

Abstract
Stroke is the leading cause of death and disability worldwide. Novel and effective therapies for ischemic stroke are urgently needed. Here, we report that melatonin receptor 1A (MT1) agonist ramelteon is a neuroprotective drug candidate demonstrated by comprehensive experimental models of ischemic stroke, including a middle cerebral artery occlusion (MCAO) mouse model of cerebral ischemia in vivo, organotypic hippocampal slice cultures ex vivo, and cultured neurons in vitro; the neuroprotective effects of ramelteon are diminished in MT1 knockout mice and MT1 knockout cultured neurons. For the first time, we report that the MT1 receptor is significantly depleted in the brain of MCAO mice and ramelteon treatment significantly recovers the brain MT1 losses in MCAO mice, which is further explained by the Connectivity Map L1000 bioinformatic analysis that shows gene-expression signatures of MCAO mice are negatively connected to melatonin receptor agonist like ramelteon; We demonstrate that ramelteon improves the cerebral blood flow (CBF) signals in ischemic stroke that is potentially mediated, at least, partly by mechanisms of activating eNOS. Our results also show that the neuroprotection of ramelteon counteracts ROS-induced oxidative stress and activates the NRF2/HO-1 pathway. Ramelteon inhibits the mitochondrial and autophagic death pathways in MCAO mice and cultured neurons, consistent with gene set enrichment analysis from a bioinformatics perspective angle. Our data suggest that ramelteon is a potential neuroprotective drug candidate and MT1 is the neuroprotective target for ischemic stroke, which provides new insights into stroke therapy. MT1 knockout mice and cultured neurons may provide the animal and cellular models of accelerated ischemic damage and neuronal cell death.
Keywords: Ischemic stroke, Ramelteon, MT1 receptor, MT1−/− mice, MT1−/− cultured neurons, MRI, CBF, p-eNOS/eNOS, Mitochondrial and autophagic death pathways, ROS, Nrf2/HO-1, Bioinformatics
Graphical Abstract

Scheme of the MT1 receptor as the target of ramelteon neuroprotection in ischemic stroke. MT1 agonist ramelteon is neuroprotective in animal and cellular experimental models of ischemic stroke. Ramelteon treatment mainly recovers the brain MT1 losses, which is further explained by the Connectivity Map L1000 bioinformatic analysis. Ramelteon inhibits mitochondrial intrinsic signaling pathway of apoptosis, autophagic death signaling pathways, oxidative stress, and inflammation in MCAO mice and cultured neurons, consistent with gene set enrichment analysis from a bioinformatics angle. Chemical structures of ramelteon and luzindole are shown.
1. INTRODUCTION
Stroke is one of the leading causes of global morbidity and mortality. Approximately 80–85% of stroke cases are ischemic stroke. Tissue plasminogen activator is the only available FDA-approved drug with limits for ischemic stroke patients. The development of novel and effective therapy for ischemic stroke is urgently needed. We and others reported the beneficial effects of melatonin and N-acetylserotonin (NAS), two full agonists of the melatonin receptors, in ischemic stroke, newborn hypoxic-ischemic brain injury, hypoxia- and/or ischemia-induced neuronal cell death1–3 and neurodegenerative diseases, Huntington’s disease (HD) and Amyotrophic Lateral Sclerosis (ALS).4, 5 We reported that melatonin-mediated neuroprotection is dependent on the presence and activation of the melatonin receptor 1A (MT1).3–5 However, melatonin has two weaknesses: a short half-life of 30 – 50 min6, 7 and multiple binding sites.8 For three decades, analogs of melatonin with a longer half-life and higher affinity for melatonin receptors have been investigated for their possibly greater therapeutic efficacy than melatonin. Ramelteon (a.k.a. Rozerem or TAK-375), a tricyclic synthetic melatonin analog, is one of the melatonergic drugs approved in 2005 by the U.S. Food and Drug Administration (FDA) for clinical use in the treatment of insomnia.9
Compared with melatonin, ramelteon has a greater affinity for the receptors and a longer half-life.10 Ramelteon has the clinical advantage with a 10-fold higher affinity for the human MT1 than the human MT2 receptors.17 The therapeutic potential of ramelteon has been investigated in Alzheimer’s disease,11, 12 Parkinson’s disease,13 traumatic brain injury,14 and importantly, in elderly patients with delirium and insomnia after acute stroke.15, 16 Wu et al. reported the potential benefits of ramelteon in the treatment of both acute and chronic ischemic brain injury by counteracting autophagic cell death via regulating AMPK/mTOR signaling pathway.18
In this study, we report the neuroprotection of ramelteon and its multiple aspects of mechanisms of action in the comprehensive experimental models of ischemic stroke, including a middle cerebral artery occlusion (MCAO) model of cerebral ischemia in vivo as well as organotypic hippocampal slice cultures (OHSCs) ex vivo and primary cerebrocortical neurons (PCNs), primary hippocampal neurons (PHNs), and HT-22 mouse hippocampal neuronal cell line (HT-22 cells) in vitro. We show that ramelteon reduces MCAO-induced infarct volume and stroke lesion volume and enhances cerebral blood flow. For the first time, we demonstrate that ramelteon improves the CBF signals in ischemic stroke that is potentially mediated, at least partly through the activation of eNOS. Furthermore, our investigation reveals that murine ischemic stroke results in the depletion of the MT1 receptors; we provide the first evidence that the neuroprotection of ramelteon is predominantly related to the activation of target MT1, which is further explained by the Connectivity Map L1000. Our results also suggest that the neuroprotection of ramelteon counteracts the mitochondrial and autophagic death and oxidative stress in experimental models of ischemic stroke.
2. MATERIALS AND METHODS
2.1. Chemicals
Ramelteon was purchased from AvaChem Scientific LLC (San Antonio, Texas). Luzindole and the 5(6)-Carboxyfluorescein diacetate (CFDA) were purchased from Sigma-Aldrich (St. Louis, MO). Rhodamine 123 (Rh 123), Tetramethylrhodamine, ethyl ester (TMRE), and propidium iodide (PI) were purchased from Life Technologies (Carlsbad, CA).
2.2. Permanent MCAO and drug treatment in wild-type and MT1 KO mice
C57BL/6 (C57) wild-type and MT1 knockout (KO) (MT1−/−) mice (5–7 weeks, 20–25 g) were housed under a natural light/dark cycle with food and water ad libitum.1–3 All experiments were conducted per the protocols approved by the Harvard Medical School Animal Care Committee. Focal cerebral ischemia was induced as we previously described1, 2. Mice were anesthetized with 2% (vol/vol) isoflurane (70% N2O/30% O2) and maintained with 1.0 to 0.5% concentrations during the entire procedure. Rectal temperature was maintained between 37.0 and 37.5°C with a heating pad. An intraluminal 7–0 nylon thread with a silicone tip (180 μm diameter) was inserted 9 ± 1.0 mm into the internal carotid artery up to the middle cerebral artery. A laser Doppler perfusion monitor (Perimed AB) was adhered to the left temporal aspect of the animal’s skull and used to confirm middle cerebral artery flow disruption. Ramelteon was prepared in normal saline containing 1.2% ethanol. “Pretreatment” took place 10 min before and 20 min after the operation by two IP injections of 10 mg/kg ramelteon. Post-treatment was the IP injection of ramelteon at doses of 10, 15, or 20 mg/kg, 1 h after the operation. Control animals were injected with equivalent volumes of saline containing 1.2% ethanol. Each animal was euthanized 24 or 72 h after surgery. The brain was removed and the blood was collected.
2.3. Determination of infarct volume
MCAO was sustained for a period of 24 h after the evaluation of the neurological score. C57 mice and MT1−/− mice were euthanized, and brains were rapidly removed, chilled for 2 minutes, and cut into coronal sections 1 mm thick. The sections were immersed in a saline solution containing 4% 2, 3, 5-triphenyltetrazolium chloride (TTC, Sigma) at 37°C for 20 minutes. After staining, each slice was scanned with a scanner. Stained areas in the ipsilateral and contralateral hemispheres were quantified with ImageJ software to calculate the infarct volume which was expressed as a percentage of the lesioned hemisphere as previously described.1, 2
2.4. MRI data acquisition
CBF by ASL and T2WI maps using 3D Slicer software
C57 mice, including the MCAO group and ramelteon treated MCAO group at 72 h after the MCAO operation, along with the control group, underwent MRI scanning under isoflurane narcosis. MRI imaging was performed on a Bruker 7 Tesla MRI scanner (Bruker Biospin). Respiration was monitored, while body temperature was maintained using a homoeothermic blanket.
The parametric map includes cerebral blood flow (CBF) performed by Arterial-spin labeling (ASL). Briefly, one slice is acquired after RF pulsing in the plane of the feeding arteries (neck) - this reduces the signal in the imaging plane proportional to the amount of blood flow. A control image is acquired after pulsing in a plane of equal distance from the imaging slice but above the imaging slice. Subtraction of the first images from the second set results in a different image that reflects the amount of blood flow. The higher the % change, the more blood flow to the region.19 Gradient-echo, echo-planar-imaging acquisition was used with 2 segments and 10 averages. CBF was obtained and analyzed by comparing the median values of the ischemic and contralateral hemispheres. The average difference was compared between different mice and translated into the relative fold-change difference. T2-weighted images (T2WI) were also acquired using the RARE pulse sequence, allowing the stroke lesions to be manually segmented. Accumulated stroke lesion volumes were quantitatively assessed with 3D slicer software (https://www.slicer.org/). The quantitative data were measured and compared, including stroke lesion volume and CBF among different groups.
