

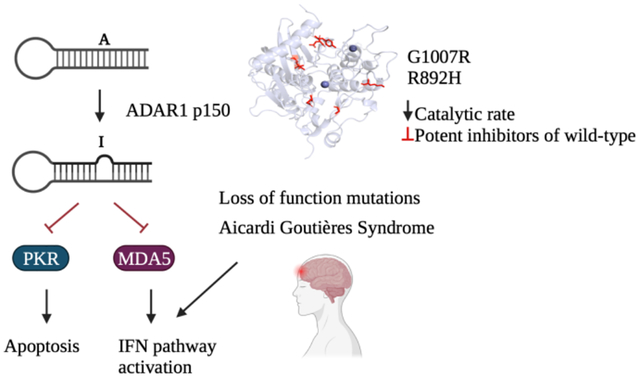
Abstract
The innate immune system relies on molecular sensors to detect distinctive molecular patterns, including viral double-stranded RNA (dsRNA), which trigger responses resulting in apoptosis and immune infiltration. Adenosine Deaminases Acting on RNA (ADARs) catalyze the deamination of adenosine (A) to inosine (I), serving as a mechanism to distinguish self from non-self RNA and prevent aberrant immune activation. Loss-of-function mutations in the ADAR1 gene are one cause of Aicardi Goutières Syndrome (AGS), a severe autoimmune disorder in children. Although seven out of the eight AGS-associated mutations in ADAR1 occur within the catalytic domain of the ADAR1 protein, their specific effects on the catalysis of adenosine deamination remain poorly understood. In this study, we carried out a biochemical investigation of four AGS-causing mutations (G1007R, R892H, K999N, and Y1112F) in ADAR1 p110 and truncated variants. These studies included adenosine deamination rate measurements with two different RNA substrates derived from human transcripts known to be edited by ADAR1 p110 (glioma-associated oncogene homolog 1 (hGli1), 5-hydroxytryptamine receptor 2C (5-HT2cR)). Our results indicate that AGS-associated mutations at two amino acid positions directly involved in stabilizing the base-flipped conformation of the ADAR-RNA complex (G1007R and R892H) had the most detrimental impact on catalysis. The K999N mutation, positioned near the RNA binding interface, altered catalysis contextually. Finally, the Y1112F mutation had small effects in each of the assays described here. These findings shed light on the differential effects of disease-associated mutations on adenosine deamination by ADAR1, thereby advancing our structural and functional understanding of ADAR1-mediated RNA editing.
Graphical Abstract
INTRODUCTION
RNA editing is an essential post-transcriptional modification process that regulates gene expression and generates protein diversity in eukaryotic systems. It involves the precise insertion, deletion, or modification of nucleotides within RNA molecules.1 Among the various types of RNA editing, the most prevalent form in metazoans is the hydrolytic deamination of adenosine (A) to inosine (I) catalyzed by the Adenosine Deaminases Acting on double-stranded RNA (ADARs).1,2,3 Inosine exhibits similar base pairing properties to that of guanosine, resulting in A to G transitions that alter RNA structure and function. Two active ADAR enzymes in humans (ADAR1 and ADAR2) exhibit a modular organization characterized by a conserved catalytic domain located at the C-terminal region and double-stranded RNA binding domains (dsRBDs) positioned within the N-terminal region.2 ADAR1 exists as two distinct isoforms: the constitutively expressed short form, ADAR1 p110, and the interferon (IFN)-inducible form, p150 which consists of an additional Z-DNA/RNA (Zα) binding domain. The Zα domain of ADAR1 is important for substrate specificity and editing of Z-RNAs.4,5,6 While both forms can shuttle between the nucleus and the cytoplasm, ADAR1 p110 is primarily localized in the nucleus, whereas p150 is predominantly found in the cytoplasm.7 Despite their high degree of similarity, ADAR1 and ADAR2 exhibit divergent substrate selectivity, which can be attributed to differences in their RNA binding domains and the distinct features of their catalytic domains.8 Notably, they possess dissimilar loops in their catalytic domains that bind RNA on the 5’ side of targeted adenosines (i.e. 5’-binding loops), and the ADAR1 catalytic domain additionally harbors a structural zinc site absent in ADAR2.9
Proper regulation of ADAR-mediated A to I editing is crucial for maintaining normal cellular function.10,11 Abnormal expression and excessive editing by ADAR1 have been observed in various cancer types, indicating its involvement in oncogenesis.12 Interestingly, the knockout of ADAR1 in tumor cells leads to increased sensitivity to immunotherapy, highlighting its potential as a therapeutic target.13,14 ADAR1 also plays a critical role in regulating cytoplasmic innate immunity at the level of double-stranded RNA (dsRNA).15,16 By introducing structural modifications to RNA, ADAR1-mediated editing prevents the recognition of cellular RNA as foreign, thereby avoiding unnecessary immune responses. Consequently, loss-of-function mutations in ADAR1 have been linked to a severe childhood immune disorder called Aicardi Goutières Syndrome (AGS).17,18,19 AGS can also be caused by mutations in six other genes involved in nucleic acid sensing and metabolism: TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and IFIH1.19 Multiple AGS-associated mutations have been identified in the ADAR1 gene, with seven occurring in the deaminase domain (A870T, I872T, R892H, K999N, G1007R, Y1112F, D1113H,) and one (P193A) in the Zα domain (Figure 1).15,16,17 Notably, among the AGS mutations found in the deaminase domain, only two (G1007R and R892H) are predicted to involve residues that directly contact RNA given the available substrate-bound ADAR2 structures (PDBID 5ED2, 5ED1, 5HP2, 5HP3, and 6VFF).20,21
Figure 1.
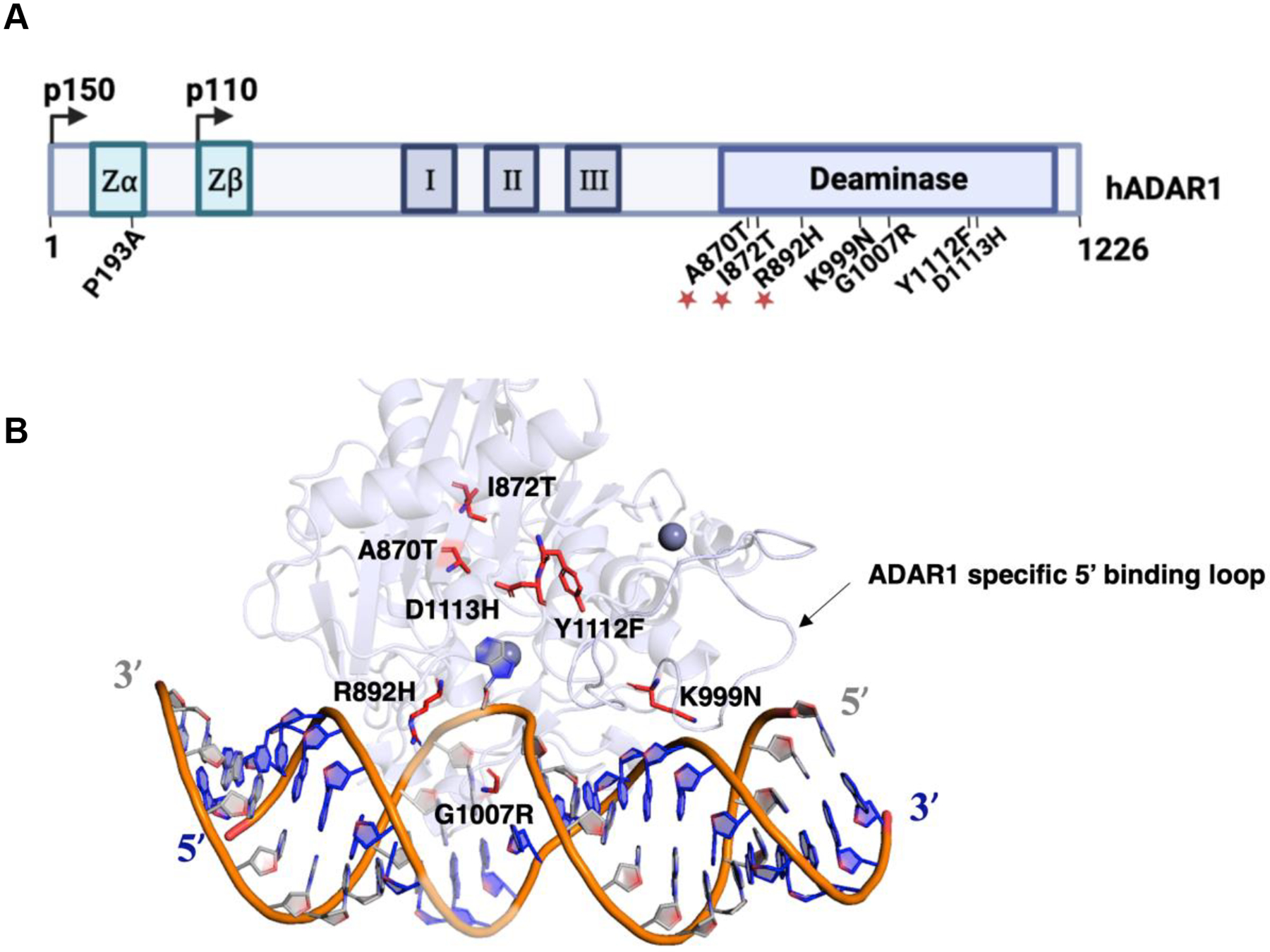
Mapping AGS mutations onto a model of ADAR1 deaminase domain bound to RNA. (A) Domain map of human ADAR1 p110 and p150. AGS mutations identified within the Zα domain and the catalytic domain of ADAR1 p150 are labeled. The mutations A870T, I872T, and R892H occur as double mutants with P193A and are denoted by asterisks. (B) Previously reported Rosetta model of ADAR1 deaminase domain bound to a dsRNA substrate.9 AGS mutations within the deaminase domain of ADAR1 are highlighted in red. The ADAR1 specific 5’ binding loop is also depicted. The active site zinc and secondary zinc are represented as dark grey spheres.
Structures of ADAR2 bound to dsRNA revealed a 20-base pair segment of the duplex RNA that interacts with the deaminase domain primarily through the phosphodiester backbone.20,21 ADARs access the target nucleotide in duplex RNA by inducing a base flipping conformational change that positions the adenosine in the active site.20,21 A loop of the protein whose sequence is conserved within the ADAR family inserts into the minor groove of the duplex. This flipping loop bears the residue E488 in ADAR2 (corresponding to E1008 in ADAR1) that plays a crucial role by contacting the “orphan base” when the reactive adenosine flips into the active site. Once in the active site, the adenosine reacts with a hydroxide ion bound to zinc. The resulting high energy tetrahedral intermediate loses ammonia forming the inosine product. Within the flipping loop, the E488 residue is flanked by two conserved glycine residues, G487 (G1007, ADAR1) and G489 (G1009, ADAR1), which allow for correct positioning of E488 in the space vacated by the flipped out base and hydrogen bonding with the orphaned base.20,22
Among the identified AGS mutations, G1007R is predicted to have the greatest effect on adenosine deamination given its proximity to E1008, an essential residue in the flipping loop, and the substantial change in the side chain structure arising from the arginine for glycine substitution.20,22 R892 corresponds to K376 in ADAR2, which contacts phosphodiesters adjacent to the edited adenosine, stabilizing the flipped-out conformation. A similar contact is predicted for R892 and the mutation to histidine could alter the enzyme’s ability to stabilize that essential conformation for catalysis.20,22 In the available homology model of ADAR1, the methylene groups of K999 are predicted to make hydrophobic contacts that stabilize the 5’-binding loop.9 Destabilization of the loop structure could result in inefficient engagement for certain RNA substrates that rely on 5’-binding loop contacts. Finally, the Y1112F mutant, categorized as a second-layer mutation given its distance from the RNA-binding interface, may also influence the stability of the adjacent ADAR1-specific 5’ binding loop.9,22
Crystal structures of ADAR2 bound to dsRNA provide insights into the possible consequences of AGS mutations. However, limitations arise from structural differences between ADAR1 and ADAR2 and the absence of a published high-resolution ADAR1 structure. Several studies have been conducted to investigate the impact of AGS mutants on ADAR1 function in cells and in animal models. Overexpression of AGS mutants of ADAR1 in HEK293 cells have provided insight into their effects on RNA editing.17,18 Mouse studies, specifically examining AGS mutants P193A, K999N, and D1113H, have demonstrated distinct effects on ADAR1 substrates and tissue-specific immune activation.23,24 Notably, the deletion of MDA5 has been shown to restore normal immune response in these mutants.16,18,24 However, despite these investigations, a comprehensive biochemical understanding of how each mutation affects ADAR catalysis of adenosine deamination is lacking. Therefore, the objective of this study was to bridge this knowledge gap by conducting biochemical characterization, including kinetic, binding, and thermal analyses of selected AGS-associated mutants.
