

Abstract
It is widely accepted that pancreatic islet β-cell failure and the onset of type 2 diabetes (T2DM) constitute an intricate interplay between the genetic expression of the disease and a host of intracellular events including increased metabolic (oxidative, endoplasmic reticulum) stress under the duress of glucolipotoxicity. Emerging evidence implicates unique roles for Caspase Recruitment Domain containing protein 9 (CARD9) in the onset of metabolic diseases, including obesity and insulin resistance. Mechanistically, CARD9 has been implicated in the regulation of p38MAPK and NFkB signaling pathways culminating in cellular dysfunction. Several regulatory factors, including B-cell lymphoma/leukemia 10 (BCL10) have been identified as modulators of CARD9 function in multiple cell types. Despite this evidence on regulatory roles of CARD9-BCL10 signalome in the onset of various pathological states, putative roles of this signaling module in islet β-cell dysfunction in metabolic stress remain less understood. This brief review is aimed at highlighting roles for CARD9 in islet β-cell function under acute (physiological insulin secretion) and long-term (cell dysfunction) exposure to glucose. Emerging roles of other signaling proteins, such as Rac1, BCL10 and MALT1 as contributors to CARD9 signaling in the islet β-cells are also reviewed. Potential avenues for future research toward the development of novel therapeutics for the prevention CARD9-BCL10-Rac1 (CBR) signalome-induced β-cell defects under metabolic stress are discussed.
Keywords: Islet β-cell, CARD9, Rac1, BCL10, MALT1, Stress kinases, Diabetes
1. Diabetes is an epidemic.
The “Diabesity” (obesity and T2DM) is the biggest epidemic in human history [1–4]. According to the International Diabetes Federation (IDF), nearly 540 million people worldwide were afflicted with diabetes in 2021, as compared to 382 million in 2019 and 317 million in 2012. Alarmingly, according to the IDF, this number might reach 643 million mark by 2030, and 783 million by 2045. Furthermore, greater than 540 million people were estimated to be glucose intolerant in 2021. The absolute global economic burden is expected to increase to $2.5 trillion in 2030, which represents a staggering increase in costs as a share of global GDP to 2.2% in 2030. These data warrant an urgent need for not only understanding the pathophysiological mechanisms underlying the metabolic dysregulation and demise of the islet β-cell, but also for increased efforts to develop novel therapeutics for halting β-cell defects and demise leading to the onset of diabetes and its associated complications .
In the chronology of events leading to the onset of T2DM, it is well established that, during the early stages of the disease, the islet β-cell works tirelessly to biosynthesize and release of excessive amounts of insulin as a compensatory response to insulin resistance in peripheral tissues. Consequential to this high insulin secretory demand, at no pre-determined time point, the islet β-cell undergoes “metabolic exhaustion” leading to relative lack of production and release of insulin culminating in hyperglycemia leading to β-cell failure and the onset of T2DM [5–8]. Existing experimental evidence further affirms that obesity, increasing age, ethnicity, and family history contribute to the onset of T2DM [9]. Despite a large number of previous and ongoing bench-to-bedside investigations in the field of pathophysiology of T2DM, putative molecular and cellular mechanisms underlying islet β-cell dysregulation and failure in T2DM remain only partially understood. Some of the known mechanisms and potential signaling mechanisms underlying the cellular events leading to β-cell failure under metabolic stress are briefly highlighted below.
2. Islet β-cell dysregulation under chronic metabolic stress
Chronic exposure of the islet β-cell to metabolic stress (e.g., elevated glucose, saturated fatty acids, such as palmitate either singly or in combination), and pro-inflammatory cytokines (e.g., IL-1β, TNFα and IFNγ) results in metabolic dysregulation and loss of functional β-cell mass. A host of signaling modules, including those involved in the induction of intracellular oxidative stress (e.g., NADPH-oxidase-derived reactive oxygen species; ROS) and endoplasmic reticulum (ER) stress have been examined and implicated in the dysfunction of the β-cell in T2DM. In this context, extant studies in human islets, rodent islets and a variety of clonal β-cells have demonstrated that a relatively poor antioxidant defense mechanism(s) renders the islet β-cell less resistant (i.e., more vulnerable) to damage under conditions of increased metabolic stress [10–13]. Earlier studies have revealed a significant increase in the intracellular ROS in β-cells exposed to metabolic stress, which, in turn, leads to activation of downstream signaling modules including stress kinase (e.g., p38MAPK, JNK1/2, p53) and mitochondrial dysregulation (defects in membrane permeability transition), and nuclear collapse (degradation of nuclear lamins) resulting in cell death [14–23]. These findings on potential roles of NADPH oxidase-derived ROS in islet dysfunction were replicated in animal models of obesity and T2DM (e.g., Zucker Diabetic Fatty rat; [24]) and inflammation and T1DM (e.g., Non Obese Diabetic mouse; [25]). It may be germane to point out that, in addition to chronic hyperglycemic and hyperlipidemic conditions, biologically-active sphingolipids (e.g., ceramides) have been shown to induce ROS-mediated metabolic stress leading to islet β-cell mass failure [26].
3. CARD9-BCL10-Rac1-MALT1 (CBRM) signalosome:
A brief description of roles of each of the members of CBRM signalosome (focus of this review) in cellular function is provided below. Caspase Recruitment Domain Containing Protein 9
(CARD9) is a scaffolding protein, which is abundantly expressed in macrophages, dendritic cells, monocytes, and neutrophils [27]. Published evidence suggests key roles for CARD9 in promoting innate immunity; its primary role is ascribed to be a signal transducer from the pattern recognition receptors localized on the cell membrane to various intracellular signaling pathways [28–31]. As will be highlighted below, CARD9 has been shown to contribute to the onset of metabolic diseases, including insulin resistance and obesity [32]. Recent evidence in animal models with CARD9 deletion demonstrated roles for CARD9 in diet-induced inflammation, obesity, and metabolic pathologies [32, 33]. B-cell lymphoma/ leukemia 10 (BCL10) is a key modulator of immune cell signaling. It has been shown to form filaments leading to aggregation into clusters for propagating signals culminating in the activation of Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) and downstream effectors, such as NFkB and JNK. Evidence is also emerging to suggest that BCL10 is functionally regulated via a variety of post-translational modifications, including phosphorylation, ubiquitination, and degradation [34]. Ras-related C3 botulinum toxin substrate 1 (Rac1) is a small G protein involved in a variety of cellular functions including cytoskeletal rearrangements, vesicular transport [35–37]. Several earlier studies in pancreatic β-cells have demonstrated key regulatory roles for Rac1 in physiological (glucose-stimulated) insulin secretion [36, 37]. Rac1 has been shown to be constitutively activated in human islets, rodent islets, and clonal β-cells following exposure to diabetogenic conditions [22, 36, 38, 39]. These findings were validated in islets derived from animal models of T2DM as well as diabetic humans [24]. Together, available evidence suggests both friendly (in physiological insulin secretion) and non-friendly (in cell dysfunction and demise) for Rac1 in islet β-cell function [40]. It is important to note that, Rac1 is a member of the cytosolic core of phagocyte-like NADPH oxidase (Nox2), activation of which leads to transient generation of ROS under conditions conducive for physiological insulin secretion. Furthermore, it has been shown that constitutive activation of Rac1 under metabolic stress conditions leads to sustained activation of Nox2 leading to increased oxidative stress in pancreatic β-cells culminating in loss in physiological insulin secretion, and subsequent dysfunction and demise of the effete β-cell [41, 42]. MALT1 is a paracaspase, which is a key component of the CBM complex. It has been implicated in the regulation of immune signaling, including tumor promotion. It has also been shown to exert dual regulatory roles in cells (e.g., lymphocytes), such as a scaffolding protein with specific roles in the functional activation of transcription factors (e.g., NFkB) as well as a regulator immune signaling and activation via proteolytic cleavage of specific substrates [43].
The following sections of this review are aimed at highlighting data accrued in studies demonstrating roles of CARD9 in cell function in general followed by emerging evidence to indicate roles of CARD9 in the onset of islet β-cell function under the duress of chronic metabolic stress.
