

Abstract
α-Sulfinyl esters can be readily prepared through thiol substitution of α-bromo esters followed by oxidation to the sulfoxide. Enzymatic resolution with lipoprotein lipase provides both the unreacted esters and corresponding α-sulfinyl carboxylic acids in high yields and enantiomeric ratios. Subsequent decarboxylative halogenation, dihalogenation, trihalogenation and cross-coupling gives rise to functionalized sulfoxides. The method has been applied to the asymmetric synthesis of a potent inhibitor of 15-prostaglandin dehydrogenase.
Keywords: decarboxylation, halogenation, lipase, resolution, sulfoxides
Graphical Abstract
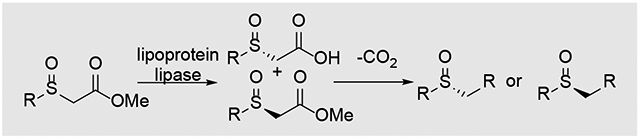
Enzymatic resolution of α-sulfinyl esters gives recovered ester and α-sulfinyl acid in high enantiomeric purity. Decarboxylative functionalization provides access to a wide range of optically active sulfoxides in both enantiomeric forms.
Introduction
Optically active sulfoxides are important components of pharmaceutical agents, natural products,[1] chemical reagents[2]chiral auxiliaries,[3] and ligands for asymmetric catalysis.4 Biologically active sulfoxides include the bicyclic octapeptide α-amanitin was isolated from the death-cap mushroom and found to potently inhibit RNA Pol II (Figure 1).[5] Carmaphycin A, produced by a marine cyanobacteria, inhibits the proteasome with IC50 = 2.5 nM,[6] while ustiloxin G is a cyclopeptide fungal metabolite that potently inhibits rice germination.[7] Stereodefined sulfoxides also appear in approved drugs, including the stimulant armodafinil[8] and the proton pump inhibitors esomeprazole and dexlansoprazole.[9] We became interested in this area in connection with our efforts to identify inhibitors of 15-prostaglandin dehydrogenase (15-PGDH), an enzyme that metabolizes several prostaglandins, including PGE2, PGF2α, and PGI2.[10] In this context, the sulfoxide SW209415 inhibits 15-PGDH with low nM IC50 values, and the R-isomer is >100-fold more potent than the S-enantiomer.[11]
Figure 1.

Representative sulfoxide containing natural products and pharmaceutical agents.
The stereospecificity of inhibition by SW209415 and the broader role of sulfoxides in biologically active small molecules highlights the importance of stereoselective syntheses of sulfoxides.12 Asymmetric, catalytic oxidations of sulfides have been developed, with several reports describing first-row transition metal complexes that use peroxide oxidants.[13] The best enantioselectivities are generally achieved with aryl thioethers, with dialkyl sulfoxides being formed with lower selectivity. Chiral phosphoric acids and their derivatives catalyze highly enantioselective oxidation of a range of sulfides. However, these catalysts require multistep syntheses, and their high molecular weight could limit their applicability. Enzymes and cell extracts have been used to kinetically resolve sulfoxides through reduction to the sulfide.[14] High enantioselectivities can be obtained with this method, but only one enantiomer is generally accessible.
We considered an approach to optically active α-sulfinyl carboxylates that would involve kinetic resolution of the corresponding ester 1 (Table 1). Recent advances in decarboxylative functionalization could provide a way to convert α-sulfinyl acids 2 to a wide variety of functionalized sulfoxides.[15] Many kinetic resolutions of sulfoxides have been described, but most of them involve selective oxidation of one sulfoxide enantiomer to the corresponding achiral sulfone.[16],[17] By contrast, lipase resolution could provide both enantiomers in easily separated, highly enantioenriched form.[18] Pioneering research from Burgess and Henderson demonstrated enzymatic hydrolysis of sulfinyl esters,[19] but the enzyme used in that work, Pseudomonas K-10, is no longer readily available.
Table 1.
