

Abstract
Introduction:
Preeclampsia (PE) is a serious hypertensive pregnancy disorder and a leading cause of maternal and perinatal morbidity and mortality. Despite the prevalence and complications, there are no approved therapeutics to relieve PE symptoms. Inflammation, oxidative stress, and angiogenic imbalance have been shown to contribute to the PE pathophysiology, though there is a lack of understanding in how best to target these pathways in PE. We recently demonstrated that the bioflavonoid luteolin is a potent inhibitor of the anti-angiogenic and pro-hypertensive soluble fms-like tyrosine kinase 1 (sFlt-1), and here we aimed to determine if luteolin was also capable of reducing inflammation and oxidative stress pathways.
Methods:
Tumor necrosis factor (TNF)-α, which is upregulated in PE, was utilized to stimulate these pathways in human placental explants and endothelial cells. Endothelin-1 (ET-1) and interleukin (IL)-6 in the media from explants and cells were measured via ELISA, and NF-κB localization and reactive oxygen species were detected via fluorescence microscopy.
Results:
Pretreatment with luteolin demonstrated significant reductions in NF-κB activation, reactive oxygen species, superoxide, and IL-6 and ET-1 expression in endothelial cells. We also saw a significant reduction in phosphorylation of NF-κB in human placental explants.
Discussion:
These data demonstrate that luteolin inhibits pathways implicated in the development of PE and should be explored further for its potential as a PE therapeutic.
Keywords: Luteolin, inflammation, ROS, ET-1, NF-κB p65, preeclampsia
Graphical Abstract

Introduction
Preeclampsia (PE) is a common hypertensive disorder of pregnancy, characterized by new-onset hypertension, proteinuria, and end-organ damage linked to endothelial and vascular dysfunction, affects approximately 2–8% of all pregnancies. Although PE is a leading cause of maternal and fetal morbidity and mortality, there remains no approved pharmacological interventions, with the only treatment option being delivery of the fetus and placenta, often occurring prematurely [1].
Although the etiology of PE is poorly understood, the disease is believed to develop as a consequence of inappropriate remodeling of uterine spiral arteries, causing decreased blood flow to the fetal-placental unit [2]. In response, the hypoxic placenta secretes pro-hypertensive factors into the maternal circulation, including pro-inflammatory cytokines tumor necrosis factor (TNF)-α and interleukin (IL)-6 [3], where they act locally on placental cells as well as on systemic vascular endothelium [4]. Signaling from inflammatory cytokines results in the activation of nuclear factor- κB (NF-κB), which is a major regulator of the inflammatory cascade [5]. Upon activation by receptors such as the TNF-α receptor, inhibitor of NF-κB proteins (IκB) is phosphorylated, releasing NF-κB [6]. This allows the p65 subunit of NF-κB to be phosphorylated and translocate to the nucleus, where it acts as a pro-inflammatory transcription factor, feeding into the upregulation of inflammatory cytokines [7]. TNF-α additionally leads to increased reactive oxygen species (ROS), which also activate NF-κB signaling, ultimately promoting inflammation and upregulation of the potent vasoconstrictor endothelin-1 (ET-1), leading to endothelial dysfunction [8, 9]. Indeed, NF-κB activation has been shown to be upregulated in both placentas [10] and vasculature [11] of PE pregnancies. Preclinical studies of antioxidants as well as targets of individual cytokines have shown poor results in alleviating the PE phenotype [12–14], perhaps due to the complexity of PE and the number of factors involved in the syndrome. Therefore, it is essential to identify treatments which are able to target multiple pathways implicated in PE pathogenesis.
Bioflavonoids are found in many plants, including their fruits and vegetables, and have been well-established for their antioxidant and anti-inflammatory effects [15–17]. Regular consumption of bioflavonoids has additionally been shown to reduce risk of cardiovascular and neurogenerative diseases [18]. Our laboratory recently demonstrated that the bioflavonoid luteolin is a potent inhibitor of the anti-angiogenic soluble fms-like tyrosine kinase 1 (sFlt-1) protein, a key pathway in the development of PE [19]. Because the pathology of PE begins with the diseased placenta secreting factors which lead to endothelial dysfunction, we chose to study the effects of luteolin on both placental tissue and endothelial cells in order to test the hypothesis that luteolin additionally targets the inflammatory and ROS pathways, preventing the increase of NF-κB activation and upregulation of inflammatory cytokines. Utilization of normotensive samples was chosen to reduce confounding from other factors in PE tissue (such as IL-6) and the effects of luteolin on those pathways. Because TNF-α is upregulated in PE, and is a known contributor to the pathways being studied, we utilized TNF-α stimulation in normotensive explants and cells to study the effects of luteolin.
Methods
Protocol for obtaining human placentas
Placental tissue was collected from normotensive (NT) patients delivered at the University of Chicago Medical Center. Patients were excluded if they had a history of diabetes, chronic hypertension, renal disease, or multiple gestations. The Institutional Review Board approved using all study-related materials at the University of Chicago (Institutional Review Board No. #14–1532).
Placental villous explant cultures
Placental villous explant tissues were cultured in a complete medium, followed by protein extraction as described previously [20, 21]. Villous biopsies of the placenta (2 cm3) were excised from the maternal surface, midway between the chorionic and basal plates, within 30 min of delivery, and decidual layers were carefully removed. Tissue was dissected into explants and thoroughly rinsed with phosphate-buffered saline (PBS) to ensure removal of maternal blood and placed in a 12-well flat-bottom plate (Falcon multi-well tissue culture plate; Becton Dickinson) containing 1 mL of conditioned medium 199. Explants were cultured under standard tissue culture conditions (room air with 5% CO2) in a humidified cell culture incubator. Samples were pre-treated with luteolin (5μM) (Sigma-Aldrich; Cat# L9283; St. Louis, MO) or dimethyl sulfoxide (DMSO) control for 6 hours before stimulation with TNF-α (50ng/mL) (Pierce Biotechnology; Cat# RTNFAI; Rockford, IL). Luteolin concentrations were chosen based on our previous studies [19]. After 24 h, the explants were removed, blotted with sterile cotton gauze, weighed, and frozen in liquid nitrogen along with corresponding conditioned media before transferred to −80°C for storage. Experiments were duplicated on explants from each patient.