MATERIALS AND METHODS
Synthesis of oligonucleotides.
RNA oligonucleotides containing 8-azanebularine (8-azaN) were synthesized using an in-house ABI 394 synthesizer at 0.2 μmol scale. The 8-azaN phosphoramidite was purchased from Berry & Associates. All other phosphoramidites were purchased from Glen research. Upon completion of oligonucleotide synthesis, columns were dried under reduced pressure overnight. Once dried, the oligonucleotides were cleaved from the solid support with the treatment of 1:3 ethanol/ 30% NH4OH at 55 °C overnight. The supernatant was collected in a screw-cap tube and dried under reduced pressure. Removal of silyl groups was performed by suspending the pellets in anhydrous DMSO and treating it with 55% (v/v) Et3N-3HF at room temperature overnight. The oligonucleotides were precipitated in solution of 65% butanol at −70 °C for 2 h. The pellets were obtained after centrifugation at 13200 × g for 20 min, washing twice with cold 70% ethanol. The RNA pellets were then desalted using a 3000 MWCO Amicon Ultra 0.5 mL centrifugal filter and purified as described below (see Table S1 for sequences). Oligonucleotide masses were confirmed by MALDI-TOF MS (Table S2).
In vitro transcription of RNA.
Substrates for deamination studies (5-HT2cR and hGlil) were transcribed from a DNA template using NEB HiScribe T7 RNA synthesis kit. The sequence of each substrate RNA can be found in Table S1.
Purification of RNA oligonucleotides.
Single-stranded transcribed and synthesized oligonucleotides were purified using denaturing urea-polyacrylamide gels and visualized by UV shadowing. Bands corresponding to desired products were excised from gels, crushed, and soaked overnight at 4 °C in 500 mM NH4OAc and 1 mM EDTA. Polyacrylamide fragments were removed with a 0.2 μm cellulose acetate filter. RNA was precipitated from the supernatant in a solution of 75% EtOH at −70 °C for 2 h. The supernatant was centrifuged 17000 × g for 20 min and supernatant was removed and washed with 70% cold ethanol. The pellet was lyophilized to dryness, dissolved in nuclease free water, and quantified by absorbance at 260 nm. Synthesized oligonucleotides were desalted using 3000 MWCO Amicon Ultra 0.5 mL centrifugal filter, and masses for synthetic oligonucleotides were confirmed by MALDI-TOF MS (Table S2).
Protein overexpression and purification.
ADAR1 p110. Human ADAR1 p110 (UniProtKB P55265–5), consisting of a C-terminal His10-tag was overexpressed in Saccharomyces cerevisiae BCY123 as previously described.25 Cells were lysed using a microfluidizer in lysis buffer containing 20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 1 M KCl, 30 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine-HCl (TCEP-HCl), 0.05% (v/v) Triton X-100, and 50 μM ZnCl2. The lysate was centrifuged at 39000 × g, 4 °C for 1 h and filtered using 0.45 μm filter. The clarified lysate was then passed over a 5 ml Ni-NTA column at a flow rate of 2 ml min−1 using an ÄKTA pure 25 FPLC system. The column was washed first with ten column volumes (10 CVs) of lysis buffer, followed with 10 CVs of wash I buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 500 mM KCl, 30 mM imidazole, 1mM TCEP-HCl, and 50 μM ZnCl2). The protein was eluted with wash I buffer with a gradient of imidazole (30 mM to 400 mM) for 10 CVs. Fractions containing the target protein were pooled, concentrated, and dialyzed against a storage buffer containing 50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, 400 mM KCl, 50 mM imidazole, 1 mM TCEP-HCl, and 0.01% (v/v) Nonidet P-40 (NP-40). Final protein concentrations were determined by running the samples alongside bovine serum albumin (BSA) standards in an SDS-PAGE gel and visualized by staining with SYPRO Orange dye (Invitrogen). ADAR1 R3D. The ADAR1 R3D protein consisting of C-terminal self-cleaving intein sequence, and a chitin binding domain (CBD) was overexpressed in Saccharomyces cerevisiae BCY123 as described above. Lysis was carried out in buffer containing 20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 750 mM NaCl, 50 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine-HCl (TCEP-HCl), 0.05% (v/v) Triton X-100, and 50 μM ZnCl2. The clarified lysate was passed over a chitin binding column using gravity flow. The bound protein was washed with 10 CVs of lysis buffer, followed by 3 CVs of cleavage buffer (20 mM Tris-HCl pH 8.0, 350 mM NaCl, 50 mM imidazole, 5% glycerol, 50 mM DTT). To ensure cleavage of the CBD domain, resin was incubated in 2 CVs of cleavage buffer overnight at 4 °C. The protein was eluted by washing columns with additional two CVs of cleavage buffer and analyzed by running on 4–13% SDS page gel. The fractions corresponding to cleaved protein were collected, diluted 5-fold with heparin equilibration buffer (20 mM Tris-HCl pH 8.0, 200 mM NaCl, 50 mM imidazole, 5% glycerol, 50 mM DTT, 1mM TCEP, 50 μM ZnCl2) and the protein was further purified through a 5 ml GE Healthcare Lifesciences Hi-Trap Heparin HP column at flow rate of 0.5 ml/min. The bound protein was washed with 10 CVs of equilibration buffer and eluted with a gradient of NaCl (200 mM – 1M NaCl) over 5 column volumes followed with step elution for 5 CVs at flow rate of 1 ml/min. Fractions were analyzed by SDS PAGE, concentrated to ~ 1 mg/ml, and dialyzed in 20 mM Tris-HCl pH 8.0, 350 mM KCl, 50 mM imidazole, 10% glycerol, 50 mM DTT, 1mM TCEP, 50 μM ZnCl2. The final protein concentration was determined as described above. ADAR1 deaminase domain. ADAR1 E1008Q deaminase domain containing an N-terminal histidine tag was purified similarly to p110 except using the following buffers: (1) lysis buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 750 mM NaCl, 30 mM imidazole, 1 mM TCEP-HCl, 0.05% (v/v) Triton X-100, and 50 μM ZnCl2. (2) wash buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 350 mM NaCl, 30 mM imidazole, 1 mM TCEP-HCl, 0.05% (v/v) Triton X-100, and 50 μM ZnCl2); and (3) storage buffer (50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, 350 mM KCl, 30 mM imidazole, 1 mM TCEP-HCl, 0.01% (v/v) NP-40). The SDS-PAGE gels displaying quality of proteins purified can be found in Figure S1.
Preparation of RNA substrates for deamination assays.
Transcribed RNA was allowed to fold in hybridization buffer (180 nM transcribed RNA target, 1X TE buffer, 100 mM NaCl), heated to 95 °C for 5 min, and slowly cooled to room temperature. The predicted secondary structure and site of reaction for each substrate RNA is found in (Figure S2, Figure S3).
Preparation of duplex substrates for EMSA with Cy5-labeled RNA.
A 38 nucleotide (nt) 8-azaN RNA was 5’ phosphorylated using T4 Polynucleotide Kinase (NEB) following manufacturer’s protocol. Ligation of the phosphorylated 38 nt RNA to a 23 nt oligonucleotide was carried out by mixing 6 nmol of each RNA and hybridized to a DNA splint at 1:1 ratio by heating at 95 °C for 5 min and slow cooling to ≤ 30 °C. T4 DNA ligase (NEB) was used to catalyze the ligation of the two RNAs at 16 °C for 16 h. The ligated 61 nt RNA was phenol:chloroform extracted, ethanol precipitated, and further purified by denaturing PAGE gel as described above. Subsequently, the ligated 61 nt 8-azaN RNA was 3’-end-labeled with fluorescent dye using pCp-Cy5 (Jena Bioscience) and T4 RNA Ligase I (NEB). The mixture was incubated at 16 °C for overnight under light-shielded conditions. Excess Cy5 dye was removed by passing the reaction mixture through a Sephadex G-25 column and the labeled RNA was purified with the phenol:chloroform extraction and ethanol precipitation. The final labeled oligonucleotide was hybridized at a 1:3 ratio to its complement in 10 mM Tris−HCl, pH 7.5, 1 mM EDTA, and 100 mM NaCl by heating at 95 °C for 5 min and slow cooling to ≤ 30 °C. The nucleotides surrounding the 8azaN are derived from the Glioma-associated Oncogene Homolog 1 (hGli1) mRNA.25 Sequences of all oligonucleotides are found in the (Table S1).
Preparation of duplex substrates for EMSA with 32P-Labeled RNA.
The 5’ end of either a 16 nt or 32 nt 8-azaN-containing RNA strand was labeled with γ-[32P]ATP (6000 Ci/mmol) using NEB polynucleotide kinase. The labeled reaction was passed through a G-25 column to remove excess ATP and further purified using a 19% denaturing PAGE gel. The labeled products were visualized using storage phosphor autoradiography. Gel bands containing labeled RNA were excised, crushed, and soaked and worked up as described for other gel-purified oligonucleotides. The dried pellet was resuspended in nuclease free water to a stock solution of approximately 300 nM and hybridized to its compliment at 1:3 ratio in 1X TE buffer, pH 7.5 and 200 mM NaCl by heating at 95 °C for 5 min and slowly cooling to 30 °C to a final concentration of approximately 50 nM. The nucleotide sequences flanking a 16 nt 8-azaN site were derived from the mRNA of Human Epidermal Growth Factor Receptor 1 (HER1).7 In addition, nucleotide sequences surrounding a 32 nt 8-azaN site were derived from hGli1 mRNA. Sequences of all oligonucleotides are found in (Table S1).
Site-directed mutagenesis.
Mutagenesis of ADAR1 p110, ADAR1 R3D, and ADAR1d E1008Q was carried out using PCR site-directed mutagenesis using a QuikChange XL Site-Directed Mutagensis Kit (Agilent) with the primers listed in Table S4. All primers were purchased from IDT and purified as described above for other oligonucleotides. Sequences for mutant plasmids were confirmed by Sanger sequencing. See Table S5 for the primer sequences.
Deamination assays.