4. Overview of roles of CARD9, and its auxiliary signaling proteins, in cell function in health and disease.
CARD9, a member of caspase recruitment domain family, has been implicated in many physiological and pathological processes including innate immune response [28–30], inflammation [27, 31], and carcinogenesis [44–46]. Emerging evidence also suggests novel roles for CARD9 in the pathology of cardiovascular [47, 48] and metabolic diseases [32]. In the context of regulatory roles of CARD9 in the onset of metabolic diseases, investigations by Cao and associates demonstrated that depletion of CARD9 ameliorates myocardial dysfunction associated with diet-induced obesity (DIO), via suppression of macrophage infiltration, inhibition of p38 MAPK phosphorylation and activation, and associated preservation of autophagy in the heart [33]. Data from the studies of Zeng and coworkers implicated significant contributory roles for CARD9 in DIO through the CARD9-MAPK pathway leading to the postulation that CARD9 knockdown might be a potential tool to improving DIO and metabolic disorders [49]. Briefly, using a CARD9 knockout animal model, these researchers have reported significantly higher insulin resistance and impaired glucose tolerance in WT animals following high fat feeding compared to those in which CARD9 is deleted [49]. Interestingly, high fat-feeding mediated increase in the expression of p38MAPK, JNK and ERK were significantly lower in the liver from CARD9 depleted animals. Based on these findings, these researchers have concluded that absence of CARD9 affords protection against DIO and related pathologies via down-regulating the CARD9-p38MAPK signaling module [49].
Recent experimental evidence identified several regulatory factors that might be involved in CARD9-mediated functions, including B-cell lymphoma/leukemia 10 (BCL10). For example, using the DIO animal model, Wang and coworkers provided compelling evidence for key regulatory roles of BCL10-CARD9-p38MAPK signaling module in obesity-related cardiac hypertrophy (ORCH) [50]. By employing a variety of complementary experimental approaches, these investigators have reported an increase in the expression of BCL10, CARD9 and p38MAPK activation in the heart in high fat-fed animals. Pharmacological inhibition of p38MAPK elicited no significant effects on the increased expression of BCL10 and CARD9. Based on these findings, these authors concluded that CARD9-BCL10 module might be upstream to p38MAPK activation. siRNA-mediated depletion of BCL10 expression in cultured cardiomyocytes inhibited saturated fatty acid (palmitate)-induced p38MAPK activation, providing further support that BCL10 regulates p38MAPK activation. It is noteworthy that supplementation of zinc rescued ORCH in these animals by suppressing the activation of BCL10-CARD9-p38MAPK signaling module, suggesting that zinc deficiency contributes to the above signaling and metabolic defects [50].
Follow-up investigations along these lines by Wang et al provided fresh insights into potential roles of zinc deficiency in ORCH. Using the DIO model fed on zinc-deficient, normal zinc and zinc supplemented diets, these investigators have reported that zinc supplementation alleviates cardiac hypertrophy in DIO mice via suppressing the p38 MAPK-dependent cardiac inflammatory and hypertrophic pathways [50]. Based on data from complementary studies, these researchers concluded that DIO and zinc deficiency synergistically induce ORCH by increasing oxidative stress-mediated activation of BCL10-CARD9-p38 MAPK signaling module [50]. A recent review by Tian and coworkers further highlighted novel roles for CARD9 in the pathology of obesity, insulin resistance and atherosclerosis [32].
In addition to BCL10, emerging evidence suggests that CARD9 can interact with mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) to form interactive protein complexes in the cells, triggering the expressions of inflammatory mediators [29, 34]. MALT1 gene encodes a caspase-like protease that plays a significant role in BCL10-induced activation of NFkB. Mechanistically, MALT1 has been shown to exert critical regulatory roles on the activation of NFkB via multiple mechanism, including tyrosine kinase receptors, G protein coupled receptors and immune receptors [51]. Experimental evidence is also suggestive that MALT1 is constitutively-activated in a variety of hematological malignancies and solid tumors culminating in NFκB target gene expression [52]. A recent review by Ruland and Hartjes highlighted regulatory regulation and signaling of CARD-BCL-MALT1 (CBM) signaling module, and their physiological roles and their pathophysiological functions in human immunodeficiency diseases, inflammatory disorders and cancers of the immune system [53]. In summary, published evidence affirms the working model that the CBM signalome regulates numerous signaling pathways (e.g., p38MAPK and NFkB) leading to the onset of multiple pathological states.
5. Modulatory roles of CARD9 in islet β-cell function in health and metabolic stress remains understudied.
Despite the aforestated advances in the field of contributory roles of CARD9 in the pathogenesis of metabolic diseases, including obesity, diabetes and insulin resistance, regulatory roles of CARD9 in islet β-cell function under acute (insulin secretion) and long-term exposure (dysfunction) to glucose remains an understudied area. Recent observations from the author’s laboratory are suggestive of novel roles for CARD9 signaling module in islet β-cell function in normal health and metabolic stress. Key findings are briefly highlighted below.
5.1. Regulatory functions of CARD9 in insulin secretion
Gamage and coworkers have recently reported that CARD9 is expressed in human islets, rat islets, mouse islets and INS-1 832/13 β-cells. siRNA-mediated knockdown of CARD9 significantly attenuated glucose-induced insulin secretion (GSIS) in insulin-secreting INS-1 832/13 cells, suggesting critical roles for this scaffolding protein in physiological insulin secretion [54]. Earlier investigations from multiple laboratories have implicated critical roles for small G proteins (e.g., Rac1) in the cascade of events leading to GSIS [36, 37, 40, 55]. Interestingly, the signaling steps involved in CARD9-mediated regulation of GSIS appear to be independent of activation of Rac1, since depletion of CARD4 exerted minimal effects on glucose-induced Rac1 activation in these cells [54]. Furthermore, insulin secretion-induced via membrane depolarization (i.e., secretion induced by KCl) or by mastoparan, a global activator of G proteins, were resistant to CARD9 knockdown in these cells. Together, these findings suggested specific roles for CARD9 in glucose-induced, Rac1-independent signaling steps leading to GSIS. Subsequent search for those signaling steps revealed key roles for CARD9 in glucose-induced p38MAPK, not ERK1/2 phosphorylation (and activation) in pancreatic β-cells leading to insulin secretion. Based on these data, it was concluded that CARD9 promotes physiological insulin secretion via a Rac1-independent and p38-dependent signaling module [54].