Immunoblotting
Tissue from placental explants (described above) were homogenized, and total protein was collected as previously described [22, 23]. Briefly, protein concentration was assessed using Pierce BCA Protein Assay Kit (ThermoFisher; Cat# 23225; Scientific, Waltham, MA), and equal quantities (30 μg total protein) were resolved on a SDS containing 4–15% polyacrylamide gel and transferred to a nitrocellulose membrane (0.2 nm). Membranes were blocked with 5% non-fat milk in tris-buffered saline-tween (TBS-T, 0.05% tween) for one hour and incubated with the primary antibodies against phosphorylated p65 (p-p65) (Cell Signaling; Cat# 3033; Danvers, MA), total p65 (Cell Signaling; Cat# 6956; Danvers, MA), or β-Actin (BD Biosciences; Cat# 612656; Sparks, MD) at 1:1000 dilution overnight at 4°C. After washing the membrane with TBS-T, goat anti-rabbit (Cell Signaling; Cat# 7074S; Danvers, MA) or anti-mouse (Cell Signaling; Cat# 7076P2; Danvers, MA) secondary antibody (1:5000 dilution) were added for one hour at room temperature. Proteins were detected using enhanced chemiluminescent reagents and quantified using ImageJ (NIH) to collect densitometry data. Data is represented as the ratio of p-p65/ total p65 in arbitrary unites (AU). Actin was measured to ensure equal protein loading.
Endothelial Cell Culture
Human umbilical vein endothelial cells (HUVECs) (ATCC; Cat# PCS-100–010; Manassas, VA) were cultured using vascular cell basal medium (ATCC; Cat# PCS-100–030; Manassas, VA) supplemented with endothelial cell growth kit (ATCC; Cat# PCS-100–041; Manassas, VA). Cells were used at fewer than 5 passages (approximately 200,000 cells per well seeded) and cultured at 37°C in a humidified incubator at 5% CO2. Cells were pre-treated with luteolin (5μM), NF-κB translocation inhibitor (referred to as SN50) (25μg/mL) (Santa Cruz Biotechnology; Cat# sc-3060; Santa Cruz, CA), or a combination for 6 hours before stimulation with TNF-α (50μg/mL) overnight. After approximately 24 hours, culture medium was collected and stored at −80°C until target proteins were measured via enzyme-linked immunosorbent assay (ELISA).
ELISA
Secreted proteins were measured in the medium using ELISA. IL-6 was measured in explant and HUVEC medium (BD Biosciences; San Jose, CA; Cat #550799), and the associated protocols followed. ProET-1 (the precursor to ET-1) was measured in HUVEC and explant medium (ThermoFisher Scientific; Carlsbad, CA; Cat #BMS2266), and the associated protocol followed. Protein expression in placental explant medium was normalized to tissue weight.
Determining Activation of NF-κB by Monitoring Nuclear Localization
Nuclear localization of NF-κB was assessed as previously described [24]. Briefly, HUVECs were seeded on 4-well chamber slides (~200,000 cells) and incubated overnight to allow adherence. Fresh medium was added to the cells, which were pre-treated with luteolin (5μM) or NF-κB inhibitor SN50 (25μg/mL) overnight before stimulation with TNF-α (50ng/mL) for one hour. Cells were fixed using chilled methanol, followed by washing with ice-cold PBS. Slides were blocked using 10% goat serum in PBS for 30 minutes and stained with mouse anti-p65 antibody (1:150 dilution) (Santa Cruz Biotechnology; Cat# sc-8008; Santa Cruz, CA) for 1 hour at 37°C. Cells were rinsed with PBS and Alexa Fluor 488 anti-mouse secondary was added for 1 hour at 37°C (Invitrogen; Cat# A21042; Eugene, OR). Antifade mounting medium with DAPI drops (Vector Laboratories; Cat# H-1200; Burlingame, CA) were added before placing the cover slip. Slides were analyzed using a fluorescent Nikon microscope (Nikon Instruments; Nikon Eclipse Ti2; Melville, NY). DAPI images were used to draw regions of interest around the nuclei, which were used to determine nuclear fluorescence intensity using ImageJ software. Nuclear intensity was then compared to cytoplasmic intensity.
Detection of ROS and superoxide
HUVECs were seeded on 4-well chamber slides as above. The ROS/superoxide detection assay kit (Abcam; Cat # ab139476) was used with the associated protocol. Briefly, cells were pre-treated with luteolin (5μM) for 1 hour before stimulation with TNF-α (50ng/mL) for 1 hour. At the same time, the oxidative stress reagent and superoxide detection reagent were added at a dilution of 1:500. Cells were gently washed, side walls removed, and cover glass was placed over the top before imaging on the Nikon microscope as above (using green and red lasers). Images were taken, and cells were randomly chosen for fluorescence quantification using ImageJ. The average of the control samples was obtained and used for normalization of all groups.
Statistical Analysis
Graphpad Prism (version 9) was used in the statistical analysis. Outliers were identified using the ROUT method (Q=10%). One- or two-way analysis of variance (ANOVA) was used to compare groups with the Tukey post-hoc test (in endothelial cells) or Dunnett’s post-hoc test (in placental explants). Statistical significance was set at p<0.05, and unless otherwise notated, significance represented is compared to groups stimulated with TNF-α.
Results
Determining luteolin’s ability to inhibit phosphorylation of NF-κB p65 in placental explants
Activation of NF-κB by TNF-α leads to phosphorylation of the p65 subunit, stimulating its translocation to the nucleus. To determine if luteolin was able to inhibit phosphorylation of p65, placental explants were pretreated with luteolin (5μM) before stimulation with TNF-α. Western blot analysis of phosphorylated p65 (p-p65) compared to total p65 showed that stimulation with TNF-α significantly increased p65 phosphorylation compared to control explants (2.51 ± 1.3 vs 0.74 ± 0.22 AU; p<0.0.001). Pretreatment with luteolin resulted in a significant decrease in p-p65 compared to TNF-α treatment alone (1.15 ± 0.24 vs 2.51 ± 1.3 AU; p<0.05). Explants treated with luteolin alone had no change in p-p65/ total p65 expression (Figure 1).
Figure 1: Luteolin inhibits phosphorylation of NF-κB subunit p65.