Enzymatic resolution of α-sulfinyl carboxylates.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | Cmpd | R | Buffer pH |
Time (h) |
% Yield 1 (% ee) |
% Yield 2 (% ee) |
| 1[b] | 1a/2a |
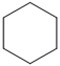
|
7.5 | 16 | 46 (>98) | 30 (99) |
| 2[b] | 1b/2b | n-Hex | 9.0 | 12 | 46 (>98) | 32 (>99) |
| 3 | 1c/2c |
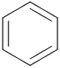
|
7.5 | 16 | 48 (>99) | 35 (>99) |
| 4 | 1d/2d |
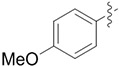
|
7.5 | 15 | 47 (>99) | 36 (96) |
| 5 | 1e/2e |
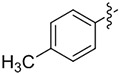
|
7.5 | 15 | 46 (>99) | 35 (>99) |
| 6 | 1f/2f |

|
8.6 | 14 | 45 (>99) | 33 (>99) |
| 7 | 1g/2g |
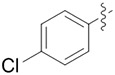
|
7.5 | 16 | 45 (>99) | 33 (98) |
| 8[b],[c] | 1h/2h |
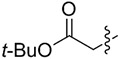
|
9.5 | 60 | 44 (>99) | 30 (89) |
Reactions on a 1 g scale with 10 wt% lipoprotein lipase from Burkholderia sp., 8:1 50 mM phosphate buffer:toluene unless otherwise noted.
30 wt% lipase.
100 mM phosphate buffer.
Results and Discussion
We evaluated lipases and esterases from various sources using the cyclohexyl sulfoxide 1a as a test substrate (Table S1).[20] Of the 16 enzymes tested, five gave >30% conversion, but only lipoprotein lipase from Burkholderia sp. (EC 3.1.1.3 ) gave >35% ee. Under optimized conditions, the ester 1a could be recovered in 46% yield on gram scale as a single enantiomer from the reaction mixture by simple extraction (Table 1). Monitoring the reaction by 1H NMR spectroscopy indicated a nearly perfect kinetic resolution with 50% conversion and selectivity factor E > 1000.21 Acidification of the aqueous phase allowed recovery of the corresponding acid 2a in 30% yield with 99% ee. A brief survey of the reaction scope showed encouraging generality. The same major enantiomer was hydrolyzed with all sulfoxides, but the R and S designations and optical rotation signs vary based on substituents’ priority and UV absorption. Both cyclic and linear alkyl sulfoxides were resolved with high efficiency and selectivity, although the acids 2a and 2b decomposed on prolonged storage. The success of dialkyl sulfoxides distinguishes this approach from many asymmetric oxidation methods. Similarly, electronic factors did not influence reactivity or selectivity with aryl-substituted sulfoxides, as illustrated by the similar yields and ee’s obtained with electron rich (1d/2d), electron neutral (1c/2c, 1e/2e) or electron poor aryl groups (1f/2f, 1g/1g). Substrate 1h represents an interesting example that is chiral by virtue of two different ester groups. The methyl ester was hydrolyzed with high chemo- and enantio-selectivity to provide diester 1h and ester/acid 2h in 99 and 89% ee, respectively.
Facile interconversion of the α-sulfinyl acid and ester allows access to either enantiomer of sulfoxide (Scheme 1), which is a distinguishing characteristic of this method compared to prior enzymatic approaches to sulfoxides. Hydrolysis of the ester with LiOH and esterification of the acid with diazomethane both proceed cleanly and without racemization. In general, ee determination was easier on the esters while many of the derivatizations highlighted below used the acid derived from the unreacted ester recovered from the enzymatic resolution.
Scheme 1.

Ester/acid interconversion allows access to both sulfoxide enantiomers.
To expand the range of optically active sulfoxides accessible through enzymatic resolution of α-sulfinyl carboxylates, we explored decarboxylative functionalization. Coupling of enantioenriched acids 2 to pyrithione (3) with a dicyclohexyl carbodiimide provided an intermediate Barton ester (Scheme 2). Irradiation of a solution of the ester in CBrCl3 or iodoform (250W, white light) promoted decarboxylative halogenation.[22] The bromomethyl sulfoxides (4) were formed in good-excellent yields while the iodomethyl sulfoxides (5) were isolated in moderate yields. In all cases, complete retention of optical purity was observed.
Scheme 2.

Decarboxylative halogenation of α-sulfinyl carboxylic acids.