5.2. Regulatory functions of CARD9-BCL10-Rac1 in the induction of metabolic stress in pancreatic β-cells under diabetogenic conditions
Given the aforementioned regulatory roles of CARD9 in the pathogenesis of a variety of metabolic diseases [32], Gamage et al have recently investigated modulatory roles of CARD9 in the cascade of events leading to metabolic dysfunction of the islet β-cell under the duress of chronic hyperglycemic conditions. They noted a significant increase in the expression of CARD9 in INS-1 832/13 cells and mouse islets following exposure to gluco- and glucolipitoxic conditions. Interestingly, however, in contrast to what was observed under acute regulatory conditions [54], siRNA-mediated knockdown of CARD9 significantly attenuated sustained activation of Rac1 in pancreatic β-cells exposed to hyperglycemic conditions, implicating specific roles for CARD9 in the activation of Rac1 under these conditions. Furthermore, siRNA-mediated depletion of CARD9 markedly suppressed high glucose induced p38 MAPK, but not JNK1/2 and ERK1/2 activation [56]. Lastly, high glucose-induced Ser-536 phosphorylation of RelA, the p65 subunit of NFκB, was inhibited in CARD9-depleted pancreatic β-cells [57]. These findings have led to the postulation that CARD9 promotes p38MAPK-NFκB signaling module in the cascade of events leading to β-cell dysregulation under metabolic stress [56, 57]. Subsequent investigations (co-immunoprecipitation approaches) to further understand the mechanisms underlying CARD9-mediated metabolic dysfunction of the islet β-cell under hyperglycemic conditions, revealed a significant increase in the interaction between CARD and LyGDI, a known GDP-dissociation inhibitor for Rac1, in β-cells exposed to hyperglycemic conditions. A weakened interaction between LyGDI and Rac1 was also noted under these conditions. Based on these observations, it was concluded that hyperglycemic conditions promote association between CARD9 and LyGDI leading to dissociation of LyGDI-Rac1 complex leading to release of “free” Rac1 for activation of downstream signaling steps including p38MAPK and NFkB under these conditions [56]. Data accrued in extant investigations further validates the above findings of Gamage and coworkers [56]. For example, Jia and coworkers have demonstrated that Dectin-1 induces phosphorylation of Ras-GRF1, a known guanine nucleotide exchange factor (GEF) for Ras activation, leading to increased association of phosphorylated Ras-GRF1 with CARD9 (Ras-GRF1-CARD9 complex) to promote ERK-NFκB signaling pathway resulting in increased production of pro-inflammatory cytokines (IL-6, IL-12, IL-1β, and TNFα) [58]. Along these lines, studies by Wu et al. demonstrated increased interaction between CARD9 and GDIs (e.g., RhoGDIβ; LyGDI) in the cascade of events leading to G protein (e.g., Rac1) activation and NADPH oxidase-mediated ROS generation [59]. Together, findings from above investigations are suggestive of key modulatory roles of small G proteins (e.g., Rac1 and Ras) and their regulatory proteins/factors (e.g., LyGDI and GRF-1) in CARD9-mediated effects on cellular function. Interestingly, however, whether CARD9 exerts direct effects on putative GEFs for Rac1 leading to its activation under metabolic stress conditions remains unclear currently. Along these lines, Roth and coworkers have implicated roles for Vav, a known GEF for Rac1, in the CARD9 signaling steps leading to C-type lectin receptor-mediated antifungal host defense [60]. In the context of the islet β-cell, earlier investigations from the author’s laboratory have demonstrated critical regulatory roles for Vav2 and Tiam1, known GEFs for Rac1, in metabolic stress-induced p38MAPK activation in pancreatic β-cell under the duress of chronic hyperglycemia. Additional investigations are needed to further assess the roles of these GEFs in CARD9-Rac1 signaling in the islet β-cell. Lastly, Gamage and coworkers have also demonstrated that metabolic stress conditions significantly increased expression of BCL10 in pancreatic β-cells, and that siRNA-mediated knockdown of CARD9, markedly attenuated high glucose-induced expression of BCL10 [56]. Altogether, these findings are suggestive of a significant crosstalk between CARD9-BCL10-Rac1 signalome in high glucose-mediated effects on islet β-cell dysregulation. Based on available evidence in other cell types, and data accrued in pancreatic β-cells, we propose key roles for CARD9-BCL10-Rac1 signaling module in the cascade of events leading to β-cell dysfunction under the duress of chronic hyperglycemic conditions (Figure 1). It is proposed that metabolic stress conditions promote the expression and association of CARD9 in pancreatic β-cells with LyGDI. This, in turn, results in dissociation LyGDI-Rac1 complex under these conditions leading to sustained activation of Rac1 culminating in the induction of intracellular oxidative stress (via activation of phagocyte-like NADPH oxidase) and ER stress (via the CHOP pathway [56]. Increased metabolic stress accelerates p38MAPK and NFκB signaling pathways leading to mitochondrial dysregulation and β-cell dysfunction. Our findings also suggest increased expression of BCL10, which has been implicated in CARD9-mediated p38MAPK activation in other cell types (see above). Putative involvement of BCL10 in islet β-cell dysregulation under diabetogenic conditions needs further investigation. Lastly, potential consequences of accelerated CARD9-BCL10 signaling in promoting alterations in the subcellular distribution of Rac1 (e.g., nuclear targeting) [61] leading to β-cell dysfunction under metabolic stress remains to verified experimentally.
Figure 1: Proposed model for CARD9-mediated metabolic dysregulation of the islet β-cell under the duress of hyperglycemic stress.
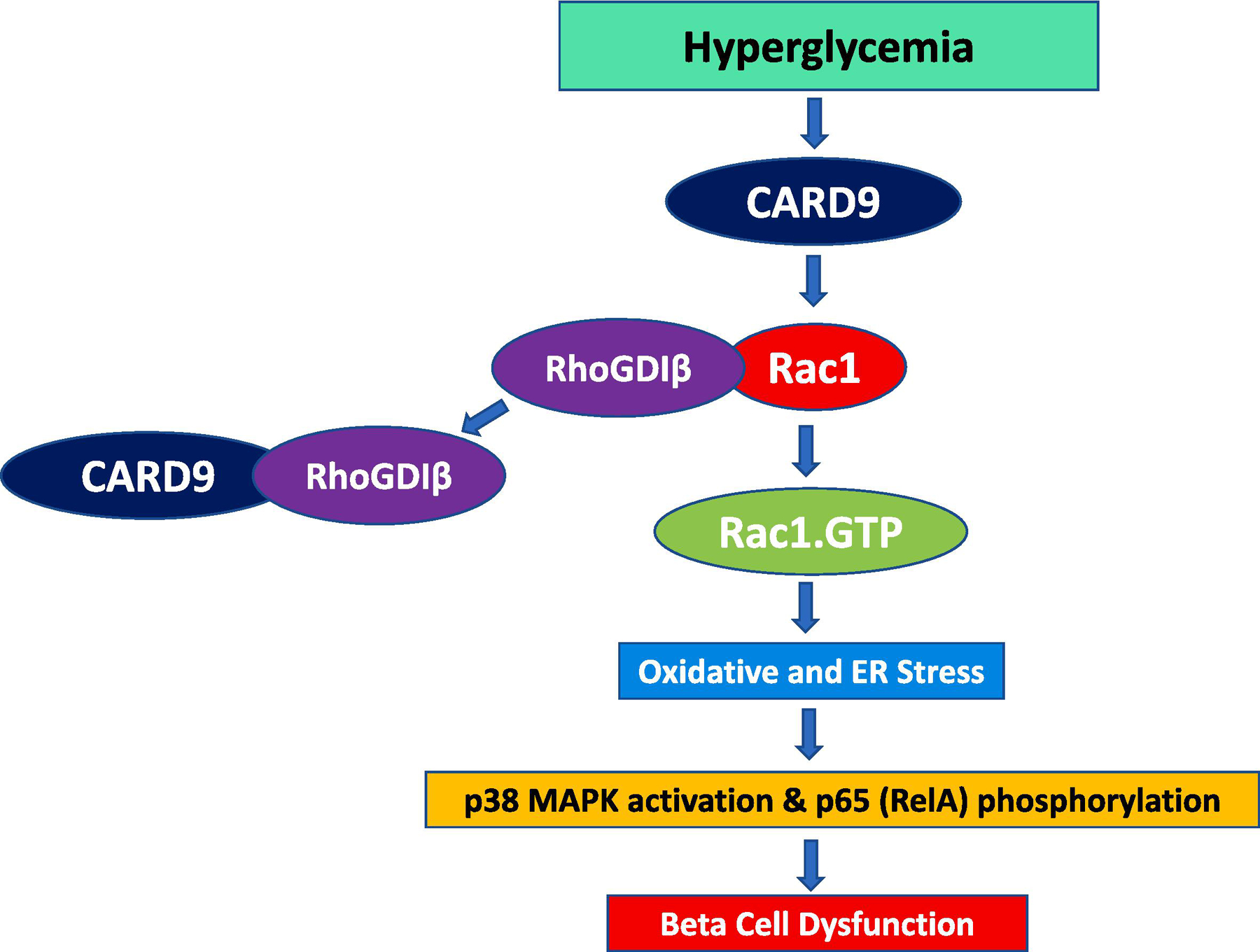
Continuous exposure of β-cells to hyperglycemic conditions promotes association between CARD9 and LyGDI and subsequent dissociation of RhoGDIβ-Rac1 complex. This, in turn, leads to the sustained activation of Rac1 and downstream signaling steps including activation of oxidative (Nox2-mediated) stress, and activation of stress kinases (p38MAPK). Data from our recent studies also demonstrated that Rac1 activation step is necessary for S536 phosphorylation of p65 (RelA), and potential translocation of p65-STAT3 complex to the nuclear fraction under these experimental conditions [57]. Collectively, increased metabolic stress, stress kinase activation and accelerated NFκB (pro-apoptotic) signaling pathway culminates in metabolic dysfunction and failure of the islet β-cell under chronic HG exposure conditions. This figure is taken, with permission from the publisher, from a recent publication from the author’s laboratory [57].
