearly instantaneous. Flowing the cells over the pattern multiple times increases the probability that a cell will flow over a given DNA spot and be captured.
-
Gently pipette PBS or serum-free media into the inlet of each flow cell to wash out excess cells. Collect the cell suspension from the outlet. Repeat 2–4 times or until a visual inspection of the slide under the microscope confirms that there are no excess cells remaining.
NOTE: It can be advantageous to save the excess cells from the first wash. If the patterning efficiency is unsatisfactory, the excess cells can be centrifuged and resuspended in a lower volume of PBS to create a more cell-dense solution, and then the process can be repeated from Step 6.2.
-
Repeat Steps 6.1–6.4 for each set of cells in the pattern. For patterns in which multiple cell types are directly patterned by the surface template, start with the least abundant cell type of the pattern and finish with the most abundant cell type.
NOTE: It is advisable to do each round of cellular assembly sequentially instead of pooling the cells, even in conditions where the cells are all labeled with orthogonal DNA sequences. Pooling the cells effectively dilutes each cell population and reduces patterning efficiency.
After the final round of cell assembly is complete, the next steps will vary based on the specific experiment. If the cells are intended to remain on the glass, add media to a Petri dish containing the slide, and then gently use forceps to nudge the PDMS flow cells off of the slide. If the cells will be embedded into a hydrogel and cultured in 3D, proceed to Step 7.
7. Transfer into hydrogel for 3D culture (optional)
-
Prepare a hydrogel precursor solution containing 2% DNase.
NOTE: The composition of the solution will vary based on experimental setup. Matrigel and mixtures of Matrigel and collagen I work well in this protocol, but other hydrogels are also possible.
Add 50 µL of hydrogel solution containing 2% DNase to the inlet of each flow cell. Aspirate the excess fluid from the outlet, driving the hydrogel solution into the flow cell. For viscous hydrogel precursors, tilting the slide slightly may be required to help the hydrogel flow into the flow cell.
Incubate the slide at 37 °C for 30–45 min (depending on hydrogel gelation kinetics) to allow the hydrogel to set and to cleave the DNA-based adhesion between the cells and the surface.
-
Remove each flow cell from the slide and place on top of hydrogel precursor solution.
Add 50 µL of hydrogel precursor to a well of a 2-well chamber slide or a 6-well plate.
Pipette 10 µL of PBS on either side of each flow cell.
-
Use a razor blade or fine-point tweezers to distribute the PBS along the full length of the flow cell, then gently lift the sides of the flow cell so that the PBS rushes underneath the hydrogel.
NOTE: This will “float” the hydrogel across the slide, allowing for transfer without distortion or tearing.
Use a razor blade to gently move the flow cell to the edge of the glass slide.
Invert the slide. With the razor blade, nudge the flow cell off the slide so that it lands on top of the razor blade.
Pick the flow cell off the razor blade using curved forceps. Invert the flow cell so that the cells are on the bottom, and then place on top of the droplet of hydrogel precursor solution.
Repeat Steps 7.4.1 – 7.4.6 for each flow cell.
Incubate for at least 30 min so that the hydrogel containing the patterned cells can bind to the hydrogel underlay, resulting in the full embedding of the patterned cells.
-
Remove the PDMS flow cell.
-
Add enough media to immerse the PDMS flow cell.
NOTE: The influx of media will loosen the adhesion between the hydrogel and the PDMS flow cell.
-
Use curved forceps, oriented along the long axis of the flow cell, to gently nudge the flow cell until it pops off and floats into the media. Collect the flow cell with forceps and discard.
NOTE: For optimal results, spread the curved forceps and apply gentle pressure to the walls of the PDMS flow cell. Apply force in the direction of the long axis of the flow cell.
-
8. Confirm successful labeling of cells with CMO (optional, for troubleshooting)
Order a fluorescently modified (FAM or AF647) oligonucleotide that is complementary to the surface adhesion sequence of the Adapter Strand being used in the experiment.
Label cells with CMO DNA and wash out excess DNA as described in Step 5. Resuspend in 200 µL of ice-cold PBS.
Make up a 4 µM solution of the fluorescently labeled complementary oligonucleotide in PBS. Add 200 µL of this solution to the cell suspension. Incubate on ice for 5 min.
Add 1 mL of ice-cold PBS. Mix. Centrifuge the cells to pellet them. Remove supernatant. Repeat this process two more times to wash out any DNA that has not hybridized.
-
Perform analytical flow cytometry to quantify the presence of DNA on the cell surface.
On a flow cytometer, analyze control cells that have not been labeled with DNA. Set up gates based on this population.
Analyze CMO-labeled cells that have been treated with a fluorescently labeled complementary oligonucleotide.
Calculate mean fluorescence intensity.
Representative Results
This protocol makes it possible to pattern cells in 2D and 3D with high precision and without the use of custom reagents or expensive cleanroom equipment. Figure 1 shows an overview of the protocol. First, DNA-functionalized slides are created through photolithography. Next, cells are labeled with CMOs. The cells are then flowed over the slide, where they attach only to the DNA-functionalized regions of the slide. After excess cells are washed away, the desired pattern of cells is revealed. These cells can be cultured on the slide or embedded in a hydrogel containing DNase and transferred off the slide for 3D cell culture.
Figure 1: Overview of CMO-DPAC protocol.