The halogenation conditions of Scheme 2 were not successful for chlorination. CCl4 and related solvents were not effective for the chlorination, and NCS and other chlorinating reagents gave no desired product. As an alternative, we considered resolving the α-chloro-α-sulfinyl esters directly (Table 2). The racemates (6) could be synthesized as a ~1:1 mixture of diastereomers by chlorinating the α-thioether with NCS prior to formation of the sulfoxide with m-CPBA (see the Supporting Information). Enzymatic resolution at pH 7.5 proceeded slowly, with recovered 6a showing only 39% ee after 72h. We hypothesized that one diastereomer might be processed more quickly than the other isomer, and that raising the pH might allow a dynamic kinetic resolution to proceed. Consistent with this expectation, 6a was isolated in >99% ee after only 18 h when the reaction was performed at pH 9.5. In general, pH 8.6 was optimal for aryl sulfoxides, and pH 9.5 worked for alkyl sulfoxides. The chlorosulfinyl esters 6 and acid 7 were isolated as ~1:1 mixtures of diastereomers with high optical purity. The exception was the cyclohexyl sulfoxide 7a, which decomposed upon attempted isolation. Heating the α-chloro-α-sulfinyl acids 7 in the presence of Cs2CO3 promoted decarboxylation and provided the chloromethyl sulfoxides 8 in good yield and excellent ee.
Table 2.
Asymmetric synthesis of chloromethyl sulfoxides.[a]
| |||||
|---|---|---|---|---|---|
| Entry | R | pH | % Yield 6 (% ee) |
% Yield 7 (% ee) |
% Yield 8 (% ee) |
| 1[b] |
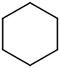
|
9.5 | 39 (>99) | unstable | -- |
| 2[b] | n-Hex | 9.5 | 41 (>99) | 28 (99) | (S)-8b 65 (99) |
| 3 |
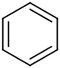
|
8.6 | 46 (>99) | 35 (>99) | (S)-8c: 81 (98) (R)-8c: 78 (99) |
| 4 |
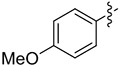
|
8.6 | 42 (>99) | 32 (90) | (S)-8d 80 (99) |
| 5 |
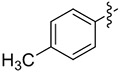
|
8.6 | 41 (>99) | 30 (93) | (S)-8e 73 (97) |
| 6 |
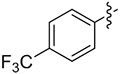
|
8.6 | 40 (>99) | 25 (95) | (S)-8f 66 (>99) |
| 7 |
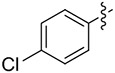
|
8.6 | 40 (>99) | 31 (98) | (S)-8g 81 (>99) |
Reactions on a 0.1-0.2 g scale with 20 wt% enzyme, 8:1 50 mM phosphate buffer : toluene unless otherwise noted.
100 mM phosphate buffer.
A similar strategy could be used to access dihalomethyl sulfoxides (Scheme 3). Resolved α-sulfinyl esters could be dichlorinated or dibrominated with N-halosuccinimide (9, 10). Hydrolysis of the ester with LiOH returned the dihalosulfoxides 11 and 12.
Scheme 3.

Synthesis of dihalosulfoxides. [a] Isolated yield over 2 steps.
The dihalogenation described in Scheme 3 proceeded cleanly from the resolved esters. However, attempts to translate these conditions to the corresponding α-sulfinyl acid generally led to complex mixtures of mono-, di- and trihalogenated products with variable levels of racemization (Table S5). Optimization efforts identified DMSO as an effective solvent for stereoretentive decarboxylative tribromination (Scheme 4). A set of alkyl and aryl sulfoxides all reacted cleanly in the presence of excess N-bromosuccinimide. Monitoring the reaction by LC/MS indicated that the reaction likely proceeds by way of dibromination followed by decarboxylation and the final bromination. Related reactions with NCS or NIS gave <10% of the desired trihalogenated products.
Scheme 4.

Tribromination of α-sulfinyl acids.
Decarboxylative cross-coupling reactions have emerged as effective strategies for C─C bond formation.[23] We explored decarboxylative cross-electrophile coupling using redox active esters derived from optically active α-sulfinyl acetates following a procedure developed by the Weix group (Table 3).[24] First, the acids were coupled N-hydroxyphthalimide. Next, the crude esters were subjected to reductive coupling conditions with iodobenzene in the presence of a Ni catalyst and a dipyridyl ligand (dtbpy). The corresponding benzyl sulfoxides were formed rapidly in high yield from aryl-substituted α-sulfinyl acids. A linear alkyl sulfoxide (2b) also participated effectively, but the phthalimide ester derived from cyclohexyl sulfoxide 2a was unstable, while the unsymmetrical ester derived from 2h could not be formed. In all cases, the decarboxylative coupling proceeded without loss of enantioenrichment.