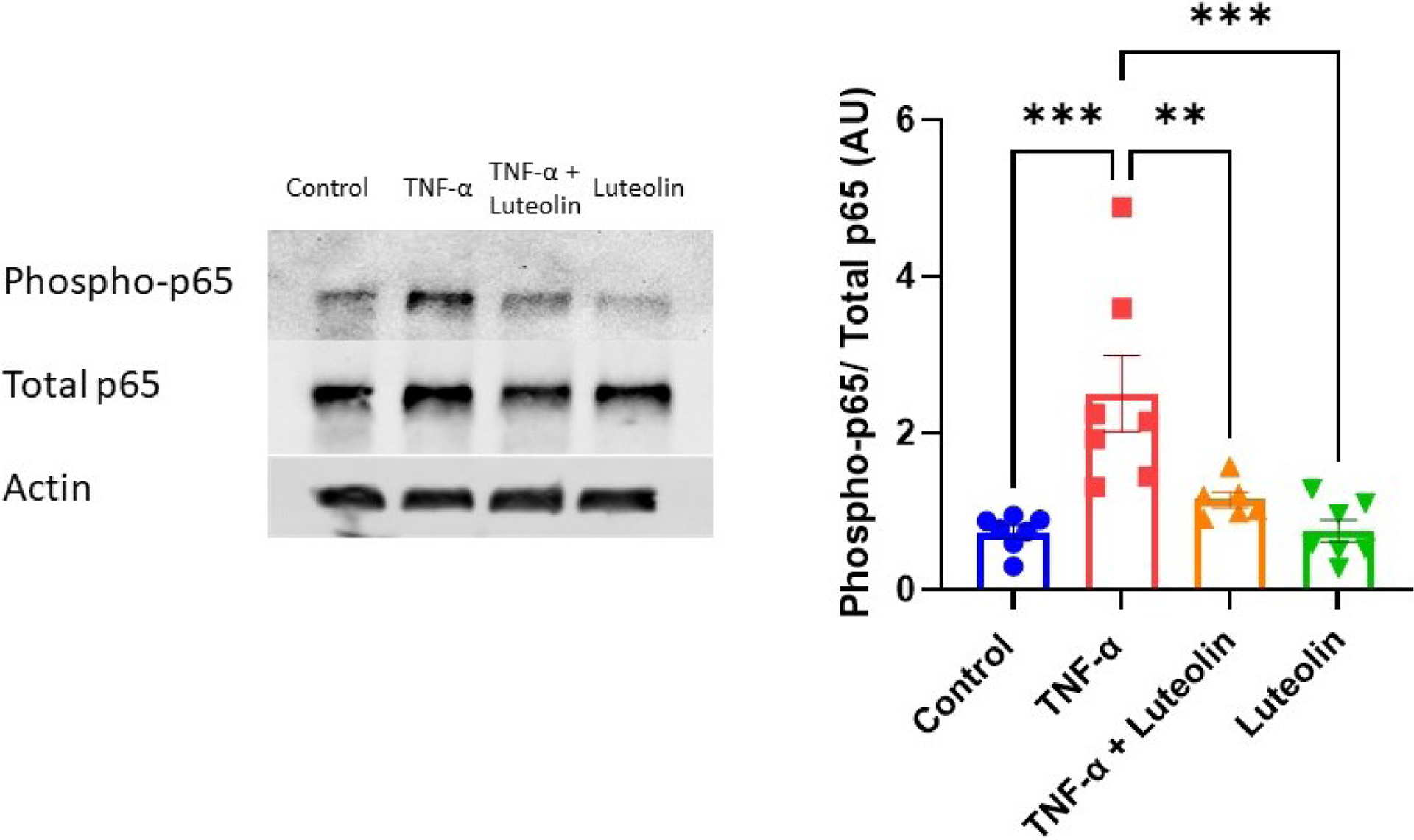
Placental explants exposed to TNF-α have a significant increase in p65 phosphorylation. Pre-treatment with luteolin prevents the increased phosphorylation, bringing levels down such that they are not different from control. Actin was not used for analysis, but to ensure equal loading. **p<0.01, ***p<0.001; N=7/ group.
Activation of NF-κB has been shown to upregulate many signaling factors promoting inflammation and endothelial dysfunction [25]. ELISAs were performed to measure IL-6 and proET-1 to examine the downstream effects of luteolin inhibition of TNF-α-induced p65 activation in placental explants. As expected, TNF-α stimulation led to significant upregulation of IL-6 (24.4 ± 10.67 vs 66.49 ± 25.39 pg/mL/μg; p<0.01). Surprisingly, however, there was no significant reduction when pretreated with luteolin (66.49 ± 25.39 vs 56.57 ± 20.49 pg/mL/μg; p=0.74). Similar trends with TNF-α-induced increase (36.28 ± 11.28 vs 152.4 ± 28.68 pg/mL/mg; p<0.001) and non-significant decrease with luteolin pretreatment (152.4 ± 28.68 vs 107.5 ± 15.98 pg/mL/mg; p=0.15) were observed when proET-1 was measured (Figure 2A and 2B).
Figure 2: Effect of luteolin on TNF-α-induced upregulation of cytokines in placental explants.