CBF by LSCI
After 24 hours of MCAO, CBF was measured in the cerebral cortex supplied by MCA using laser speckle contrast image (LSCI) (SIM BFI HR Pro, SIMOPTO) in the indicated group. Mouse was anesthetized and placed in the prone position. A midline scalp incision exposed the skull, and the LSCI device was appropriately positioned above the skull’s surface. A whole-brain scan was performed by using LSCI. The random speckle pattern, which is exploited by laser speckle contrast analysis, can be generated by irradiating the tissue with laser light and varies as blood cells move in the selected area20. The LSCI system quantified regional tissue blood flow (rTBF) within the ischemic area. The degree of perfusion was represented on a spectrum from blue (low perfusion) to red (high perfusion). Subsequent quantitative data collection and analysis were executed.
2.5. Neurological score
C57 mice and MT1−/− mice were assessed for neurological deficits 24 h after the MCAO procedure. Each animal was assigned a neurological score based on the following scale as we previously described1, 2: 0, no observable deficits; 1, forelimb flexion when lifted by the tail; 2, forelimb flexion and consistently reduced resistance to lateral push; 3, forelimb flexion, reduced resistance to lateral push, and unilateral circling toward the paretic side; and 4, forelimb flexion and ambulation inability or difficulty.
2.6. Digi Gait analysis
The DigiGait automated gait analysis system (Mouse Specifics), one of the most sensitive tools for the evaluation of animal locomotion and gait impairment,21–23 was used to assess gait parameters post-ischemia. C57 mice were assessed for computerized gait analysis 2 days after the MCAO procedure ± ramelteon. The animals were trained twice before MCAO surgery. Digital video of an animal running on a treadmill set at 15 cm/second and run duration of 3 seconds was acquired with a high-speed camera. The ratio of left hind paw area CV (cm2)/right hind paw area CV (cm2) and distance difference between two hind paws from the body in horizontal midline were analyzed.
2.7. Wild-type and MT1−/− cultured neurons and induction of cell death
The cerebral cortex or hippocampus was isolated from E14 to E16 C57 wild-type mice, MT1−/− mice, or Swiss Webster mice (Charles River Laboratories). The cells were dissociated by 0.25% trypsin and cultured in poly-D-lysine-coated dishes in a neurobasal medium supplemented with 2% B27, 2 mM glutamine, 100 U/mL penicillin, and streptomycin sulfate 10,000 μg/mL. Experiments on PCNs or PHNs were performed after 5–7 days in culture. HT-22 mouse hippocampal neuronal cell line was purchased from The Salk Institute. PCNs, PHNs, or HT-22 cells were subjected to various apoptotic inducers.1, 2, 24 For Oxygen-glucose deprivation (OGD) experiment, the culture medium was replaced with glucose-free Earle’s balanced salt solution, and cells were incubated for 2 h with defined concentrations of ramelteon and/or luzindole. The cells were then placed in an anaerobic chamber with a BBL GasPak Plus (Becton Dickinson), lowering the oxygen concentration to 100 ppm within 90 minutes. After 3 h, OGD was terminated by a return to normal culture conditions. Control cells were incubated for the same length of time in Earle’s balanced salt solution with glucose in a normoxic incubator. Alternatively, PCNs, PHNs, or HT-22 cells were treated with 100 μM hydrogen peroxide (H2O2) or 0.5 mM NMDA for 18 h. Cells were pre-incubated for 2 h or post-treated 2 h with ramelteon. Cell death of PCNs and PHNs was measured by LDH assay, and HT-22 cell death was measured by MTS assay.
2.8. Organotypic hippocampal slice cultures and oxygen-glucose deprivation treatment
OHSCs were prepared from 5–9 day-old mice as we previously described.2 Pups of C57 or Swiss Webster mice were decapitated, and the brains were removed and transversely cut into 400-μm thick slices using the Vibroslice (World Precision Instruments, Inc.). Slices were transferred into ice-cold Hank’s balanced salt solution supplemented with 5 mg/ml D-Glucose and placed onto porous transparent membrane inserts (Millipore Corporation), which were prior plated into six-well tissue culture plates containing culture medium (50% Eagle’s minimal essential medium with Earle’s salt, 25% Hank’s balanced salt solution, 25% horse serum, 25 mM HEPES, 1 mM glutamine, and 1% antibiotic/antimycotic solution supplemented with 6.5 mg/ml D-Glucose). OHSCs were maintained at 37 °C in a humidified incubator with 95% air and 5% CO2 atmosphere, and the culture medium was changed every three days. Experiments were carried out after 12–14 days. OGD was induced in OHSCs with a serum-free and glucose-free medium in an airtight chamber at 37°C for 2 h. Inserts with slice cultures were rinsed and transferred into 35-mm dishes in which culture medium had been completely replaced by a serum-free and glucose-free balanced salt solution containing (in mM): NaCl, 124; KCl, 5; MgCl, 1.3; NaH2PO4, 1.25; NaHCO3, 26; CaCl2, 2 (pH 7.4). Dishes were sealed in an airtight chamber with anaerobic system envelopes with a palladium catalyst (BD BBL GasPak Plus, Becton Dickinson). The whole tank was kept in a gassed incubator at 37°C for 3 h. Control slice cultures in a balanced salt solution containing 5 mg/ml glucose were put into the incubator at 37°C for 3 h, but not exposed to anaerobic conditions. Following hypoxia and hypoglycemia, cultures were transferred to a normal serum-free medium (containing 75% MEM, 25% Hanks’ balanced salt solution, 5 mg/ml glucose) with 0.25 mg/ml PI and returned to the incubator under normoxic conditions. PI imaging was carried out 18–24 h in normal culture conditions after 3 h induction of OGD. Images were taken using a Nikon ECLIPSE TE 200 fluorescence microscope and processed by IP LAB software (Spectra Services).
2.9. Lactate dehydrogenase assay
The extent of cell death of wild-type and MT1−/− PCNs was determined by measuring the amount of lactate dehydrogenase (LDH) released into the extracellular fluid, per the manufacturer’s instructions (Roche Applied Science, IN). Briefly, the culture medium was collected 18 h or 24 h later and centrifuged at 1000 ×g for 10 min. The supernatant (100 μl) was then mixed with the reaction mixture (100 μl). The absorbance of samples at 490 nm was measured in an ELISA reader after 30 min of incubation at room temperature.
2.10. MTS assay
Cell death was determined by performing the 3-(4,5)-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt assay as described previously3, 4 and per the manufacturer’s instructions (Promega). HT-22 cells were subjected to 2 h of OGD with or without pre-incubated ramelteon for 2 hours. Then HT-22 cells were returned to normal culture conditions for 24 h. The absorbance of cells at 490 nm was measured in an ELISA reader.
2.11. Propidium iodide and DAPI staining
Cell death induced by OGD in OHSCs was detected using PI. PI (0.25 μg/mL) was added to the culture medium during both exposure to OGD and the following recovery period. Following 18–24 h incubation in normal culture conditions after 3 h of OGD, slice cultures were observed and images were analyzed. The intensity of PI fluorescence in regions of interest (CA1, CA3, and DG) was used as an index of cell death. Cell death induced by H2O2 in PCNs ± ramelteon was detected using DAPI, a nuclear counterstain. Digital images were taken with a Nikon ECLIPSE TE 200 fluorescence microscope and processed with IP LAB Software.
2.12. Determination of mitochondrial transmembrane potential
PCNs and PHNs were treated as indicated ± ramelteon and/or luzindole. Rh 123, a green-fluorescent dye, is a probe of mitochondrial membrane potential (ΔΨm) and TMRE is a red-orange dye that readily accumulates in active mitochondria. Living PCNs and PHNs were stained with 2 μM Rh 123 for 5 min at room temperature2 or 25 nM TMRE in PCNs for 10 min at RT, followed by rinsing by PBS. Reduced Rh 123 or TMRE fluorescence indicates the dissipated ΔΨm. Digital images were taken with a Nikon ECLIPSE TE 200 fluorescence microscope and processed with IP LAB Software.
2.13. Western blot
OHSCs or PCNs/PHNs were exposed to OGD or H2O2 ± ramelteon. Cells were collected in ice-cold lysis buffer (20 mM Tris, pH 8.0/137 mMNaCl/10% glycerol/1% Nonidet P-40/2 mM EDTA with 5 mM Na2VO4, a protease inhibitor mixture, supplemented with 0.2 mM phenylmethylsulfonyl fluoride). The lysate was cleared by centrifugation at 19,720 × g for 10 minutes at 4°C, and the supernatant was analyzed by Western blot (WB). Samples of mouse brains were lysed in ice-cold RIPA buffer with protease inhibitors2. Protein concentration was assayed by the Bradford dye-binding procedure. Proteins were run on SDS-PAGE and transferred to PNDF membranes. After blocking with 5% milk-Tris Buffered Saline Tween-20 for 1 h, membranes were blotted with anti-caspase-3, or anti-active caspase-3, or Beclin 1 antibodies (Cell Signaling Technology), LC3 antibody (Novus Biologicals, CO), p62 antibody (Santa Cruz Biotechnology, Inc.), melatonin receptor 1A antibody (Millipore) (The specificity of the antibody has not been verified in MT knockout mice), eNOS (ABclonal Technology), phosphorylated eNOS Ser1177 (ABclonal Technology), Nrf2 (Zenbio), HO-1 (ABclonal Technology), and β-actin antibody (Sigma). The reaction was followed by a horseradish conjugated secondary anti-mouse (or rabbit, or rat) antibody (Amersham Pharmacia Biotech, NJ), or anti-goat antibody (Santa Cruz Biotechnology, Inc.) and detected with enhanced chemiluminescence reagents by exposure to Kodak film. Densitometry was conducted using the Quantity One software (Bio-Rad).