Table 3.
Decarboxylative cross couplings of α-sulfinyl acids.
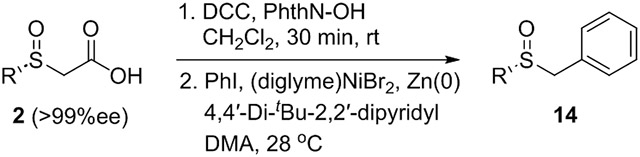
| |||||
|---|---|---|---|---|---|
| Entry | Cmpd | R | Time (h) | Yield 14 (%) | Ee 14 (%) |
| 1[a] | 14a |
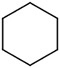
|
-- | -- | -- |
| 2 | 14b | n-Hex | 4 | 68 | >99 |
| 3 | 14c |
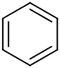
|
2 | 70 | >99 |
| 4 | 14d |
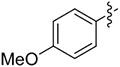
|
2 | 71 | >99 |
| 5 | 14e |
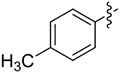
|
2 | 67 | >99 |
| 6 | 14f |
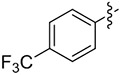
|
2 | 65 | >99 |
| 7 | 14g |
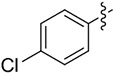
|
2 | 67 | >99 |
| 8[b] | 14h |
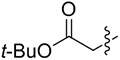
|
-- | -- | -- |
Phthalimide ester unstable.
Phthalimide ester not formed.
To demonstrate the utility of the enzymatic resolution and decarboxylative functionalization, we synthesized a novel inhibitor of 15-PGDH. Previous syntheses of this inhibitor class relied on preparative HPLC over a chiral stationary phase, which was both time consuming and wasteful.[11a] As shown in Scheme 5, aldol condensation of imidazole aldehyde 15 with methyl ketone 16 provided the enone 17. Annulation with cyanothioacetamide generated the thiopyridine 18. Alkylation with either (R)- or (S)-4a provided the enantiomeric sulfoxides 19. Final cyclization constructed the thienopyridine core without racemization. The two enantiomers were tested for their ability to inhibit recombinant human 15-PGDH. (R)-20 was found to be a potent inhibitor with IC50 values approaching the limit of detection of this assay.[10a] By contrast, (S)-20 was more than 100-fold less active, highlighting the importance of preparing the sulfoxides in single isomer form.
Scheme 5.

Asymmetric synthesis of sulfoxide inhibitors of 15-prostaglandin dehydrogenase (15-PGDH).
Conclusion
Lipase resolutions are scalable and operationally simple. The resolved ester can be isolated in nearly pure form through extraction, and the acid of the opposite enantiomer can similarly be obtained through extraction after acidification. Finally, decaroboxylative functionalization can proceed with retention of stereochemistry, thereby providing access to a wide range of optically active sulfoxides.
Experimental Section
Representative procedure: To a solution of ester (±)-1a (1.5 g, 7.35 mmol) in toluene (7.5 mL) were added phosphate buffer (60 mL of pH 7.5, 0.05 M) and lipoprotein lipase from Burkholderia sp. (EC 3.1.1.3, 150 mg, 10 wt%). This heterogeneous mixture was stirred at 25 °C for 18 h. The mixture was filtered through celite to remove the enzyme and extracted with EtOAc (3 × 40 mL). Evaporation of the solvent and flash chromatography gave enantioenriched ester derivative (R)-1a (0.69 g, 46%). Ee was determined to be >99% by HPLC (See SI). Acetic acid (15.0 mL) was added and aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over Na2SO4 and removal of the solvent gave acid (S)-2a (0.42 g, 30%). Ee was determined following conversion to the methyl ester, as described below.
Under a nitrogen atmosphere, a solution of (trimethylsilyl)diazomethane (2.0 M solution in diethyl ether, 21 mmol) was added to a solution of the acid derivative 2a (2.1 mmol) in dry MeOH (3.0 mL) at 0 °C. The resulting mixture was warmed slowly to rt and stirred overnight. After completion of the reaction, the solvent was removed in vacuo. Purification by ISCO flash column chromatography afforded the ester derivative (S)-2a in quantitative yield.
Supplementary Material
Acknowledgements
Financial support provided by the Welch Foundation (I-1612) and the NIH (RM1GM142002).