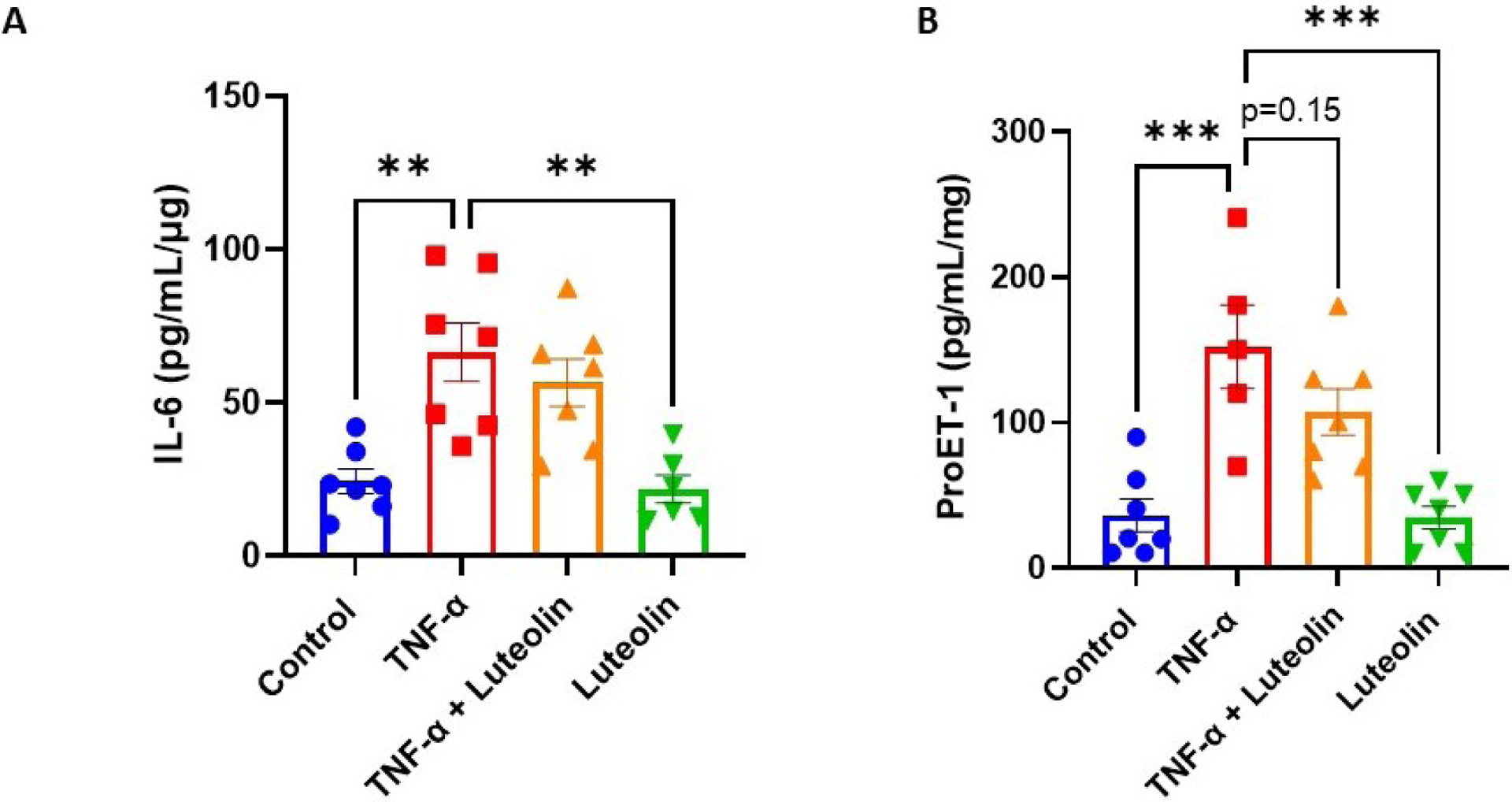
Stimulation of placental explants with TNF-α led to significant upregulation of IL-6 (A). Luteolin pretreatment did not significantly decrease IL-6 expression. Stimulation of placental explants with TNF-α led to significant upregulation of vasoconstrictor proET-1. Pretreatment with luteolin resulted in a non-significant trend for decrease in proET-1 expression, and luteolin treatment alone had no effect on proET-1 expression (B). Samples were normalized to explant weight. **p<0.01, ***p<0.001; N=7/group.
Examining luteolin’s ability to inhibit NF-κB translocation in endothelial cells
In order to determine if luteolin treatment could inhibit immune modulator NF-κB, HUVECs were stimulated with TNF-α to promote the activation and translocation of the p65 subunit of NF-κB. Figure 3 demonstrates how under control (untreated) conditions, p65 is found in the cytosol of the cells. Upon stimulation with TNF-α, the p65 subunit is translocated to the nucleus, such that the nuclear: cytoplasmic p65 is significantly increased (0.69 ± 0.22 vs 1.35 ± 0.54 AU; p<0.0001). When the cells were pretreated with luteolin, however, the nuclearization of p65 is ameliorated, and the protein is once again observed in the cytosol (0.69 ± 0.24 vs 1.35 ± 0.54 AU; p<0.0001). Similarly, cells pretreated with SN50 demonstrated an increase in cytosolic p65 (0.94 ± 0.19 vs 1.35 ± 0.54 AU; p<0.05). Cells treated with luteolin or SN50 in the absence of TNF-α did not result in a further increase of cytosolic p65 compared to control.
Figure 3: Luteolin prevents the nuclearization of NF-κB p65 subunit in HUVECs.
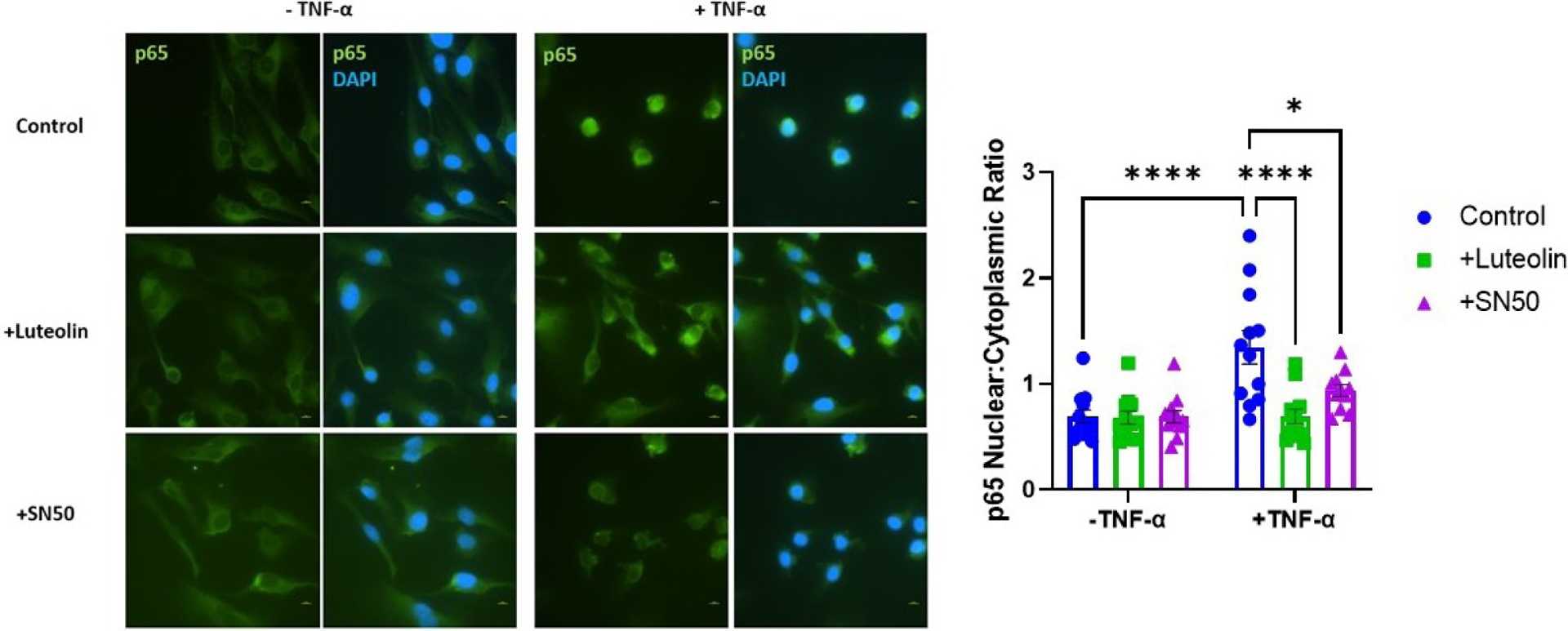
Cells exposed to TNF-α have significantly increased localization of the p65 subunit of NF-κB, with a lesser amount being present in the cytosol. When the cells are pretreated with luteolin, the translocation of p65 is inhibited, and is found in the cytosol, similar to control conditions. Cells were viewed under 60x magnification and scale bars represent 10μm *p<0.05, ****p<0.0001; N=12/ group.
