

Abstract
Background.
Mitochondrial dysfunction is a well-established result of acute kidney injury (AKI). Previously, we identified that 5-hydroxytryptamine 1F (5-HT1F) receptor agonism with lasmiditan induces mitochondrial biogenesis (MB) and improves renal vasculature and function in an AKI mouse model. We hypothesize that lasmiditan also modulates mitochondrial dynamics and mitophagy in a mouse model of AKI.
Methods.
Male mice were subjected to renal ischemia/reperfusion (I/R) and treated daily with lasmiditan (0.3 mg/kg) or vehicle beginning 24h after injury for 3 or 6d. Serum creatinine was measured to estimate glomerular filtration. Electron microscopy was used to assess mitochondrial morphology and mitophagy. Mitochondrial-related protein were confirmed with immunoblotting. Mitochondrial function was assessed with ATP measurements.
Results.
Lasmiditan treatment improved mitochondrial and kidney recovery as early as 3d post-AKI, as evidenced by increased ATP, and decreased serum creatinine, respectively. Electron micrographs of renal cortices revealed that lasmiditan also decreased mitochondrial damage and increased mitochondrial area and size by 6d after I/R injury. Additionally, lasmiditan treatment increased mitolysosomes by 3d, indicating induction of mitophagy. Phosphorylation of mitophagy-related proteins were also increased in the renal cortices of lasmiditan-treated AKI mice 3d after I/R injury, whereas fusion-related proteins were increased at 6d after I/R injury.
Conclusion.
These data reveal that lasmiditan accelerates renal recovery, restores normal mitochondrial membrane and cristae morphology, decreases excessive mitochondrial fission, and accelerates mitophagy post-AKI in a time-dependent manner, establishing mitochondrial function and recovery from AKI.
Keywords: Mitophagy, mitochondrial dynamics, mitochondrial biogenesis, mitochondrial dysfunction, acute kidney injury
1. Introduction
The kidney is the second most energy-demanding organ in the body, with the greatest number of mitochondria being located in the proximal tubules (1). This high energy demand and mitochondrial abundance make the kidneys particularly susceptible to mitochondrial dysfunction after injuries such as ischemia/reperfusion (I/R) (2). I/R insult is one of the main causes of acute kidney injury (AKI), defined as the rapid loss of kidney function characterized by increased serum creatinine and decreased urinary output (3–5). AKI increases morbidity and severe cases have mortality incidences of 40–60% (6). Currently, there is no FDA-approved pharmacological treatment for AKI, forcing patients to rely on alternative approaches such as kidney transplants or dialysis (7–9).
The pathogenesis of AKI can vary depending on the severity of the injury; however, many studies indicate that mitochondrial dysfunction is a critical factor for the progression of AKI (10–12). Mitochondrial dysfunction leads to ROS accumulation, ATP depletion, and increased release of cytochrome c (13–16). Additionally, AKI has been associated with altered mitochondrial quality control mechanisms: Mitochondrial biogenesis (MB), mitochondrial fusion and fission, and mitophagy (17–20). In recent years, mitophagy and the clearance of defective mitochondria has gained attention for their role in normal kidney physiology and in kidney disease (21).
Mitophagy is a selective autophagy specifically for damaged or redundant mitochondria and this process is regulated by a multitude of enzymes such as AMP-activated protein kinase (AMPK) and Unc-51 like autophagy activating kinase 1 (ULK1). Mitophagy, along with fission/fusion and MB, helps to maintain a healthy mitochondrial network which is essential for proper kidney function (11, 22–25). Restoring mitochondrial homeostasis via regulation of MB, fusion/fission, and mitophagy after AKI may decrease injury progression and restore kidney function (14, 26, 27). In support of this concept, mitophagy has been found to be renoprotective after I/R-induced AKI and chronic kidney disease (CKD) (28–30).
Lasmiditan, a selective 5-hydroxytryptamine 1F (5-HT1F) receptor agonist is an FDA-approved drug for the treatment of migraines (31). Our group has reported that lasmiditan induces MB, accelerates renal recovery, decreases vascular leakage, and prevents fibrosis in an AKI mouse model (32). Thus, we hypothesize that lasmiditan modulates other mitochondrial quality control mechanisms, including mitochondrial dynamics and mitophagy, and their roles in injury progression and recovery after I/R-induced AKI.
2. Methods
2.1. Experimental design
Male C57BL/6NCrl mice (8 weeks-of-age, ~20 g) were purchased from Charles River (Hollister, CA) and acclimated one week prior to surgery. All animals were individually housed in temperature-controlled conditions under a light/dark photo-cycle with food and water supplied ad libitum until euthanasia by cervical dislocation. All experiments were approved by the University of Arizona in accordance with the guidelines set forth by the NIH Guide for the Care and Use of Laboratory Animals. Lasmiditan was purchased from ChemBlock (Burlingame, CA; Cat#L22337) and dissolved in saline/0.1% DMSO. Mice were treated with lasmiditan (0.3 mg/kg, intraperitoneally (IP), a dose that induces MB, or vehicle (32). Mice were assigned to one of six groups: 1) sham, 2) I/R 24 h, 3) 3d I/R + vehicle, 4) 3d I/R + 0.3 mg/kg lasmiditan, 5) 6d I/R + vehicle, 6) 6d I/R + 0.3 mg/kg lasmiditan and dosed for 2 or 5 consecutive days starting 24 h after I/R injury and euthanized on 3d or 6d, respectively. Every group is reported as N=6.
I/R was established using bilateral renal pedicle clamping as described by Funk and Collier (33, 34). Briefly, buprenorphine (Patterson Veterinary, Loveland, CO; Cat#07–892-5235; 0.05 mg/kg) was administered, and the renal artery and vein were isolated. Blood flow was occluded with a vascular clamp for 18 min while body temperature was maintained at 36.5 °C. Sham mice underwent surgery without renal pedicle clamping. Kidney cortices were collected and snap-frozen in liquid nitrogen. Blood was collected by retro-orbital puncture and kept at room temperature for 30 min. Serum creatinine (SCr) was measured using a creatinine enzymatic reagent assay kit (Diazyme; Poway, CA; Cat# 7548–120) based on the manufacturer’s directions. Sham mice did not undergo surgical procedures or receive drug treatment.
2.2. Transmission electron microscopy (TEM) analysis.
Kidney cortex samples were fixed in 2.5% glutaraldehyde and then PBS (Thermo-Fisher Scientific; Waltham, MA; Cat# 50–366-997 & 13–151-014), stored overnight, and submitted to the TEM core facility at the University of Arizona. Images were obtained with a FEI Tecnai Spirit Transmission Electron Microscope (Hillsboro, OR) at 100 kV. TIFF images (8-bit) were captured with an XR41 CCD digital camera (Woburn, MA) at 6,000x. For all cases, 5–6 images were analyzed per sample and mitochondrial morphology per field were calculated. Each image represents 2–3 cells per field. Each cell contained ~50–100 mitochondria. Mitochondria length was obtained using the major axis as reported previously (35). TEM images were analyzed utilizing MathLab2020b software.
Criteria for proximal tubule identification was selected based on mitochondrial and brush border abundance as well as cell size and morphology (36). Mitochondrial damage score criteria are as follows: 0 = no injury; 1 = minimal injury, minimal loss of cristae; 2 = moderate injury, moderate loss of cristae and moderate membrane swelling; 3 = severe injury, severe to complete loss of cristae, severe mitochondrial swelling, and mitochondrial membrane rupture (37–40). Mitochondria length were obtained using the major axis. Autolysosomes represented autophagic vacuoles with clear membranes and any cargo. Mitolysosomes represented an autophagic vacuole with mitochondria exclusively as cargo.