Deamination assays were performed under single-turnover conditions in 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, 0.16 U/μL RNAse inhibitor, 10 nM RNA, and 100 nM protein at 30 °C. Reactions were quenched at 1, 5, 15, 30, 60 and 90 min for hGli1 RNA and 5, 15, 30, 60, 90, and 120 min for 5-HT2CR with 20 ul water at 95°C and heating at 95°C for 5 min. Reaction products were used to generate cDNA using RT-PCR (Promega Access RT-PCR System). The DNA product was purified with DNA Clean & Concentrator kit (Zymo) and subjected to Sanger sequencing via GeneWiz (Azenta). Sequencing peak heights at the edit site was quantified using 4Peaks (Nucleobytes). Data were fit to the equation [P]t = [P]f [1−e(−kobs.t)] where [P]t is percent edited at time t, [P]f is the final endpoint of editing, and kobs is the observed rate constant. All statistical analyses and nonlinear fits were conducted in Microsoft Excel and GraphPad Prism.
Competition Assay.
Samples containing 50 nM wild type ADAR1 p110, and 5, 10, 25, 50, 100 or 200 nM of mutant p110 proteins were incubated in 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, 0.16 U/μL RNAse inhibitor for 5 min. Reaction was initiated with addition of 10 nM of substrate and quenched after 30 min with 100 ul of water at 95°C and heating at 95°C for 5 min. Reaction products were used to generate cDNA and worked up as described above.
Qualitative EMSA using Cy5-labeled duplex RNA and ADAR1 p110.
Samples containing 20 nM Cy5 labeled duplex RNA and 0 to 100 nM enzyme (ADAR1 p110 WT, K999N, R892H and Y1112F) and 0 to 120 nM enzyme (ADAR1p110 G1007R) were incubated respectively in 20 mM Tris−HCl pH 7.4, 140 mM KCl, 10 mM NaCl, 1mM MgCl2, 0.5 mM EDTA and 0.003% (v/v) NP-40 at room temperature for 30 min. Samples were loaded onto a 4 to 16% Bis-Tris, NativePAGE mini protein gel (Invitrogen) and electrophoresed under nondenaturing conditions in 1× Native PAGE running Buffer (Invitrogen) at 4 °C for 1.5 h under light-shielded conditions. The gels were scanned using a Bio-Rad GelDoc Imaging system.
Quantitative EMSA using 32P duplex RNA and ADAR1 R3D.
Samples containing ≤ 1 nM 32 base pair (bp) RNA duplex and varying concentrations of the ADAR1 R3D proteins (0, 0.25, 0.5, 1, 2, 4, 8, 16, 32, 64, 128 nM) were incubated together in 20 mM Tris-HCl, pH 7.0, 3.5% glycerol, 0.5 mM DTT, 60 mM KCl, 20 mM NaCl, 0.1 mM β-mercaptoethanol, 1.5 mM EDTA, 0.003% Nonidet P-40, 0.16 U/μL RNAse inhibitor, 0.2 mg/mL BSA, and 1 μg/mL yeast tRNA for 30 min at 30 °C. Samples were loaded onto a 6% gel, electrophoresed in nondenaturing polyacrylamide gel (79:1 acrylamide:bisacrylamide) in 1× TBE buffer at 4 °C for 90 min. The gels were dried on a Bio-Rad gel dryer for 90 min at 80 °C under vacuum followed by exposure to storage phosphor imaging plates (Kodak) for 24 h in the dark. After exposure, the gels were removed, and the phosphor imaging plates were scanned by Typhoon Trio Variable Mode Imager (GE Healthcare). Dissociation constants were measured by calculating the fraction of RNA bound by the protein and using the equation fraction bound = A*([protein]/[protein]+Kd), where the Kd is the fitted apparent disassociation constant and A is the fitted maximum fraction of RNA bound.14,16 The sequences for the 32 bp duplex used in the assay are described in Table 1. EMSA for ADAR1d E1008Q and mutants with the 8-azaN-modified 23 bp duplex was performed as described above for ADAR1 R3D except with ≤ 5 nM RNA and in buffer containing 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, 0.2 mg/ml BSA and 0.16 U/μL RNAse inhibitor.
Table 1.
Adenosine deamination rate constants measured under single-turnover conditions for ADAR1 p110 and AGS mutants on hGli1 and 5-HT2cR B site RNAsa.
| Substrate | Enzyme | k obs (min −1 ) b | k rel c |
|---|---|---|---|
| Gli1 | ADAR1 p110 | 1.3 ± 0.1 | 1 |
| ADAR1 p110, G1007R | 0.042 ± 0.003 | 0.03 | |
| ADAR1 p110, K999N | 0.4 ± 0.1 | 0.3 | |
| ADAR1 p110, R892H | 0.09 ± 0.01 | 0.1 | |
| ADAR1 p110, Y1112F | 0.6 ± 0.3 | 0.5 | |
| 5-HT2c | ADAR1 p110 | 0.05 ± 0.01 | 1.0 |
| ADAR1 p110, G1007R | - | - | |
| ADAR1 p110, K999N | 0.04 ± 0.01 | 0.7 | |
| ADAR1 p110, R892H | - | - | |
| ADAR1 p110, Y1112F | 0.02 ± 0.01 | 0.5 |
hGli1 and 5-HT2cR pre-mRNA substrate sequences are shown in Table S1. ADAR1 p110 reactions were carried out with 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% (v/v) glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, and 0.16 U/μL RNAse inhibitor.
kobs was calculated by fitting product formed at different time points to the equation: [P]t = α[1 −e−kobst] where [P]t is percent edited, α is the end point fitted to 95%, and kobs is the observed rate constant.
krel = kobs for mutant/kobs for ADAR1 p110.
Protein melting temperature analysis.
Solutions containing 2X SYPRO orange dye, 5 μM ADAR1d E1008Q or mutants were mixed under the following conditions: 50 mM Tris-HCl, pH 8.0, 10% (v/v) glycerol, 350 mM KCl, 30 mM imidazole, 1 mM TCEP-HCl, 0.01% (v/v) NP-40, and 50 μM ZnCl2. To a 96 well plate, 20 μl of each solution was added, wells were sealed with PCR plate sealing film. Fluorescence was measured as the solutions were heated from 5 °C to 90 °C at a rate of 2 °C/min. Spectra were obtained using a Bio-Rad CFX Connect Real-Time PCR Detection System. The derivative of fluorescence signal as a function of temperature (–dF/dT) was exported, and the background values of the buffered solution without protein was subtracted from each sample. Melting temperature was determined as the temperature where the derivative of fluorescence signal was at a minimum. Measurements were performed in triplicate. Melting temperature values reported are the average of each replicate ± standard deviation (SD).
RESULTS
Effects of AGS mutations on ADAR1 p110.
Earlier work demonstrated the effects on editing of a model RNA substrate when AGS mutations were introduced into the p110 and p150 isoforms of ADAR1 overexpressed in HEK293 cells.17,18 Among the mutations tested, G1007R showed the largest effect on editing activity for both isoforms. These experiments were an important demonstration of the effect of the different AGS mutations on RNA editing with ADAR1 overexpressed in human cells. However, changes in RNA editing activity observed in these cell-based experiments could arise from several sources including changes in the protein’s catalytic activity, RNA binding affinity, protein-protein interactions with ADAR1 binding partners, sensitivity to cellular proteases, etc. To gain additional understanding of the consequences of AGS-causing mutations on specific biochemical properties of ADAR1, we aimed to characterize their effects using purified proteins. Mutations in ADAR1 are believed to cause AGS through decreased ADAR1 p150 editing activity in the cytosol, resulting in elevated levels of dsRNA sensed to induce interferon production.17 However, due to challenges associated with expression and purification of ADAR1 p150 and its mutants, we introduced select AGS associated mutations into ADAR1 p110 and two truncated forms of the protein. One of these shorter forms contains dsRBDIII and the deaminase domain (ADAR1 R3D aa711–1226) while the other constitutes only the deaminase domain (ADAR1d, aa886–1226). We suggest that the magnitude of effects observed for certain mutations may also depend on the substrate RNA used. The immunogenic RNAs in the cytosol that are edited by ADAR1 to prevent INF production are not yet defined. ADARs prefer to edit in AC mismatches or AU pairs and favor adenosines adjacent to 5’U or A and 3’G.30 Important structural features such as thermodynamic stability of RNA (mostly duplexed) can affect editing efficiency. Therefore, we tested the effect of AGS mutations using two well-established ADAR1 substrates from the human transcriptome: the Glioma-associated Oncogene Homolog 1 (hGli1) mRNA26, and 5-hydroxytryptamine receptor 2C (5-HT2cR) pre-mRNA.27 By examining the impact of AGS-associated mutations on catalysis of adenosine deamination across various protein constructs (ADAR1 p110, ADAR1 R3D, and ADAR1d) and these distinct substrate RNAs, we aimed to uncover the impact of these mutations on properties of ADAR1’s catalytic domain.
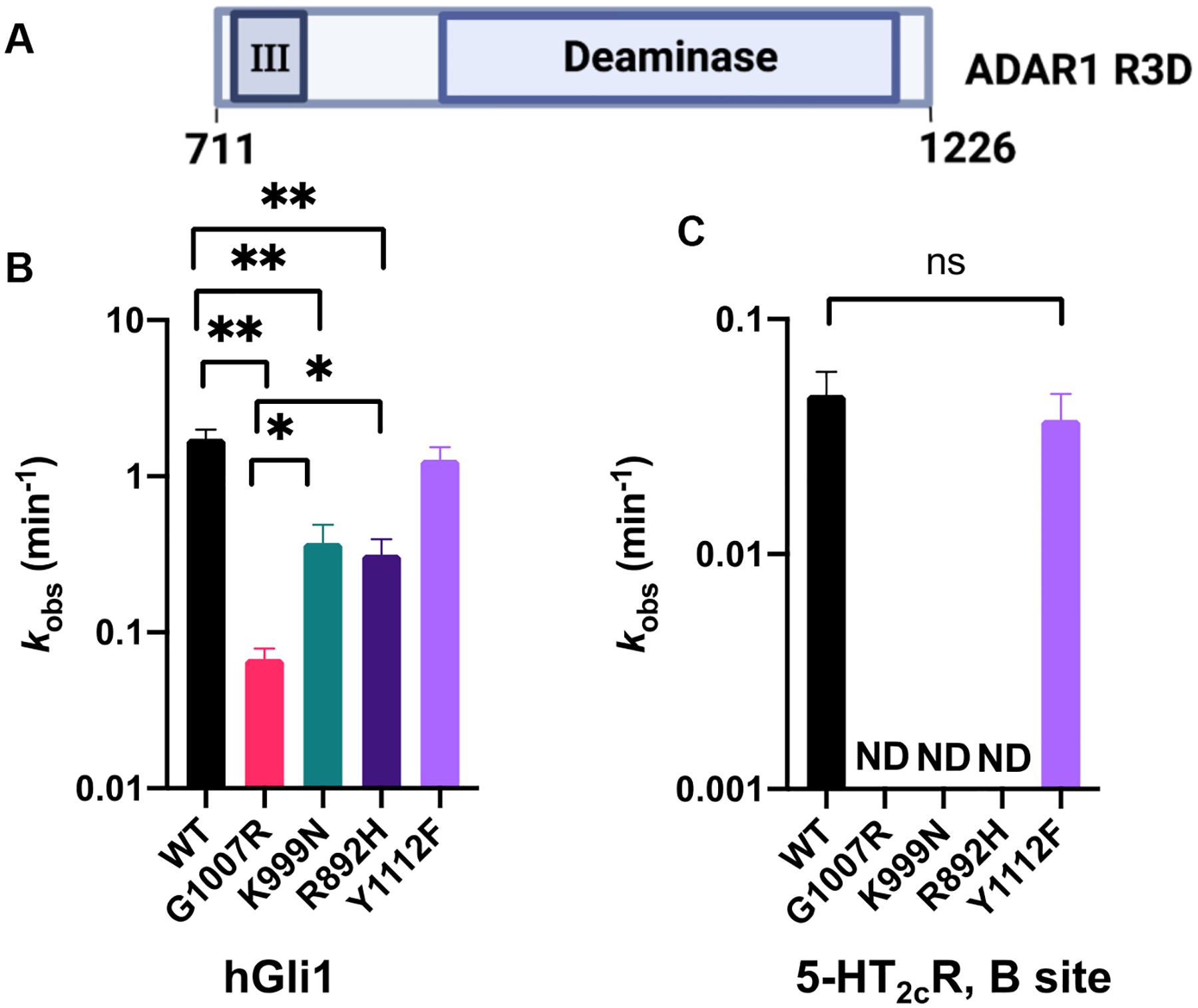
We initiated our analysis by introducing mutations into the C-terminal histidine-tagged p110 protein. Expression and purification of the wild-type and mutant variants (G1007R, K999N, R892H, and Y1112F) enabled us to investigate the impact of these mutations within and outside the RNA binding interface. Initially, we assessed the functional consequences of these variants on catalysis by measuring deamination rate constants under single turnover conditions (Figure 2, Figure S4). Consistent with the findings from cellular studies, the G1007R mutant exhibited the slowest reaction rate, showing a 30-fold decrease compared to the wild-type enzyme with the hGli1 substrate RNA (Figure 2B; Table 1).17,18 Interestingly, the R892H mutation also resulted in a 15-fold reduction in reaction rate, while the Y1112F and K999N mutations caused only 2–3-fold decreases. Notably, while rates and reaction end points measured on the more slowly edited 5-HT2cR substrate were slightly reduced for Y1112F and K999N mutants, we could not detect any editing activity on this substrate with the G1007R and R892H p110 mutants (Figure 2C, Figure S4). Thus, the deamination rate measurements for p110 mutants on the two RNA substrates analyzed showed that two AGS mutations (G1007R and R892H) have severe defects in catalysis while the other two mutations tested (K999N and Y1112F) resulted in much smaller effect on catalysis.