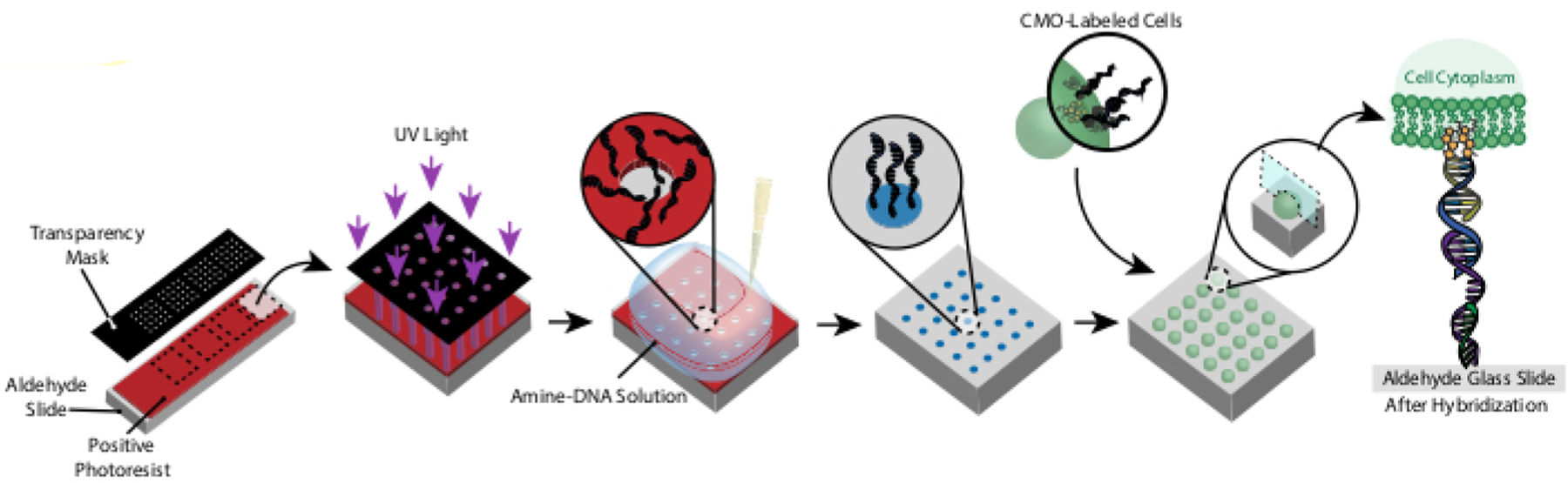
First, a DNA-patterned slide is created by coating an aldehyde-functionalized glass slide with a positive photoresist, covering it with a transparency mask in the desired pattern, and exposing it to UV light. The UV-exposed photoresist is washed away with developer, leaving exposed regions of the aldehyde slide and allowing the binding of amine-functionalized DNA to the surface. Cells are then labeled with CMOs and flowed over the surface. The DNA on the cell membrane hybridizes to the DNA on the surface, resulting in adhesion.
Labeling of cells with CMOs allows for their attachment to the DNA patterned slide (Figure 2). First, the cholesterol-modified Universal Anchor Strand is pre-hybridized with the Adapter Strand. Next, the Universal Anchor + Adapter solution is mixed 1:1 with the cell suspension. The cholesterol on the Universal Anchor + Adapter complex inserts into the cell membrane. Addition of the cholesterol-modified Universal Co-Anchor Strand, which hybridizes with the Universal Anchor Strand, improves the stability of the CMO complex in the cell membrane by increasing the net hydrophobicity of the complex26. After washing out the excess DNA from the cell suspension, the cells are flowed over the slide. Hybridization between the Adapter Strand and the Surface DNA Strand results in attachment of cells to the DNA-patterned regions of the slide.
Figure 2: Cells are labeled with CMOs in a stepwise process.
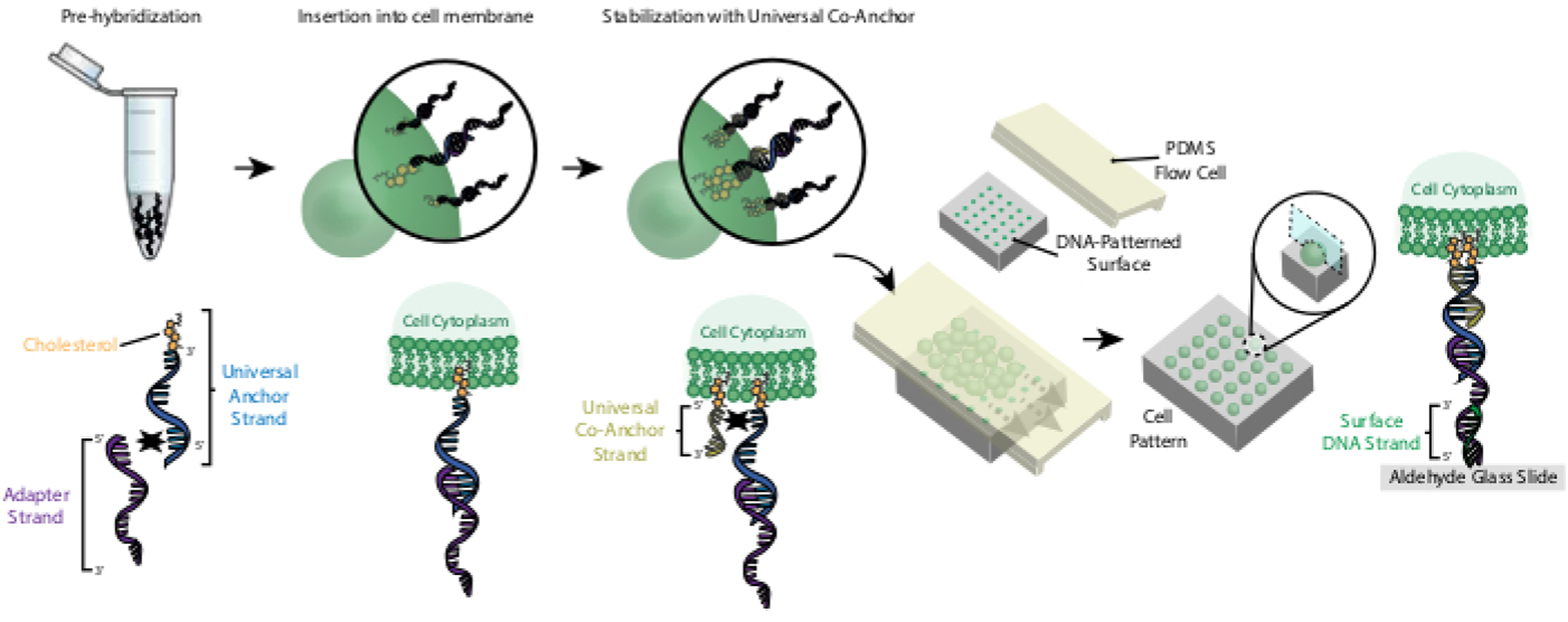
First, the cholesterol-modified Universal Anchor Strand is pre-hybridized with the Adapter Strand. Next, the Universal Anchor + Adapter solution is mixed with the cell suspension. The cholesterol on the Universal Anchor + Adapter complex inserts into the cell membrane. After incubation, the cholesterol-modified Universal Co-Anchor Strand is added to the cell suspension, where it hybridizes with the Universal Anchor Strand and inserts into the cell membrane. The addition of the second cholesterol molecule increases the net hydrophobicity of the DNA complex and stabilizes it within the membrane26. After washing out the excess DNA, the cells are concentrated and added to a PDMS flow cell on top of the patterned surface. The 3’ end of the Adapter Strand hybridizes with the Surface DNA Strand on the glass slide, resulting in adhesion to the slide specifically in regions functionalized with complementary DNA.