2.3. Immunoblotting.
Protein was extracted from kidney cortices using RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, pH 7.4) with protease inhibitor cocktail (1:100 Millipore Sigma; Burlington, MA, Cat# P8340), 1 mM sodium fluoride, and 1 mM sodium orthovanadate (Thermo-Fisher Scientific; Waltham, MA; Cat# S299100 and AC205330500, respectively) as previously described (18). Membranes were visualized using chemiluminescence (Thermo-Fisher Scientific; Waltham, MA; Cat# PI34076) on a GE ImageQuant LAS4000 (GE Life Sciences; Pittsburgh, PA). Optical density was quantified with ImageJ software. Primary antibodies were purchased from Abcam (Cambridge, MA): Mfn1 (1:000, Cat#ab221661), Mfn2 (1:1,000, Cat#ab56889), TFAM (ab131607). R and D Systems (Minneapolis, MN): KIM-1 (1:1000, Cat# AF1817). NOVUS (Centennia, CO): LC3B, (1:1000, Cat# NB6001384). Cell Signaling (Danvers, MA): DRP1 (1:1000, Cat# 5391S), p-AMPK-T172 (1:1,000, Cat#2535S), p-mTOR-S2448 (1:1,000, Cat#2971S), pULK1-S555 (1:1,000, Cat#5869), P62 (1:1,000, Cat#5114s), Beta-actin (1:1,000, Cat#3700S; Danvers, MA). R&D (Minneapolis,MN): KIM1 (1:1,000, Cat# AF1817). Secondary antibodies: Goat Anti-Rabbit IgG H&L (HRP) (1:10,000, Abcam, Cat# ab6721), Rabbit Anti-Mouse IgG H&L (HRP) (1:10,000, Abcam, Cat# 6728), and Donkey Anti-Goat IgG H&L (HRP) (1:10,000, Abcam, Cat# ab97110) were used as secondary antibodies. All antibodies were validated for their respective targets. See their respective websites using the provided catalog numbers for a detailed description of their validation processes.
2.4. Nucleic Acid Isolation and Quantitative Polymerase Chain Reaction.
To measure mitochondrial DNA (mtDNA) content, DNA was extracted from frozen renal cortices using a DNeasy Blood and Tissue kit (QIAGEN; Valencia, CA). PCR products were amplified from 5 ng of cellular DNA using a real-time SYBR Green quantitative PCR (BioRad). Mitochondrial DNA (mtDNA) copy number was calculated by comparing the mitochondrial D loop to nuclear ApoB apolipoprotein B (ApoB) forward 5′-CGT GGG CTC CAG CAT TCT-3′, ApoB reverse 5′-TCA CCA GTC ATT TCT GCC TTT G-3′, and D loop forward 5′-CCCAAG CAT ATA AGC TAG TA-3′, D loop reverse 5′-ATA TAA GTC ATA TTT TGG GAA CTA C-3′.
2.5. ATP measurements.
ATP content of renal cortices were measured using an ATP Assay Kit (Colorimetric/Fluorometric) (Abcam; Cambridge, MA, Cat# ab83355) and the manufacturer’s protocol.
2.6. Statistical analysis.
For experimental groups (N = 6), N represents one animal. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Vehicle and lasmiditan groups were normalized to their corresponding sham group, either 3d or 6d. Immunoblot analyses were merged into a single graph per marker. Data were analyzed using GraphPad Prism software (La Jolla, CA and p<0.05 was considered statistically significant. Letters at the tops of the bars represent different statistical changes.
3. Results
3.1. Lasmiditan decreases injury markers and increases survival after I/R-AKI
Serum creatinine was measured to estimate glomerular filtration. Based on maximal serum creatinine 24 h post-AKI, mice were assigned to either vehicle (I/R+V) or lasmiditan (I/R+L) treatment groups and treated daily for 3 or 6d. Mice treated with lasmiditan post-AKI had decreased serum creatinine compared to vehicle-treated mice post-AKI at 3d (Fig. 1A) and 6d (Fig. 1B). Importantly, 100% survival was observed with lasmiditan treatment, whereas only 66% survival was observed with vehicle (Fig. 1C). Kidney injury molecule 1 (KIM-1) is a proximal tubule injury marker only expressed upon injury, so data were normalized to KIM1 protein levels 1 d after AKI. Renal cortex KIM-1 remained elevated at 3d regardless of treatment (Fig. 1 D, E). By 6d, however, KIM-1 was increased 1.6-fold in I/R+V mice compared to 1d and remained elevated and was restored to sham levels in the I/R+L group (Fig. 1D). These results show that lasmiditan decreases kidney injury markers and enhances survival after I/R injury.
Figure 1. Treatment with lasmiditan (L) increases renal recovery and survival after ischemia reperfusion injury.

(A) Serum creatinine was assessed at 1 and 3 days after renal IR surgery. (B) Serum creatinine at 6 days after renal IR surgery. (C) Kaplan-Meier survival analysis of mice that underweant IR surgery. (D) Densitometry analysis of KIM-1. (E) Representative immunoblot if KIM-1. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
3.2. Lasmiditan restores mitochondrial morphology, mitochondrial DNA, and ATP in the renal cortex after I/R-AKI
Mitochondrial morphology and cristae integrity were measured using electron microscopy. mtDNA and ATP were measured with PCR and a colorimetric assay, respectively. Electron microscopy revealed swollen mitochondria and loss of cristae in the renal cortex beginning 1d after AKI and continuing through 6d (Fig. 2A). Mitochondrial damage increased 1.6-fold 1 d after AKI (Fig. 2B). By 3d, mitochondrial damage was increased by 2.1-fold in the I/R+V group compared to sham, remaining elevated until 6d (Fig. 2B). In contrast, lasmiditan treatment decreased mitochondrial damage 0.5-fold at 3d and 0.75-fold by 6d compared to vehicle (Fig. 2B).
Figure 2. Treatment with lasmiditan (L) decreases mitochondrial damage score after ischemia reperfusion injury.
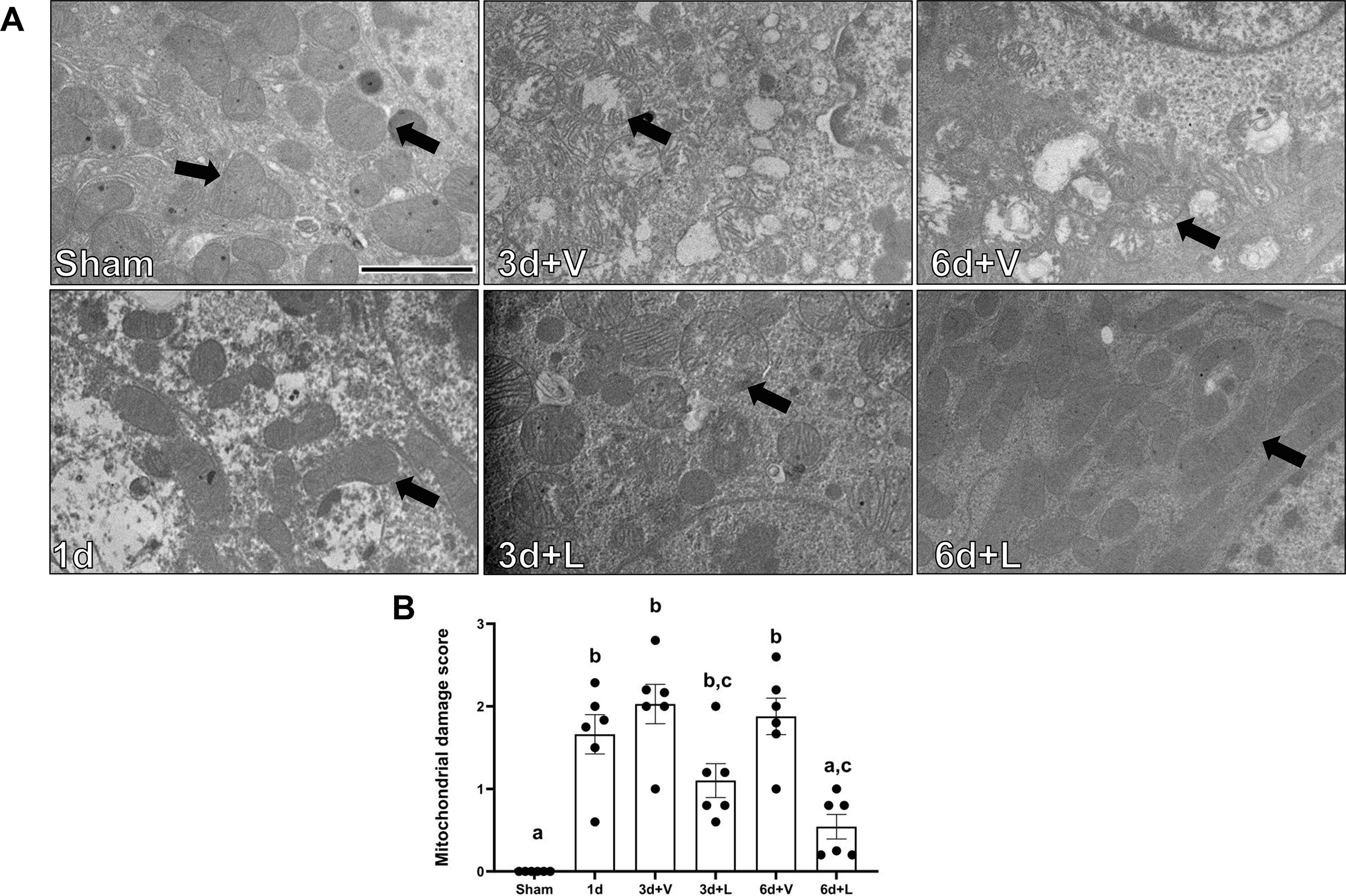
(A) Representative electron micrographs. Black arrows point at mitochondria. Scale bar=2μm. (B) Average mitochondria damage score. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
Mitochondrial transcription factor A (TFAM) protein levels decreased starting at 1d after I/R injury and remained down six days after I/R injury in both lasmiditan and vehicle groups. Lasmiditan treatment seemed to increase its levels compared to vehicle, however it did not reach statistical significance (Fig. 3A). Renal cortex mitochondrial copy number was increased 1.8-fold in the I/R+L group compared to vehicle 6d after AKI (Fig. 3B). ATP content decreased 0.85-fold compared to sham 1 d after injury and remained decreased in the I/R+V group through 6d. Total ATP concentration in I/R+L mice, however, increased compared to vehicle by 0.65-fold at 3d and 0.70-fold by 6d (Fig. 3C). These data confirm that lasmiditan restores mitochondrial morphology and increases mitochondrial DNA and ATP in renal cortices after I/R injury.