6. Does MALT1 contribute to CARD9-BCL10-Rac1 mediated islet β-cell dysregulation under metabolic stress?
As stated above, we recently reported evidence suggesting expression of CARD9 in rodent islets, human islets and clonal INS-1 832/13 cells [56] and BCL10 in INS-1 832/13 cells [56]. Composite Western blot data depicted in Figure 2 demonstrate expression of CARD9, BCL10 and MALT1 in INS-1 832/13 cells, rodent islets and human islets. To our knowledge, this is the first evidence for expression of MALT1 in insulin-secreting cells. It should be emphasized that while our earlier findings highlighted above provide evidence key regulatory roles for CARD9-BCL10-Rac1 (CBR) signaling axis in islet β-cell dysregulation under metabolic stress conditions [56], putative roles of MALT1, more specifically the CBMR signalome, in islet dysregulation remain to be investigated further. Based on the available evidence in the islet β-cell and other cell types, we propose a working model for CBMR signalome in islet dysregulation under metabolic stress conditions (Figure 3).
Figure 2: Immunological evidence for the expression of CARD9, BCL-10 and MALT-1 in INS-1 832/13 cells, rat islets and human islets:
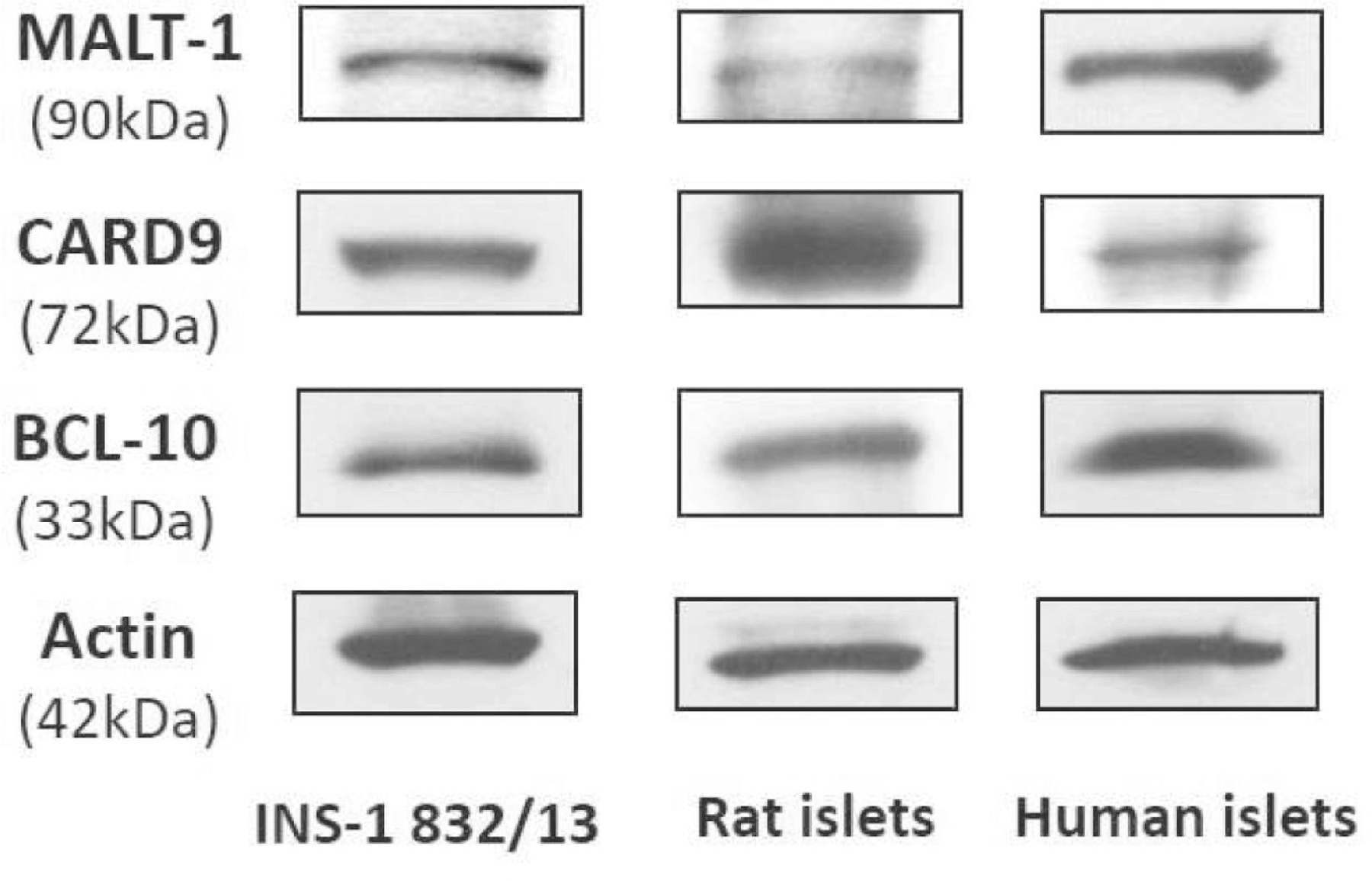
Western blot data depicting expression of CARD9, BCL-10 and MALT-1 in INS-1 832/13 cells, rat islets and human islets is shown here. Methods: INS-1 832/13 cells were propagated in RPMI-1640 medium consisting of 10% FBS supplemented with 100 IU/ ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50μM 2-mercaptoethanol, and 10 mM HEPES. Prior to a given study, cells were treated overnight with low serum/ low glucose media [79, 80]. Rat (Sprague- Dawley) islets were isolated using the collagenase digestion method as we described in [22, 81]. Human islets (from a 54-year-old Caucasian male, 75”, 172 lbs., with a BMI of 21.6, non-diabetic donor pancreas) were obtained from Prodo Labs (Aliso Viejo, CA, USA). Protocols involving use of rat and human islets received approvals from the appropriate committees at Wayne State University and the JDD VA Medical Center, Detroit. Antibody directed against MALT1 (TA807326S; 1:1,000 dilution) was from OriGene Technologies, Inc. (Rockville, MD, USA). Antibodies against CARD9 (sc-374569; 1:1,000 dilution) and BCL-10 (sc-5273 (1:500 dilution) were from Santa Cruz Biotechnology (Dallas, TX, USA). β-actin antibody (A1978) was from Sigma Aldrich (St. Louis, MO, USA) and used at 1:5,000 dilution. Anti-mouse IgG HRP-conjugated secondary antibody was from Cell Signaling Technology, Inc. (Danvers, MA, USA) used at 1:1,500 dilution. Collectively, these findings suggest expression of CARD9, BCL10 and MALT1 in a variety of insulin secreting cells, including human islets.
Figure 3: A proposed model for potential roles of CBMR signalome in the cascade of events leading to β-cell dysfunction under diabetogenic conditions.