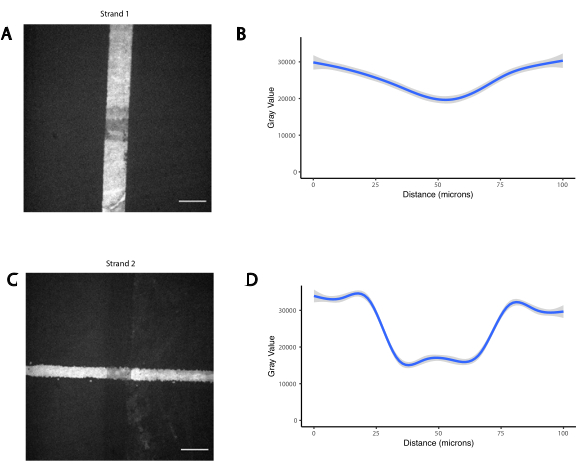
The pattern of the cells is created by using photolithography to restrict the attachment of amine-modified DNA oligos to specific regions of an aldehyde-modified glass slide29 (Figure 3A). Positive photoresist is spin-coated onto an aldehyde-functionalized slide. A transparency photomask is then placed on top of the slide and the slide is exposed to UV light. After developing, the regions of the slide that were exposed to UV light are no longer coated in photoresist and thus have exposed aldehyde groups. A 20 µM solution of amine-modified DNA oligos is then dropped onto the slide and spread to cover the patterned regions. Baking followed by reductive amination results in a covalent bond between the amine-modified DNA and the slide. Remarkably, this process can be repeated to pattern multiple oligos without any loss of functionality of the previously patterned oligos (Figure 3B). However, care should be taken to avoid overlapping patterns, which results in the presence of both oligos at a reduced concentration (Supplemental Figure 3). Multiple cell populations can be patterned sequentially by using Adapter Strands that differ in their modular domain (the 20 bases closest to the 3’ end).
Figure 3: Photolithography is used to create the DNA-patterned slides that will ultimately dictate the placement of cells.
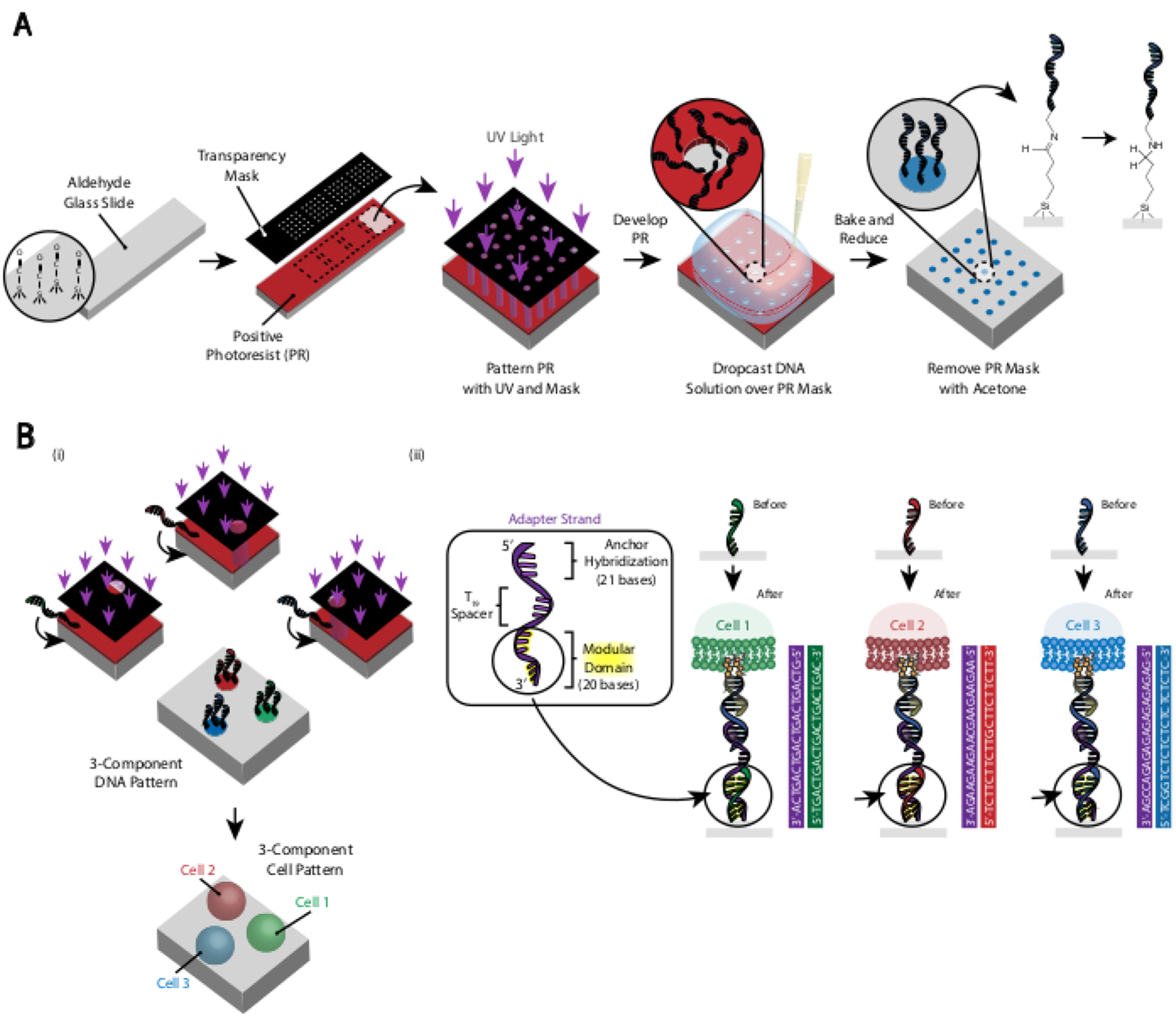
(A) Overview of photolithography process. An aldehyde-functionalized slide is spin-coated with a positive photoresist. UV light shines onto the slide through a transparency photomask that is transparent where cell adhesion is desired. After the slide is developed, the regions that were previously exposed to UV light now have exposed aldehyde groups. A 20 µM solution of an amine-functionalized DNA oligo is then dropped onto the slide and spread over the patterned regions. The slide is then baked to induce the formation of Schiff bonds (C=N) between the amine and aldehyde groups, a reversible covalent bond29. Subsequent reductive amination with 0.25% sodium borohydride in PBS converts the Schiff base to a secondary amine by reductive amination, resulting in an irreversible bond between the DNA and the slide. The remaining photoresist can then be removed by rinsing with acetone. (B) This process can be repeated to create multi-component DNA patterns and therefore perform experiments with multiple cell populations. (i) After the first oligo is patterned, the slide is again coated in photoresist and the protocol proceeds as before. Alignment of the photomasks using fiduciary markers is necessary for patterning multiple DNA strands. (ii) Each cell type being patterned differs in the 20-base modular domain of the Adapter Strand. By using orthogonal sets of complementary oligos, multiple cell types can be patterned without cross-adhesion.