Figure 3. Treatment with lasmiditan (L) increases mitochondria DNA and ATP levels after ischemia reperfusion injury.
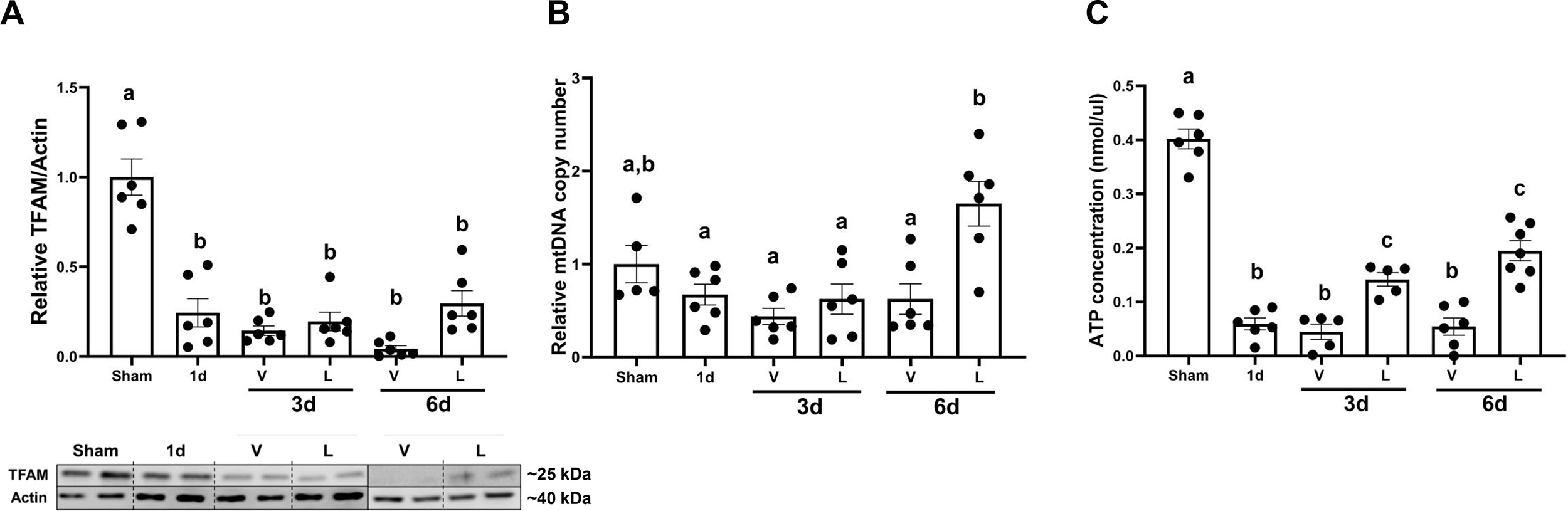
(A)Densitometry analysis of TFAM (B) Relative mitochondrial DNA content in the renal cortex. (C) Renal cortical ATP analysis. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
3.3. Lasmiditan restores mitochondrial area, perimeter and length in the renal cortex after I/R-AKI
Electron microscopy was used to assess mitochondrial area, perimeter, and length. (Fig. 4A). Whereas mitochondrial number did not change across groups or timepoints (Fig. 4B), mitochondrial area decreased in I/R+V group 0.58-fold at 6d compared to sham and was restored to sham levels in the I/R+L group (Fig. 4C). Similarly, mitochondrial length decreased by 0.38-fold at 6d in the I/R+V group compared to sham and was restored to sham levels in the I/R+L group (Fig. 4D).
Figure 4. Treatment with lasmiditan (L) restores mitochondrial area, perimeter, and length after ischemia reperfusion injury.
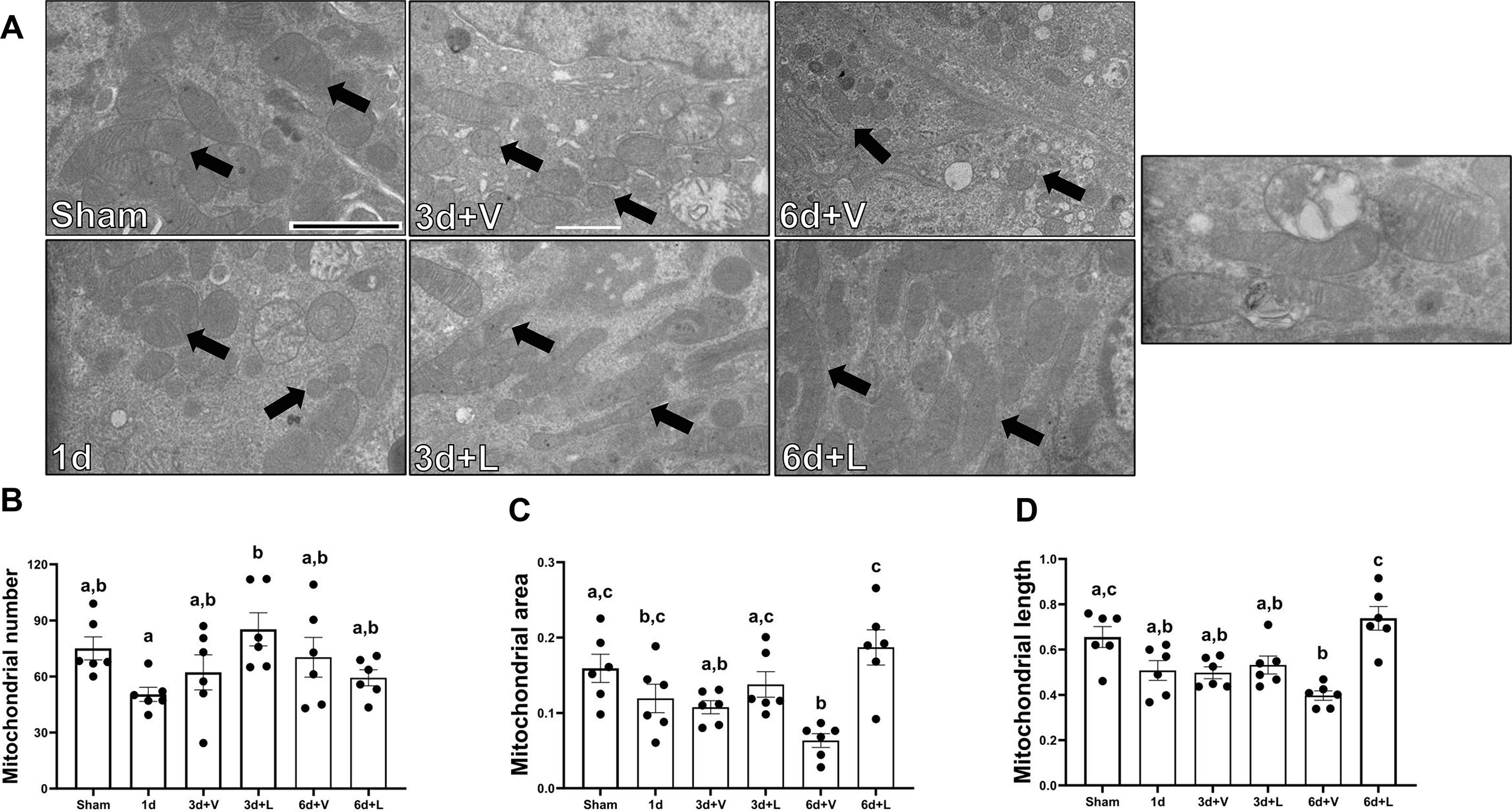
(A) Representative electron micrographs. Black arrows point at mitochondria. Scale bar=2μm. (B) Average mitochondria number. (C) Average mitochondria area analysis. (D) Average mitochondrial lenght analysis. (D) Average mitochondrial length analysis. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
Also, mitochondrial fusion protein mitofusin 1 (Mfn1) decreased in the I/R+V group by 0.40-fold at 3d compared to sham and remained decreased until 6d. Then, it was restored to sham levels in the I/R+L group (Fig. 5A). Mitofusin 2 (Mfn2) decreased in the I/R+V group by 0.52-fold at 3d and by 0.70-fold at 6d compared to sham. However, Mfn2 increased by 0.35-fold in the I/R+L group at 