Figure 2.
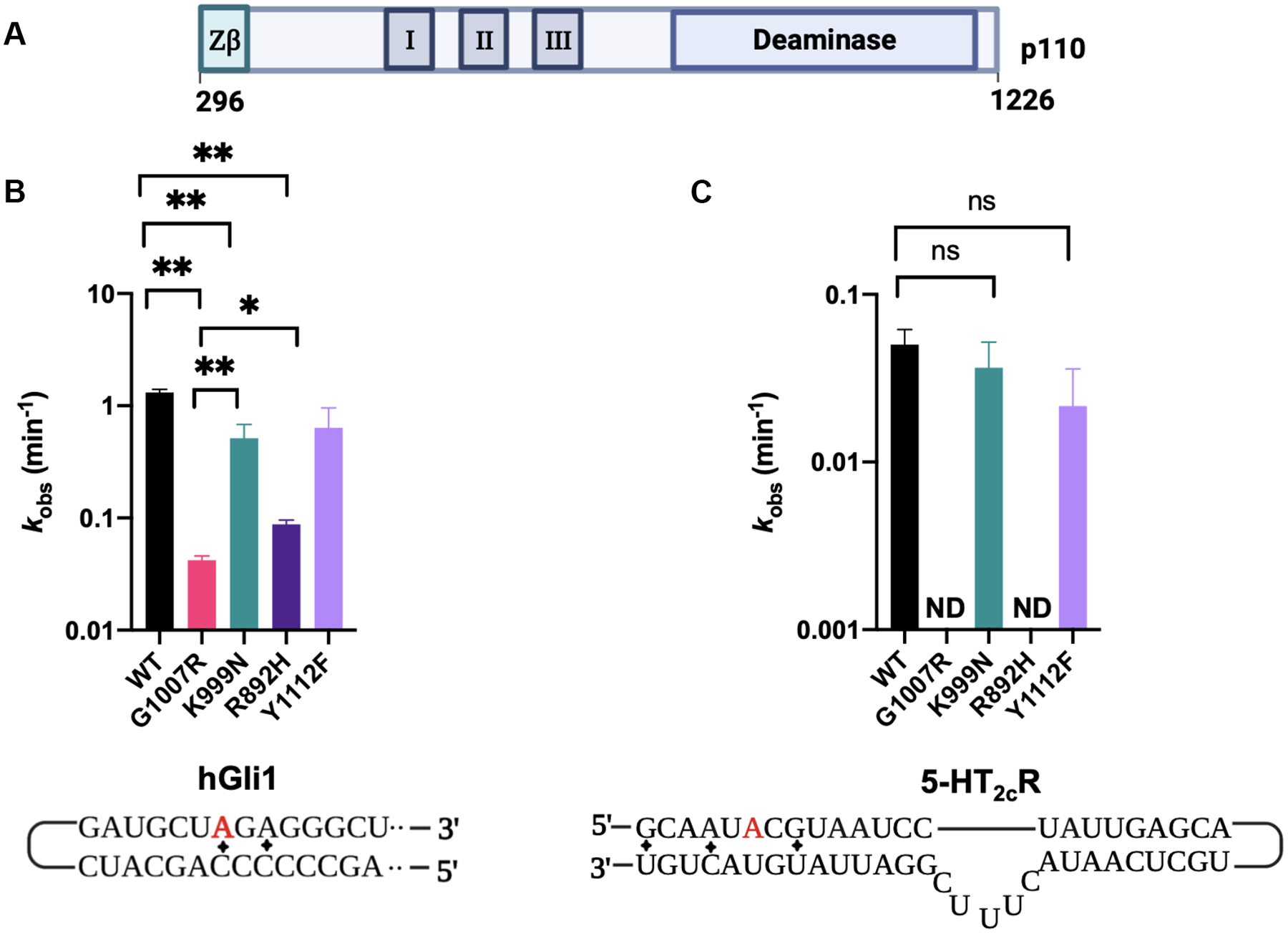
Adenosine deamination rate constants measured with different AGS mutants in the context of ADAR1 p110. (A) ADAR1 p110 domain map. (B) Comparison of rate constants for reaction with 10 nM hGli1 RNA substrate and 100 nM ADAR1 p110. (C) Comparison of rate constants for reaction with 10 nM 5-HT2cR RNA (B site) and 100 nM ADAR1 p110. Editing site monitored for each substrate is highlighted in red. ND: no product detected. Mismatches in the substrates are represented by diamonds.
We have previously shown that introduction of the nucleoside analog 8-azanebularine (8-azaN) into ADAR substrate RNAs allows for the mechanistic trapping of ADAR-RNA complexes for binding and structural studies.20,28,29 The 8-azaN nucleobase is hydrated by ADAR to form a product that is a structural mimic of the ADAR reaction intermediate.28,29 Since the 8-azaN hydrate lacks a good leaving group, the 8-azaN reaction cannot proceed forward, preventing catalytic turnover. The resulting complexes can then be analyzed by various techniques, including electrophoretic mobility shift assays (EMSAs).20,21 Here we tested the effects of different AGS mutations on the formation of ADAR1-RNA complexes stabilized by 8-azaN-modification of the RNA. We carried out EMSAs using a 61-bp RNA duplex with sequence derived from hGli1 bearing 8-azaN near the center, a 3’ Cy5 tag for visualization in the gels and varying concentrations of ADAR1 p110 and AGS mutants (Figure 3, Figure S5). Interestingly, the G1007R mutant required a significantly higher protein concentration to observe a stable, shifted complex than the wild-type p110 or other AGS mutants. Whereas the wild-type p110, K999N, Y1112F, and R892H each showed > 30% complex formation at 10 nM enzyme, 100 nM G1007R was required to observe a shifted band in these experiments (Figure 3B, Figure 3C). Unfortunately, due to the high RNA concentrations required to visualize the protein-RNA complexes using fluorescently labeled RNA (20 nM), we were unable to quantify the apparent dissociation constants using this approach.
Figure 3.
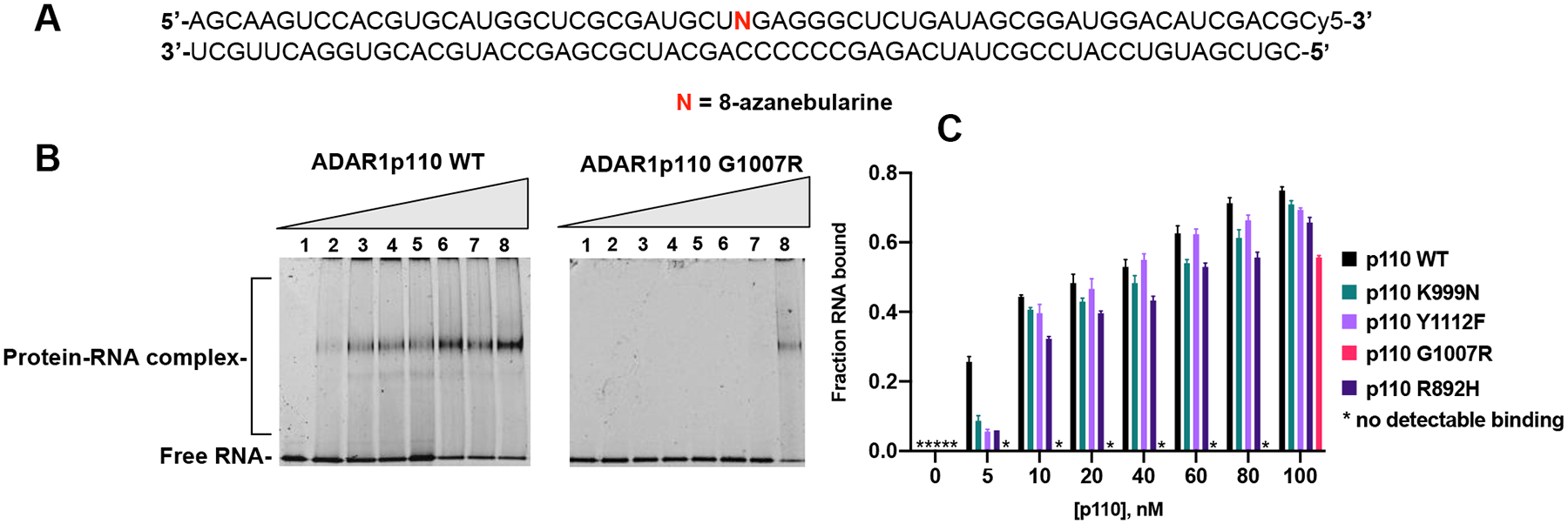
Binding of AGS mutants of ADAR1 p110 to a 61 bp duplex bearing 8-azanebularine (N) analyzed by EMSA. (A) Sequence of 61 bp RNA duplex with the adenosine analog 8-azanebularine (N). N allows for trapping of the protein-RNA complex in the base-flipped conformation18,19. The sequence is labeled with Cy5 at the 3’ end. (B) Representative EMSA gels of p110 WT and AGS mutant G1007R with the 61 bp duplex. (C) Quantification of fraction RNA bound at various concentrations (lanes 1–8: 0, 5, 10, 20, 40, 60, 80 and 100 nM) of WT or mutant (G1007R, K999N, R892H and Y1112F) p110 and 20 nM 61 bp duplex.