2.14. Subcellular fractionation
Cytosolic fractionation was performed2. Briefly, OHSCs or PCNs and PHNs, treated or untreated with OGD or H2O2 in the presence or absence of ramelteon, were scraped from dishes and homogenized in a Dounce homogenizer for 15 strokes on ice in a homogenization buffer (10 mM HEPES, pH 7.4, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT plus a protease inhibitor cocktail), followed by 2000×g centrifugation for 5 min at 4 °C; the supernatant was centrifuged at 15,000×g for 25 min at 4 °C and used as the cytosolic component. Cytochrome c (Cyto. c) was analyzed by WB with anti-Cyto. c antibody (PharMingen).
2.15. Immunocytochemistry
TOM20, a mitochondrial marker, is used to track mitochondrial fragmentation of PCNs as previously described2, 3. PCNs were exposed to H2O2 or OGD ± ramelteon. Cells were fixed in 4% paraformaldehyde overnight, incubated with blocking solution (normal goat serum 1:20 [v/v]) for half an hour at RT, and anti-TOM20 (1: 300) overnight at 4 °C, and then with secondary antibody FITC conjugate (1:200) for 1 hour at room temperature, and analyzed by fluorescence microscopy. Digital images were taken. For the quantitative measurement of mitochondrial length, ImageJ (v. 1.43) software was used to set a scale per the picture pixel and size. Nano Measurers (v.1.2.5) software was used to track mitochondria and measure mitochondrial length. Mitochondria were classified into different categories as to length: < 1, 1–2, 2–3, 3–4, and ≥ 5 μm. A minimum of 200 mitochondria for each picture was counted and the percentage of each length of mitochondria in each picture was measured. The data represent the mean and SEM from three independent experiments, and p values were obtained by One Way ANOVA.
2.16. Assays of caspase activity
Cell extracts and enzyme assays were performed as previously described1, 2 and per the manufacturer’s instructions (Clontech). PCNs were exposed to H2O2 ± ramelteon. Cells were homogenized with a Dounce homogenizer in cell lysis buffer, one component of the ApoAlertCaspase 3 Assay Kit (Clontech), for 10 strokes. The protein concentration of lysate of each sample was determined and lysate samples were adjusted to have an equal concentration with the extraction buffer. The lysates were incubated with 1 μM caspase-3-like substrate Ac-DEVD-AFC. The final volume of reaction mixtures was 50 μL. Enzyme activity was determined from the fluorescence of the AMC product using a TecanGENios microplate reader (excitation at 400 nm and emission at 505 nm).
2.17. Detection of intracellular ROS
The reactive oxygen species (ROS) ELISA assay was performed as previously described,25 and per the manufacturer’s instructions (MyBioSource, Inc). PCNs were exposed to H2O2 either ± ramelteon for 18 h. Mouse ROS antibody was pre-coated onto an ELISA plate. Harvested cells were washed with and suspended in PBS. PCNs were collected by trypsinization and washed with HBSS and then trypsinized and centrifuged for 10 min at 3000 rpm. The supernatants were added into ELISA plate wells and incubated for 90 min at 37 °C in an incubator; the biotin-labeling polyclonal antibody was added to the ELISA plate and incubated for 60 min at 37 °C. Avidin-peroxidase conjugates were then added to the ELISA plate and incubated for 30 min at 37 °C. 3,3’,5,5’-Tetramethylbenzidine substrates for coloring were added to the ELISA plate for 30 min. The absorbance of PCNs samples at 450 nm was measured using a Tecan GENios Microplate Reader, and intercellular ROS (IU/ml) was measured.
2.18. Mature interleukin-1L-1β (IL-1β) Determination
PCNs were exposed to H2O2 either ± ramelteon for 18 h. Supernatants from PCN cultures were assayed and mature IL-1β quantification was measured using an ELISA kit (R&D Systems).
2.19. Measurement of mitochondrial permeability transition in isolated mouse brain mitochondria
Non-synaptosomal brain mitochondria were isolated from mice using a discontinuous Ficoll gradient and the mPTP assay was performed.2, 26 Brain mitochondria of C57 mice were incubated in the buffer containing 100 mM sucrose, 65 mM KCl, 10 mM HEPES, pH 7.4, 2 mM KH2PO4, 150 M μATP, 150 μM MgCl2, 3 μM EDTA, and 5 mM glutamate/malate at a concentration of 0.25 mg/ml mitochondria.26 Ca2+ uptake capacity, membrane potential (ΔΨ), NAD(P)H/NADH oxidation, and swelling of brain mitochondria were measured simultaneously on a multichannel dye fluorimeter (C&L Instruments, Inc.). Mitochondrial Ca2+ uptake and release capacities were measured as changes in CaGreen-5N (125 nM) fluorescence intensity at excitation and emission wavelengths of 485 and 535 nm, respectively. ΔΨm changes were estimated by measuring changes in the fluorescence intensity of TMRM (60 nM) at excitation and emission wavelengths of 543 and 590 nm, respectively. Mitochondrial swelling was measured as a function of light scattering at excitation and emission wavelengths of 587 nm or by a standard spectroscopic assay on a plate reader at 540 nm. Mitochondria were challenged with sequential Ca2+ additions, each was 25 nmolCa2+/mg mitochondrial protein. Original fluorimeter records were analyzed using OriginPro v.8.0 (OriginLab Corp.) software.
2.20. Bioinformatic analysis
Connectivity Map (CMap) L1000 platform is a valuable tool in understanding drug mechanism of action and discovery of connections between genes, drugs, and diseases,27 which was used in this study to find connections between MT1 levels and MCAO mice gene expression signatures. A heatmap was used to present the L1000 results. Gene Set Enrichment Analysis was used to analyze the roles of ramelteon based on the genes ramelteon differentially expressed.28
2.21. Statistical analysis
Group data were presented as mean values ± SEM. GraphPad InStat (GraphPad Software, Inc.) or Statview (SAS Institute Inc.) was used to analyze the quantitative data. Non-parametric tools, either the Mann-Whitney test for group comparisons or the Spearman correlation test, were used for quantitative data analysis. When data allowed, parametric tools, either group comparisons by t-test or one-way ANOVA or Pearson correlation test, were used. The two-tail method was used and a value of p < 0.05 was considered significant. The annotation * indicates a p-value < 0.05; ** p < 0.01; *** p < 0.001. Densitometry was conducted using the Quantity One software (Bio-Rad).
3. RESULTS
3.1. Ramelteon mitigates ischemic stroke
The MCAO mouse model was developed to mimic human ischemic stroke and serves as an indispensable tool in drug discovery research, in particular for studies of the molecular mechanisms of neuroprotective agents in stroke.29, 30 We evaluated the potential of ramelteon to reduce ischemic damage in an MCAO mouse model. Infarct volume was quantified by staining with 4% TTC. As shown in Figure 1A and 1B, we found that mice pretreated with ramelteon (10 mg/kg) by intraperitoneal (IP) injections had significantly smaller infarcts after MCAO compared to mice treated with vehicle alone (Figure 1A, 1B, upper panel). Blood-brain barrier (BBB) dysfunction has been reported in MCAO animal models of ischemic stroke.31 Our data showed that ramelteon significantly alleviated BBB disruption in MCAO mice (Figure 1C). Therapeutically, post-treatment of neuroprotective agents can closely mimic the human conditions of stroke patients. Ramelteon was administered as a “post-stroke” treatment after MCAO surgery. Of several doses tested, 20 mg/kg showed the best protection. As shown in Figure 1D and 1E, mice post-treated with 20 mg/kg ramelteon, but not 10 mg/kg or 15 mg/kg ramelteon, significantly reduced MCAO-induced infarct volume. Taken together, our results demonstrate that ramelteon ameliorates ischemia injury in MCAO mice.
Figure 1. Ramelteon provides neuroprotection preventively and therapeutically in an MCAO mouse model of cerebral ischemia.