6d compared to vehicle-treated animals (Fig. 5B). Mitochondrial fission protein dynamin-related protein 1 (DRP1) increased by 3-fold in the I/R+V group compared to sham whereas in the I/R+L group, DRP1 was restored to sham levels (Fig. 5C). Thus, lasmiditan restored mitochondrial area and length and increased Mfn2 but decreased DPR1 protein at 6d. These findings are consistent with increased mitochondrial fusion and decreased fission.
Figure 5. Treatment with lasmiditan (L) restores Mfn1 and Mfn2 and de creases DRP1 after ischemia reperfusion injury.
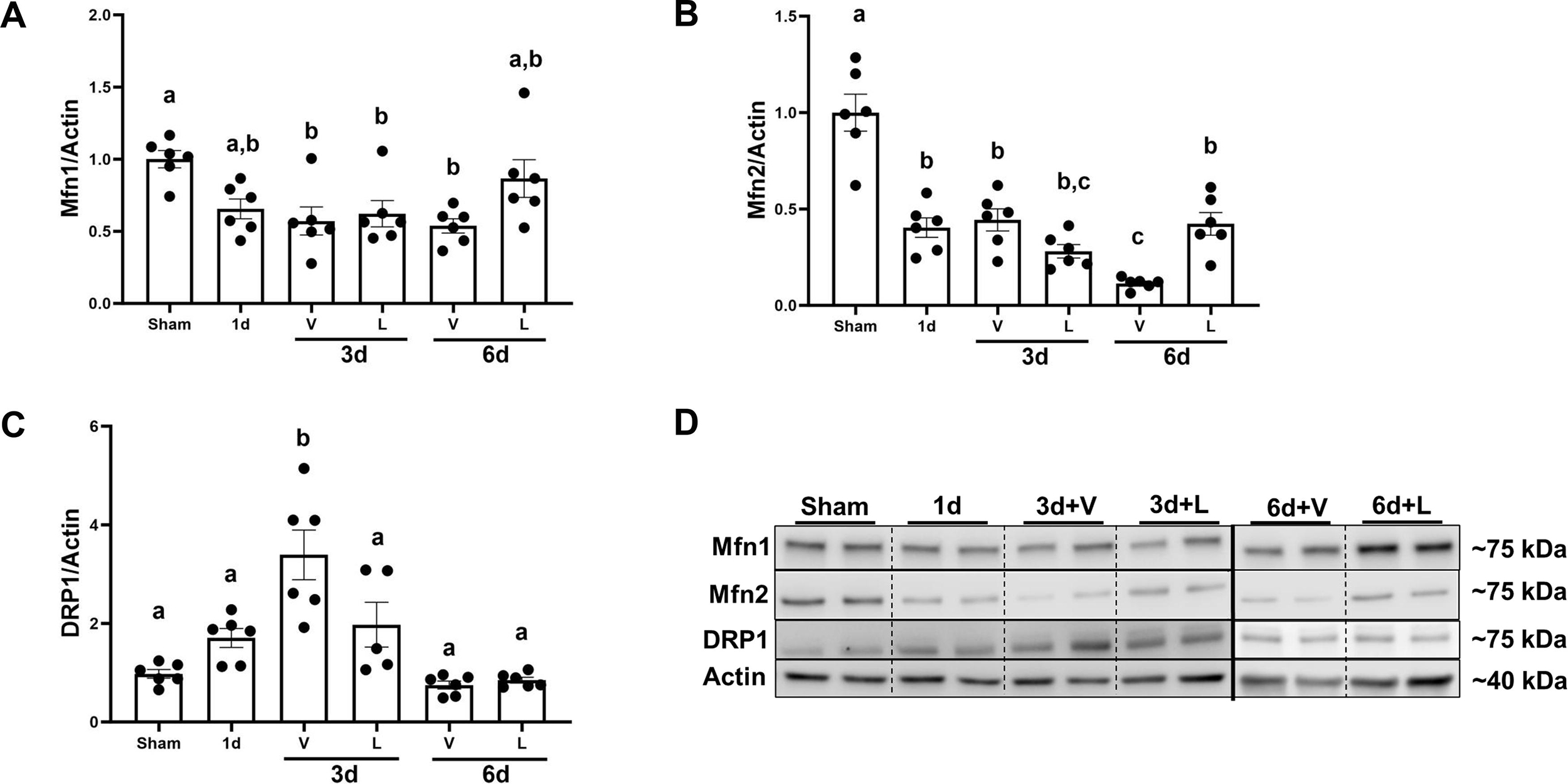
(A) Densitometry analysis of Mfn1. (B) Densitometry analysis of Mfn2. (C) Densitometry analysis of DRP1. (D) Representative immunoblots. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
3.4. Lasmiditan increases clearance of damaged mitochondria renal cortex following I/R-AKI
To determine whether lasmiditan increases mitophagy in renal cortices, electron microscopy was used to count total mitolysosomes per field. Autolysosomes number per field increased by 1-fold compared to sham starting at 3d after I/R injury in both lasmiditan and vehicle group. No significant increase of autolysosomes was observed among treatment groups. Autolysosomes number per field was restored to sham levels at 6d in both lasmiditan and vehicle group. No significant increase of mitolysosomes was observed in the I/R+V group independent of the timepoint. However, mitolysosomes per field increased 1.5-fold per field in the I/R+L group compared to the I/R+V group at 3d (Fig. 6B). Mitolysosomes per field did not change at the 6d timepoint independent of treatment.
Figure 6. Treatment with lasmiditan (L) restores increases mitolysosomes formation after ischemia reperfusion injury.