References
- [1].a) Wang N, Saidhareddy P, Jiang X, Nat. Prod. Rep 2020, 37, 246–275; [DOI] [PubMed] [Google Scholar]; b) Hai Y, Wei M-Y, Wang C-Y, Gu Y-C, Shao C-L, Mar. Life Sci. Technol 2021, 3, 488–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Carreno MC, Chem. Rev 1995, 95, 1717–1760; [Google Scholar]; b) Casoni G, Kucukdisli M, Fordham JM, Burns M, Myers EL, Aggarwal VK, J. Am. Chem. Soc 2017, 139, 11877–11886; [DOI] [PubMed] [Google Scholar]; c) Aiken SG, Bateman JM, Liao H-H, Fawcett A, Bootwicha T, Vincetti P, Myers EL, Noble A, Aggarwal VK, Nat. Chem 2023, 15, 248–256. [DOI] [PubMed] [Google Scholar]
- [3].a) Satoh T, Oohara T, Ueda Y, Yamakawa K, J. Org. Chem 1989, 54, 3130–3136; [Google Scholar]; b) Lanfranchi DA, Hanquet G, J. Org. Chem 2006, 71, 4854–4861; [DOI] [PubMed] [Google Scholar]; c) Carmen Carreño M, Hernández-Torres G, Ribagorda M, Urbano A, Chemical Commun. 2009, 6129–6144; [DOI] [PubMed] [Google Scholar]; d) Antczak MI, Cai F, Ready JM, Org. Lett 2011, 13, 184–187; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Batisse C, Panossian A, Hanquet G, Leroux FR, Chemical Commun. 2018, 54, 10423–10426; [DOI] [PubMed] [Google Scholar]; f) Salom-Roig X, Bauder C, Synthesis 2020, 52, 964–978. [Google Scholar]
- [4].a) Trost BM, Rao M, Angew. Chem. Int. Ed 2015, 54, 5026–5043; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 5112–5130; [Google Scholar]; b) Otocka S, Kwiatkowska M, Madalińska L, Kiełbasiński P, Chem. Rev 2017, 117, 4147–4181; [DOI] [PubMed] [Google Scholar]; c) Jia T, Wang M, Liao J, in Sulfur Chemistry (Ed.: Jiang X), Springer International Publishing, Cham, 2019, pp. 399–427. [Google Scholar]
- [5].Block SS, Stephens RL, Barreto A, Murrill WA, Science 1955, 121, 505–506. [DOI] [PubMed] [Google Scholar]
- [6].Pereira AR, Kale AJ, Fenley AT, Byrum T, Debonsi HM, Gilson MK, Valeriote FA, Moore BS, Gerwick WH, ChemBioChem 2012, 13, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang X, Wang J, Lai D, Wang W, Dai J, Zhou L, Liu Y, Toxins, 2017, 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Prisinzano T, Podobinski J, Tidgewell K, Luo M, Swenson D, Tetrahedron: Asymmetry 2004, 15, 1053–1058; [Google Scholar]; b) Cao J, Prisinzano TE, Okunola OM, Kopajtic T, Shook M, Katz JL, Newman AH, ACS Med. Chem. Lett 2011, 2, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sachs G, Shin JM, Howden CW, Ailment. Pharmacol. Ther 2006, 23, 2–8. [DOI] [PubMed] [Google Scholar]
- [10].a) Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, Tiwari S, Willis JE, Williams NS, Dawson DM, Wald D, Chen W-D, Wang Z, Kasturi L, Larusch GA, He L, Cominelli F, Di Martino L, Djuric Z, Milne GL, Chance M, Sanabria J, Dealwis C, Mikkola D, Naidoo J, Wei S, Tai H-H, Gerson SL, Ready JM, Posner B, Willson JKV, Markowitz SD, Science 2015, 348, aaa2340; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smith JNP, Otegbeye F, Jogasuria AP, Christo KF, Antczak MI, Ready JM, Gerson SL, Markowitz SD, Desai AB, Biol. Blood and Marrow Transplant 2020, 26, 1552–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Antczak MI, Zhang Y, Wang C, Doran J, Naidoo J, Voruganti S, Williams NS, Markowitz SD, Ready JM, J. Med. Chem 2017, 60, 3979–4001; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Desai A, Zhang Y, Park Y, Dawson DM, Larusch GA, Kasturi L, Wald D, Ready JM, Gerson SL, Markowitz SD, Haematologica 2018, 103, 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fernández I, Khiar N, Chem. Rev 2003, 103, 3651–3706; [DOI] [PubMed] [Google Scholar]; b) Haimeng Zhu CW, Zong Lili, Chinese J. Org. Chem 2021, 41, 3431–3447. [Google Scholar]