To examine the downstream effects of luteolin’s inhibition of NF-κB in HUVECs, ELISAs were performed on IL-6 and ProET-1. Cells stimulated with TNF-α had significant upregulation of IL-6 compared to untreated controls (741 ± 446 vs 204 ± 152 pg/mL; p<0.01). When pretreated with luteolin, the levels of IL-6 were normalized (741 ± 446 vs 164 ± 134 pg/mL; p<0.001) (Figure 4A). Similarly, pretreatment with NF-κB inhibitor SN50 led to normalization of IL-6 expression (741 ± 446 vs 146 ± 162 pg/mL; p<0.001). Treatment with luteolin or SN50 alone caused substantial reductions in IL-6, such that levels were nearly undetectable in these samples, and when treatments of luteolin and SN50 were combined in the presence or absence of TNF-α, levels of IL-6 were undetectable. Similar trends were observed in measuring HUVEC production of proET-1, the precursor to ET-1. Stimulation with TNF-α significantly upregulated proET-1 compared to control samples (46.4 ± 4.7 vs 18.7 ± 10.8 pg/mL; p<0.01). Normalization of proET-1 expression was observed in cells pretreated with luteolin or SN50 (46.4 ± 4.7 vs 18.9 ± 16.7 pg/mL; p<0.01 and 46.4 ± 4.7 vs 19.5 ± 12.7 pg/mL; p<0.01, respectively) (Figure 4B). Unlike expression of IL-6, which decreased further when cells were treated with luteolin or SN50 alone, proET-1 levels did not decrease below control with treatment of luteolin or SN50, but the additive effect of luteolin with SN50 was still observed in the presence or absence of TNF-α.
Figure 4: Luteolin prevents TNF-α-induced cytokine production in cultured endothelial cells.
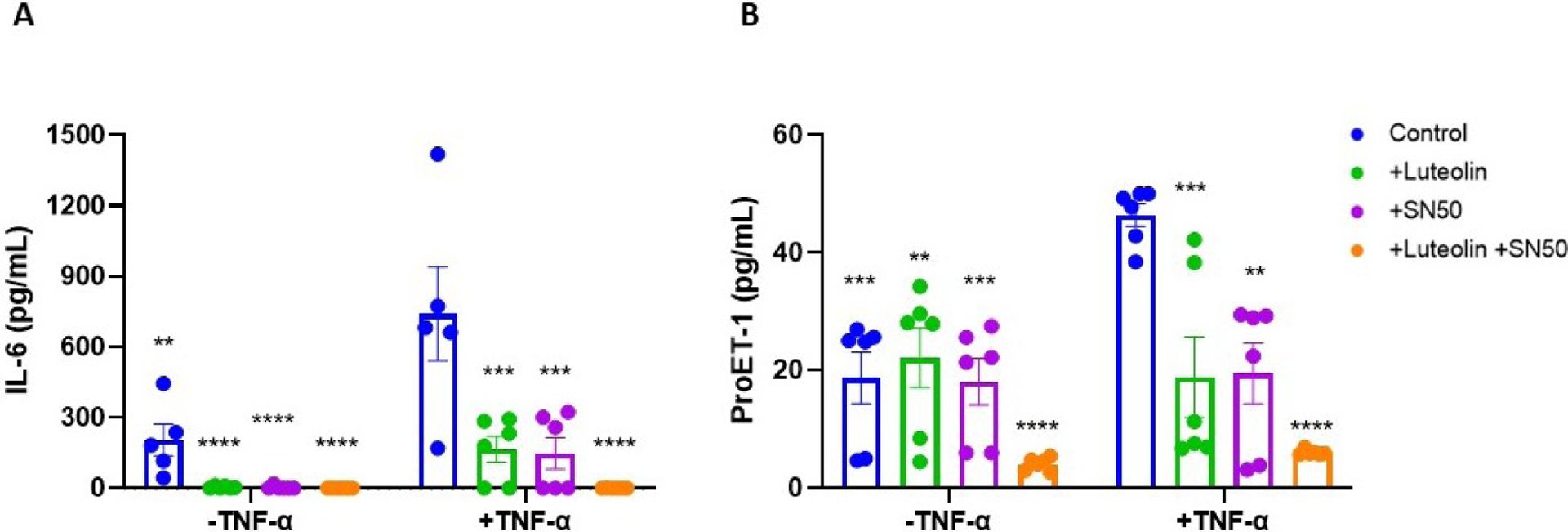
HUVECs stimulated with TNF-α have significantly upregulated expression of inflammatory cytokine IL-6 (A) and vasoconstrictor ET-1 (B). Pretreatment with either luteolin or NF-κB inhibitor SN50 returned levels to normal, and treatment with luteolin or SN50 alone cause IL-6 levels to fall such that they were undetectable. For both IL-6 and proET-1, there appeared to be an additive effect with treatment utilizing both luteolin and SN50, as the reduction was greater with dual treatment. Significance compared to TNF-α-stimulated group: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; N=6/group.