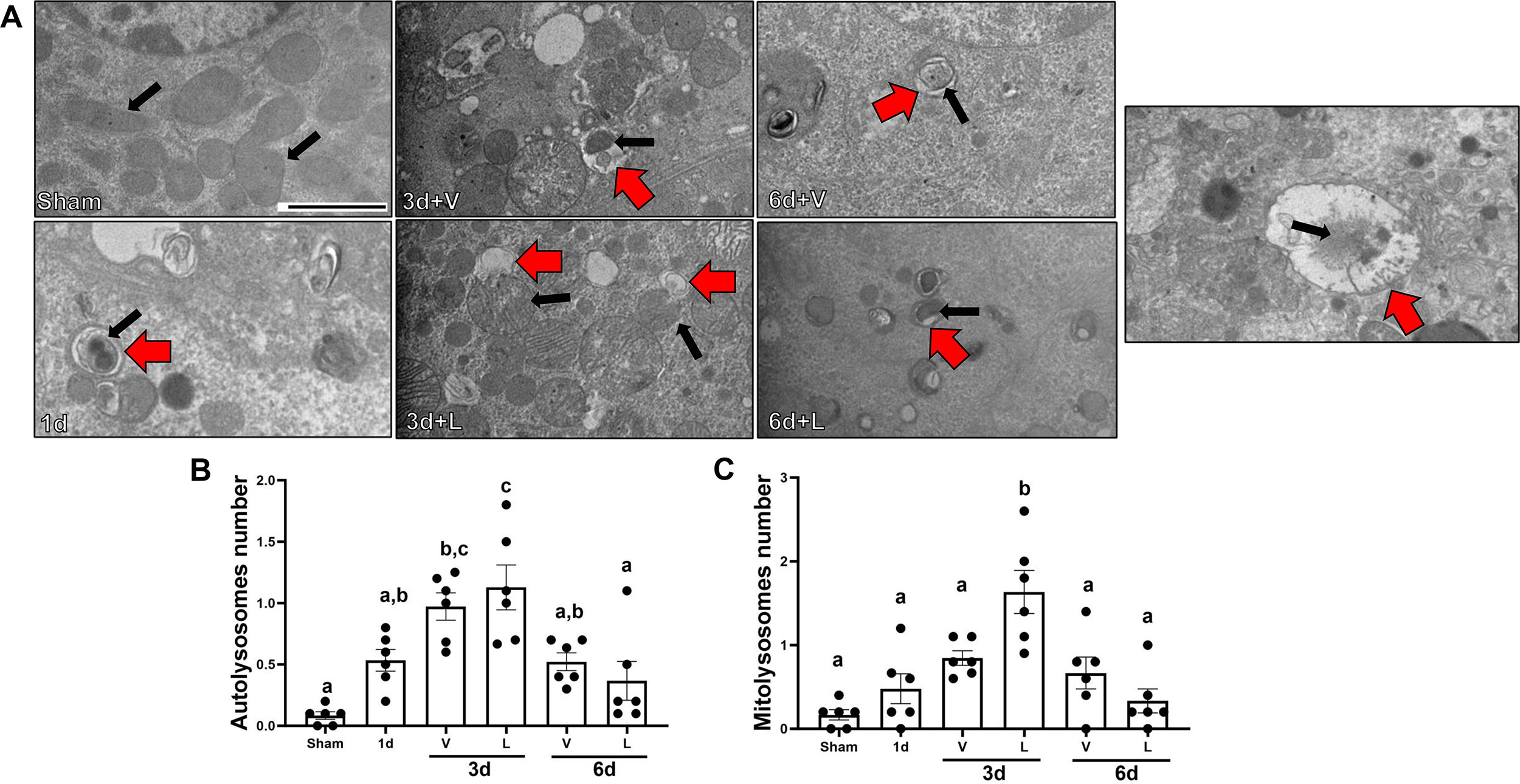
(A) Representative electron micrographs. Black arrows point at mitochondria and red arrows point at autophagic vacuoles. Scale bar=2μm. (B) Average number of mitolysosomes per field. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
It has been reported that AMPKα regulates autophagy by phosphorylating ULK1 (41, 42). Protein levels of the mammalian target of rapamycin (mTOR) decreased by 0.5-fold starting at 1d and continuously decreased by 0.80-fold in I/R+V and I/R+L groups at 6d compared to sham (Fig. 7A). AMPKα protein levels did not change from 1d or in I/R+V and I/R+L groups at 3d. However, levels of AMPKα increased by 1.53-fold in the I/R+L group compared to I/R+V group (Fig. 7B). Protein levels of ULK1 increased in the I/R+V and I/R+L groups at 3d by 20-fold and 23-fold, respectively (Fig. 7C). P62 protein increased 15-fold at 1 d compared to sham and then returned to sham levels in I/R+V and I/R+L groups at 3d and 6d (Fig. 7D). Finally, the light chain 3 B (LC3BII:I) decreased by 0.40-fold in the I/R+V and I/R+L groups at 3d and then was restored to sham levels at 6d (Fig. 7E). Interestingly, the phosphorylation levels of AMPKα increased by 2.53-fold in the I/R+L group compared to I/R+V group at 3d and phosphorylation levels were restored to sham group levels by 6d (Fig. 8B). This densitometry analysis along with electron microscopy data suggest that lasmiditan increases mitophagy at 3d post-I/R injury.
Figure 7. Treatment with lasmiditan (L) increases levels of AMPKα and ULK1 after ischemia reperfusion injury.
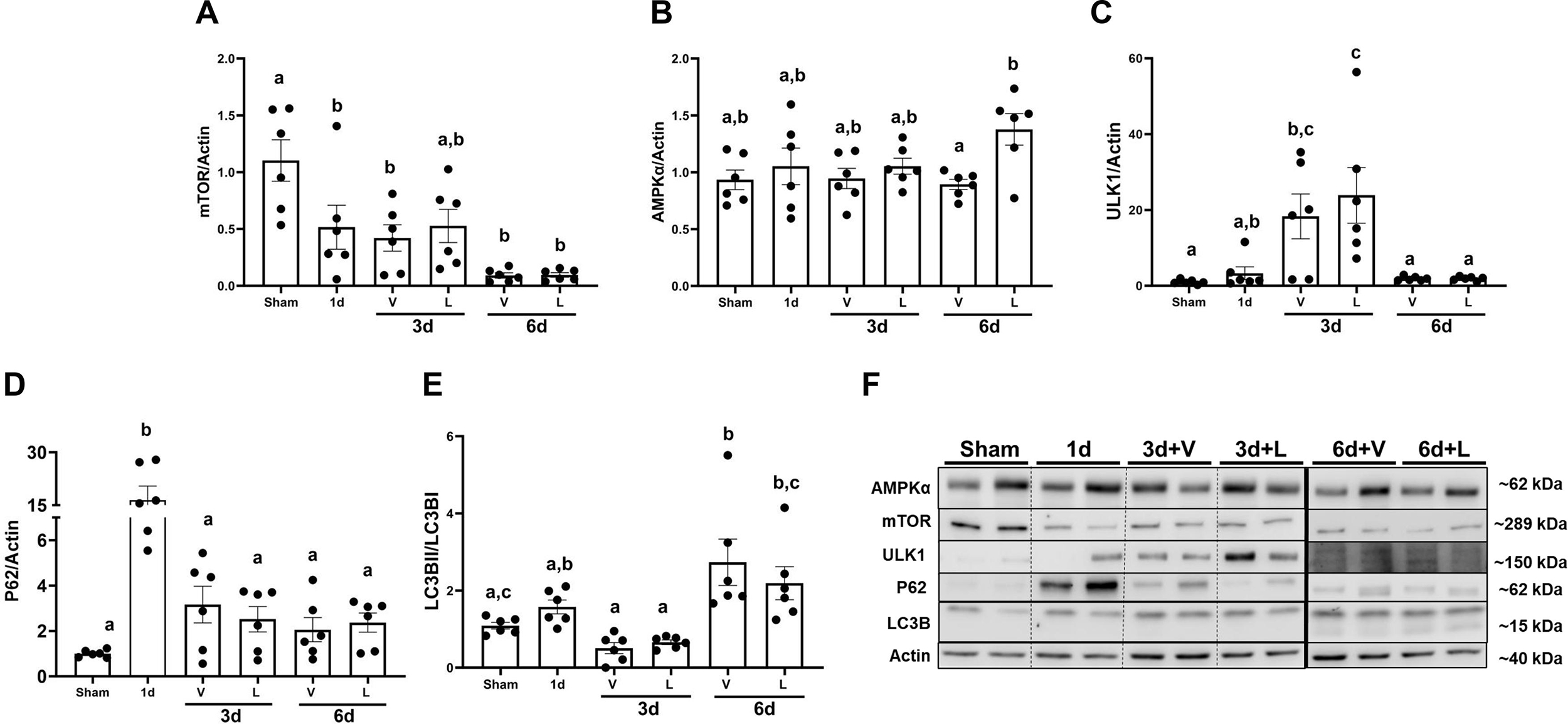
Densitometry analysis of the following genes (A) mTOR (B) AMPKα (C) ULK1 (D) P62 (E) LC3B (F) Representative immunoblots. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
Figure 8. Treatment with lasmiditan (L) increases phosphorylation of AMPKα.