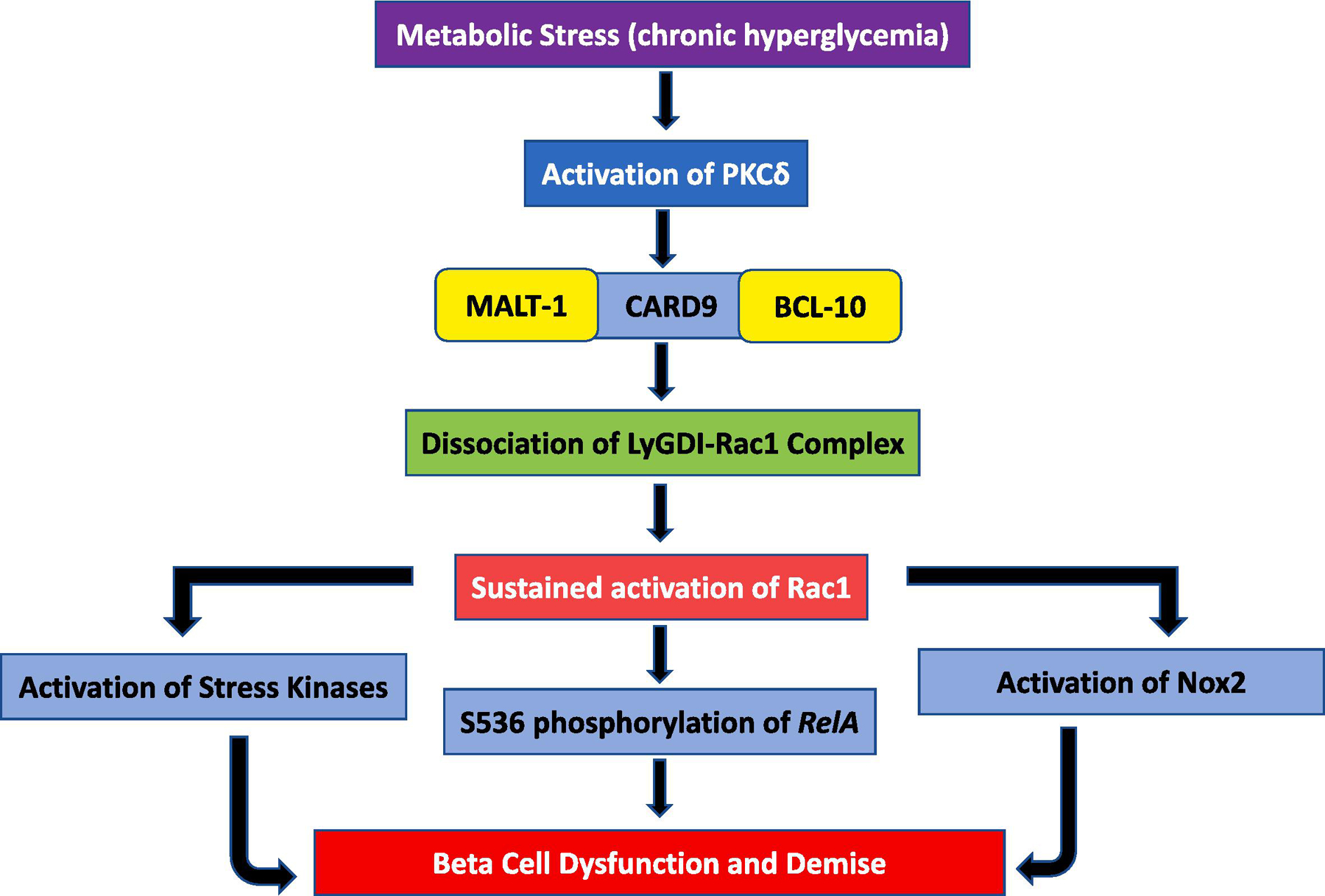
Metabolic stress (e.g., hyperglycemia, hyperlipidemia or both ) promotes functional activation of PKCδ, which, in turn, has been shown to phosphorylate CARD9 (Thr 231) culminating in functional activation of CARD9 [62]. Activation of CARD9 leads to sustained activation of several metabolic axes (NADPH oxidase, p38MAPK) culminating in the demise of the effete β-cell. More recent studies from our laboratory have also demonstrated increased S536 phosphorylation of RelA mediated via the CARD9-Rac1 signaling module in pancreatic β-cells exposed to hyperglycemic milieu; a signaling step implicated in cellular apoptosis [57]. Potential upstream regulatory roles of BCL10 and MALT1 in functional activation of NADPH oxidase, p38MAPK and NFkB in β-cells exposed to diabetogenic conditions remains to be determined.
Long-term exposure of pancreatic β-cells to metabolic stress conditions (e.g., hyperglycemia, hyperlipidemia or both) leads to functional activation of protein kinase δ (PKCδ), which, in turn, has been shown to phosphorylate CARD9 at Thr-231 culminating in functional activation of CARD9 [62]. As depicted in Figure 1, activation of CARD9 leads to dissociation of LyGDI-Rac1 complex, and increased interaction (complexation) between CARD9 and LyGDI. Dissociation of Rac1 from LyGDI promotes its activation directly through the intermediacy of a variety of guanine nucleotide exchange factors (e.g., Tiam1 and Vav2) or through yet an unidentified mechanism(s). Sustained activation of Rac1 under these conditions leads to activation of several metabolic axes including activation of stress kinases (e.g., p38 MAPK) and NADPH oxidases (e.g., Nox2) culminating in mitochondrial dysfunction and nuclear collapse and demise of the effete β-cell. Published evidence in pancreatic β-cells on CARD9-Rac1-mediated phosphorylation of RelA (at Ser-536) under the duress of hyperglycemic conditions further affirm key roles for this signaling cascade in β-cell demise [57]. In the context of pancreatic β-cell, it is likely that MALT1 (Figure 2) might work in tandem with CARD9-BCL10 in promoting downstream signaling events, including p38MAPK and NFkB activation. Lastly, the postulation that PKCδ mediates activation of Nox2 via phosphorylation and activation of CARD9 under specific experimental conditions remains to be verified in the islet β-cell. Together, we propose that CARD9-BCL10-MALT1-Rac1 signaling events might contribute to islet β-cell dysregulation under metabolic stress conditions (Figure 3). Future studies will further validate this model.
7. Translational impact of CARD9 signaling on the pathogenesis of islet β-cell defects under metabolic stress
Even though the filed is in its infancy, the author envisions a significant translational impact of these early observations on the involvement of CARD9 and the CBMR signalome islet β-cell function in health and metabolic stress. Albeit limited, recent experimental evidence affirms support to our postulation. First, using a dual systems genetic approach, Kaur and coworkers have recently reported CARD9 as one of the nine genes identified in the “T1D-T2D islet expression quantitative trait locus interaction network” in human islets. Furthermore, extended network analyses of these nine genes have identified several signaling modules that CARD9 might potentially be involved, including CLR, CLEC7A (Dectin-1), NLR, immune system and NOD1/2 signaling pathways. It should be noted that Dectin 1 and Dectin 2 have been shown to be upstream to CARD signaling pathway [27], and investigations aiming to understand regulatory roles of Dectin-CARD9 signaling in the onset of islet β-cell dysregulation will shed much needed insights in this field. Indeed, earlier studies have suggested roles for toll like receptors (e.g., TLR2) and Dectin-1 in the induction of immune response and prevention of type 1 diabetes [63–65]. Studies of Castoldi and coworkers have shown critical regulatory roles Dectin-1 in adipose tissue inflammation in obesity and insulin resistance [66]. Compatible with these conclusions, studies of Al Madhoun and coworkers have proposed Dectin-1 as a biomarker of metabolic inflammation in obesity [67]. Lastly, Ren and coworkers have demonstrated prolongation of murine islet allograft survival following inhibition of Dectin-1 on dendritic cells [68]. The reader is referred to a recent review by Kalia et al highlighting potential contributory roles of Dectin-1 in pathology of various diseases [69]. Second, given the regulatory roles of CARD9 in the regulation of sustained activation of Rac1 via dissociation of LyGDI-Rac1 complex to promote functional activation of “free” Rac1 under metabolic stress conditions makes CARD9-Rac1 signaling pathway more critical for the pathogenesis of islet dysfunction under metabolic stress conditions. We envision that this might be at the level of promoting activation of Nox2 and associated generation of ROS leading to modulation of downstream signaling steps, including activation of stress kinases culminating in mitochondrial and nuclear dysregulation leading to β-cell failure.
It is noteworthy that, in the context of regulatory roles Rac1-induced, NADPH oxidase mediated production of ROS and oxidative stress contributing to the pathogenesis of T2DM, Azarova et al examined whether single-nucleotide polymorphisms (SNP) at the RAC1 gene, a member of NADPH oxidase holoenzyme, are associated with the risk of T2DM, glucose metabolism and redox homeostasis [70]. These researchers genotyped DNA samples from 1579 T2DM patients and 1627 controls were for six common SNPs and reported that the SNP rs7784465 was associated with an increased risk of T2DM. Interestingly, these investigations revealed associations of Rac1 polymorphisms with T2D were modified by environmental factors (e.g., sedentary lifestyle, psychological stresses etc.). Based on these observations, the authors concluded that polymorphisms in the Rac1 gene represent novel genetic markers of T2DM, and their link with glucose homeostasis and the pathogenesis of T2DM, potentially associated with the changes in redox homeostasis [70].
8. Conclusions and future directions to further affirm roles of CBMR signalome in islet β-cell. dysregulation under metabolic stress
A growing body of evidence affirms novel roles for the CBM signaling module in the pathogenesis of immune and cardiac disorders. Mechanistic evidence implicates activation of p38MAPK and NFkB might represent the downstream signaling steps, leading to cellular dysfunction and the onset of pathology. Studies in CARD9 knockout animal models have yielded a wealth of information on critical roles of this scaffolding protein in the pathogenesis of metabolic disorders, including obesity and insulin resistance. Emerging evidence for the roles of CARD9 in islet β-cell function in health and metabolic stress is encouraging. Additional studies are needed, however, to further validate models that we highlighted in this review (Figures 1 and 2). Potential areas for future research in conclusively demonstrate regulation of β-cell (dys)function by the CBRM signalome. They are briefly highlighted below.