The inability of the G1007R mutant to form a stable complex with 8-azaN RNA was somewhat surprising given its three unaltered dsRBDs and the previous report of this mutant’s ability to inhibit the activity of the wild type protein in HEK293 cells.17 To test directly the ability of the AGS mutants to influence the reaction of wild type ADAR1, we performed a competition experiment using the wild-type enzyme to edit the 5-HT2cR substrate RNA in the presence of varying concentrations of added AGS mutant p110 proteins. Interestingly, despite being unable to form a stable complex with 8-azaN-modified RNA, the G1007R mutant strongly inhibited wild-type activity on the 5-HT2cR substrate (Figure S6). At a concentration of 200 nM G1007R p110, editing of the wild-type protein was completely abolished on this substrate. Also, R892H p110 and, to a lesser degree, K999N p110 inhibited the activity of the wild-type protein. No inhibition was observed in this competition experiment with the Y1112F mutant. We suggest the inhibition observed is caused by competitive binding to the substrate RNA by the deaminase–defective mutant enzyme with the most potent inhibition observed for the mutant least capable of catalyzing adenosine deamination itself on the RNA substrate (i.e. G1007R).
Effects of AGS mutations on the N-terminal deletion mutant ADAR1 R3D.
Subsequently, we investigated the effects of the AGS mutants in the ADAR1 R3D protein (Figure 4). Overall, the trends in editing were similar to those observed in the p110 isoform. Notably, the G1007R mutation had a substantially larger impact on the deamination rate than the other mutations with a 28-fold decrease in editing activity with the hGli1 substrate (Figure 4; Table 2). The other mutations reduced the deamination rate from 1.3–5.6-fold with this substrate (Table 2). It is interesting to note that impact of the K999N mutation appears to be greater in ADAR1 R3D than in p110 since editing of the hGli1 substrate is slowed by 4.2-fold compared to wild type (vs 3.3-fold compared to wild type with p110) (Table 1 and Table 2). This effect is much more pronounced with the 5-HT2cR substrate. With this RNA, no editing could be detected for the K999N mutant (or with G1007R and R892H). Wild type ADAR1 R3D and the Y1112F mutant deaminated the 5-HT2cR substrate with nearly identical rates.
Figure 4.
Adenosine deamination rate constants measured with different AGS mutants in the context of ADAR1 R3D. (A) Domain map for ADAR1 R3D. (B) Comparison of rate constants of reactions with 10 nM hGli1 substrate and 100 nM ADAR1 R3D. (C) Comparison of rate constants of reactions with 10 nM 5HT2cR (B site) and 100 nM ADAR1 R3D. Plotted values are the means of three technical replicates ± standard deviation. ND: no product detected.
Table 2.
Adenosine deamination rate constants measured under single-turnover conditions for ADAR1 R3D and AGS mutants on hGli1 and 5-HT2cR B site RNAsa.
| Substrate | Enzyme | k obs (min −1 ) b | k rel c |
|---|---|---|---|
| Gli1 | ADAR1 R3D | 1.7 ± 0.2 | 1 |
| ADAR1 R3D, G1007R | 0.07 ± 0.01 | 0.04 | |
| ADAR1 R3D, K999N | 0.4 ± 0.1 | 0.2 | |
| ADAR1 R3D, R892H | 0.3 ± 0.1 | 0.2 | |
| ADAR1 R3D, Y1112F | 1.3 ± 0.2 | 0.7 | |
| 5-HT2c | ADAR1 R3D | 0.05 ± 0.01 | 1.0 |
| ADAR1 R3D, G1007R | - | - | |
| ADAR1 R3D, K999N | - | - | |
| ADAR1 R3D, R892H | - | - | |
| ADAR1 R3D, Y1112F | 0.04 ± 0.01 | 0.8 |
hGli1 and 5-HT2cR pre-mRNA substrate sequences are shown in Table S1. ADAR1 R3D reactions were carried out with 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% (v/v) glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, and 0.16 U/μL RNAse inhibitor.
kobs was calculated by fitting fitting product formed at different time points to the equation: [P]t = α[1 −e−kobst] where [P]t is percent edited, α is the end point fitted to 95%, and kobs is the observed rate constant.
krel = kobs for mutant/kobs for ADAR1 R3D.
The truncated ADAR1 R3D construct studied in conjunction with a 32P-labeled 32 bp duplex derived from hGli1, facilitated quantification of the effect of these mutations on 8-azaN RNA binding by EMSA (Figure 5). We had previously used this duplex and a similar truncated variant of human ADAR2 to measure Kd values.21 Consistent with our observations for the p110 protein, the G1007R mutation within ADAR1 R3D showed a detrimental effect on 8-azaN RNA binding with a Kd value of 49 ± 8 nM compared to 8 ± 2 nM for wild type R3D (6.3-fold difference) (Figure 5B, Figure 5C, Figure S7). Importantly, we noticed that the complex formed with the G1007R mutant moved to a position in the gel consistent with the binding of a protein monomer21, which subsequently transitioned to a higher-order complex at elevated concentrations (Figure 5B). This suggested the possibility that this mutation influenced ADAR1 dimerization in addition to adenosine deamination. The measured dissociation constants for each of the other mutations tested (R892H, K999N, Y1112F) were approximately 3-fold higher than for wild type ADAR1 R3D. Considering results from the qualitative gel shift analysis with the p110 mutants and the quantitative binding analysis using the ADAR1 R3D, it appears the G1007R mutation is the most detrimental to 8-azaN RNA binding of all the AGS mutations tested here.
Figure 5.
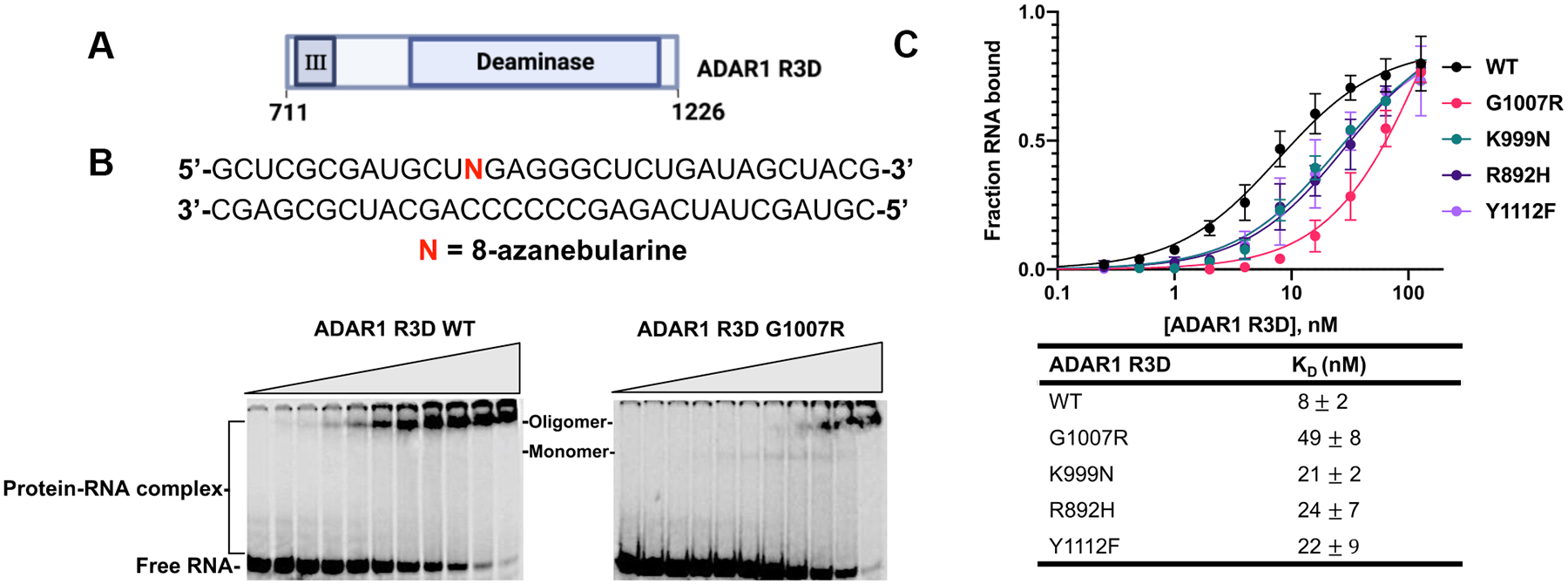
Binding of AGS mutants of ADAR1 R3D to a 32 bp duplex bearing 8-azanebularine (N) analyzed by quantitative EMSA. (A) Domain map of ADAR1 R3D. (B) Sequence of 32 bp duplex bearing 8-azanebularine (N) and representative EMSA gels for ADAR1 R3D WT and AGS mutant G1007R. Reaction was performed at 1.1 nM 32 bp duplex and protein concentration was varied from 0 to 128 nM. (C) Quantitative analysis of the gel shift data. The binding data obtained from the gel shifts were plotted using the equation: y = A × [x/ (KD + x)], where y represents the fraction of RNA bound, x corresponds to the concentration of ADAR1 R3D, A denotes the binding endpoint, and KD represents the dissociation constant. The dissociation constant values were calculated from the plot and reported as the means of three technical replicates ± standard deviation.
Effects of AGS mutants on the ADAR1 deaminase domain.
To explore the functional consequences of the AGS mutations further, we decided to truncate the protein to solely the deaminase domain (ADAR1d). In addition to the AGS-causing mutations, we introduced a hyperactive mutation, E1008Q (Figure 6). The corresponding mutation in ADAR2 is known to enhance base flipping and increase deamination rate.30 We evaluated the impact of each mutation by measuring deamination rates for editing the hGli1 substrate RNA. Again, the G1007R mutation had the greatest effect on editing as no product was observed when this mutation was introduced into the ADAR1 deaminase domain, even with the hyperactive E1008Q variant also present. In addition, upon removal of the RNA binding domains, a large decrease in the rate of editing (100-fold) was observed for the R892H mutation (kobs = 0.02 min−1, Table 3). Consistent with our findings using p110, the Y1112F and K999N mutations led to small reductions in rate for the hyperactive mutant of the deaminase domain. EMSAs performed with an 8-azaN-bearing 23 bp duplex derived from HER1 mRNA, showed the G1007R double mutant exhibited weaker binding to 8-azaN RNA and primarily existed as a monomer, while the E1008Q mutant shifted to a higher order complex at significantly lower concentrations (Figure S8). This provides further evidence supporting the hypothesis that G1007R disrupts ADAR1 dimerization in addition to adenosine deamination. Finally, we examined the impact of each mutation on the thermal stability of the ADAR1 deaminase domain. Most mutations did not affect protein stability, including the E1008Q mutation, as indicated by comparable TM values to the wild-type deaminase domain. However, the G1007R double mutant exhibited a notable deviation with a TM value lower by 5 °C compared to the protein with the E1008Q mutation alone (Table 4).
Figure 6.