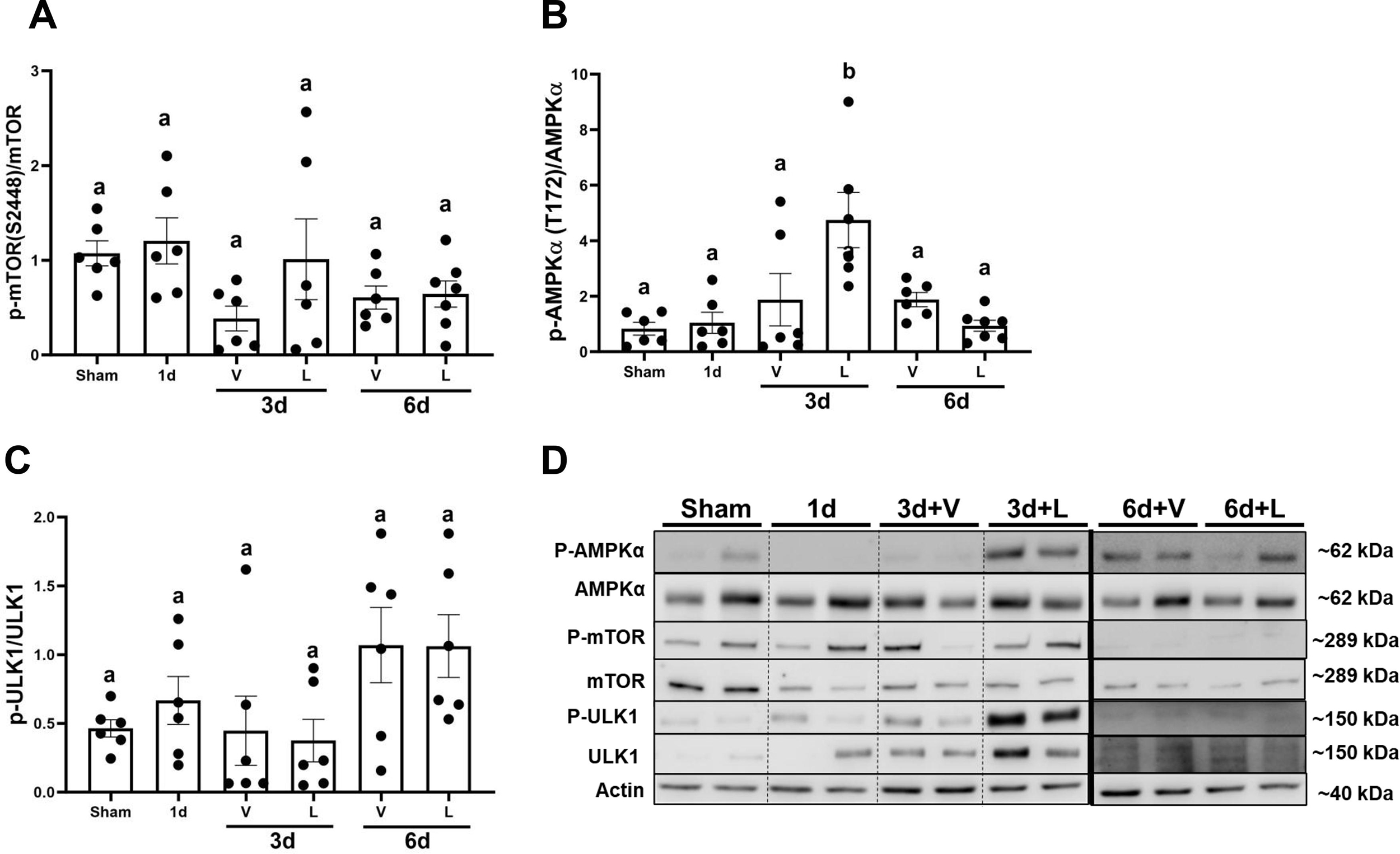
Densitometry analysis of the following genes (A) AMPKα (T172) (B) mTOR (S2448). (C) ULK1 (S555). (D) Representative immunoblots. Data represents n=6 and are expressed as means ± SEM; p<0.05. Data represents n=6 and are expressed as means ± SEM; p<0.05. Statistical significance was determined by two-way ANOVA followed by a Tukey’s post-hoc test. Different letters on top of bars represent different statistical significance.
4. Discussion
We measured the effects of lasmiditan on mitochondrial quality control mechanisms, specifically MB, fusion/fission and mitophagy in an I/R-induced AKI mice model. Previous work from our laboratory indicated that daily administration of lasmiditan, a selective 5-HT1F receptor agonist, beginning 24 h post-I/R injury accelerated renal recovery and restored levels of the master regulator of MB, Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α, and mitochondrial proteins. (32). Additionally, we have reported that loss of the 5-HT1F receptor exacerbates injury in an AKI mouse model and that lasmiditan restores endothelial cell vascular integrity (15, 43). Pharmacological activation of PGC-1α and restoration of mitochondrial homeostasis has been proposed as a novel therapeutic target for treatment of both acute and chronic diseases characterized by mitochondrial dysfunction (11, 14). However, limited pharmacological agents have been proposed to induce MB without toxicity (44–46). Repurposing lasmiditan, an FDA-approved drug, may foster the discovery of a drug safe for treating AKI.
We observed decreases in several injury markers, as well as restoration of mitochondrial morphology in the renal cortices of mice treated with lasmiditan compared to vehicle. Lasmitidan treatment decreased serum creatinine and KIM-1 levels and increased probability of survival after I/R injury compared to vehicle (Fig. 1A-E). Lasmiditan treatment increased the phosphorylation of AMPKα, resulting in increased autophagy after I/R injury (Fig. 9A).
Figure 9. Treatment with lasmiditan (L) modules mitochondrial quality controls.