Although this photopatterning protocol was developed by Scheideler et al. in the context of a clean room, we have demonstrated that it is possible to achieve similar results with an inexpensive, “home-brew” photolithography setup that fits easily within a chemical fume hood. The setup includes a $400 spin coater made of a DC motor, digital controller, and CD cake box, as well as a UV lamp that was assembled from individual components and housed in a repurposed sharps container (Supplemental Figure 1). The main advantage of the home-brew photolithography setup is that it is very affordable (<$1000 for all of the equipment) while still being able to create single-cell-sized features. However, the use of inexpensive equipment does have its limitations - for example, it is more challenging to precisely align fiducial markers to pattern multiple DNA oligos without use of a mask aligner. We recommend this inexpensive photolithography setup for labs that do not have convenient access to a clean room or that want to try this method without a large investment.
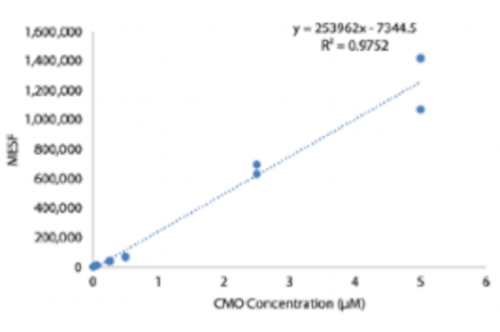
To identify optimal conditions for DNA-programmed cell adhesion, we systematically varied the concentrations of DNA strands on cell surfaces and measured the efficiency of cell adhesion to DNA-modified glass surfaces. The concentration of Universal Anchor + Adapter Strand and Universal Co-Anchor in labeling solutions were varied across several orders of magnitude (Figure 4A,B), resulting in 104 - 106 DNA complexes per cell (Supplemental Figure 4). Cell adhesion was dose dependent, with minimal cell adhesion to the DNA pattern when cells were labeled with CMOs at a concentration of 0.05 µM or less, and high occupancy at a concentration of 2.5 µM and higher. We, therefore, used a 2 µM solution of Universal Anchor + Adapter Strand and 2 µM solution of Universal Co-Anchor in most experiments. Cell adhesion would also be expected to decrease if the amount of DNA used on the glass surface decreased29 or if mismatches between the Adapter Strand and surface strand increased. More information about Adapter Strand sequence design is provided in Supplemental File 2. CMO labeling using Adapter Strands without CpG repeats did not stimulate TLR9 in HEK cells expressing mouse TLR9 (Supplemental Figure 5).
Figure 4: Adhesion of CMO-labeled cells to DNA patterns increases as a function of CMO concentration during labeling.
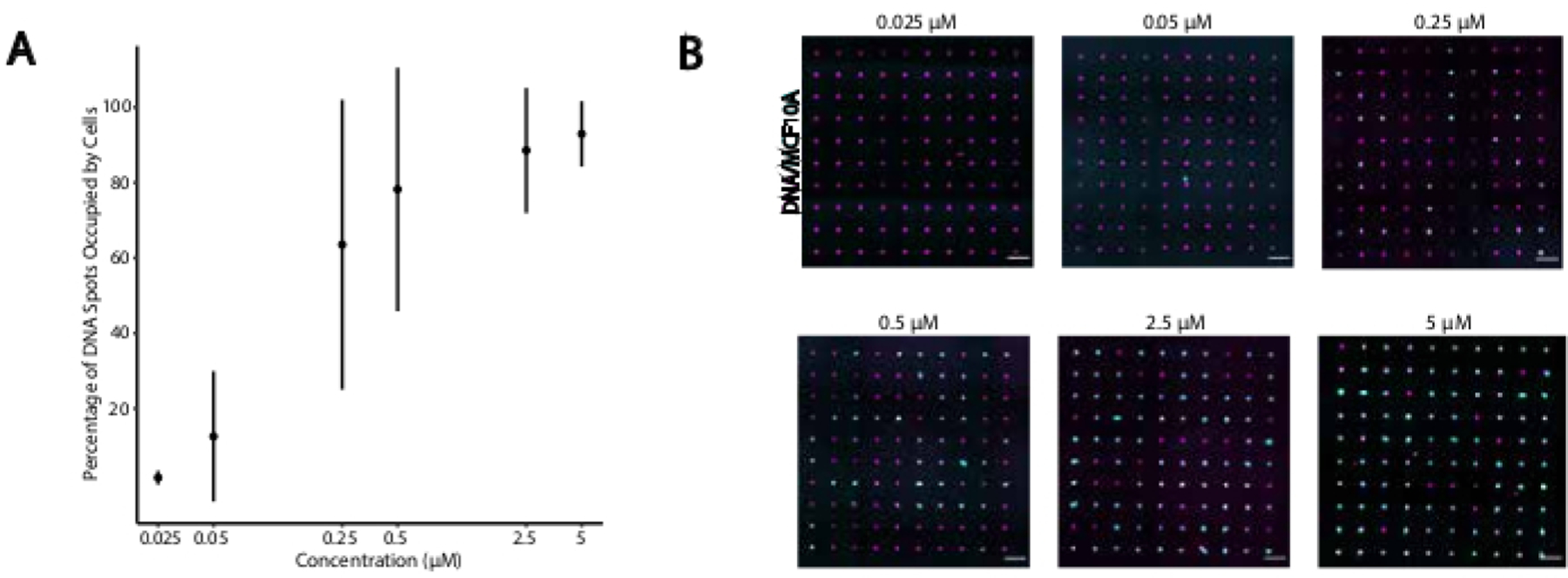
In this experiment, the Universal Anchor + Adapter Strand (pre-hybridized) and the Universal Co-Anchor were used at equal concentrations. Concentration refers to the concentration of CMO in the cell suspension during CMO labeling of cells. (A) Quantification of the percentage of 15 µm diameter DNA spots that were occupied by CMO-labeled MCF10A cells as a function of CMO concentration during cell labeling. Data represented as the mean ± standard deviation from three experiments. (B) Representative images of the DNA patterns (magenta) and adhered MCF10As (cyan) at different concentrations of CMO. Scale bar = 100 µm.