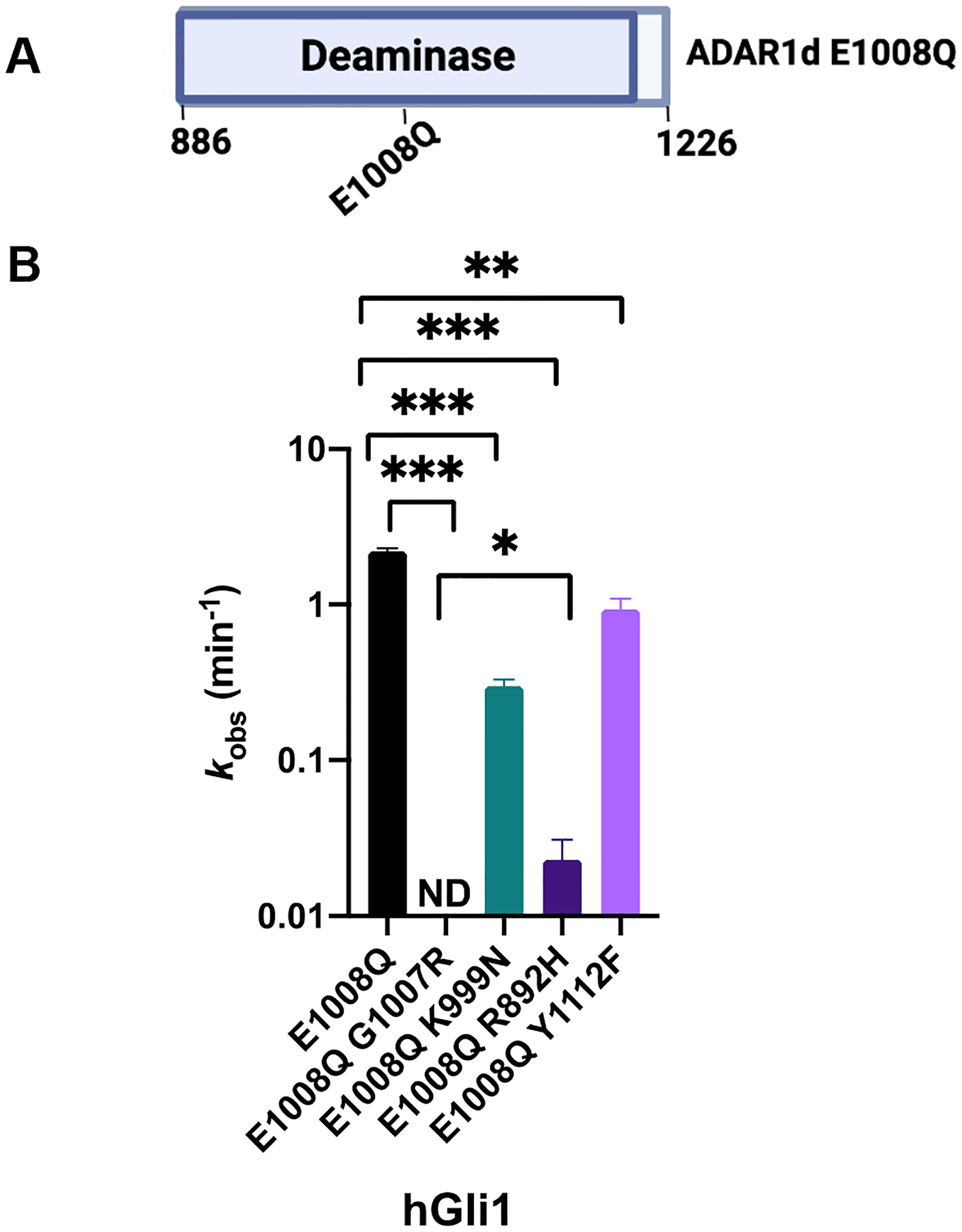
Adenosine deamination rate constants measured with different AGS mutants in the context of ADAR1d E1008Q. (A) Domain map of ADAR1d E1008Q. (B) Comparison of rate constants of 10 nM hGli1 substrate and 100 nM ADAR1d E1008Q. Plotted values are the means of three technical replicates ± standard deviation. ND: no product detected.
Table 3.
Adenosine deamination rate constants measured under single-turnover conditions for ADARld E1008Q and AGS mutants on hGlil RNAa.
| Substrate | Enzyme | k obs (min −1 ) b | k rel c |
|---|---|---|---|
| Gli1 | ADAR1d E1008Q | 2.0 ± 0.1 | 1 |
| ADAR1d E1008Q, G1007R | - | - | |
| ADAR1d E1008Q, K999N | 0.30 ± 0.03 | 0.1 | |
| ADAR1d E1008Q, R892H | 0.02 ± 0.01 | 0.01 | |
| ADAR1d E1008Q,Y1112F | 0.4 ± 0.1 | 0.4 |
Gli1 substrate sequence is shown in Table S1. ADAR1d E1008Q reactions were carried out with 15 mM Tris–HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% (v/v) glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, and 0.16 U/μL RNAse inhibitor.
kobs was calculated by fitting fitting product formed at different time points to the equation: [P]t = α[1 −e−kobst] where [P]t is percent edited, α is the end point fitted to 95%, and kobs is the observed rate constant.
krel = kobs for mutant/kobs for ADAR1d E1008Q.
Table 4.
Experimental thermal melting temperatures for ADAR1d E1008Q and AGS mutants a.
| Enzyme | TM(°C)b * |
|---|---|
| ADAR1d WT | 50 ± 1 |
| ADAR1d E1008Q | 51 ± 1 |
| ADAR1d E1008Q G1007R | 46 ± 0 |
| ADAR1d El008Q K999N | 49 ± 0 |
| ADAR1d E1008Q R892H | 52 ± 0 |
| ADAR1d E1008O Y1112F | 50 ± 2 |
Values reported are the average of three independent measurements ± standard deviation
DISCUSSION
Recent studies have highlighted the regulatory roles of RNA editing by ADAR1 in preventing recognition of self RNA as viral or non-self.10,16,18,31 Given its role in immune regulation, loss of function mutations within the ADAR1 gene are associated with several immune disorders, including AGS.17,18,31 There are a few reports in the field investigating the consequences of these mutations on RNA editing by ADAR1 in mammalian cell lines and animal models.6,7,18,23,24 However, there is no published reports of the effects of these mutations on the biochemical properties of ADAR1 evaluated under well-defined solution conditions with purified proteins. Here we measured rates of adenosine deamination in two different RNA substrates and binding to 8-azaN RNAs for four different AGS-causing mutants of ADAR1. In addition, we tested the effect of these mutations in N-terminal deletion variants that lack RNA-binding domains. The results of these experiments have highlighted differences among these mutations suggesting the following trend of impact on adenosine deamination: G1007R >> R892H > K999N > Y1112F. The effects observed were also context dependent. For instance, the G1007R and R892H p110 mutants exhibited no detectable editing activity on the 5-HT2cR substrate, and the truncation to ADAR1 R3D was enough to nullify the activity of the K999N mutation on this substrate. In contrast, these mutants exhibited significant editing on hGli1, a substrate considered ideal for ADAR editing due to its 5’ and 3’ nearest neighbor nucleotides and an AC mismatch at the editing position.21,27,30 In fact, the deaminase domain is sufficient for hGli1 editing.8 In contrast, the 5-HT2cR substrate presents structural complexities, including bulges and mismatches, necessitating the engagement of double-stranded RNA-binding domains (dsRBDs).21
G1007R is highly detrimental to catalysis and binding to 8-azaN RNA.
G1007R emerged as the most potent mutation studied here. Interestingly, it is also consistently observed in a heterozygous form among all AGS patients carrying this mutation.17,32 Given the impact of this mutation on adenosine deamination by ADAR1, a G1007R homozygote would likely be similar to an ADAR1 null mutation, which is known to be embryonic lethal.17,18,32 Positioned within the GEG triplet in the protein’s flipping loop, G1007 plays a crucial role in facilitating the intercalation of E1008 into duplex RNA substrate, thereby ensuring the stability of the flipped-out adenosine conformation for editing. Perturbation of the adjacent glycine residues would be expected to profoundly disrupt the enzyme’s ability to induce base flipping.20,21 Our kinetic analyses, employing both full-length and truncated ADAR1 proteins, illustrates the significant impairment of catalysis caused by the G1007R mutation. In addition, in EMSAs with 8-azaN RNAs using various ADAR1 constructs, we consistently observed the highly detrimental effect of the G1007R mutation. It should be noted here that tight binding of an ADAR protein to 8-azaN RNA requires the ability to flip the 8-azaN into the ADAR active site20,21,28,29 so a mutation that substantially alters the flipping loop, like G1007R does, would be expected to impair binding to 8-azaN RNA. However, the G1007R mutation does not prevent the mutant protein from blocking the activity of the wild- type enzyme as indicated in the competition experiments with p110, suggesting a mode of RNA binding not captured in our EMSAs with 8-azaN RNA (Figure S6). This is consistent with previous reports that described G1007R can function as a dominant-negative mutant capable of inhibiting the activity of the wild-type enzyme.17
Our results also suggest that G1007R may disrupt ADAR1 dimerization, as indicated by EMSAs with ADAR1 R3D and ADAR1d constructs (Figure 5B, Figure S8). The recently reported crystal structure of an ADAR2 dimer bound to RNA identified a dimer interface including an α-helix composed of a conserved sequence shared between ADAR1 and ADAR2.21 In one monomer of the ADAR2 dimer, G487 is positioned near the dimerization interface and mutation of this residue to arginine would lead to steric clashes with neighboring residues (Figure 7A). We suggest that these clashes may disrupt dimerization in the G1007R mutant of ADAR1. Indeed, the AlphaFold model of the ADAR1 dimer also shows clashes with neighboring residues for the G1007R mutant (Figure 7B). Therefore, it is plausible that the inhibitory effects of G1007R are exerted by preventing dimerization, while still retaining the ability to bind substrates at elevated concentrations. In previous studies, we demonstrated the editing of the 5-HT2cR substrate is dependent on an intact dimerization interface, while hGli1 is minimally affected by dimerization mutations in ADAR1 or ADAR2.21 Considering the potential role of the G1007 residue in ADAR1 dimerization, this provides an explanation for the observed slower editing in the serotonin substrate and the significant editing of the G1007R mutant in the hGli1 substrate. It is likely robust substrates that resemble hGli1 will demonstrate some activity with the G1007R mutant. Finally, G1007R exhibits noticeable deviations in thermal stability compared to both the wild-type and other mutants (Table 4).
Figure 7.
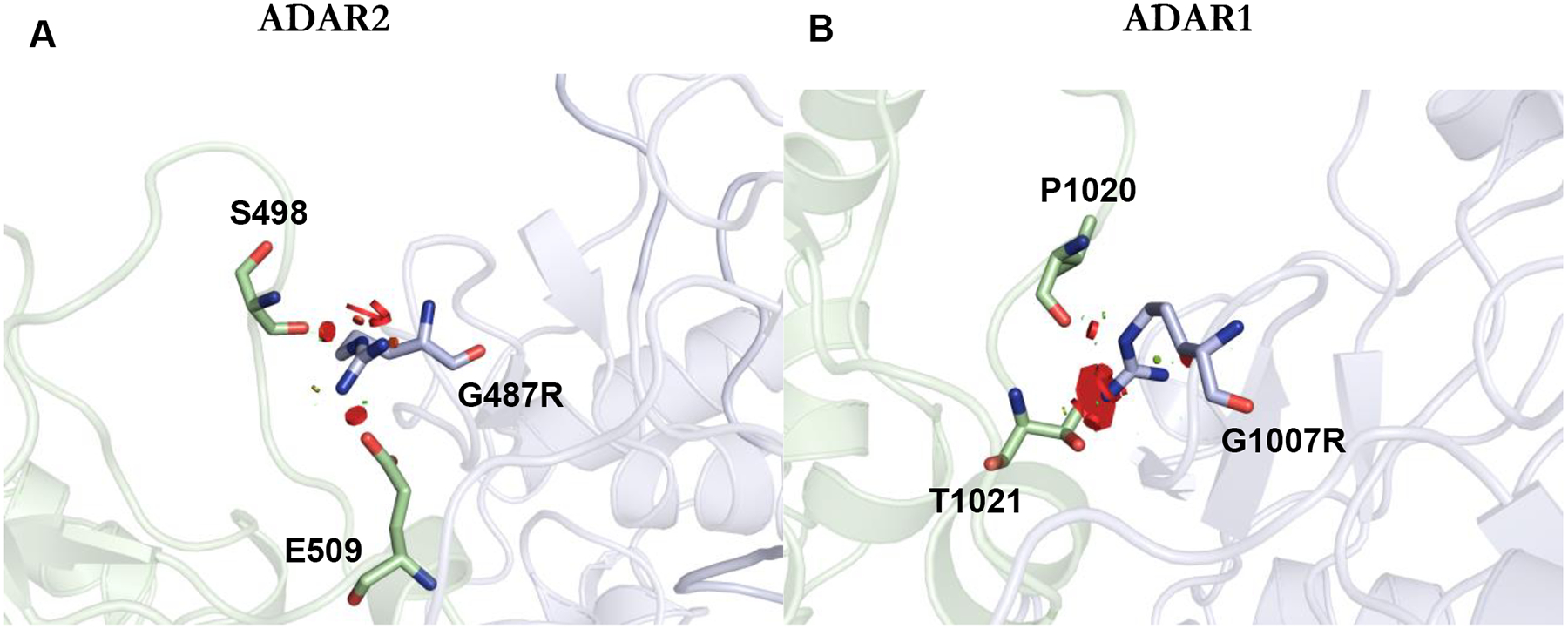
G1007R effect on the ADAR dimer interface. (A) Close up view mapping the location of G487R (analogous to G1007R mutation in ADAR1) on the non-catalytic monomer (light blue) of hADAR2 R2D dimer (PDBID 6VFF). The red discs represent a significant Van der Waals overlap, suggesting a clash between atoms. At the dimer interface, G487R clashes with E509 and S498. (B) An AlphaFold model of the ADAR1 dimer interface. In the model, G1007R clashes with T1021 and P1020.