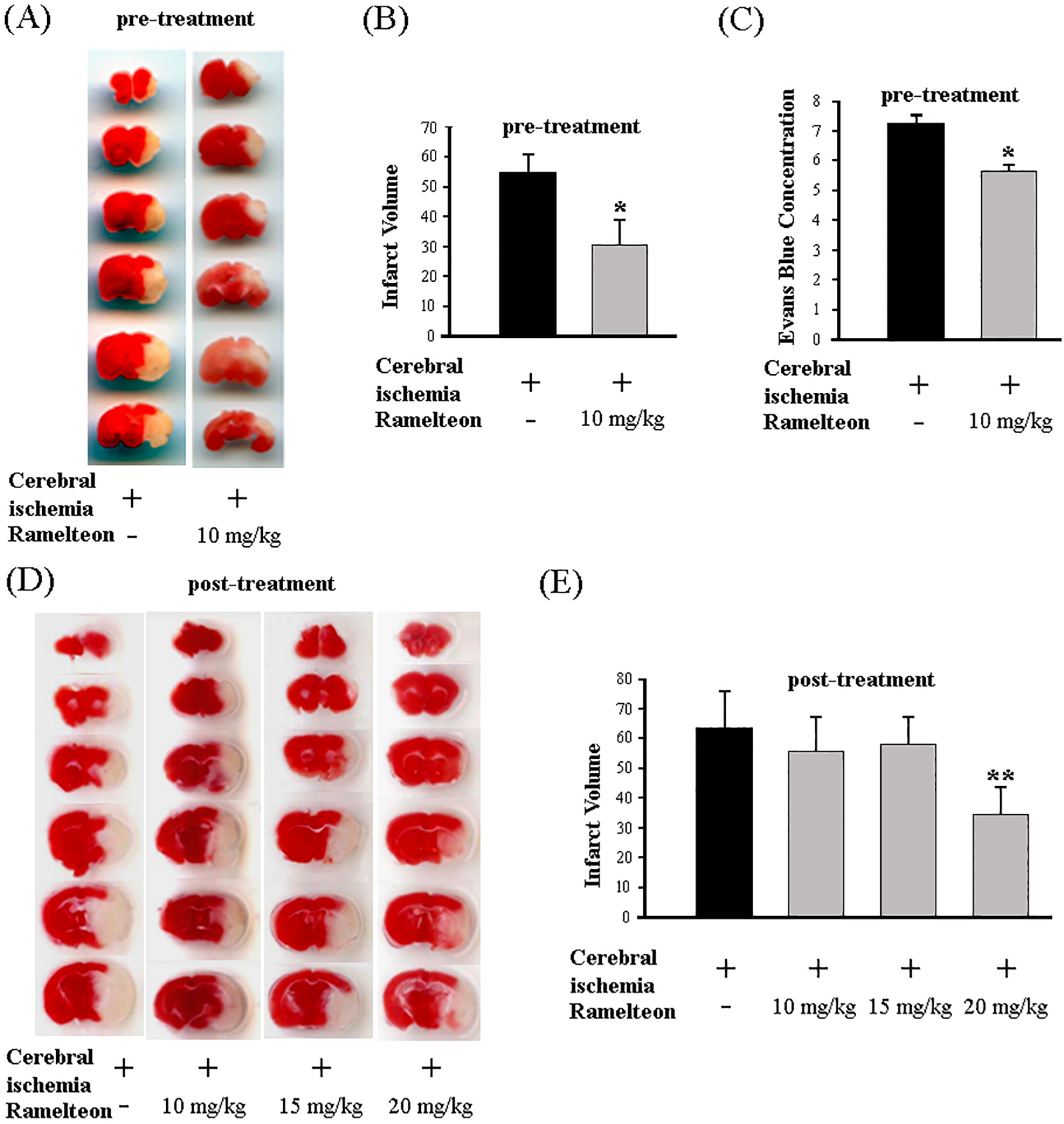
Ramelteon (10 mg/kg) was administered by IP injection 10 min before and 20 min after the onset of MCAO (preventive pre-treatment in A-C). Brains were removed after 24 h of ischemia, cut into coronal sections, and stained with 2% TTC. Lesion size (A, B) was determined for mice injected with saline (MCAO group, n = 10) and ramelteon (MCAO + ramelteon group, n = 10). Ramelteon (10, 15, 20 mg/kg) was administered by IP injection 1 h after the onset of MCAO (therapeutic post-treatment, n = 5–8) (D, E). Lesion size (B, E) was determined for mice injected with saline and ramelteon (n = 6 – 8). The data are presented as mean ± SEM, *p < 0.05, **p < 0.01. Deficits in BBB permeability were induced in MCAO-exposed vehicle mice, and ramelteon significantly reduced BBB injury in MCAO-exposed mice (C). Evans blue dye (2% in saline, 4 ml/kg body weight) was injected intravenously as a BBB permeability tracer via the tail vein 0.5 h before the animals were sacrificed. Cortex tissue (100 mg) was dissected under a stereomicroscope and incubated in formamide at 60°C for 7–8 hours and extracted Evans blue dye was quantitatively measured at a wavelength of 620 nm and calculated using a standard curve. The Evans blue leakage was expressed as micrograms per gram of brain tissue and Evans blue concentration is shown (C), * p < 0.01.
3.2. Ramelteon reduces stroke lesion volume and enhances cerebral blood flow via the activation of eNOS
MRI imaging has been a very valuable method for the evaluation of stroke lesion volume32 and CBF.32, 33 The quantitative stroke lesion volume/infarct area on T2WI maps was calculated using 3D Slicer software and compared between different groups. Our MRI analysis showed that the stroke lesion volume in the MCAO group was significantly increased compared with the control group mice (Figure 2A and 2B), whereas ramelteon significantly reduced the infarcted area (Figure 2A, 2B). Our results on the evaluation of stroke lesion volume using MRI image and 3D Slicer software analysis further confirm the finding of TTC staining of infarct volume.
Figure 2. Ramelteon reduces stroke lesion volume and enhances CBF in MCAO mice.

Experimental mice, including the control group, MCAO group, ramelteon (20 mg/kg) treated MCAO group at 72 h after the MCAO operation, were scanned on a Bruker 7 Tesla MRI scanner. T2WI was generated, and accumulated stroke lesion volume was quantitatively assessed with 3D Slice software (A, B). CBF was analyzed, and representative photos of three groups of mice were shown (C). The average difference of CBF (D) for an ischemic region/ipsilateral hemisphere was compared with their anatomical parallel in the contralateral hemisphere among the three groups of mice (n = 3–4/group). Furthermore, the control group, MCAO group, ramelteon (20 mg/kg) treated MCAO group at 24 h after the MCAO operation were scanned, and the representative LSCI images were presented (E). The blood flow index (PU) was compared (F). Brains of the three groups of mice were quickly removed, and whole cell lysates were extracted for analysis by WB using antibodies to p-eNOS (Ser1177) (upper panel, G) or eNOS (middle panel, G). β-actin was used as a loading control (lower panel, G). These blots are representative of three independent experiments. Densitometry was performed to quantify the intensity of the p-eNOS (Ser1177) or eNOS bands compared to that from β-actin (H or I, respectively, *p < 0.05, **p < 0.01).
Cerebral ischemia is a common mechanism of brain injury that results from impaired blood flow to the brain. The initial CBF interruption in ischemic stroke causes local brain infarct at the acute phase. The cumulative CBF within the ischemic area holds the potential as a predictive indicator for the extent of acute neurological impairment. Among these same animals in the evaluations of stroke lesion volume, we continue to observe their CBF signals (Figure 2C showing the representative photos of three groups of mice) and compare ischemic region/ipsilateral hemisphere with their anatomical parallel in the contralateral hemisphere by ASL quantitative measurement (Figure 2D). CBF showed high signals in the control mice (Figure 2C, left panel). Interestingly, we found a significant CBF signal reduction in MCAO mice compared with the control mice by calculating the median CBF values in the ipsilateral hemisphere/the median CBF values in the contralateral hemisphere (Figure 2C and 2D, respectively), while ramelteon enhances CBF signals in the ramelteon treated MCAO mice comparing with vehicle-treated MCAO mice (Figure 2C, right panel). The average difference of CBF for an ischemic region/ipsilateral hemisphere was increased, although not significantly, compared with their anatomical parallel in the contralateral hemisphere and further confirms that ramelteon enhances CBF signals in stroke mice (Figure 2D). Thus, ramelteon partly recovers CBF signals in the brain of stroke animals.
Moreover, we further validated the CBF observations through LSCI. Post 24 hours of MCAO, the representative images of CBF, and blood flow index analysis in the indicated groups in Figure 2E and 2F, respectively, are shown. We also observed a notable decline in CBF signals in MCAO mice compared to the control group. Our data showed that administration of ramelteon to MCAO mice significantly enhances CBF signals compared to vehicle-treated MCAO mice, as evidenced by the observed improvement in ramelteon-treated MCAO mice (Figure 2E and 2F).
Nitric oxide (NO) is pivotal as a mediator in the modulation of CBF. Specifically, endothelial NO emerges as a critical signaling molecule overseeing CBF regulation. The production of endothelial NO primarily depends on endothelial nitric oxide synthase (eNOS) activity. The eNOS is a potential therapeutic target for cerebrovascular diseases34 and impairment of eNOS activity by ischemic stroke has been implicated in many cellular mechanisms of neuronal injury.35 Protective agent ticagrelor improves CBF in MCAO mice and attenuates ischemia reperfusion injury via phosphorylation of eNOS.36 Short-term acute preconditioning exercise improves ischemic stroke through eNOS activation (active eNOS expression by p-S1177-eNOS) and eNOS inhibitor abolishes the exercise-induced improvement in outcomes.37 Given ramelteon’s capacity to enhance CBF post-ischemic stroke, we explored this phenomenon at the cellular level by assessing phosphorylation of eNOS and expression of eNOS. Our WB analysis demonstrates that there are significant reductions in the level of phosphorylation of eNOS at Ser1177 (down to 0.22-fold, Figure 2G, 2H) and the expression of eNOS (down to 0.60-fold, Figure 2G, 2I) in the infarct area of brain samples of MCAO mice compared with the control mice, while upregulation of phosphorylation eNOS and nNOS in the ramelteon treated MCAO mice is observed comparing with vehicle-treated MCAO mice (6.72 folds in Figure 2H and 2.85 folds in Figure 2I). In summary, ramelteon improves the CBF signals and offers neuroprotective effects against ischemic stroke, which may be, at least partly through the molecular mechanisms of increasing phosphorylation of eNOS and eNOS expression.