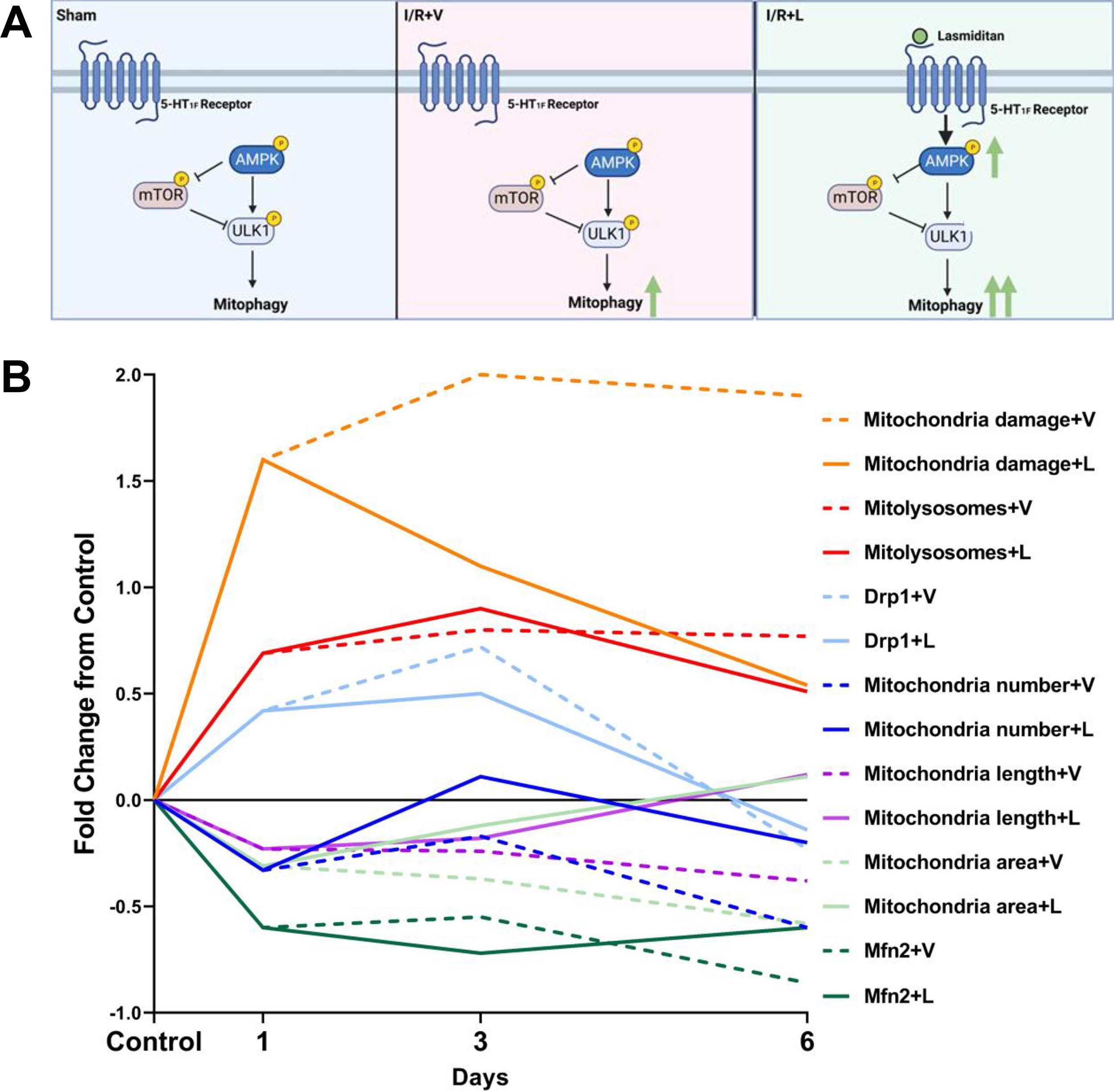
(A) Signaling pathway representation of regulation of mitophagy. (B) Analysis of fold change of mitochondrial makers after I/R injury compared to sham group. Solid lines represent lasmiditan treated group and dash lines represent vehicle group.
One day after I/R injury, mitochondria damage, mitolysosomes number, and DRP1 increased. At the same time, mitochondrial number, length, and area and Mfn2 decreased compared to control. By the third day after I/R injury, mitochondrial damage and DRP1 increased compared to 1d and mitochondria area and length remained decreased compared to 1d in the vehicle group. By the sixth day, mitochondrial damage remained elevated; mitochondrial fission marker DRP1 went back to control levels and mitochondrial fusion markers mitochondrial area and length and Mfn2 remained decreased in the vehicle group (Fig. 9B). In summary, without treatment, MB and mitochondrial fusion decreased and mitochondrial fission increased over six days after I/R.
Lasmiditan, by the third day after I/R, decreased mitochondrial damage and DRP1 and increased mitolysosomes number, mitochondrial area, and length compared to 1d and vehicle. By the sixth day after I/R injury, mitochondria damage decreased almost to control levels and mitochondrial fusion/fission markers DRP1, Mfn2, mitochondria number, area, and length were restored to control levels (Fig. 9B). In summary, with lasmiditan treatment, MB and mitochondrial fusion increased and mitochondrial fission decreased over six days after I/R.
In addition to MB, mitochondrial dynamics (fusion and fission), are key for maintenance of mitochondrial metabolism. Abnormal fission can lead to an imbalance in mitochondrial homeostasis (22). Pharmacological shifting into mitochondrial fusion and decreased mitochondrial fission might be a potential target for mitochondrial dysfunction-related diseases (47–49). Defects in mitochondrial dynamic proteins such as Mfn1, Mfn2, and Drp1 can lead to mitochondrial dysfunction, which has been shown to be associated with tubular damage at the onset stages of diseases such as AKI. (12, 50). Here, we present evidence that mitochondrial size and length are significantly reduced after I/R injury, which is consistent with other published studies (51). Importantly, lasmiditan-treated mice restored mitochondrial size and mitochondrial fusion protein levels returned to control levels.
Mitophagy has been reported to be renoprotective after AKI and it has been linked to reduced cell death, suppressed inflammation, and decreased fragmented mitochondrial in AKI models (51–53). A complication with mitophagy research is the mechanisms that orchestrate the autophagic machinery depend on the tissue, the timepoint, and the disease in question. Reports indicate that phosphorylation of AMPK at Thr-172 leads to mTOR inhibition and increased phosphorylation of ULK1 at Ser-555, followed by autophagic processes (41, 54, 55). Interestingly, we observed an increase in the same AMPKα phosphorylation site and number of mitolysosomes after injury with lasmiditan treatment compared to vehicle. This could be due to a need to rapidly degrade damaged mitochondria to prevent mitochondrial dysfunction as well as an effort to restore mitochondrial homeostasis. The acceleration of damaged mitochondria degradation in the lasmiditan treatment groups could explain fewer damaged mitochondria and increased ATP and MtDNA we observed in the I/R+L group. The occurrence of mitophagy events at earlier timepoints allows for a faster induction of fusion and MB, resulting in restoration of mitochondrial homeostasis and ultimately renal recovery.
The goal of this study was to explore the therapeutic benefits of lasmiditan treatment after AKI, specifically with regard to mitochondrial quality control mechanisms. Our data show that lasmiditan treatment after I/R surgery is beneficial starting at 3d as it decreases serum creatinine up to 6d of continuous dosing. Similarly, lasmiditan-induced MB decreased mitochondria damage score and reduced kidney injury marker serum creatinine starting at 3d. These data are similar to those reported previously using the same AKI- mice model at 6d, suggesting an earlier mechanism that facilitates recovery. This may indicate that mitochondrial fission is essential for the exclusion of damaged sections of mitochondria for its further degradation through mitophagy. Thus, our data suggest that lasmiditan has clinical importance as a treatment strategy for AKI.
New and Noteworthy Statement.
Poor clearance of damaged mitochondria, mitochondrial fission, and altered mitochondrial morphology and function have been attributed to the progression of AKI and other kidney diseases. Currently, there is no FDA-approved treatment for AKI. Our group previously reported that treatment with the FDA-approved drug, lasmiditan, accelerated renal recovery, decreased vascular leakage, decreased fibrosis, and MB induction after I/R injury. Here, we describe the effect of lasmiditan on integral mitochondrial control mechanisms such as mitochondrial fusion and fission, and mitophagy using electron microscopy in an AKI mice model.
6. Acknowledgments
We thank the University of Arizona Animal Facility for Animal Care and the University of Arizona Imaging Core – Electron (RRID: SCR_023279) members, Dr. Paula Tonino and Binh Chau for the electron microscopy sample preparation. The graphical abstract created with BioRender.com.
9. Funding
This study was funded by VA Merit Grant BX000851 and T32 ES007091.
Abbreviations
- (5-HT1F)
5-hydroxytryptamine 1F receptor
- (AKI)
Acute kidney injury
- (AMPK)
AMP-activated protein kinase
- (CKD)
Chronic kidney disease
- (DRP1)
dynamin-related protein 1
- (I/R)
Ischemia/Reperfusion
- (IP)
intraperitoneally
- (KIM-1)
Kidney injury molecule 1
- (LC3B)
light chain 3 B
- (MB)
Mitochondrial biogenesis
- (Mfn1/2)
mitofusin 1/2
- (mtDNA)
mitochondrial DNA
- (mTOR)
Mammalian target of rapamycin
- (PGC1α)
Peroxisome proliferator-activated receptor-gamma coactivator
- (SCr)
Serum creatinine
- (TFAM)
Mitochondrial transcription factor A
- (ULK1)
Unc-51 like autophagy activating kinase 1
Footnotes
Disclosures
All authors have nothing to disclose.
Conflict of interest
The authors have no conflict of interest.