We provide several demonstrations that the revised protocol provides reproducible and efficient DNA-programmed cell adhesion. For example, human umbilical vein endothelial cells (HUVECs) labeled with CMOs adhered to DNA patterns with high efficiency. CMO-labeled HUVECs adhered as well as LMO-labeled HUVECs (Figure 5A). Cells patterned using CMO-DPAC retained their viability and functionality. Cells labeled with CMOs were stained by calcein AM and ethidium homodimer to assess viability (Supplemental Figure 6). Differences in viability compared to unlabeled control cells were small (94% vs 97%). Single MDCKs patterned via CMO-DPAC and transferred into Matrigel were able to proliferate and polarize correctly after 5 days of culture (Figure 5B). DPAC also provides a means of elaborating patterns of cells into the third dimension (Figure 5C). For example, multilayered, multicellular aggregates can be created by alternating layers of cells labeled with complementary CMOs (Figure 5C). These experiments demonstrate that the protocol is reproducible, does not negatively affect cell viability or functionality, and yields cellular patterns that can be successfully cultured within a single imaging plane in a 3D ECM.
Figure 5: CMO-DPAC can be used to create two-dimensional cell patterns that can subsequently be embedded into a three-dimensional hydrogel for culture and/or layered to create multilayered structures.
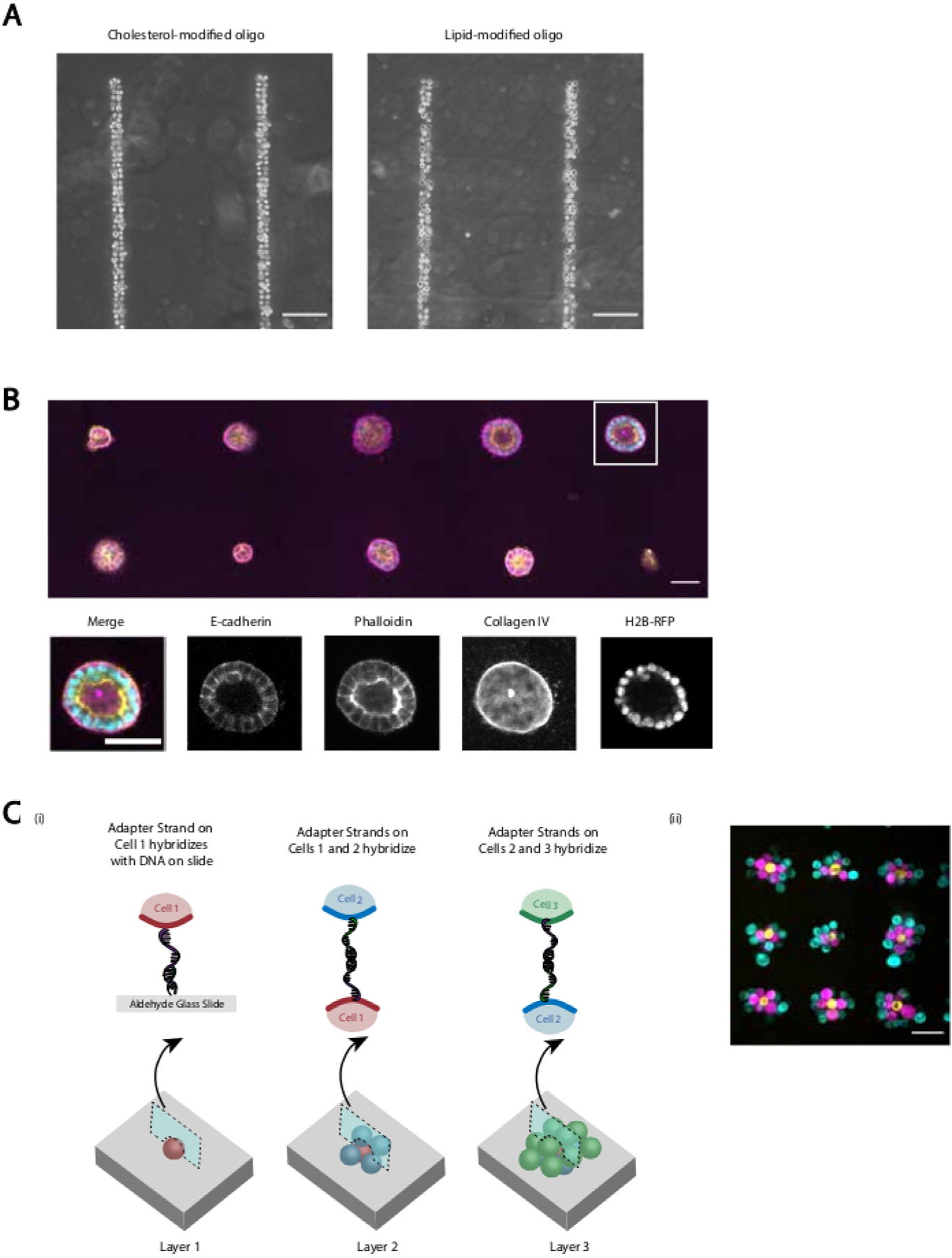
(A) Direct comparison between CMO-labeled human umbilical vein endothelial cells (HUVECs) and LMO-labeled HUVECs adhered to a linear DNA pattern. Both methods of cell labeling result in nearly 100% occupancy of the DNA pattern. (B) Single Madin-Darby Canine Kidney cells (MDCKs) expressing H2B-RFP were patterned onto 15 µm diameter spots spaced 200 µm apart and subsequently embedded in Matrigel. After 120 h of culture, the resulting epithelial cysts were fixed and stained for E-cadherin, actin, and collagen IV. Spheroid in white box is shown in detail. Scale bar = 50 µm. (C) Multilayered cellular structures can be created by labeling separate cell populations with complementary Adapter Strands and patterning sequentially so that each new addition of cells adheres to the cell layer before it. (i) A schematic of the sequential patterning of cell populations to create multilayered structures. (ii)Three-layered cell aggregates of MCF10As (visualized using dyes) were created using this process. Scale bar = 50 µm.
By providing orthogonal DNA sequences to direct cell adhesion, DPAC provides a means of patterning multiple cell types on a single surface. To implement this feature of DPAC, DNA patterns generated by photolithography must be aligned with respect to one another. Metal fiduciary markers deposited onto the slide allowed for the alignment of multiple photomasks and therefore the patterning of multiple cell types at once. MCF10As stained with different unique dyes were labeled with orthogonal CMOs and patterned to create a visualization of the UC Berkeley and UCSF logos (Figure 6). This experiment demonstrates that multiple unique cell populations can be patterned together with high precision and without cross-contamination.
Figure 6: Multiple cell types can be patterned without cross-contamination or loss of adhesion.

Multiple amine-modified DNA oligos were patterned sequentially onto an aldehyde slide and aligned through use of metal fiduciary markers. Three populations of MCF10As (cyan, magenta, yellow) were stained with unique dyes labeled with complementary CMOs, and patterned onto the slide, resulting in an image of the UC Berkeley and UCSF logos. Scale bar 1 mm.