Effect of luteolin on ROS production in endothelial cells
TNF-α has been shown to stimulate production of ROS, both of which are elevated in preeclamptic patients. HUVECs stimulated with TNF-α for 1 hour demonstrated significant upregulation of total ROS (1.57 ± 0.43 vs 1.08 ± 0.41 fold change; p<0.01) compared to untreated cells. Pretreatment with luteolin, however, returned these levels to normal (1.57 ± 0.43 vs 1.17 ± 0.20 fold change; p<0.05) (Figure 5A and 5B). Cells which were treated with luteolin alone had a further reduction in ROS, approximately 30% of what was observed under normal conditions (0.29 ± 0.06 vs 1.08 ± 0.41 fold change; p<0.0001), demonstrating the power of luteolin to reduce oxidative stress (Figure 5A and 5B).
Figure 5: Luteolin treatment reduces oxidative stress in HUVECs.
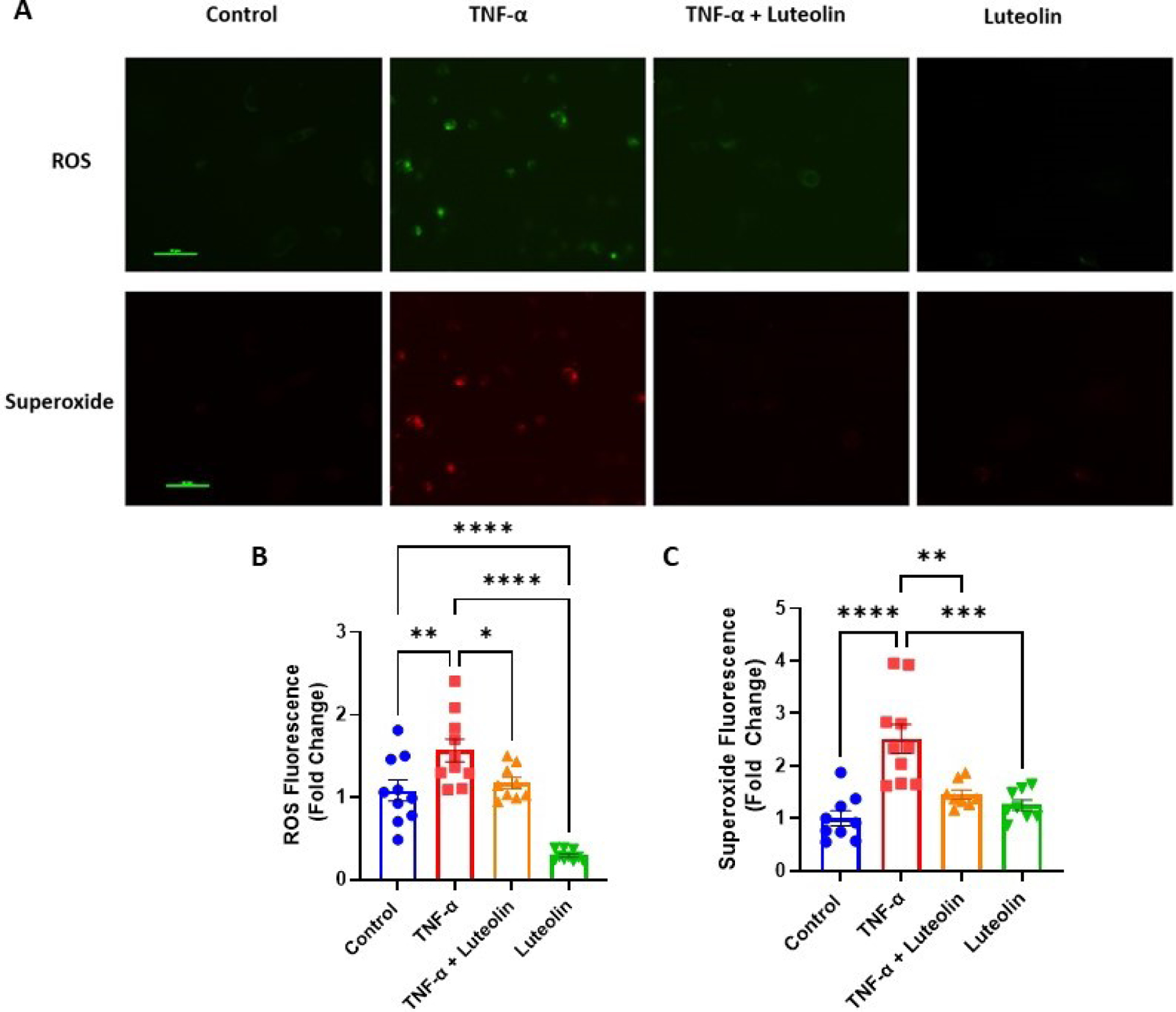
Treatment with TNF-α leads to significant increases in reactive oxygen species (ROS) and superoxide. Pretreatment with luteolin significantly reduced production of ROS and superoxide. While treatment with luteolin alone did not have an effect on superoxide production, ROS levels decreased such that levels were significantly lower than control. Cells were viewed under 40x magnification and scale bars represent 50μm. Fold change was calculated by normalizing to the average of the control group. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; N=10/group.
Similarly, superoxide production was significantly increased in TNF-α-treated cells compared to control (2.52 ± 0.87 vs 1.00 ± 0.43 fold change; p<0.0001). Again, there was a significant reduction in superoxide when the cells were pretreated with luteolin (2.52 ± 0.87 vs 1.45 ± 0.25 fold change; p<0.01). Unlike measurements of total ROS, however, superoxide levels did not decrease below control levels when cells were treated with luteolin alone (1.25 ± 0.28 vs 1.00 ± 0.43 fold change; p= 0.78) (Figure 5A and 5C).
Discussion
We recently identified luteolin as a novel compound to target sFlt-1 production in PE, leading to significant reductions in placental sFlt-1 expression in vitro [19]. As an extension and because PE is a multifaceted disease, this study aimed to determine the ability of luteolin to inhibit pathways which contribute to the inflammation and oxidative stress observed in PE. In this study, we report several key findings: (1) luteolin inhibits NF-κB activation by preventing phosphorylation of the p65 subunit, (2) luteolin inhibits the translocation of p65 to the nucleus, (3) luteolin inhibits NF-κB-induced production of cytokines, and (4) luteolin inhibits production of ROS, including superoxide.