Credit author statement
Conceptualization by R.G.S and K.H.; Data curation by J.J. and K.H.; Formal analysis by K.H.; Funding acquisition by R.G.S. and K.H.; Investigation by K.H.; Methodology by K.H.; Project administration by R.G.S. and K.H.; Resources by R.G.S.; Software by R.G.S and K.H.; Supervision by R.G.S.; Validation by R.G.S.; Visualization by R.G.S and K.H.; Roles/Writing original draft by K.H.; and Writing - review & editing by R.G.S and K.H..
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhatia D, Capili A, Choi ME. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res Clin Pract. 2020;39(3):244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malek M, Nematbakhsh M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Inj Prev. 2015;4(2):20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pavkov ME, Harding JL, Burrows NR. Trends in Hospitalizations for Acute Kidney Injury - United States, 2000–2014. MMWR Morb Mortal Wkly Rep. 2018;67(10):289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawhney S, Fraser SD. Epidemiology of AKI: Utilizing Large Databases to Determine the Burden of AKI. Adv Chronic Kidney Dis. 2017;24(4):194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiva N, Sharma N, Kulkarni YA, Mulay SR, Gaikwad AB. Renal ischemia/reperfusion injury: An insight on in vitro and in vivo models. Life Sci. 2020;256:117860. [DOI] [PubMed] [Google Scholar]
- 6.Birkelo BC, Pannu N, Siew ED. Overview of Diagnostic Criteria and Epidemiology of Acute Kidney Injury and Acute Kidney Disease in the Critically Ill Patient. Clin J Am Soc Nephrol. 2022;17(5):717–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cote JM, Murray PT, Rosner MH. New drugs for acute kidney injury. Curr Opin Crit Care. 2020;26(6):525–35. [DOI] [PubMed] [Google Scholar]
- 8.Pickkers P, Murray PT, Ostermann M. New drugs for acute kidney injury. Intensive Care Med. 2022;48(12):1796–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goyal A, Daneshpajouhnejad P, Hashmi MF, Bashir K. Acute Kidney Injury. StatPearls. Treasure Island (FL)2023. [PubMed] [Google Scholar]
- 10.Dong Y, Zhang Q, Wen J, Chen T, He L, Wang Y, et al. Ischemic Duration and Frequency Determines AKI-to-CKD Progression Monitored by Dynamic Changes of Tubular Biomarkers in IRI Mice. Front Physiol. 2019;10:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13(10):629–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant. 2016;31(7):1062–9. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Agborbesong E, Li X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int J Mol Sci. 2021;22(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrett SM, Whitaker RM, Beeson CC, Schnellmann RG. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther. 2014;350(2):257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbs WS, Collier JB, Morris M, Beeson CC, Megyesi J, Schnellmann RG. 5-HT(1F) receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am J Physiol Renal Physiol. 2018;315(4):F1119–F28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholpa NE, Simmons EC, Crossman JD, Schnellmann RG. Time-to-treatment window and cross-sex potential of beta(2)-adrenergic receptor-induced mitochondrial biogenesis-mediated recovery after spinal cord injury. Toxicol Appl Pharmacol. 2021;411:115366. [DOI] [PubMed] [Google Scholar]
- 17.Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol. 2016;27(11):3356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakabayashi T, Karbowski M. Structural changes of mitochondria related to apoptosis. Biol Signals Recept. 2001;10(1–2):26–56. [DOI] [PubMed] [Google Scholar]
- 19.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowak G, Bakajsova D, Samarel AM. Protein kinase C-epsilon activation induces mitochondrial dysfunction and fragmentation in renal proximal tubules. Am J Physiol Renal Physiol. 2011;301(1):F197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhatia D, Choi ME. The Emerging Role of Mitophagy in Kidney Diseases . J Life Sci (Westlake Village). 2019;1(3):13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83(4):568–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Zhu J, Liu Z, Shu S, Fu Y, Liu Y, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark AJ, Parikh SM. Targeting energy pathways in kidney disease: the roles of sirtuins, AMPK, and PGC1alpha. Kidney Int. 2021;99(4):828–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22(6):1041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14(5):880–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82(12):1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czajka A, Ajaz S, Gnudi L, Parsade CK, Jones P, Reid F, et al. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine. 2015;2(6):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger AA, Winnick A, Popovsky D, Kaneb A, Berardino K, Kaye AM, et al. Lasmiditan for the Treatment of Migraines With or Without Aura in Adults. Psychopharmacol Bull. 2020;50(4 Suppl 1):163–88. [PMC free article] [PubMed] [Google Scholar]
- 32.Hurtado KA, Janda J, Schnellmann RG. Lasmiditan promotes recovery from acute kidney injury through induction of mitochondrial biogenesis. Am J Physiol Renal Physiol. 2023;324(1):F56–F63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funk JA, Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol. 2012;302(7):F853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collier JB, Schnellmann RG. Extracellular Signal-Regulated Kinase 1/2 Regulates Mouse Kidney Injury Molecule-1 Expression Physiologically and Following Ischemic and Septic Renal Injury. J Pharmacol Exp Ther. 2017;363(3):419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam J, Katti P, Biete M, Mungai M, AshShareef S, Neikirk K, et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells. 2021;10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cong J, Chang SJ, Thomsen JS, Andreasen A, Chen X, Xing J, et al. Ultrastructural identification of developing proximal tubules based on three-dimensional reconstruction. Vet Med Sci. 2021;7(5):1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben Mosbah I, Alfany-Fernandez I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1(7):e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Zhou L, Ruan Z, Mi S, Jiang M, Li X, et al. Chlorogenic acid ameliorates intestinal mitochondrial injury by increasing antioxidant effects and activity of respiratory complexes. Biosci Biotechnol Biochem. 2016;80(5):962–71. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X, Zhang E, Ren X, Bai X, Wang D, Bai L, et al. Edaravone alleviates cell apoptosis and mitochondrial injury in ischemia-reperfusion-induced kidney injury via the JAK/STAT pathway. Biol Res. 2020;53(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayhan M, Vouillamoz J, Rodriguez DG, Bugarski M, Mitamura Y, Gschwend J, et al. Intrinsic TGF-beta signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat Commun. 2023;14(1):3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holczer M, Hajdu B, Lorincz T, Szarka A, Banhegyi G, Kapuy O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci Rep. 2020;10(1):17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma H, Guo X, Cui S, Wu Y, Zhang Y, Shen X, et al. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 2022;101(2):315–30. [DOI] [PubMed] [Google Scholar]
- 43.Dupre TV, Jenkins DP, Muise-Helmericks RC, Schnellmann RG. The 5-hydroxytryptamine receptor 1F stimulates mitochondrial biogenesis and angiogenesis in endothelial cells. Biochem Pharmacol. 2019;169:113644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297(1):H13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55(1):120–7. [PubMed] [Google Scholar]
- 46.Pagel-Langenickel I, Bao J, Joseph JJ, Schwartz DR, Mantell BS, Xu X, et al. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J Biol Chem. 2008;283(33):22464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cleveland KH, Schnellmann RG. Pharmacological Targeting of Mitochondria in Diabetic Kidney Disease. Pharmacol Rev. 2023;75(2):250–62. [DOI] [PubMed] [Google Scholar]
- 48.Cleveland KH, Schnellmann RG. The beta(2)-adrenergic receptor agonist formoterol restores mitochondrial homeostasis in glucose-induced renal proximal tubule injury through separate integrated pathways. Biochem Pharmacol. 2023;209:115436. [DOI] [PubMed] [Google Scholar]
- 49.Cleveland KH, Brosius FC 3, Schnellmann RG. Regulation of mitochondrial dynamics and energetics in the diabetic renal proximal tubule by the beta(2)-adrenergic receptor agonist formoterol. Am J Physiol Renal Physiol. 2020;319(5):F773–F9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369(23):2236–51. [DOI] [PubMed] [Google Scholar]
- 51.Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013;305(4):F495–509. [DOI] [PubMed] [Google Scholar]
- 52.Tang C, Han H, Liu Z, Liu Y, Yin L, Cai J, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019;10(9):677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sulkshane P, Ram J, Thakur A, Reis N, Kleifeld O, Glickman MH. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021;45:102047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108(12):4788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holczer M, Hajdu B, Lorincz T, Szarka A, Banhegyi G, Kapuy O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. Int J Mol Sci. 2019;20(22). [DOI] [PMC free article] [PubMed] [Google Scholar]