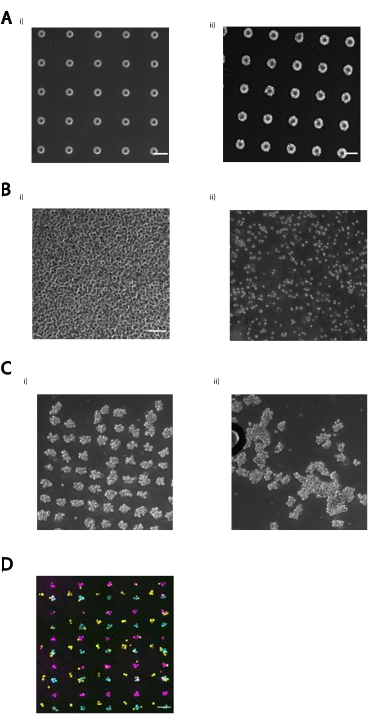
Successful patterning of cells using CMO-DPAC requires high-quality photolithography, sufficient concentration of oligo on the cell surface, a high density of cells over the pattern, and sufficient washing. Failure of any one of these steps affects the final result. Supplemental Figure 2 includes example images of correct and incorrect photolithography (Supplemental Figure 2A), the desired cell density over the pattern to create fully occupied patterns (Supplemental Figure 2B), the loss of patterned cells due to overly vigorous pipetting during subsequent steps of DPAC (Supplemental Figure 2C), and undesired clumping of cells (Supplemental Figure 2D). Table 1 includes a list of common failure points and the suggested troubleshooting. The use of fluorescent complementary oligos is recommended as a tool for troubleshooting to confirm the presence of patterned DNA on the slide and the presence of CMOs on the cell surface by flow cytometry (see Step 8 of protocol).
Discussion
In this article, we present a detailed protocol for high-resolution patterning of cells in 2D and 3D for in vitro cell culture experiments. Unlike previously published versions of this method, the protocol presented here focuses on usability: it does not require highly specialized equipment and all reagents can be purchased from vendors instead of requiring custom synthesis. Unlike other cell micropatterning methods, this method is rapid and cell-type agnostic: it does not require specific adhesion to extracellular matrix proteins15. Cells patterned by CMO-DPAC can be embedded within an extracellular matrix such as Matrigel or collagen, resulting in 3D cultures with much higher spatial resolution than is currently possible with extrusion printing-based methods22. CMO-DPAC can be used to create hundreds to thousands of microscopic features per slide, allowing for many replicates to be performed at the same time.
One of the most important parameters in the success of this protocol is the density of cells added to the flow cells on top of the patterned slide. Ideally, the density should be at least 25 million cells/mL. When loaded into the flow cells, this density of cells results in a nearly close-packed monolayer of cells above the pattern (Supplemental Figure 2B). These high cell densities maximize the probability that a cell will settle directly on top of a DNA spot and adhere. Reducing the cell density will decrease the overall patterning efficiency. Another critical step in this protocol is thoroughly re-suspending the cells in PBS or serum-free media before adding the CMO solution. The CMOs partition very rapidly into cell membranes and adding the CMO solution directly to a cell pellet will result in heterogeneous labeling of cells. After adding the CMO solution to the cell suspension, it is important to mix thoroughly by pipetting so that the cells are uniformly labeled with the CMOs. After the incubations, it is necessary to thoroughly wash out the excess CMOs through multiple centrifugation and wash steps. Excess free CMO present in the cell suspension will bind to the patterned amine-modified DNA on the glass slide, blocking hybridization and adhesion of the CMO-modified cells in suspension. Time is also a key consideration for this protocol. It is important to work as quickly as possible when using CMOs and to keep the cells on ice in order to minimize internalization of the CMOs and maximize cell viability. Flow cytometry experiments have shown that CMOs do not persist as long on the cell surface as LMOs, with 25% loss of CMO complexes over two hours of incubation on ice36. Furthermore, the viability of cells will decrease as the cell handling time increases. Viability can be maximized by working quickly, keeping cells on ice, using ice-cold reagents, and using serum-free media to provide some nutrients.
Although CMO-DPAC can be a powerful way of studying cell biology by patterning cells with high precision, it does have its limitations. CMO-DPAC experiments can be challenging, particularly as the experimental complexity is added with multiple cell types, layers, or 3D cell culture (Supplemental File 1). Experimental failures can be common when starting this protocol, as described in Table 1. Therefore, we recommend that users institute quality control checks (confirming that DNA is present on the slide, confirming that cells are sufficiently labeled with DNA (Step 8), confirming that excess cells have been thoroughly washed away, etc.) to make sure that the experiment succeeds and to identify steps that may require further optimization. We hope that the information provided in this manuscript and its supplemental files will help facilitate any required troubleshooting.
Cholesterol is a bioactive molecule whose internalization may influence cell metabolism, gene expression, and membrane fluidity37 , 38. A previous study compared the effects on gene expression of CMO- and LMO-labeled cells using single cell RNA sequencing. CMO-labeled HEK cells had altered gene expression compared to unlabeled and LMO-labeled cells36. Labeling cells with CMOs resulted in the differential expression (> 1.5-fold) of eight genes relative to unlabeled controls, including AP2B1, which has been linked to cholesterol and sphingolipid transport (GeneCards), and MALAT1, a long non-coding RNA that regulates cholesterol accumulation39. While minor, these transcriptional responses may nevertheless be of concern if the experiment in question is studying metabolism, membrane dynamics, or other cholesterol-associated pathways in cells.