R892H inhibits adenosine deamination by ADAR1.
ADAR sequence alignments suggest R892 of human ADAR1 plays the same role as K376 in human ADAR2.21 In structures of ADAR2 bound to RNA, K376 is shown stabilizing the base-flipped conformation by hydrogen bonding to both the 5’ and 3’ phosphodiesters of the nucleotide immediately adjacent to the editing site on the 3’ side (Figure 8).20,21 The guanidinium group of R892 could also simultaneously hydrogen bond to both 5’ and 3’ phosphodiesters of this nucleotide in the substrate RNA. Mutation of R892 to histidine would disrupt this contact. Our observation of dramatic reductions in catalytic rate for R892H ADAR1 p110 and R892H ADAR1d (E1008Q) with both RNA substrates and R892H ADAR1 R3D with the 5-HT2cR substrate is consistent with this prediction. In addition, R892H was a strong inhibitor of ADAR1 p110 wild-type activity with the 5-HT2cR substrate. The R892H mutation is not as detrimental to catalysis of adenosine deamination as is the G1007R mutation as indicated by a comparison of reaction rates for R892H ADAR1 R3D and R892H ADAR1d (E1008Q) with the hGli1 RNA substrate and the corresponding G1007R mutants (Table 2, Table 3). This is understood when one considers that a histidine at position 892 could maintain a hydrogen bonding contact and, in its protonated form, even a salt bridge with at least one of the adjacent phosphodiesters in the substrate RNA. It is noteworthy that the R892H mutant was identified in conjunction with the Z domain mutation P193A17,33. It is possible that the presence of both the P193A mutation and a catalytic mutant is necessary to observe a disease phenotype. However, homozygous mice carrying the P193A mutation alone exhibit a normal phenotype.6,33,34 Since our analysis revealed that the R892H mutation alone can substantially disrupt catalysis of adenosine deamination by ADAR1, it is possible that the R892H mutation is the key change leading to disease for the P193A, R892H double mutant found in the AGS patient population. Additionally, a recent study by Jantsch et al. has indicated that the specificity of the ADAR1 p150 isoform is predominantly influenced by cellular localization31. This suggests that the p150 isoform’s function can be complemented by any active ADAR isoform by altering its cellular localization, hinting at the potential role of the mutations within the catalytic domain of ADAR1 as the leading cause of AGS phenotype31.
Figure 8.
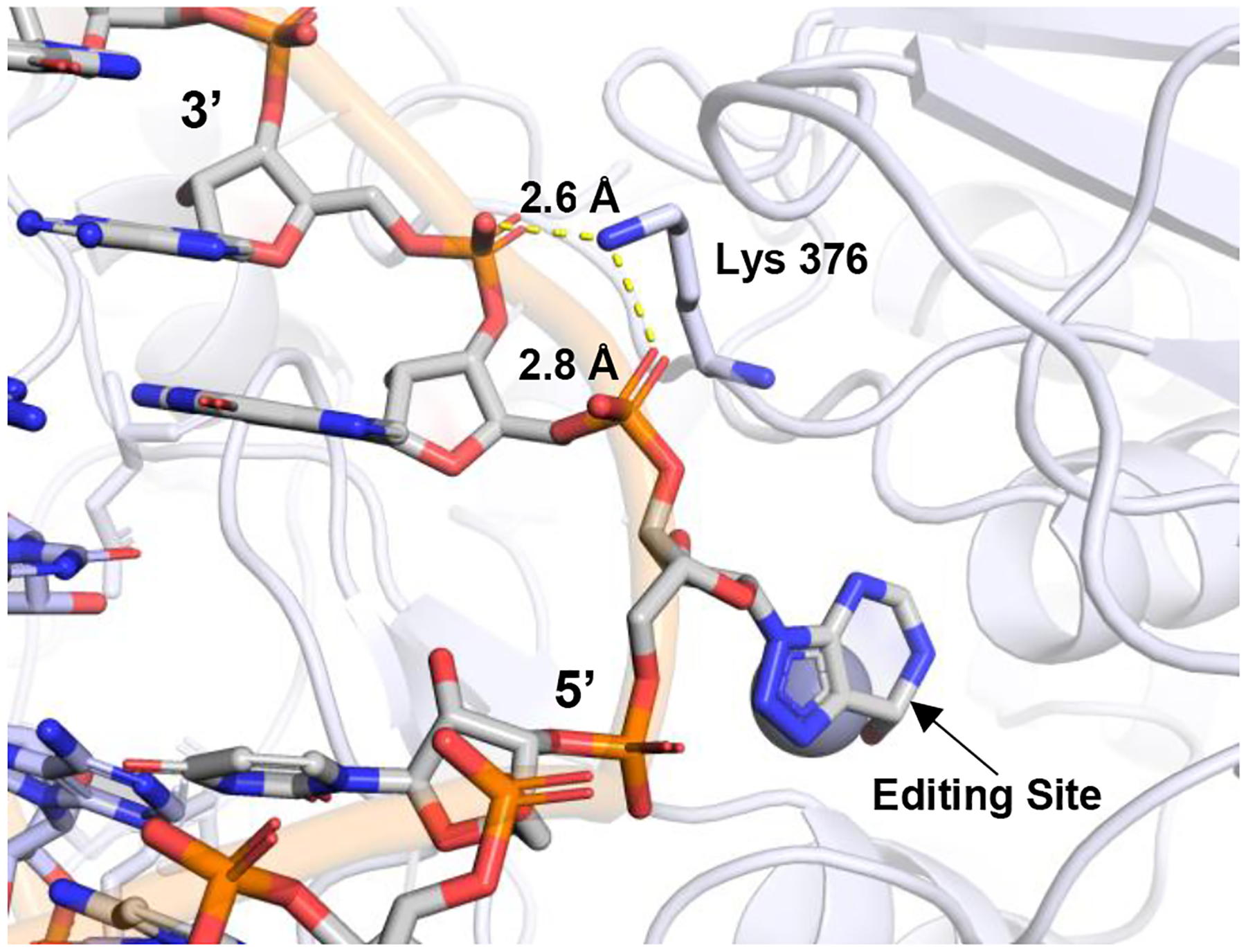
Close up view mapping the location of R892H mutation onto hADAR2 R2D structure (PDBID 6VFF). Residue Lys 376 in ADAR2 makes a contact with phosphates of G14 and A15 3’ to the edited base.
K999N and Y1112F exert mild effects on ADAR1.
Overall, the K999N and Y1112F mutations had more mild effects on the ADAR1 properties evaluated in this study than did G1007R or R892H. The K999N and Y1112F mutations, located adjacent to an important RNA binding loop in the catalytic domain, are observed as homozygous mutations in AGS patients consistent with a less severe impact on catalytic activity.17,18,23,24 In the context of p110, we measured only a slight reduction in deamination rate with K999N and Y1112F variants (1–3-fold changes in rate). We did note that the K999N mutation had a greater effect in ADAR1 R3D, particularly with the 5-HT2cR substrate, and with ADAR1d (E1008Q) and the hGli1 substrate, highlighting the context-dependent effect of this mutation. Interestingly, a recent mouse model study by Guo et al. also observed a substrate-dependent effect of the ADAR1 p150 K999N mutant.23,24,24 They found a significant reduction in editing of the 5-HT2cR substrate at sites A, B, and C, while site D, which is a preferred ADAR2 site, showed an increase. In contrast, editing of glutamate ionotropic receptor AMPA type subunit 2 and 3 (GRIA2 and 3) RNAs remained comparable to the wild-type. These changes in editing patterns were sufficient to hinder the growth of mice, elevate levels of several immune signature genes, and increase inflammatory cytokine levels in the brain.24 In contrast to K999N, our studies showed only small effects for the Y1112F mutation in each of our assays. Given its location in the ADAR1 catalytic domain, Y1112F is predicted to indirectly effect substrate recognition by interrupting interactions with the 5’ binding loop. Like K999N, this mutation may exert substrate-dependent effects that were not observed in our studies. Clearly more studies with ADAR1 Y1112F will be necessary to identify properties of the protein that are substantially altered by this mutation.
CONCLUSIONS
While the association between mutations within ADAR1 and AGS is established, the underlying mechanisms through which these mutations alter ADAR1 activity are still being unraveled. Our biochemical studies provide mechanistic insights into select mutants and their interference with catalysis of adenosine deamination at the protein level. Further investigations using other model systems will be important to determine if specific substrates are affected by each mutation. Unraveling the effects of ADAR1 mutations deepens our understanding of RNA editing’s regulatory roles and expands our knowledge of ADAR1-substrate recognition.
Supplementary Material
Funding Sources
P.A.B. acknowledges financial support from National Institutes of Health in the form of grant R35GM141907.
Footnotes
ACCESSION CODES
ADAR1 p110: P55265-5
ADAR1 p150: P55265-1
SUPPORTING INFORMATION
Oligonucleotide sequences and masses; predicted secondary structures for substrate RNAs; gel images for purified proteins; kinetic plots; representative EMSA gel images; competitive inhibition data.