Inflammatory cytokines, such as TNF-α, have repeatedly been shown to be elevated in preeclamptic patients [3, 26–28]. Animal models have similarly shown the relevance of TNF-α in the pathophysiology of PE. Indeed, infusion of TNF-α into pregnant rats has been shown to significantly increase mean arterial pressure and circulating sFlt-1 [29], similar to what is observed in PE pregnancies. Additionally, the widely used reduced uterine perfusion pressure (RUPP) model of PE has been shown to have elevated TNF-α levels, and TNF-α blockade in RUPP led to an improvement in symptoms [30]. Furthermore, studies have demonstrated that TNF-α originating from the placenta leads to upregulation of endothelial cell inflammatory cytokines [31]. Therefore, the effects of TNF-α should be examined in both placental and endothelial cells. In the present study, we examined the ability of luteolin to inhibit the effects of TNF-α in normotensive placental explants and endothelial cells. Utilization of normotensive samples, where we can isolate the TNF-α pathway, was chosen due to the other pathways which could act as confounding factors in PE tissue. As expected, stimulation by TNF-α led to increases in NF-κB activation, expression of IL-6 and ET-1, and ROS. Pretreatment with luteolin demonstrated significant anti-inflammatory and antioxidant effects in preventing these increases in HUVECs. In the presence of TNF-α, both luteolin and NF-κB inhibitor SN50 demonstrated reductions of IL-6 and proET-1 such that they returned to control levels. When combined, there was an additive effect, and levels were further reduced, suggesting that luteolin is capable of inhibiting additional pathways.
TNF-α has been shown to increase production of ROS in endothelial cells, contributing to the development of endothelial dysfunction [9]. There are a variety of pathways through which TNF-α can elicit its actions to increase oxidative stress. One of these is through uncoupling of mitochondrial complexes, causing disruption of the electron transport chain, ultimately leading to production of reactive oxygen species [32]. We found that stimulation with TNF-α indeed significantly increased production of total ROS and superoxide. Pretreatment with luteolin successfully attenuated the actions of TNF-α, but further studies will need to be performed in order to determine luteolin’s mechanism of action in preventing oxidative stress.
Similar to HUVECs, TNF-α stimulation in placental explants led to significant upregulation of NF-κB activation, in the form of phosphorylated p65, and expression of IL-6 and proET-1. Pretreatment with luteolin significantly decreased levels of phosphorylated p65 to control levels. As SN50 is a competitive inhibitor of p65 translocation and does not play a role in the p65 phosphorylation, it was not utilized in the placental explant experiments. Unlike what was observed in HUVECs, there was not a significant reduction in IL-6 and proET-1 with luteolin pretreatment. Because activation of NF-κB was attenuated, it is likely that TNF-α stimulates alternate pathways in placental explants [33] which may not be inhibited by luteolin.
While some may believe that the actions of ET-1 are limited to the vascular endothelium, ET-1 has also been shown to play a role in the PE placenta. Indeed, ET-1 has been shown to inhibit the proliferation and vitality of placental trophoblasts. Additionally, ET-1 was shown to promote oxidative stress in placental explants and trophoblasts, having elevated levels of lipid peroxidation metabolites and decreased levels of antioxidant mechanisms [34]. Although the reduction in proET-1 in the presence of luteolin did not reach statistical significance, there was a reduction of approximately 30%, which may help to improve placental health.
Our current study is limited to luteolin’s ability to reduce TNF-α-induced inflammation and oxidative stress pathways in vitro. Our data demonstrates that luteolin is capable of inhibiting NF-κB activation in human endothelial cells and placental explants, as well as reduce production of ROS and cytokines in endothelial cells. Further studies need to be done in order to determine if luteolin is capable of inhibiting oxidative stress in the placenta and elucidate the pathways through which ROS production is inhibited. We have previously shown that luteolin is capable of targeting HIF-1α and sFlt-1 pathways in vitro, and we have ongoing studies to determine the effects of luteolin on these pathways in vivo.
Data Availability
The data that support the findings of this study are available in the Methods section of this article.
Supplementary Material
Highlights:
Inflammation and oxidative stress pathways are activated in preeclampsia
Luteolin inhibits activation of NF-κB in placental tissue and endothelial cells
Inflammatory cytokine upregulation in endothelial cells is inhibited by luteolin
Production of reactive oxygen species and superoxide is ameliorated by luteolin
Acknowledgements
This study was primarily funded by SR (NIH/NHLBI: 1R56HL157579-01). JPG is funded through several NIH awards (NIH/NIGMS: U54GM115428; NIH/NHLBI: T32HL105324; NIH/ NIGMS: 1P20 GM104357; NIH/NHLBI: R01HL148191). FTS is funded through NIH/NHLBI R00130577; NIH/NIGMS P20 GM121334; Industry: AWD-001111.
We want to acknowledge Drs. Ernst Lengyel and Ayman Al-Hendy for use of laboratory space and equipment.
Conflict of Interest
SR reports serving as a consultant to Roche Diagnostics and ThermoFisher Scientific and has received funding from Roche Diagnostics and Siemens for studies related to the use of angiogenic factors in pregnancy which is unrelated to the present work.
DECLARATION OF INTEREST -
SR reports serving as a consultant for Roche Diagnostics, ThermoFisher, Siemens and has received research funding from Roche Diagnostics and Siemens for work related to angiogenic biomarkers unrelated to this work. All other authors have no conflict of interest.