This protocol is flexible and can be adjusted to meet the needs of each experiment. Because the CMO inserts itself into the lipid membrane instead of using any specific receptor, the method is cell type agnostic (HUVECs, MCF10As, HEKs, and MDCKs have been demonstrated here). Although cholesterol is a different hydrophobic anchor than our previously published LMOs, we have thus far found them to behave similarly. Thus, we would expect the CMOs to work with any of the wide variety of cell types that we have previously published with LMOs, including but not limited to neural stem cells, fibroblasts, peripheral blood mononuclear cells, tumor cells, and primary mammary epithelial cells6 , 23 , 27 , 29 , 36. CMO labeling does not stimulate TLR9, suggesting that the protocol is compatible with immune cells. Membrane incorporation of the CMO is a function of total cell size and the degree of negative charge in the cell glycocalyx35. Thus, we have included a protocol (Step 8) for testing the extent of membrane incorporation that is amenable to rapid optimization. The specific features of each cell pattern will inevitably vary based on the experimental design (see Supplemental File 1 for more guidance). Although the photopatterning protocol described above for patterning the DNA is recommended, any method of spatially confining droplets of amine-DNA solution should work, such as the use of high-resolution droplet printers. The pattern resolution and minimum feature spacing will vary based on the method used. It is also theoretically possible to combine the DNA-photopatterning sections of this protocol with other methods that have been used to label cells with DNA, such as with DNA hybridized to membrane-expressed zinc fingers40, using NHS-conjugated DNA41, and reacting azido sialic acid residues on the cell surface with phosphine-conjugated DNA42. CMO-DPAC can be applied to a variety of experiments that require tight control over cell-cell spacing, including studies of the interactions between pairs of cells, co-culture experiments looking at the transfer of signals from “sender” cells to “receiver” cells, and investigations of the effect of nearby extracellular cues on stem cell differentiation6 , 29. The method can also be used to create microtissues that can be used to study cell migration in three dimensions, the self-organization of cells into tissues23 , 27, and the dynamic interplay between cells and the ECM27. We hope that this protocol will provide researchers with an accessible platform to explore new applications of high-resolution DNA-based cell patterning in their own labs.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to thank Jeremy Garcia for testing this protocol and Bhushan Kharbikar for providing training on the equipment at the UCSF Biomedical Micro and Nanotechnology Core. This research was supported in part by grants from the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221), NIH (U01CA199315, DP2 HD080351-01, 1R01CA190843-01, 1R21EB019181-01A, and 1R21CA182375-01A1), the NSF (MCB1330864), and the UCSF Center for Cellular Construction (DBI-1548297), an NSF Science and Technology Center. O.J.S was funded by an NSF Graduate Research Fellowship, a Siebel scholarship, and a P.E.O. Scholarship. Z.J.G and A.R.A. are Chan-Zuckerberg BioHub Investigators.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/61937.
Disclosures
Z.J.G. is an advisor and equity holder in Provenance Biosciences.
Supplemental Figure 1: Example images of the benchtop photolithography setup. (A) Slide on spin coater, covered with positive photoresist, before spin coating. (B) Picture of transparency photomask. (C) During exposure, the photomask is sandwiched between the photoresist-coated slide and a glass disc. (D) Housing for UV lamp was made out of a re-purposed sharps container. (E) Slide immersed in developer solution. (F) Developed slide. (G) Amine-modified DNA solution spread on patterned regions of the slide. (H) PDMS flow cells placed on top of patterned regions of the slide.
Supplemental Figure 2: Some examples of common failures of this protocol. (A) (i) Under-baking before UV exposure or over-developing features post-exposure can result in features that have jagged edges and may be irregular in size. (ii) An example of a correctly photopatterned slide that has clean edges around features, uniform feature size, and no obvious cracks in the pattern. Scale bar = 50 µm. (B) Cell density is critical to patterning efficiency. When observing the cells on top of the pattern under a microscope, few gaps should exist between cells, as evidenced by the example image on the left. Scale bar = 50 µm. (C) Patterned cells can be sensitive to fluid forces arising from overly vigorous pipetting, which can damage and dislodge the patterned cells. Multilayered cell aggregates are particularly vulnerable, as one cell at the bottom is supporting a structure of multiple cells. (i) An array of cell aggregates successfully embedded into Matrigel. (ii) A grid of cell aggregates that dislodged as a result of pipetting viscous Matrigel too vigorously. (D) Clumping of cells can occur, particularly with epithelial cells. These clumps are usually homotypic but can be heterotypic (cells adhering to already patterned cells of a different type) if the cells are particularly sticky. Image shows three different populations of MCF10As were patterned onto an array composed of three different single-cell sized DNA spots (15 µm). Most DNA spots have 2–4 cells attached. Clumping can be resolved by EDTA treatment or by filtering out the clumps before patterning. Scale bar = 100 µm.
Supplemental Figure 3: Overlapping photopatterns results in presence of both oligos at reduced concentration. Two orthogonal amine-modified oligos were photopatterned sequentially, first a vertical line (Strand 1), followed by a horizontal line that overlapped it (Strand 2). The oligos were then visualized by hybridization with fluorescent complementary oligos. (A) Fluorescence image of Strand 1. (B) Quantification of the fluorescence profile of Strand 1 over a 100 µm vertical line spanning the overlap. (C) Fluorescence image of Strand 2. (D) Quantification of the fluorescence profile of Strand 2 over a 100 µm horizontal line spanning the overlap. Scale bar = 50 µm.
Supplemental Figure 4: Quantification of DNA complexes on the cell surface as a function of CMO labeling concentration. HUVECs were labeled with different concentrations of CMO solution, washed, and then incubated with a fluorescent complementary strand. An MESF (Molecules of Equivalent Soluble Fluorochrome) microsphere kit was used to do quantitative flow cytometry and estimate the number of DNA complexes on the cell surface as a function of CMO concentration during labeling.
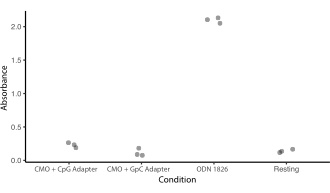
Supplemental Figure 5: CMO labeling does not stimulate TLR9 response. An experiment was carried out to see whether CMO labeling would trigger the DNA-detection mechanism of TLR9 and whether this would be affected by CpGs in the Adapter Strand sequence. HEK cells expressing mouse TLR9 were incubated overnight with 0.2 µM of either ODN 1826 (a CpG-containing TLR9 agonist), CMO Universal Anchor + Universal Co-Anchor + Adapter Strand containing the same sequence as ODN 1826 (CMO-CpG), or CMO Universal Anchor + Universal Co-Anchor + Adapter Strand containing a similar sequence but with replacement of the CpGs with GpCs (CMO-GpC). TLR9 stimulation would result in the production of SEAP (secreted embryonic alkaline phosphatase). SEAP secretion was quantified by a colorimetric assay (absorbance). Treatment conditions were compared to resting cells that were only treated with PBS. Incubation with CMO-GPC did not stimulate TLR9 expression. Incubation with CMO-CpG was slightly higher than resting cells but much lower than ODN-1826.
Supplemental Figure 6: Viability of cells after CMO labeling process. To assess how the protocol impacts viability, HUVECs were split into four populations: one remained on ice for 1 h, one was mock-labeled with PBS but otherwise taken through all centrifuge and wash steps, one was labeled with CMOs, and one was labeled with CMOs and filtered through a 40 µm filter to remove clumps. The cells were then stained with calcein AM and ethidium homodimer to assess the number of alive and dead cells. All treatments resulted in significantly decreased viability than the ice control (one-way ANOVA with Tukey post-hoc analysis), but median viability for CMO-labeling (with or without filtering) was about 94%. Data collected from three independent experiments. * = p < 0.05. **** = p < 0.0001.