3.3. Ramelteon improves cerebral ischemia-induced neurological and behavioral deficits
We reported that melatonin and NAS improve neurological scores in the MCAO mice1, 2. Here, we evaluate neurological scores, as reflected in neurological outcomes, and demonstrate that MCAO mice pre-treated with ramelteon had significantly better behavior than vehicle-treated MCAO mice (Figure 3A). Furthermore, mice post-treated with 20 mg/kg ramelteon, but not 10 and 15 mg/kg ramelteon, significantly improved post-ischemic behavior compared with vehicle-treated MCAO neurological impairment mice (Figure 3B).
Figure 3. Ramelteon improves MCAO-caused neurological and behavioral deficits.
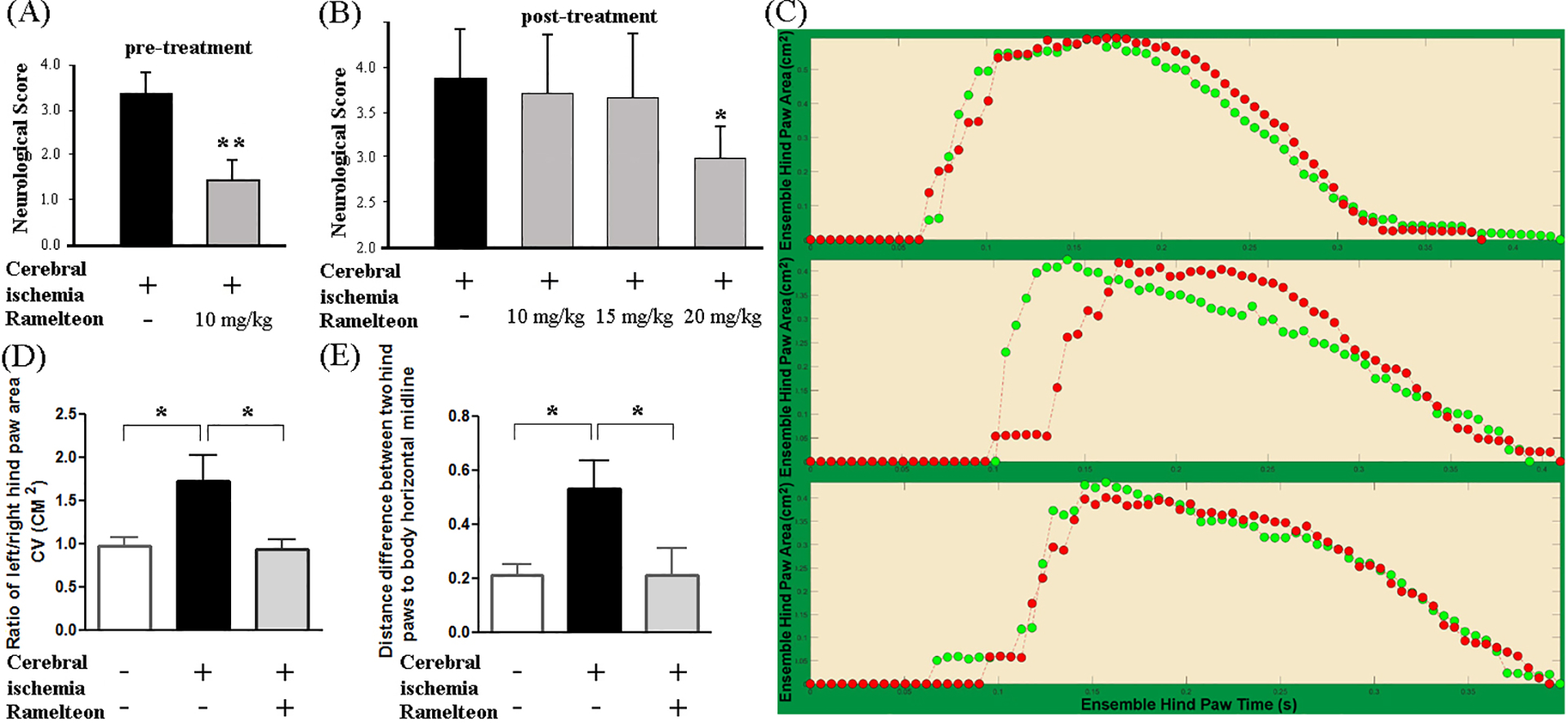
Animals were assessed for neurological deficits 24 h (A, B) or Digi Gait analysis 48 h (D) after the MCAO procedure in indicated groups of mice. Ramelteon (10, 15, or 20 mg/kg) was administered preventively (A) and therapeutically (B-D). Each mouse was assigned a neurological score, and the neurological score was compared (A, n = 10; B, n = 5 – 8). Mice were submitted to the Digi Gait automated gait analysis system on a treadmill set at 15 cm/second. A representative gait signal in the control group, MCAO group, and MCAO + ramelteon group were shown by the ensemble paw area (cm2) (C). Behavioral quantitative gait parameters measured post-ischemia include a ratio of left/right hind paw area and distance difference between two hind paws to the body horizontal midline. The ratio of left/right hind paw area and distance difference between two hind paws to the body horizontal midline in different groups of mice (D and E, respectively, n = 3 – 8) were compared. Statistically significant differences are indicated with *p < 0.05 and **p < 0.01.
MCAO causes global brain injury and gait disturbance (e.g., hemiparetic gait). The loss of coordination, change of dynamic gait signals such as the spatial paw statistics, including paw area CV (cm2), and gait asymmetry occur in ischemic stroke.38 DigiGait is a powerful tool to characterize behavioral deficits in MCAO mice.38 We perform a quantitative gait analysis by DigiGait to detect the effects of ramelteon on behavioral outcomes based on the main categories of gait parameters.22, 38, 39 The paw area (cm2) provides a dynamic gait signal by representing the temporal record of paw placement relative to the treadmill belt23. Gait analyses revealed the ratio of dynamic gait signal left hind paw area CV/right hind paw area CV were significantly increased in MCAO mice compared with the control mice (Figure 3C, 3D, 1.78-fold), while ramelteon significantly ameliorated this impaired gait signal (Figure 3C, 3D). Gait deficits also include gait asymmetry or differences in the bilateral behavior of the legs during walking, which significantly contribute to functional disability after stroke.40 Our results show that there was remarked enhancement for the distance difference between two hind paws from the body horizontal midline in MCAO mice vs. the control mice (Figure 3E, 2.48-fold), while ramelteon significantly recovered the impaired gait asymmetry (Figure 3E). Our results show that ramelteon effectively recovers the impaired gait signals and may provide promising neurorehabilitation in ischemic stroke.
3.4. Ramelteon inhibits cell death in the primary cerebrocortical neuron model of ischemic stroke in vitro
We reported that melatonin is protective in cellular and animal models of neuronal hypoxia.1 As ramelteon is a potent and highly selective agonist of MT1/MT2 melatonin receptors with a 3–16-fold higher affinity for MT1 and MT2 than melatonin, and also has a longer duration of action,17 it could act more efficiently than melatonin in inhibiting neurotoxicity. Next, we extend the above in vivo findings of the neuroprotective effect of ramelteon in the MCAO animal model to in vitro and ex vivo models. The primary cortical neuron (PCN) model of ischemic stroke provides the possibility to study the role of isolated neuronal cells of one particular cortical neuron type (about 95% purity) in an environment that simulates ischemic stroke. OGD is an ischemic-like experimental model in vitro.41 Lactate dehydrogenase (LDH) data showed that the incubating PCNs with ramelteon results in statistically significant inhibition of OGD-induced PCN cell death (Figure 4A). Ramelteon rescued OGD-mediated PCNs with an IC50 of 11.5 nM (nM range), much higher than the melatonin IC50 of 0.49 μM1 (μM range, a 43-fold decrease) and maximum protection of 53.0%, a 1.31-fold increase over melatonin’s 40.6%1. Thus, ramelteon acts more efficiently than melatonin in inhibiting OGD-mediated PCN cell death. Exposing PCNs to NMDA or H2O2 ± ramelteon (0.01 μM to 200 μM, 2 h before NMDA or H2O2) resulted in dose-dependent inhibition of PCN cell death (Figure 4B and Figure 4C). Plotting the resulting semilogarithmic curve as a function of drug concentrations, the NMDA-induced death curve defined the IC50 and Maximum afforded by ramelteon (8.5 nM (nM range) and 48.2%) (Figure 4B), and the H2O2-induced death curve defined the IC50 and Maximum afforded by ramelteon (1.48 nM (nM range) and 55.8%) (Figure 4C).
Figure 4. Neuroprotective effects of ramelteon on the cell death of PCNs and PHNs in vitro and OHSCs ex vivo.