Abbreviations:
- PE
Preeclampsia
- TNF-α
Tumor necrosis factor-α
- IL
Interleukin
- NF-κB
Nuclear factor-κB
- ROS
Reactive oxygen species
- ET-1
Endothelin-1
- sFlt-1
Soluble fms-like tyrosine kinase 1
- NT
Normotensive
- ACOG
American College of Obstetricians and Gynecologists
- PBS
Phosphate-buffered saline
- DMSO
Dimethyl sulfoxide
- HUVEC
Human umbilical vein endothelial cells
- TBS-T
Tris-buffered saline- tween
- ELISA
Enzyme linked immunosorbent assay
- ANOVA
Analysis of variance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.ACOG Practice Bulletin No. 202 Summary: Gestational Hypertension and Preeclampsia. Obstet Gynecol, 2019. 133(1): p. 1. [DOI] [PubMed] [Google Scholar]
- 2.Bates DO, Pre-eclampsia and the microcirculation: a novel explanation. Clin Sci (Lond), 2003. 104(4): p. 413–4. [DOI] [PubMed] [Google Scholar]
- 3.Rolfo A, et al. , Pro-inflammatory profile of preeclamptic placental mesenchymal stromal cells: new insights into the etiopathogenesis of preeclampsia. PLoS One, 2013. 8(3): p. e59403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zárate A, et al. , Early disturbed placental ischemia and hypoxia creates immune alteration and vascular disorder causing preeclampsia. Arch Med Res, 2014. 45(7): p. 519–24. [DOI] [PubMed] [Google Scholar]
- 5.Lee JI and Burckart GJ, Nuclear factor kappa B: important transcription factor and therapeutic target. The Journal of Clinical Pharmacology, 1998. 38(11): p. 981–993. [DOI] [PubMed] [Google Scholar]
- 6.Yu H, et al. , Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduction and Targeted Therapy, 2020. 5(1): p. 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayden MS and Ghosh S, Regulation of NF-κB by TNF family cytokines. Semin Immunol, 2014. 26(3): p. 253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang MW, et al. , Nafamostat Mesilate Inhibits TNF-α-Induced Vascular Endothelial Cell Dysfunction by Inhibiting Reactive Oxygen Species Production. Korean J Physiol Pharmacol, 2015. 19(3): p. 229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X, et al. , Role of Reactive Oxygen Species in Tumor Necrosis Factor-alpha Induced Endothelial Dysfunction. Curr Hypertens Rev, 2008. 4(4): p. 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan J and Walsh S, Activation of NF-κB in placentas of women with preeclampsia. Hypertens Pregnancy, 2012. 31(2): p. 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah TJ and Walsh SW, Activation of NF-kappaB and expression of COX-2 in association with neutrophil infiltration in systemic vascular tissue of women with preeclampsia. Am J Obstet Gynecol, 2007. 196(1): p. 48 e1–8. [DOI] [PubMed] [Google Scholar]
- 12.Winship A, et al. , Interleukin 11 blockade during mid to late gestation does not affect maternal blood pressure, pregnancy viability or subsequent fertility in mice. Reproductive biomedicine online, 2018. 36(3): p. 250–258. [DOI] [PubMed] [Google Scholar]
- 13.Stratta P, et al. , Vitamin E supplementation in preeclampsia. Gynecol Obstet Invest, 1994. 37(4): p. 246–9. [DOI] [PubMed] [Google Scholar]
- 14.Gulmezoglu AM, Hofmeyr GJ, and Oosthuisen MM, Antioxidants in the treatment of severe pre-eclampsia: an explanatory randomised controlled trial. Br J Obstet Gynaecol, 1997. 104(6): p. 689–96. [DOI] [PubMed] [Google Scholar]
- 15.Aziz N, Kim MY, and Cho JY, Anti-inflammatory effects of luteolin: A review of in vitro, in vivo, and in silico studies. J Ethnopharmacol, 2018. 225: p. 342–358. [DOI] [PubMed] [Google Scholar]
- 16.Lin Y, et al. , Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr Cancer Drug Targets, 2008. 8(7): p. 634–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu JF, et al. , Reduction of lipid accumulation in HepG2 cells by luteolin is associated with activation of AMPK and mitigation of oxidative stress. Phytother Res, 2011. 25(4): p. 588–96. [DOI] [PubMed] [Google Scholar]
- 18.Kris-Etherton PM, et al. , Bioactive compounds in foods: their role in the prevention of cardiovascular disease and cancer. Am J Med, 2002. 113 Suppl 9B: p. 71s–88s. [DOI] [PubMed] [Google Scholar]
- 19.Eddy AC, et al. , Bioflavonoid luteolin prevents sFlt-1 release via HIF-1α inhibition in cultured human placenta. Faseb j, 2023. 37(8): p. e23078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajakumar A, et al. , Impaired oxygen-dependent reduction of HIF-1alpha and −2alpha proteins in pre-eclamptic placentae. Placenta, 2003. 24(2–3): p. 199–208. [DOI] [PubMed] [Google Scholar]
- 21.Rana S, et al. , Ouabain inhibits placental sFlt1 production by repressing HSP27-dependent HIF1alpha pathway. FASEB J, 2014. 28(10): p. 4324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieman KM, et al. , Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med, 2011. 17(11): p. 1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenny HA, et al. , The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest, 2008. 118(4): p. 1367–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eddy AC, et al. , Biopolymer-Delivered, Maternally Sequestered NF-κB (Nuclear Factor-κB) Inhibitory Peptide for Treatment of Preeclampsia. Hypertension, 2020. 75(1): p. 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierce GL, et al. , Nuclear factor-{kappa}B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation, 2009. 119(9): p. 1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cackovic M, et al. , Fractional excretion of tumor necrosis factor-alpha in women with severe preeclampsia. Obstet Gynecol, 2008. 112(1): p. 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khodadadi A, et al. , The TNF-α−308G/A Gene Polymorphism and Serum TNF-α Levels in Women With Preeclampsia. J Family Reprod Health, 2022. 16(3): p. 205–211. [PMC free article] [PubMed] [Google Scholar]
- 28.Lau SY, et al. , Tumor necrosis factor-alpha, interleukin-6, and interleukin-10 levels are altered in preeclampsia: a systematic review and meta-analysis. Am J Reprod Immunol, 2013. 70(5): p. 412–27. [DOI] [PubMed] [Google Scholar]
- 29.George EM, et al. , Heme oxygenase induction attenuates TNF-alpha-induced hypertension in pregnant rodents. Front Pharmacol, 2015. 6: p. 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaMarca B, et al. , Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: effect of tumor necrosis factor-alpha blockade. Hypertension, 2008. 52(6): p. 1161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw J, et al. , Inflammatory processes are specifically enhanced in endothelial cells by placental-derived TNF-α: Implications in preeclampsia (PE). Placenta, 2016. 43: p. 1–8. [DOI] [PubMed] [Google Scholar]
- 32.Kastl L, et al. , TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett, 2014. 588(1): p. 175–83. [DOI] [PubMed] [Google Scholar]
- 33.Tanabe K, et al. , Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J Neuroinflammation, 2010. 7: p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiore G, et al. , Endothelin-1 triggers placental oxidative stress pathways: putative role in preeclampsia. J Clin Endocrinol Metab, 2005. 90(7): p. 4205–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the Methods section of this article.