First, it will be interesting to further validate the roles of CARD9 in animal models in which CARD9 is conditionally deleted in the pancreatic β-cell. These investigations are critical since CARD9 has been implicated in a variety of metabolic disorders, including obesity and insulin resistance [32]. Second, it would be worthwhile to design small molecule inhibitors for each of these signaling proteins in the CBM complex to further assess their regulatory roles in cell dysfunction, including the islet β-cell. Original investigations by Leshchiner et al. have identified BRD5529 as a selective inhibitor of CARD9 activation. These researchers have shown that BRD5529 binds to CARD9, thereby preventing interaction between CARD9 and TRIM62, a known E3 ubiquitin ligase for CARD9, resulting in functional inactivation of CARD9 [71]. Recent investigations from the author’s laboratory have demonstrated no significant effects of BRD5529 on GSIS [54], or on high glucose-induced p38MAPK activation in pancreatic β-cells [56]. Based on these data, we concluded that CARD9-TRIM62 signaling pathway may not be involved in CARD9 mediated effects on β-cells under acute or chronic exposure to high glucose conditions. However, additional investigations may be necessary not only to validate these observations, but also develop specific inhibitors for the CBM signaling module to further assess its roles in islet β-cell function in health and metabolic stress. Martin et al have described the results of a series of toxicology studies in rat and dog species using MLT-943, a novel potent and selective MALT1 protease inhibitor [72]. Testing of small molecule compounds, such as MLT-943 might prove useful to further evaluate roles of MALT1 protease in the pathogenesis of islet dysregulation under metabolic stress. Third, as highlighted above, evidence in other cell types suggest critical roles for this signaling protein in the functional regulation of p38MAPK and NFkB. Future investigations might reveal regulatory roles of MALT1 in promoting specific signaling steps that might be involved in islet β-cell dysregulation in models of impaired insulin secretion and diabetes. Fourth, published evidence suggests critical regulatory roles for phosphorylation of CARD9 represents a key signaling step in its functional regulation. For example, it has been shown that Dectins (Dectin-1 and Dectin-2) elicit direct regulatory effects via Syk-phospholipase Cγ-mediated effects on PKCδ, which, in turn, phosphorylates CARD9 at Thr-234 leading to its functional activation [62, 73] Despite this evidence, potential involvement of PKCδ in the regulation of CARD9 signaling module in the pancreatic islet β-cell remains to be verified. It may be germane to point out that, extant data from multiple laboratories implicates PKCδ in the onset of dysregulation of the islet β-cell under a variety of metabolic stress conditions (e.g., exposure to saturated fatty acids and proinflammatory cytokines). Interestingly, however, data from studies of Welters and Morgan have suggested that activation of PKCδ is not required for palmitate-induced cytotoxicity in insulin-secreting in [74]. In addition to the aforestated in vitro observations, earlier investigations in in vivo model systems have also highlighted contributory roles of PKCδ in pancreatic islet β-cell dysregulation. For example, studies by Hennige and coworkers have demonstrated that overexpression of kinase-negative PKCδ in pancreatic β-cells protects mice from diet-induced glucose intolerance and β-cell dysfunction [75]. Cantley and Biden reported that deletion of PKCδ in mice modulates stability of inflammatory genes and protects against cytokine-stimulated β-cell death in vitro and in vivo [76]. Taken together, data from these investigations lend support for contributory roles for PKCδ in islet β-cell dysregulation. While phosphorylation of CARD9 at Thr-234 leads to its activation, phosphorylation at T531/T533 has been shown to attenuate CARD9 function [27]. Taken together, potential roles of CARD9 phosphorylation (and its associated signaling proteins) functions in the islet β-cell under metabolic stress remains a fertile area of investigation. Lastly, it will be interesting to investigate signaling mechanisms/ pathways involved in increased expression of CARD9 and BCL10 in pancreatic β-cells chronically exposed to gluco- and glucolipotoxic conditions. Potential regulatory roles of these conditions on transcriptional and/or epigenetic pathways leading to increased expression of these proteins need further examination. Along these lines, recent investigations have demonstrated transcriptional regulation of Rac1 by high glucose in retinal endothelial cells [77, 78]. Therefore, it will be interesting to undertake methodical investigations on transcriptional/epigenetic regulation of CARD9 expression in pancreatic β-cell exposed to diabetogenic conditions.
9. Acknowledgements
The author wishes to thank past and current members of his laboratory, specifically, Dr. Suhadinie Gamage and Ms. Mirabela Hali for their contributions to the field of CARD9’s roles in islet β-cell function in health and metabolic stress. The research work highlighted in this review is supported by Merit Review (I01 BX004663) and Senior Research Career Scientist (IK6 BX005383) awards from the US Department of VA and an R01 grant from the NIH/NEI (EY022230). The author would like to thank the Office of the President of Wayne State University for the Distinguished Professorship.
Abbreviations used are:
- BCL-10
B-cell lymphoma/ leukemia 10
- BRD5529
A known inhibitor of CARD9-E3 ubiquitin ligase
- CARD9
Caspase Recruitment Domain Containing Protein 9
- CLR
C-type lectin receptor
- DIO
Diet-induced obesity
- ERK
Extracellular signal-regulated kinase
- ER stress
Endoplasmic reticulum stress
- GDI
Guanine nucleotide dissociation inhibitor
- GEF
Guanine nucleotide exchange factor
- GSIS
Glucose-stimulated insulin secretion
- LyGDI
Rho-GDP dissociation inhibitor-β
- MALT-1
Mucosa-associated lymphoid tissue lymphoma translocation protein 1
- Nox2
Phagocyte-like NADPH oxidase
- ORCH
Obesity-related cardiac hypertrophy
- p38MAPK
p38 mitogen-activated protein kinase
- PKCδ
Protein kinase Cδ
- PPRs
Pattern recognition receptors
- Rac1
Ras-related C3 botulinum toxin substrate 1
- RelA
ReLA Proto-Oncogene NF-KB Subunit
- ROS
Reactive oxygen species
- S536
Serine-536
- siRNA
Small interfering RNA
- SNP
Single-nucleotide polymorphisms
- T2DM
Type 2 diabetes mellitus
- Tiam1
T-lymphoma invasion and metastasis-inducing protein 1
- TRIM62
Tripartite motif containing 62
- Vav2
Vav guanine nucleotide exchange factor 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Michaelidou M, Pappachan JM, Jeeyavudeen MS, Management of diabesity: Current concepts, World J Diabetes 14(4) (2023) 396–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Farag YM, Gaballa MR, Diabesity: an overview of a rising epidemic, Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 26(1) (2011) 28–35. [DOI] [PubMed] [Google Scholar]
- [3].Ortega MA, Fraile-Martínez O, Naya I, García-Honduvilla N, Álvarez-Mon M, Buján J, Asúnsolo Á, de la Torre B, Type 2 Diabetes Mellitus Associated with Obesity (Diabesity). The Central Role of Gut Microbiota and Its Translational Applications, Nutrients 12(9) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kumar A, Gangwar R, Ahmad Zargar A, Kumar R, Sharma A, Prevalence of diabetes in India: A review of IDF Diabetes Atlas 10th edition, Current diabetes reviews (2023). [DOI] [PubMed]
- [5].Prentki M, Peyot ML, Masiello P, Madiraju SRM, Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell, Diabetes 69(3) (2020) 279–290. [DOI] [PubMed] [Google Scholar]
- [6].Wysham C, Shubrook J, Beta-cell failure in type 2 diabetes: mechanisms, markers, and clinical implications, Postgrad Med 132(8) (2020) 676–686. [DOI] [PubMed] [Google Scholar]
- [7].Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, Reinecke M, Mechanisms of beta-cell death in type 2 diabetes, Diabetes 54 Suppl 2 (2005) S108–13. [DOI] [PubMed] [Google Scholar]