Supplemental file 1.
Supplemental file 2.
Supplemental file 3.
Supplemental file 4.
References
- 1.Kreeger PK, Strong LE, Masters KS Engineering approaches to study cellular decision-making. Annual Review of Biomedical Engineering (December 2017), 49–72 (2018). [DOI] [PMC free article] [PubMed]
- 2.Goubko C. a., Cao X Patterning multiple cell types in co-cultures: A review. Materials Science and Engineering C 29 (6), 1855–1868 (2009). [Google Scholar]
- 3.Sun W et al. The bioprinting roadmap. Biofabrication 12 (2), 022002 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Liu WF, Chen CS Cellular and multicellular form and function. Advanced Drug Delivery Reviews 59 (13), 1319–1328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy RM, Sun Y, Feinberg AW Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle. Annals of Biomedical Engineerin.g 44 (6), 2076–2089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S et al. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nature Communications 7, 10309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaya O et al. Cell-cell contact area affects notch signaling and notch-dependent patterning. Developmental Cell 40 (5), 505–511.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao N et al. A co-culture device with a tunable stiffness to understand combinatorial cell–cell and cell–matrix interactions. Integrative Biology 5 (11), 1344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sriraghavan V, Desai RA, Kwon Y, Mrksich M, Chen CS Micropatterned dynamically adhesive substrates for cell migration. Langmuir 26 (22), 17733–17738 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Wong L, Pegan JD, Gabela-Zuniga B, Khine M, McCloskey KE Leaf-inspired microcontact printing vascular patterns. Biofabrication 9 (2), 021001 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Chen TH et al. Directing tissue morphogenesis via self-assembly of vascular mesenchymal cells. Biomaterials 33 (35), 9019–9026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laurent J et al. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nature Biomedical Engineering 1 (12), 939–956 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Lin C, Khetani SR Micropatterned co-cultures of human hepatocytes and stromal cells for the assessment of drug clearance and drug-drug interactions. Current Protocols in Toxicology 2017 (May), 1–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hui EE, Bhatia SN Micromechanical control of cell-cell interactions. Proceedings of the National Academy of Sciences of the United States of America 104 (14), 5722–5726 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Arcangelo E, McGuigan AP Micropatterning strategies to engineer controlled cell and tissue architecture in vitro. BioTechniques 58 (1), 13–23 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Rivas A, González-Quijano GK, Proa-Coronado S, Séverac C, Dague E Methods of micropatterning and manipulation of cells for biomedical applications. Micromachines 8 (12), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S et al. Simple lithography-free single cell micropatterning using laser-cut stencils. Journal of Visualized Experiments (158), e60888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strale PO et al. Multiprotein printing by light-induced molecular adsorption. Advanced Materials 28 (10), 2024–2029 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Melero C et al. Light-induced molecular adsorption of proteins using the primo system for micro-patterning to study cell responses to extracellular matrix proteins. Journal of Visualized Experiments 2019 (152) e60092 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Reid JA, Mollica PM, Bruno RD, Sachs PC Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Research 1–13 (2018). [DOI] [PMC free article] [PubMed]
- 21.Wang Z, Lee SJ, Cheng H-J, Yoo JJ, Atala A 3D bioprinted functional and contractile cardiac tissue constructs. Acta Biomaterialia 70, 48–56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miri AK et al. Effective bioprinting resolution in tissue model fabrication. Lab on a Chip 19 (11), 2019–2037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todhunter ME et al. Programmed synthesis of three-dimensional tissues. Nature Methods 12 (10), 975–981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Todhunter ME, Weber RJ, Farlow J, Jee NY, Gartner ZJ Fabrication of 3D microtissue arrays by DNA programmed assembly of cells. Current Protocols in Chemical Biology 8 (3), 147–178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Csizmar CM, Petersburg JR, Wagner CR Programming cell-cell interactions through non-genetic membrane engineering. Cell Chemical Biology 25 (8), 931–940 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber RJ, Liang SI, Selden NS, Desai TA, Gartner ZJ Efficient targeting of fatty-acid modified oligonucleotides to live cell membranes through stepwise assembly. Biomacromolecules 15 (12), 4621–4626 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes AJ et al. Engineered tissue folding by mechanical compaction of the mesenchyme. Developmental Cell 44 (2), 165–178.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weber RJ et al. Rapid organoid reconstitution by chemical micromolding. ACS Biomaterials Science & Engineering 2 (11), 1851–1855 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Scheideler OJ et al. Recapitulating complex biological signaling environments using a multiplexed, DNA-patterning approach. Science Advances 6 (12), eaay5696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viola JM et al. Guiding cell network assembly using shape-morphing hydrogels. Advanced materials (Deerfield Beach, Fla.) 2002195, e2002195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohammad A, Davis M, Aprelev A, Ferrone FA Note: Professional grade microfluidics fabricated simply. Review of Scientific Instruments 87 (10), 1–4 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Lee OJ, Chuah HS, Umar R, Chen SK, Yusra AFI Construction of cost effective homebuilt spin coater for coating amylose-amylopectin thin films. Journal of Fundamental and Applied Sciences 9 (2S), 279 (2018). [Google Scholar]
- 33.Webb K, Hlady V, Tresco PA Relative importance of surface wettability and charged functional groups on NIH 3T3 fibroblast attachment, spreading, and cytoskeletal organization. Journal of Biomedical Materials Research 41 (3), 422–430 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Microchem SU-8 2000 Permanent Expoxy Negative Photoresist. Processing Guidelines for: SU-8 2025, SU-8 2035, SU-8 2050, SU-8 2075 1–5, at <https://kayakuam.com/wp-content/uploads/2019/09/SU-82000DataSheet2025thru2075Ver4.pdf> (2019).
- 35.Palte MJ, Raines RT Interaction of nucleic acids with the glycocalyx. Journal of the American Chemical Society 134 (14), 6218–6223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGinnis CS et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods 16 (7), 619–626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maxfield FR, van Meer G Cholesterol, the central lipid of mammalian cells. Current Opinion in Cell Biology 22 (4), 422–429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo J, Yang H, Song BL Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology 21 (4), 225–245 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Tan L, Yao J, Yang L Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Molecular Medicine Reports 21 (4), 1761–1770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mali P, Aach J, Lee JH, Levner D, Nip L, Church GM Barcoding cells using cell-surface programmable DNA-binding domains. Nature Methods 10 (5), 403–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsiao SC et al. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir 25 (12), 6985–6991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gartner ZJ, Bertozzi CR Programmed assembly of 3-dimensional microtissues with defined cellular conductivity. Proceedings of the National Academy of Sciences (17), 1–5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.