Cell death of PCNs (A-C), PHNs (D-F), HT-22 cells (G), and OHSCs (H, I) was induced by 3 h exposure to OGD (A, D, G, H, I) or 18 h exposure to 0.5 mM NMDA (B, E) and 1000 μM H2O2 (C, F) ± a series of concentrations of ramelteon. Cultured cells and OHSCs were preincubated with ramelteon for 2 h. Cell death was evaluated by LDH assay (A-F, I) or MTS assay (G). Data from three independent experiments are presented, and statistically significant differences are indicated with *p < 0.05, **p < 0.01, and ***p < 0.001. The resulting curves (plotted semi-logarithmically) define the IC50 and maximum protection calculated by the GraphPad Prism program. PI fluorescence images were obtained (H). Hippocampal slices under normal control conditions displaying background PI fluorescence (H, left). Intense PI labeling in OHSCs exposed to OGD mainly occurred in the CA1, CA3 pyramidal cell fields as well as dentate gyrus (H, middle). Ramelteon significantly attenuated PI labeling, demonstrating neuroprotective effects (H, right). Scale bars: upper lane, 0.5 mm; lower lane, 0.1 mm.
3.5. Ramelteon inhibits cell death in cellular models of ischemic stroke in vitro and ex vivo
To determine whether ramelteon-mediated neuroprotection could be extended to other neuronal types, primary hippocampal neurons (PHNs) (Figure 4D–F) and HT-22 hippocampal cells (Figure 4G) were subjected to OGD ± ramelteon. LDH tests also demonstrate the neuroprotection of ramelteon in rescuing hippocampal neurons after OGD (Figure 4D). The resulting curves define the IC50 and maximum protection afforded by ramelteon, like PCNs does, in nM range (9.9 nM and 47.0%). Next, PHNs were subjected to NMDA or H2O2 ± ramelteon. Ramelteon rescued NMDA-challenged PHNs with an IC50 of 11.7 nM (nM range) and maximum protection of 33% (Figure 4E), while the resulting curves define the IC50 and maximum protection afforded by ramelteon in H2O2-mediated cell death (9.8 nM (nM range) and 38.1%) (Figure 4F). In addition to PHNs, we tested whether ramelteon also significantly inhibits cell death in HT-22 cells. MTS data demonstrated that exposing HT-22 cells to OGD induced significant cell death, while ramelteon results in statistically significant inhibition of OGD-induced cell death (Figure 4G). Ramelteon rescued OGD-mediated HT-22 cell death with an IC50 of 1.6 nM (nM range) and maximum protection of 47.3%. Compared to the culture of dissociated neurons, organotypic cultures made from slices of the hippocampus can live for weeks, thus, representing a more complex multi-cellular environment and providing many parallels with the MCAO animal model.42 In particular, OGD-treated OHSC cultures mimic ischemic conditions in animals and provide a good model of ischemic stroke ex vivo.43 We and others reported that NAS inhibits OGD- and H2O2-induced cell death and melatonin reduces kainic acid-induced oxidative stress in OHSCs.2, 44 Here, we found that compared to controls (Figure 4H, left panel), slice cultures exposed to OGD significantly enhanced PI fluorescence uptake (Figure 4H, middle panels). Neurons in the hippocampus CA1 subregion are more vulnerable to apoptosis, and the apoptosis rate increases after cerebral ischemia-reperfusion.45 Consistent with a previous report2, densitometric analysis of PI uptake in OHSCs exposed to OGD followed by 24 h of reoxygenation revealed that cell viability was mainly affected in the CA1 subregion (Figure 4H, middle panels). Interestingly, the intensity of PI fluorescence in the CA1 subregion was significantly reduced by ramelteon, demonstrating the protective capability and therapeutic potential of ramelteon against OGD-induced cell death (Figure 4H, right panels). We further demonstrated that OGD induced cell death in OHSCs by LDH assay. LDH data showed ramelteon significantly inhibited OGD-mediated OHSC cell death, leading us to conclude that ramelteon was protective in OHSCs (Figure 4I). Thus, ramelteon inhibits cell death mediated by OGD, NMDA, or H2O2 in PCNs, PHNs, or HT-22 cell lines in vitro as well as in OHSCs ex vivo. Apoptosis and neurogenesis are two important but opposing aspects of ischemic stroke and interventions that can regulate the two aspects are believed to have therapeutic potential.45 Ramelteon not only inhibits apoptosis (Figure 4) but also augments neurogenesis (Figure 5A). Neurite outgrowth is a key process during neurogenesis that can be imaged using the CFDA green fluorescent probe to stain cells. Because CFDA does not stain dead cells, photomicrographs demonstrate H2O2-mediated loss of PHN and the loss of neurite outgrowth, while neurite outgrowth can be recovered, at least partially, by ramelteon, further confirming the neuroprotective effect of ramelteon in PHNs (Figure 5A).
Figure 5. Ramelteon inhibits the dissipation of mitochondrial ΔΨm, mitochondrial fragmentation, and morphology alteration.
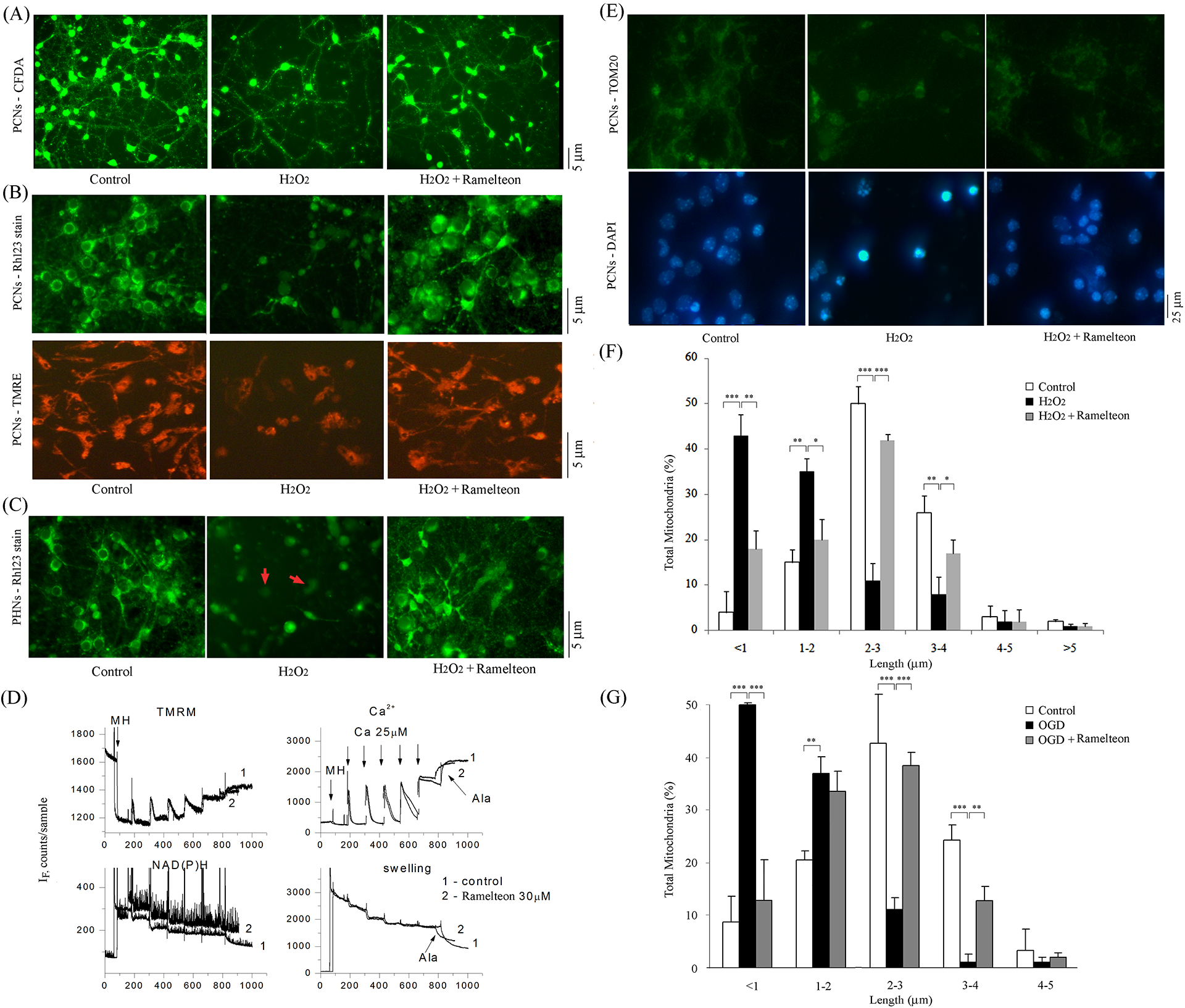
PCNs (B, E, F, G) and PHNs (A, C) were subjected to 1000 μM H2O2 (A-C, E, F) for 18 h or 3 h OGD (G) with or without 10 μM ramelteon. Neurite growth in PHNs was observed and analyzed by CFDA stain (A). PHNs under normal control conditions display regular CFDA fluorescence (A, left). Remarked reduction of CFDA stain in PHNs exposed to H2O2 (A, middle). Ramelteon significantly attenuated the loss of CFDA and the loss of neurite outgrowth, demonstrating its effect on augmenting neurogenesis (A, right). Living cells were stained with 2 μM Rh 123 (green in B, upper panel, and C) or TMRE (red-orange in B, lower panel). Brain mitochondria (0.25 mg/ml) energized with glutamate/malate 5 mM were challenged with a series of Ca 2+ additions (25 mM each) until they began to spontaneously release Ca 2+ (D). Changes ΔΨm and absorbance, indicators of mitochondrial swelling and induction of mPT, were monitored. Alamethicin (Ala) was added. Ramelteon (30 μM) was added 1 min before Ca 2+ addition. Representative images of TOM20 immunostaining (green) and DAPI staining (blue) were shown (E). ImageJ software was used to set a scale and Nano Measurers software to measure the percentage of various mitochondrial lengths (F, G). A minimum of 200 mitochondria/per picture was counted. Mitochondria were classified into different categories from a length ranging from < 1, 1–2, 2–3, 3–4, 4–5, to > 5 μm. The quantitative measurement represents three independent experiments. White bars: control PCNs. Black bars: H2O2- or OGD-treated PCNs. Grey bars: PCNs with H2O2 or OGD ± ramelteon. *p < 0.05, **p < 0.01, and ***p < 0.001. Scale bars correspond to 5 μm (A-C) or 25 μm (E).