- [13].a) Wojaczyńska E, Wojaczyński J, Chem. Rev 2010, 110, 4303–4356; [DOI] [PubMed] [Google Scholar]; b) Han J, Soloshonok VA, Klika KD, Drabowicz J, Wzorek A, Chem. Soc. Rev 2018, 47, 1307–1350. [DOI] [PubMed] [Google Scholar]
- [14].a) Nosek V, Míšek J, Angew. Chem 2018, 130, 9997–10000; [Google Scholar]; Angew. Chem. Int. Ed 2018, 57, 9849–9852; [DOI] [PubMed] [Google Scholar]; b) Bierbaumer S, Schmermund L, List A, Winkler CK, Glueck SM, Kroutil W, Angew. Chem. Int. Ed 2022, 61, e202117103; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2022, 134, e202117103; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Peng T, Tian J, Zhao Y, Jiang X, Cheng X, Deng G, Zhang Q, Wang Z, Yang J, Chen Y, Angew. Chem. Int. Ed 2022, 61, e202209272; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2022, 134, e202209272. [DOI] [PubMed] [Google Scholar]
- [15].a) Chen H, Liu YA, Liao X, Synthesis 2020, 53, 1–29; [Google Scholar]; b) Li L, Yao Y, Fu N, E. J. Org. Chem 2023, 26, e202300166. [Google Scholar]
- [16].a) Komatsu N, Hashizume M, Sugita T, Uemura S, J. Org. Chem 1993, 58, 7624–7626; [Google Scholar]; b) Sun J, Zhu C, Dai Z, Yang M, Pan Y, Hu H, J. Org. Chem 2004, 69, 8500–8503; [DOI] [PubMed] [Google Scholar]; c) Drago C, Caggiano L, Jackson RFW, Angew. Chem 2005, 117, 7387–7389; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2005, 44, 7221–7223; [DOI] [PubMed] [Google Scholar]; d) Zeng Q, Wang H, Wang T, Cai Y, Weng W, Zhao Y, Adv. Synth. Catal 2005, 347, 1933–1936; [Google Scholar]; e) Wang J, Frings M, Bolm C, Chem Eur. J 2014, 20, 966–969. [DOI] [PubMed] [Google Scholar]
- [17].a) Represenative non-oxidative kinetic resolutions of sulfoxides: Lao JR, Fernández-Pérez H, Vidal-Ferran A, Org. Lett 2015, 17, 4114–4117; [DOI] [PubMed] [Google Scholar]; b) Zhu Y-C, Li Y, Zhang B-C, Zhang F-X, Yang Y-N, Wang X-S, Angew. Chem 2018, 130, 5223–5227; [Google Scholar]; Zhu Y-C, Li Y, Zhang B-C, Zhang F-X, Yang Y-N, Wang X-S, Angew. Chem. Int. Ed 2018, 57, 5129–5133. [DOI] [PubMed] [Google Scholar]
- [18].Keith JM, Larrow JF, Jacobsen EN, Adv. Synth. Catal 2001, 343, 5–26. [Google Scholar]
- [19].a) Burgess K, Henderson I, Tetrahedron Lett. 1989, 30, 3633–3636; [Google Scholar]; b) Liu Z, Burgess K, Tetrahedron Lett. 2011, 52, 6325–6327 [Google Scholar]
- [20].Santaniello E, Ferraboschi P, Grisenti P, Manzocchi A, Chem. Rev 1992, 92, 1071–1140. [Google Scholar]
- [21].Chen CS, Fujimoto Y, Girdaukas G, Sih CJ, J. Am. Chem. Soc 1982, 104, 7294–7299. [Google Scholar]
- [22].Barton DHR, Crich D, Motherwell WB, Tetrahedron 1985, 41, 3901–3924. [Google Scholar]
- [23].a) Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS, Science 2016, 352, 801–805; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DWC, Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]
- [24].Huihui KMM, Caputo JA, Melchor Z, Olivares AM, Spiewak AM, Johnson KA, DiBenedetto TA, Kim S, Ackerman LKG, Weix DJ, J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.