REFERENCES
- (1).Eisenberg E; Levanon EY A-to-I RNA Editing — Immune Protector and Transcriptome Diversifier. Nat Rev Genet 2018, 19 (8), 473–490. [DOI] [PubMed] [Google Scholar]
- (2).Bass BL RNA Editing by Adenosine Deaminases That Act on RNA. Annu Rev Biochem 2002, 71 (1), 817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang Y; Zheng Y; Beal PA Adenosine Deaminases That Act on RNA (ADARs). Enzymes 2017, 41, 215–268 [DOI] [PubMed] [Google Scholar]
- (4).Nakahama T; Kato Y; Shibuya T; Inoue M; Kim JI; Vongpipatana T; Todo H; Xing Y; Kawahara Y Mutations in the Adenosine Deaminase ADAR1 That Prevent Endogenous Z-RNA Binding Induce Aicardi-Goutières-Syndrome-like Encephalopathy. Immunity 2021, 54 (9), 1976–1988.e7. [DOI] [PubMed] [Google Scholar]
- (5).de Reuver R; Dierick E; Wiernicki B; Staes K; Seys L; De Meester E; Muyldermans T; Botzki A; Lambrecht BN; Van Nieuwerburgh F; Vandenabeele P; Maelfait J ADAR1 Interaction with Z-RNA Promotes Editing of Endogenous Double-Stranded RNA and Prevents MDA5-Dependent Immune Activation. Cell Rep 2021, 36 (6), 109500. [DOI] [PubMed] [Google Scholar]
- (6).Guo X; Liu S; Sheng Y; Zenati M; Billiar T; Herbert A; Wang Q ADAR1 Zα Domain P195A Mutation Activates the MDA5-Dependent RNA-Sensing Signaling Pathway in Brain without Decreasing Overall RNA Editing. Cell Rep 2023, 42 (7), 112733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nie Y; Zhao Q; Su Y; Yang J-H Subcellular Distribution of ADAR1 Isoforms Is Synergistically Determined by Three Nuclear Discrimination Signals and a Regulatory Motif. Journal of Biological Chemistry 2004, 279 (13), 13249–13255. [DOI] [PubMed] [Google Scholar]
- (8).Wang Y; Park S; Beal PA Selective Recognition of RNA Substrates by ADAR Deaminase Domains. Biochemistry 2018, 57 (10), 1640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Park S; Doherty EE; Xie Y; Padyana AK; Fang F; Zhang Y; Karki A; Lebrilla CB; Siegel JB; Beal PA High-Throughput Mutagenesis Reveals Unique Structural Features of Human ADAR1; 2020; Vol. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nakahama T; Kawahara Y The RNA-Editing Enzyme ADAR1: A Regulatory Hub That Tunes Multiple DsRNA-Sensing Pathways. Int Immunol 2023, 35 (3), 123–133. [DOI] [PubMed] [Google Scholar]
- (11).Song B; Shiromoto Y; Minakuchi M; Nishikura K The Role of RNA Editing Enzyme ADAR1 in Human Disease. WIREs RNA 2022, 13 (1), 1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu J; Wang F; Zhang Y; Liu J; Zhao B ADAR1-Mediated RNA Editing and Its Role in Cancer. Front Cell Dev Biol 2022, 10, 956649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gannon HS; Zou T; Kiessling MK; Gao GF; Cai D; Choi PS; Ivan AP; Buchumenski I; Berger AC; Goldstein JT; Cherniack AD; Vazquez F; Tsherniak A; Levanon EY; Hahn WC; Meyerson M Identification of ADAR1 Adenosine Deaminase Dependency in a Subset of Cancer Cells. Nat Commun 2018, 9 (1), 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bhate A; Sun T; Li JB ADAR1: A New Target for Immuno-Oncology Therapy. Mol Cell 2019, 73 (5), 866–868. [DOI] [PubMed] [Google Scholar]
- (15).Heraud-Farlow JE; Walkley CR What Do Editors Do? Understanding the Physiological Functions of A-to-I RNA Editing by Adenosine Deaminase Acting on RNAs. Open Biol 2020, 10 (7), 200085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liddicoat BJ; Piskol R; Chalk AM; Ramaswami G; Higuchi M; Hartner JC; Li JB; Seeburg PH; Walkley CR RNA Editing by ADAR1 Prevents MDA5 Sensing of Endogenous DsRNA as Nonself. Science (1979) 2015, 349 (6252), 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rice GI; Kasher PR; Forte GMA; Mannion NM; Greenwood SM; Szynkiewicz M; Dickerson JE; Bhaskar SS; Zampini M; Briggs TA; Jenkinson EM; Bacino CA; Battini R; Bertini E; Brogan PA; Brueton LA; Carpanelli M; De Laet C; de Lonlay P; del Toro M; Desguerre I; Fazzi E; Garcia-Cazorla À; Heiberg A; Kawaguchi M; Kumar R; Lin J-PS-M; Lourenco CM; Male AM; Marques W; Mignot C; Olivieri I; Orcesi S; Prabhakar P; Rasmussen M; Robinson RA; Rozenberg F; Schmidt JL; Steindl K; Tan TY; van der Merwe WG; Vanderver A; Vassallo G; Wakeling EL; Wassmer E; Whittaker E; Livingston JH; Lebon P; Suzuki T; McLaughlin PJ; Keegan LP; O’Connell MA; Lovell SC; Crow YJ Mutations in ADAR1 Cause Aicardi-Goutières Syndrome Associated with a Type I Interferon Signature. Nat Genet 2012, 44 (11), 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lamers MM; van den Hoogen BG; Haagmans BL ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol 2019, 10, 453024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Crow YJ; Chase DS; Lowenstein Schmidt J; Szynkiewicz M; Forte GMA; Gornall HL; Oojageer A; Anderson B; Pizzino A; Helman G; Abdel-Hamid MS; Abdel-Salam GM; Ackroyd S; Aeby A; Agosta G; Albin C; Allon-Shalev S; Arellano M; Ariaudo G; Aswani V; Babul-Hirji R; Baildam EM; Bahi-Buisson N; Bailey KM; Barnerias C; Barth M; Battini R; Beresford MW; Bernard G; Bianchi M; Billette de Villemeur T; Blair EM; Bloom M; Burlina AB; Luisa Carpanelli M; Carvalho DR; Castro-Gago M; Cavallini A; Cereda C; Chandler KE; Chitayat DA; Collins AE; Sierra Corcoles C; Cordeiro NJV; Crichiutti G; Dabydeen L; Dale RC; D’Arrigo S; De Goede CGEL; De Laet C; De Waele LMH; Denzler I; Desguerre I; Devriendt K; Di Rocco M; Fahey MC; Fazzi E; Ferrie CD; Figueiredo A; Gener B; Goizet C; Gowrinathan NR; Gowrishankar K; Hanrahan D; Isidor B; Kara B; Khan N; King MD; Kirk EP; Kumar R; Lagae L; Landrieu P; Lauffer H; Laugel V; La Piana R; Lim MJ; Lin J-PS-M; Linnankivi T; Mackay MT; Marom DR; Marques Lourenço C; McKee SA; Moroni I; Morton JEV; Moutard M-L; Murray K; Nabbout R; Nampoothiri S; Nunez-Enamorado N; Oades PJ; Olivieri I; Ostergaard JR; Pérez-Dueñas B; Prendiville JS; Ramesh V; Rasmussen M; Régal L; Ricci F; Rio M; Rodriguez D; Roubertie A; Salvatici E; Segers KA; Sinha GP; Soler D; Spiegel R; Stödberg TI; Straussberg R; Swoboda KJ; Suri M; Tacke U; Tan TY; te Water Naude J; Wee Teik K; Mary Thomas M; Till M; Tonduti D; Maria Valente E; Noel Van Coster R; van der Knaap MS; Vassallo G; Vijzelaar R; Vogt J; Wallace GB; Wassmer E; Webb HJ; Whitehouse WP; Whitney RN; Zaki MS; Zuberi SM; Livingston JH; Rozenberg F; Lebon P; Vanderver A; Orcesi S; Rice GI Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015, 167 (2), 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Matthews MM; Thomas JM; Zheng Y; Tran K; Phelps KJ; Scott AI; Havel J; Fisher AJ; Beal PA Structures of Human ADAR2 Bound to DsRNA Reveal Base-Flipping Mechanism and Basis for Site Selectivity. Nat Struct Mol Biol 2016, 23 (5), 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Thuy-Boun AS; Thomas JM; Grajo HL; Palumbo CM; Park S; Nguyen LT; Fisher AJ; Beal PA Asymmetric Dimerization of Adenosine Deaminase Acting on RNA Facilitates Substrate Recognition. Nucleic Acids Res 2020, 48 (14), 7958–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fisher AJ; Beal PA Effects of Aicardi-Goutières Syndrome Mutations Predicted from ADAR-RNA Structures. RNA Biol 2017, 14 (2), 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Guo X; Wiley CA; Steinman RA; Sheng Y; Ji B; Wang J; Zhang L; Wang T; Zenatai M; Billiar TR; Wang Q Aicardi-Goutières Syndrome-Associated Mutation at ADAR1 Gene Locus Activates Innate Immune Response in Mouse Brain. J Neuroinflammation 2021, 18 (1), 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guo X; Steinman RA; Sheng Y; Cao G; Wiley CA; Wang Q An AGSAssociated Mutation in ADAR1 Catalytic Domain Results in Early-Onset and MDA5-Dependent Encephalopathy with IFN Pathway Activation in the Brain. J Neuroinflammation 2022, 19 (1), 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mendoza HG; Matos VJ; Park S; Pham KM; Beal PA Selective Inhibition of ADAR1 Using 8-Azanebularine-Modified RNA Duplexes. Biochemistry 2023, 62 (8), 1376–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shimokawa T; Rahman MF-U; Tostar U; Sonkoly E; Ståhle M; Pivarcsi A; Palaniswamy R; Zaphiropoulos PG RNA Editing of the GLI1 Transcription Factor Modulates the Output of Hedgehog Signaling. RNA Biol 2013, 10 (2), 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Eggington JM; Greene T; Bass BL Predicting Sites of ADAR Editing in Double-Stranded RNA. Nat Commun 2011, 2 (1), 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Haudenschild BL; Maydanovych O; Véliz EA; Macbeth MR; Bass BL; Beal PA A Transition State Analogue for an RNA-Editing Reaction. J Am Chem Soc 2004, 126 (36), 11213–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Phelps KJ; Tran K; Eifler T; Erickson AI; Fisher AJ; Beal PA Recognition of Duplex RNA by the Deaminase Domain of the RNA Editing Enzyme ADAR2. Nucleic Acids Res 2015, 43 (2), 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kuttan A; Bass BL Mechanistic Insights into Editing-Site Specificity of ADARs. Proceedings of the National Academy of Sciences 2012, 109 (48), 3925–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kleinova R; Rajendra V; Leuchtenberger AF; Lo Giudice C; Vesely C; Kapoor U; Tanzer A; Derdak S; Picardi E; Jantsch MF The ADAR1 Editome Reveals Drivers of Editing-Specificity for ADAR1-Isoforms. Nucleic Acids Res 2023, 51 (9), 4191–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rice G; Kitabayashi N; Barth M; Briggs T; Burton A; Carpanelli M; Cerisola A; Colson C; Dale R; Danti F; Darin N; De Azua B; De Giorgis V; De Goede C; Desguerre I; De Laet C; Eslahi A; Fahey M; Fallon P; Fay A; Fazzi E; Gorman M; Gowrinathan N; Hully M; Kurian M; Leboucq N; Lin J-P; Lines M; Mar S; Maroofian R; Martí-Sanchez L; McCullagh G; Mojarrad M; Narayanan V; Orcesi S; Ortigoza-Escobar J; Pérez-Dueñas B; Petit F; Ramsey K; Rasmussen M; Rivier F; Rodríguez-Pombo P; Roubertie A; Stödberg T; Toosi M; Toutain A; Uettwiller F; Ulrick N; Vanderver A; Waldman A; Livingston J; Crow Y Genetic, Phenotypic, and Interferon Biomarker Status in ADAR1-Related Neurological Disease. Neuropediatrics 2017, 48 (03), 166–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liang Z; Chalk AM; Taylor S; Goradia A; Heraud-Farlow JE; Walkley CR The Phenotype of the Most Common Human ADAR1p150 Zα Mutation P193A in Mice Is Partially Penetrant. EMBO Rep 2023, 24 (5), 55835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Langeberg CJ; Nichols PJ; Henen MA; Vicens Q; Vögeli B Differential Structural Features of Two Mutant ADAR1p150 Zα Domains Associated with Aicardi-Goutières Syndrome. J Mol Biol 2023, 435 (8), 168040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.