3.6. Ramelteon mitigates H2O2- and OGD-induced mitochondrial dysfunction
Proper ΔΨm is critical for appropriate cellular bioenergetic homeostasis, and its dissipation is an important event associated with the progression of mitochondrial dysfunction leading to cell death. In healthy PCNs and PHNs, Rh 123 and TMRE stainings demonstrated a high intensity/punctuate pattern of uniform green (Figure 5B, upper level, and Figure 5C, left panel) or red-orange (Figure 5B, lower level, left panel) fluorescence, H2O2 produces a more-diffuse and lower-intensity staining pattern in both PCNs and PHNs (Figure 5B, middle panels) and PHNs (Figure 5C, middle panel). Like our reports that melatonin, NAS, methazolamide, dipyrone, and nortriptyline forestall the loss of ΔΨm in H2O2-induced death of primary cultured neurons,1, 2, 46, 47 Ramelteon counteracted H2O2-associated loss of mitochondrial ΔΨm in PCNs and PHNs (increased green dots in Figure 5B and increased red-orange dots in Figure 5C, right panels). Rh 123 staining and TMRE staining confirm each other; thus, we conclude that ramelteon-mediated neuroprotection involves the preservation of ΔΨm in primary cultured neurons.
The opening of mPTP stimulates neuronal apoptosis in ischemic stroke.48 We evaluated the effects of ramelteon on the mPTP by simultaneous measurement of membrane potential (following TMRM fluorescence), Ca2+ flux (using CaGreen-5N), NAD+/NADH redox status (using autofluorescence of the NAD+/NADH couple), and swelling (via light scatter). The addition of ramelteon (30 μM) to isolated brain mitochondria did not have any significant effect on ΔΨm, calcium transport, NAD+/NADH redox status, or swelling (Figure 5D). Our results indicated that ramelteon is not a direct mPT inhibitor in isolated mitochondria.
The mitochondria are the key target organelles affected by apoptotic proteins, which may cause mitochondrial swelling through the formation of membrane pores or increase the mPT induction and then cause the activation of apoptotic effectors. Using TOM20, a mitochondrial marker, to track mitochondrial fragmentation, we and others reported that mitochondrial fragmentation correlates with H2O2- or OGD- mediated primary neuronal death.2, 3, 49 The mitochondrial length varied in PCNs, and mitochondria were classified into different length categories: < 1, 1–2, 2–3, 3–4, 4–5, and > 5 μm. By TOM20 immunofluorescence staining and quantifying mitochondria length, data showed that, in healthy controls of PCNs, 96% of mitochondria had a length of > 1 μm (Figure 5E, left, upper panel, and Figure 5F, white bars). Of these, 15% were 1–2 μm; 50% 2–3 μm, 26% 3–4 μm, 3% 4–5 μm, and 2% > 5 μm. Induction by H2O2 caused mitochondria in the majority of PCNs to undergo clear fragmentation/breaking and swelling (Figure 5E, middle, bottom panel, and Figure 5F, black bars) as well as to develop morphologic features indicative of apoptosis (i.e., chromatin condensation and cell shrinkage; blue in Figure 5E, middle, bottom panel, cell death–associated nuclear fragmentation, and chromatin condensation). These changes left 43% of mitochondria < 1 μm and 35% 1–2 μm, whereas only 11% of mitochondria were 2–3 μm, 8% 3–4 μm, 2% 4–5 μm, and 1% > 5 μm. These alterations in mitochondrial fragmentation/morphology were significantly reduced by ramelteon (Figure 5E, right and Figure 5F, gray bars). Data show that 82% of mitochondria had a length of > 1 μm among PCNs incubated with ramelteon combined with H2O2. Of these, 20% ranged 1–2 μm; 42% 2–3 μm, 17% 3–4 μm, 2% 4–5 μm, and 1% > 5 μm. Thus, ramelteon restored H2O2-mediated mitochondrial fragmentation in PCNs.
We next used OGD as an apoptotic inducer. Similarly, we found that 92% of mitochondria in control PCNs had lengths of > 1 μm (Figure 5G, white bars). Of these, 21% ranged 1–2 μm; 43% 2–3 μm, 24% 3–4 μm, and 3% 4–5 μm. OGD caused the mitochondria to become clearly fragmented/broken (Figure 5G, black bars). These changes made 50% of mitochondria < 1 μm, 37% 1–2 μm, 11% 2–3 μm, 1% 3–4 μm, and 1% 4–5 μm. Ramelteon restored OGD-mediated mitochondrial fragmentation (Figure 5G, gray bars). Data showed that among PCNs incubated with ramelteon and subjected to OGD, 87% of mitochondria had a length of > 1 μm. Of these, 34% were 1–2 μm; 38% 2–3 μm, 13% 3–4 μm, and 2% 4–5 μm. Thus, ramelteon repaired OGD-mediated mitochondrial fragmentation. We conclude that ramelteon-mediated neuroprotection in primary neurons might not only preserve ΔΨm for appropriate cellular energetics but also influence mPTP opening and restore normal mitochondrial morphology in intact cells.
3.7. Ramelteon inhibits mitochondrial death pathways
Secondary injury mechanisms after stroke encompass mitochondrial dysfunction and apoptosis. We have reported that hypoxic injury to primary neurons causes mitochondria to release apoptogenic factors cyto. c, apoptosis-inducing factor, and Smac/DIABLO, which in turn activate caspases.1, 2, 41, 46, 47 Here we find that ramelteon significantly blocked the H2O2-mediated release of cyto. c in PCNs demonstrated by WB (Figure 6A). Moreover, WB results also showed elevated cyto. c release in the cytosolic fraction of the stroke brain, whereas it was barely detected in control animals. Ramelteon-treated mice have significantly reduced cyto. c release (Figure 6B).
Figure 6. Ramelteon inhibits mitochondrial and autophagic cell death.

PCNs (A, C, D, G, I, J) and OHSCs (E) were induced by subjecting PCNs to 1000 μM H2O2 for 18 h (A, C, D, G, I, J) and subjecting OHSC to 1500 μM H2O2 for 18 h (E) ± 10 μM ramelteon. Ramelteon (10 mg/kg) was administered by IP injection 10 min before and 20 min after the onset of MCAO (B, F, H, K, L). PCNs were extracted, and either cytosolic components (A) or whole-cell lysates (C, D, G, I) were analyzed by WB. Lysates of brain tissue were resolved into cytosolic fractions (B) or whole-cell lysates (F, H, K, L) for analysis by WB. Each sample (50 μg of protein) was analyzed by antibodies to cyto. c (A, B), or caspase-3 (C, E, F), or LC-3/Beclin 1/P62 (G, H) or Nrf2/HO-1 (J, K, L). β-actin was used as a loading control. Densitometry was performed to quantify the intensity of the bands from the three independent experiments. Caspase-3 activity was also quantified using a fluorogenic assay in lysed PCNs (D). PCN cell lysates in indicated treatments were centrifuged for 10 min at 3000 rpm, and generated supernatants were submitted for intercellular ROS (IU/ml) measurement (I). Conditioned media was collected and assayed for mature IL-1β release after the completion of H2O2 induction (M). The quantitative analyses come from three independent experiments (D, I, J). In all graphs, data are presented as mean ± SEM, statistically significant effects are marked with *p < 0.05, **p < 0.01, and ***p < 0.001. White bars correspond to control PCNs (A, C, D, E, G, I, J) or brain samples from animals (B, F, H, K, L) that neither underwent MCAO nor received ramelteon. Black bars correspond to samples from H2O2-treated PCNs (A, C, D, E, G, I, J) or saline-injected animals (B, F, H, K, L) that did undergo MCAO. Grey bars correspond to samples from H2O2- and ramelteon-treated PCNs (A, C, D, E, G, I, J) or test mice (B, F, H, K, L) that were both treated with ramelteon and underwent MCAO.
The release of cyto. c and other apoptogenic factors trigger sequential maturation of caspase-9, and mature caspase-9 activates executioner caspase-3 and caspase-independent cell death events.1, 2, 41, 46, 47 WB analysis demonstrated that caspase-3 was significantly activated in PCNs after H2O2 insult, and ramelteon effectively inhibited caspase-3 activation (Figure 6C). Moreover, the fluorogenic assay also revealed the activation of caspase-3 in H2O2-induced PCNs, whereas ramelteon significantly inhibited the activation (Figure 6D). Furthermore, WB analysis showed the activation of caspase-3 in H2O2-induced OHSCs, whereas ramelteon significantly inhibited the activation (Figure 6E). Next, we measured the activity of caspase-3 in ischemic and nonischemic tissues. Data indicate that ramelteon significantly diminished caspase-3 activation in the brain of MCAO mice (Figure 6F). Taken together, these findings suggest that ramelteon inhibits mitochondrial death pathways in primary neurons in vitro, OHSCs ex vivo, and in MCAO mice in vivo.