- [8].Esser N, Utzschneider KM, Kahn SE, Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia, Diabetologia 63(10) (2020) 2007–2021. [DOI] [PubMed] [Google Scholar]
- [9].Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, Martín C, Pathophysiology of Type 2 Diabetes Mellitus, Int J Mol Sci 21(17) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Acharya JD, Ghaskadbi SS, Islets and their antioxidant defense, Islets 2(4) (2010) 225–35. [DOI] [PubMed] [Google Scholar]
- [11].Lenzen S, Drinkgern J, Tiedge M, Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues, Free Radic Biol Med 20(3) (1996) 463–6. [DOI] [PubMed] [Google Scholar]
- [12].Lenzen S, Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic β-cells, Biochim Biophys Acta Gen Subj 1861(8) (2017) 1929–1942. [DOI] [PubMed] [Google Scholar]
- [13].Kowluru A, Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns, Metabolites 10(12) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kulkarni A, Muralidharan C, May SC, Tersey SA, Mirmira RG, Inside the β Cell: Molecular Stress Response Pathways in Diabetes Pathogenesis, Endocrinology 164(1) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mukherjee N, Lin L, Contreras CJ, Templin AT, β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances, Metabolites 11(11) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lytrivi M, Castell AL, Poitout V, Cnop M, Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes, J Mol Biol 432(5) (2020) 1514–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Swisa A, Glaser B, Dor Y, Metabolic Stress and Compromised Identity of Pancreatic Beta Cells, Front Genet 8 (2017) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krause M, Bock PM, Takahashi HK, Homem De Bittencourt PI Jr., Newsholme P, The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes, Clinical science (London, England : 1979) 128(11) (2015) 789–803. [DOI] [PubMed] [Google Scholar]
- [19].Montane J, Cadavez L, Novials A, Stress and the inflammatory process: a major cause of pancreatic cell death in type 2 diabetes, Diabetes, metabolic syndrome and obesity : targets and therapy 7 (2014) 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Potter KJ, Westwell-Roper CY, Klimek-Abercrombie AM, Warnock GL, Verchere CB, Death and dysfunction of transplanted β-cells: lessons learned from type 2 diabetes?, Diabetes 63(1) (2014) 12–9. [DOI] [PubMed] [Google Scholar]
- [21].Poitout V, Robertson RP, Glucolipotoxicity: fuel excess and beta-cell dysfunction, Endocr Rev 29(3) (2008) 351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sidarala V, Kowluru A, Exposure to chronic hyperglycemic conditions results in Ras-related C3 botulinum toxin substrate 1 (Rac1)-mediated activation of p53 and ATM kinase in pancreatic beta-cells, Apoptosis 22(5) (2017) 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khadija SG, Chen F, Hadden T, Commissaris RL, Kowluru A, Biology and Regulatory Roles of Nuclear Lamins in Cellular Function and Dysfunction, Recent Pat Endocr Metab Immune Drug Discov 9(2) (2015) 111–20. [DOI] [PubMed] [Google Scholar]
- [24].Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, Kowluru A, Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet, Diabetes 60(11) (2011) 2843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Veluthakal R, Sidarala V, Kowluru A, NSC23766, a Known Inhibitor of Tiam1-Rac1 Signaling Module, Prevents the Onset of Type 1 Diabetes in the NOD Mouse Model, Cell Physiol Biochem 39(2) (2016) 760–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kowluru A, Kowluru RA, RACking up ceramide-induced islet beta-cell dysfunction, Biochem Pharmacol 154 (2018) 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu X, Jiang B, Hao H, Liu Z, CARD9 Signaling, Inflammation, and Diseases, Front Immunol 13 (2022) 880879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ruland J, CARD9 signaling in the innate immune response, Ann N Y Acad Sci 1143 (2008) 35–44. [DOI] [PubMed] [Google Scholar]
- [29].Roth S, Ruland J, Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation, Trends in immunology 34(6) (2013) 243–50. [DOI] [PubMed] [Google Scholar]
- [30].Zhong X, Chen B, Yang L, Yang Z, Molecular and physiological roles of the adaptor protein CARD9 in immunity, Cell Death Dis 9(2) (2018) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luo P, Yang Z, Chen B, Zhong X, The multifaceted role of CARD9 in inflammatory bowel disease, J Cell Mol Med 24(1) (2020) 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tian C, Tuo YL, Lu Y, Xu CR, Xiang M, The Role of CARD9 in Metabolic Diseases, Curr Med Sci 40(2) (2020) 199–205. [DOI] [PubMed] [Google Scholar]
- [33].Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, Lin X, Nair S, Ren J, He G, CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity, J Mol Cell Cardiol 92 (2016) 185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gehring T, Seeholzer T, Krappmann D, BCL10 - Bridging CARDs to Immune Activation, Front Immunol 9 (2018) 1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kowluru A, Small G proteins in islet beta-cell function, Endocr Rev 31(1) (2010) 52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kowluru A, GPCRs G Proteins, and Their Impact on β-cell Function, Compr Physiol 10(2) (2020) 453–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Veluthakal R, Thurmond DC, Emerging Roles of Small GTPases in Islet β-Cell Function, Cells 10(6) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kowluru A, Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dysfunction, Biochem Pharmacol 132 (2017) 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kowluru A, Roles of GTP and Rho GTPases in pancreatic islet beta cell function and dysfunction, Small GTPases (2020) 1–13. [DOI] [PMC free article] [PubMed]
- [40].Kowluru A, Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches, Biochem Pharmacol 81(8) (2011) 965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kowluru A, Kowluru RA, Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes, Biochem Pharmacol 88(3) (2014) 275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Newsholme P, Morgan D, Rebelato E, Oliveira-Emilio HC, Procopio J, Curi R, Carpinelli A, Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell, Diabetologia 52(12) (2009) 2489–98. [DOI] [PubMed] [Google Scholar]
- [43].O’Neill TJ, Tofaute MJ, Krappmann D, Function and targeting of MALT1 paracaspase in cancer, Cancer treatment reviews 117 (2023) 102568. [DOI] [PubMed] [Google Scholar]
- [44].Zhong X, Chen B, Yang L, Yang Z, Card9 as a critical regulator of tumor development, Cancer Lett 451 (2019) 150–155. [DOI] [PubMed] [Google Scholar]
- [45].Zhong X, Chen B, Liu M, Yang Z, The Role of Adaptor Protein CARD9 in Colitis-Associated Cancer, Molecular therapy oncolytics 15 (2019) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sheng R, Yang Z, CARD9 as a potential therapeutic target in lung cancer, Front Biosci (Landmark Ed) 26(12) (2021) 1621–1626. [DOI] [PubMed] [Google Scholar]
- [47].Peterson MR, Haller SE, Ren J, Nair S, He G, CARD9 as a potential target in cardiovascular disease, Drug Des Devel Ther 10 (2016) 3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang Y, Vandestienne M, Lavillegrand JR, Joffre J, Santos-Zas I, Lavelle A, Zhong X, Le Goff W, Guérin M, Al-Rifai R, Laurans L, Bruneval P, Guérin C, Diedisheim M, Migaud M, Puel A, Lanternier F, Casanova JL, Cochain C, Zernecke A, Saliba AE, Mokry M, Silvestre JS, Tedgui A, Mallat Z, Taleb S, Lenoir O, Vindis C, Camus SM, Sokol H, Ait-Oufella H, Genetic inhibition of CARD9 accelerates the development of atherosclerosis in mice through CD36 dependent-defective autophagy, Nat Commun 14(1) (2023) 4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zeng X, Du X, Zhang J, Jiang S, Liu J, Xie Y, Shan W, He G, Sun Q, Zhao J, The essential function of CARD9 in diet-induced inflammation and metabolic disorders in mice, J Cell Mol Med 22(6) (2018) 2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang S, Gu J, Xu Z, Zhang Z, Bai T, Xu J, Cai J, Barnes G, Liu QJ, Freedman JH, Wang Y, Liu Q, Zheng Y, Cai L, Zinc rescues obesity-induced cardiac hypertrophy via stimulating metallothionein to suppress oxidative stress-activated BCL10/CARD9/p38 MAPK pathway, J Cell Mol Med 21(6) (2017) 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gomez Solsona B, Schmitt A, Schulze-Osthoff K, Hailfinger S, The Paracaspase MALT1 in Cancer, Biomedicines 10(2) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Imbert V, Peyron JF, NF-κB in Hematological Malignancies, Biomedicines 5(2) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ruland J, Hartjes L, CARD-BCL-10-MALT1 signalling in protective and pathological immunity, Nat Rev Immunol 19(2) (2019) 118–134. [DOI] [PubMed] [Google Scholar]
- [54].Gamage S, Hali M, Kowluru A, CARD9 mediates glucose-stimulated insulin secretion in pancreatic beta cells, Biochem Pharmacol 192 (2021) 114670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Asahara S, Shibutani Y, Teruyama K, Inoue HY, Kawada Y, Etoh H, Matsuda T, Kimura-Koyanagi M, Hashimoto N, Sakahara M, Fujimoto W, Takahashi H, Ueda S, Hosooka T, Satoh T, Inoue H, Matsumoto M, Aiba A, Kasuga M, Kido Y, Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin, Diabetologia 56(5) (2013) 1088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gamage S, Hali M, Chen F, Kowluru A, CARD9 Mediates Pancreatic Islet Beta-Cell Dysfunction Under the Duress of Hyperglycemic Stress, Cell Physiol Biochem 56(2) (2022) 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kowluru A, Gamage S, Hali M, Gleason N, Hyperglycemic Conditions Promote Rac1-Mediated Serine536 Phosphorylation of p65 Subunit of NFκB (RelA) in Pancreatic Beta Cells, Cell Physiol Biochem 56(4) (2022) 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jia XM, Tang B, Zhu LL, Liu YH, Zhao XQ, Gorjestani S, Hsu YM, Yang L, Guan JH, Xu GT, Lin X, CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity, J Exp Med 211(11) (2014) 2307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wu W, Hsu YM, Bi L, Songyang Z, Lin X, CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex, Nat Immunol 10(11) (2009) 1208–14. [DOI] [PubMed] [Google Scholar]
- [60].Roth S, Bergmann H, Jaeger M, Yeroslaviz A, Neumann K, Koenig PA, Prazeres da Costa C, Vanes L, Kumar V, Johnson M, Menacho-Márquez M, Habermann B, Tybulewicz VL, Netea M, Bustelo XR, Ruland J, Vav Proteins Are Key Regulators of Card9 Signaling for Innate Antifungal Immunity, Cell Rep 17(10) (2016) 2572–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Baidwan S, Chekuri A, Hynds DL, Kowluru A, Glucotoxicity promotes aberrant activation and mislocalization of Ras-related C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet β-cells: reversal of such metabolic defects by metformin, Apoptosis 22(11) (2017) 1380–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP, Brombacher F, Urlaub H, Baier G, Brown GD, Leitges M, Ruland J, Syk kinase-coupled C-type lectin receptors engage protein kinase C-δ to elicit Card9 adaptor-mediated innate immunity, Immunity 36(1) (2012) 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Karumuthil-Melethil S, Gudi R, Johnson BM, Perez N, Vasu C, Fungal β-glucan, a Dectin-1 ligand, promotes protection from type 1 diabetes by inducing regulatory innate immune response, J Immunol 193(7) (2014) 3308–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Karumuthil-Melethil S, Perez N, Li R, Vasu C, Induction of innate immune response through TLR2 and dectin 1 prevents type 1 diabetes, J Immunol 181(12) (2008) 8323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Karumuthil-Melethil S, Sofi MH, Gudi R, Johnson BM, Perez N, Vasu C, TLR2- and Dectin 1-associated innate immune response modulates T-cell response to pancreatic β-cell antigen and prevents type 1 diabetes, Diabetes 64(4) (2015) 1341–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Castoldi A, Andrade-Oliveira V, Aguiar CF, Amano MT, Lee J, Miyagi MT, Latância MT, Braga TT, da Silva MB, Ignácio A, Carola Correia Lima JD, Loures FV, Albuquerque JAT, Macêdo MB, Almeida RR, Gaiarsa JW, Luévano-Martínez LA, Belchior T, Hiyane MI, Brown GD, Mori MA, Hoffmann C, Seelaender M, Festuccia WT, Moraes-Vieira PM, Câmara NOS, Dectin-1 Activation Exacerbates Obesity and Insulin Resistance in the Absence of MyD88, Cell Rep 19(11) (2017) 2272–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Al Madhoun A, Kochumon S, Al-Rashed F, Sindhu S, Thomas R, Miranda L, Al-Mulla F, Ahmad R, Dectin-1 as a Potential Inflammatory Biomarker for Metabolic Inflammation in Adipose Tissue of Individuals with Obesity, Cells 11(18) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ren A, Li Z, Zhang X, Deng R, Ma Y, Inhibition of Dectin-1 on Dendritic Cells Prevents Maturation and Prolongs Murine Islet Allograft Survival, Journal of inflammation research 14 (2021) 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kalia N, Singh J, Kaur M, The role of dectin-1 in health and disease, Immunobiology 226(2) (2021) 152071. [DOI] [PubMed] [Google Scholar]
- [70].Azarova I, Klyosova E, Polonikov A, Association between RAC1 gene variation, redox homeostasis and type 2 diabetes mellitus, Eur J Clin Invest 52(8) (2022) e13792. [DOI] [PubMed] [Google Scholar]
- [71].Leshchiner ES, Rush JS, Durney MA, Cao Z, Dančík V, Chittick B, Wu H, Petrone A, Bittker JA, Phillips A, Perez JR, Shamji AF, Kaushik VK, Daly MJ, Graham DB, Schreiber SL, Xavier RJ, Small-molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease, Proc Natl Acad Sci U S A 114(43) (2017) 11392–11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Martin K, Junker U, Tritto E, Sutter E, Rubic-Schneider T, Morgan H, Niwa S, Li J, Schlapbach A, Walker D, Bigaud M, Beerli C, Littlewood-Evans A, Rudolph B, Laisney M, Ledieu D, Beltz K, Quancard J, Bornancin F, Zamurovic Ribrioux N, Calzascia T, Pharmacological Inhibition of MALT1 Protease Leads to a Progressive IPEX-Like Pathology, Front Immunol 11 (2020) 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Staal J, Driege Y, Haegman M, Kreike M, Iliaki S, Vanneste D, Lork M, Afonina IS, Braun H, Beyaert R, Defining the combinatorial space of PKC::CARD-CC signal transduction nodes, Febs j 288(5) (2021) 1630–1647. [DOI] [PubMed] [Google Scholar]
- [74].Welters HJ, Smith SA, Tadayyon M, Scarpello JH, Morgan NG, Evidence that protein kinase Cdelta is not required for palmitate-induced cytotoxicity in BRIN-BD11 beta-cells, J Mol Endocrinol 32(1) (2004) 227–35. [DOI] [PubMed] [Google Scholar]
- [75].Hennige AM, Ranta F, Heinzelmann I, Düfer M, Michael D, Braumüller H, Lutz SZ, Lammers R, Drews G, Bosch F, Häring HU, Ullrich S, Overexpression of kinase-negative protein kinase Cdelta in pancreatic beta-cells protects mice from diet-induced glucose intolerance and beta-cell dysfunction, Diabetes 59(1) (2010) 119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cantley J, Boslem E, Laybutt DR, Cordery DV, Pearson G, Carpenter L, Leitges M, Biden TJ, Deletion of protein kinase Cδ in mice modulates stability of inflammatory genes and protects against cytokine-stimulated beta cell death in vitro and in vivo, Diabetologia 54(2) (2011) 380–9. [DOI] [PubMed] [Google Scholar]
- [77].Kowluru RA, Radhakrishnan R, Mohammad G, Regulation of Rac1 transcription by histone and DNA methylation in diabetic retinopathy, Sci Rep 11(1) (2021) 14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kowluru RA, Mishra M, Kumar B, Diabetic retinopathy and transcriptional regulation of a small molecular weight G-Protein, Rac1, Exp Eye Res 147 (2016) 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hali M, Wadzinski BE, Kowluru A, Alpha4 contributes to the dysfunction of the pancreatic beta cell under metabolic stress, Mol Cell Endocrinol 557 (2022) 111754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Thamilselvan V, Kowluru A, Paradoxical regulation of glucose-induced Rac1 activation and insulin secretion by RhoGDIbeta in pancreatic beta-cells, Small GTPases (2019) 1–8. [DOI] [PMC free article] [PubMed]
- [81].Sidarala V, Veluthakal R, Syeda K, Vlaar C, Newsholme P, Kowluru A, Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1), Biochem Pharmacol 95(4) (2015) 301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]