3.8. Ramelteon inhibits activation of the autophagy pathway
Protein LC3-II is encoded by gene MAP1LC3A/MAP1LC3B. Gene BECN1 is a member of the nanoparticle-triggered autophagic cell death gene set. Protein P62 is encoded by SQSTM1, a member of the hallmark apoptosis gene set. Autophagic cell death is induced with increased levels of two pacemakers in the autophagic cascade LC3-II and Beclin 1 and reduced levels of p62 in apoptotic PCNs and mice after cerebral ischemia.2, 50, 51 To explore the contribution of autophagy to neuroprotection by ramelteon, Using WB analysis, we measured the levels of LC3, Beclin 1, and p62 in H2O2-induced PCNs ± ramelteon. The levels of LC3-II, but not LC3-I, were remarkably higher upon H2O2 insult compared with the control group. Ramelteon significantly inhibited the H2O2-induced increase in LC3-II. In parallel, upregulation of Beclin 1 was found in PCNs upon H2O2 insult, whereas ramelteon elicited a remarked Beclin 1 decline. Furthermore, there was extensive downregulation of p62 in PCNs upon H2O2-challenge, while ramelteon significantly restored p62 levels (Figure 6G). Thus, ramelteon inhibits autophagy activation in environmentally stressed PCNs.
We assessed the effects of ramelteon on autophagy in MCAO mice. Levels of LC3-II and Beclin 1 were greatly increased in vehicle-treated MCAO mice, whereas ramelteon significantly reduced their levels, while the expression of p62 decreased greatly in MCAO mice, and ramelteon significantly blocked the downregulation of p62 (Figure 6H). It is interesting to note that ramelteon’s role in the inhibition of Beclin 1 and thus apoptosis was also demonstrated in prior research.52 Our findings that ramelteon inhibits autophagic activation in MCAO mice and environmentally stressed PCNs are consistent with each other, and the inhibition of the autophagic pathway is an important factor in neuroprotection by ramelteon in vivo and in vitro.
To get more insights on our experimental findings, we further looked at the CMap connections of the aforementioned genes differentially expressed by ramelteon using CMap’s online tool L1000 and additional ramelteon gene expression profile from prior research.53 CMap is a collection of genome-wide transcriptional expression data used for the discovery of functional connections between drugs, genes, and diseases through the transitory feature of common gene-expression changes. Three interesting connections of ramelteon are listed in Figure 7.
Figure 7. Bioinformatic analysis of ramelteon.

(A). Ramelteon’s differential expression profile was used as input to the online CMap tool L1000. L1000 generated three connections, Anti-Amyloidogenic Agents, JAK Inhibitor and ATPase Inhibitor. Green bars are positive connections and the red bar is a negative connection. These connections, especially the strong negative connection with ATPase inhibitor at 97%, are consistent with ramelteon’s neuroprotection role observed in vivo and in vitro. (B). The bar graph of the CMap connections of the differentially expressed genes by ramelteon. (C). MCAO mouse’s differential expressed genes were used as input to the online CMap tool L1000. L1000 generated five interesting connections, Aurora Kinase Inhibitor, JAK Inhibitor, Mitochondrial Complex I Inhibitor, Src Inhibitor and Melatonin Receptor Agonist. A heatmap representing the connection name and connection score, instances in red area and yellow area indicate negative connection score and instances in green area indicate positive connection score. The negative connection with melatonin receptor agonists is consistent with MT1 deficiency in the brains of MCAO mice. (D). The bar graph of the CMap connections of MCAO mice differential expressions, with the green bar being a positive connection and the red bars being negative connections. (E). The heatmap of the CMap connections. Legends: AKI (Aurora Kinase Inhibitor), JI (JAK Inhibitor), MCII (Mitochondrial Complex I Inhibitor), SI (Src inhibitor), MRA (Melatonin Receptor Agonist), and CS (Connection Score).
Administration gelsolin could reduce the amyloid load in the transgenic mouse model of Alzheimer’s disease.54 JAK inhibitors are increasingly being developed and tested for the treatment of chronic inflammatory conditions.55 Two drugs, ruxolitinib and baricitinib, have been approved by FDA to treat autoimmune diseases Rheumatoid arthritis (RA) and Psoriatic arthritis (PsA). The sarco/endoplasmic reticulum calcium ATPase (SERCA) plays a key role in the maintenance of Ca2+ ion homeostasis with considerable impacts on cell life and death decision.56 One of the ATPase inhibitors, thapsigargin, was shown to perturb the intracellular Ca2+ and direct cells toward apoptosis regardless of the cell cycle phase.57 From a bioinformatics angle, ramelteon’s positive connection with anti-amyloidogenic agents and JAK inhibitor, and negative connection with ATPase inhibitors again corroborated its neuroprotection role observed in our experiment (Figure 7A, B).
3.9. Ramelteon ameliorates ROS-induced oxidative stress associated with the activation of the NRF2/HO-1 pathway and inflammation in models of ischemic stroke
The ischemic brain is highly vulnerable to free radicals-mediated secondary neuronal damage.29 Reactive oxygen species (ROS), free radicals, arachidonic acid, nitric oxide, and cytokines cause damage that leads to inflammation.29 Oxidative stress is important in the pathological process of ischemic stroke and ROS describes several reactive molecules and free radicals.
Mathes et al. claim that ramelteon displays no relevant antioxidant capacity in a radical cation assay in vitro, as compared to melatonin or luzindole.58 Other researchers report that ramelteon ameliorates oxidative stress in methotrexate-induced cerebral toxicity,59 and free fatty acid-induced oxidative stress in brain vascular endothelial cells by reducing the levels of intracellular ROS60 as well as suppress the generation of mitochondrial ROS in human brain microvascular endothelial cells.61 Moreover, ramelteon supplementation can significantly decrease ROS and improve oocyte maturation and subsequent embryo development.62
Our present study, consistent with our previous report,25 showed that H2O2 increased intracellular ROS levels in PCNs compared with normal controls (Figure 6i). Importantly, ramelteon significantly ameliorated the ROS elevation (Figure 6I), thus, providing supportive evidence that ramelteon decreases ROS levels in accordance with the reports that ramelteon ameliorates oxidative stress.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator in the transcriptional activation of genes involved in antioxidation, antioxidant biosynthesis, and metabolic shift.63 The activation of Nrf2 is caused by excessive ROS generation after cerebral ischemia.64 Heme oxygenase-1 (HO-1) is a corresponding oxidative stress marker, and HO-1 expression leads to decreased ROS generation.65 Studies confirm that antioxidant protein HO-1 expression increased after cerebral ischemia.66
In the responses to oxidative stress, such as ROS-mediated oxidative stress, the Nrf2/HO-1 is a key signaling pathway in ischemia stroke.67, 68 Nrf2 and HO-1 are upregulated in the hippocampus of focal cerebral ischemia-reperfusion injury rats67 and MCAO brain tissues of animals.68 Pharmacological manipulation of the Nrf2/HO-1 pathway plays an important role in ischemic stroke. For instance, β-caryophyllene attenuates focal cerebral ischemia-reperfusion injury by Nrf2/HO-1 pathway activation in rats67, and MCAO brain tissue68 as well as mangiferin upregulates the expression of the Nrf2/HO-1 axis in the brain of stroke animals.69
To address the mechanism by which ramelteon decreases ROS levels, we further investigate whether ramelteon acts on the Nrf2/HO-1 pathway. Using WB analysis, upregulations of both Nrf2 and HO-1 are found in brain tissues of MCAO mice compared with the control mice (Figure 6J–L), whereas ramelteon markedly upregulates the Nrf2 and HO-1 expressions (Figure 6J–L). Our findings that treatment with ramelteon significantly increases the expression of Nrf2 and HO-1 in the ischemic region are consistent with other reports.67, 69, 70 Taken together, our present study, for the first time, suggests that the mechanism of action of ramelteon in protecting against ischemic stroke is associated with the reduction of ROS and the activation of the NRF2/HO-1 pathway.
HO-1 protects cells by diminishing oxidative stress and inflammation and maintaining mitochondrial integrity, thereby promoting cell survival.71 Oxidative stress and inflammation play critical roles in ischemic stroke.72, 73 Ramelteon administration remarkably reversed the excessively released inflammatory factors in brain vascular endothelial cells.60 IL-1β is a proinflammatory cytokine. Our data, consistent with our previous findings,1, 46 demonstrated that H2O2 induced the release of mature IL-1β (Figure 6M), while ramelteon significantly ameliorated mature IL-1β release (Figure 6M).
Furthermore, H2O2 induced Il-1β secretion in primary cortical neurons, and ramelteon significantly reduces the Il-1β level (Figure 6j). Thus, our evidence supports the contention that ramelteon prevents oxidative stress and inflammation.
Interestingly, our findings are consistent with gene expression analysis with ramelteon using whole-genome microarray analysis.53 Gene IL-1β, a member of the apoptosis/inflammation gene set, was reported to be downregulated by ramelteon, while ramelteon reversed the upregulation of the gene caused by the hemorrhagic shock. As IL-1 is considered a proinflammatory factor, ramelteon likely reversed the inflammatory effect, helping to inhibit the H2O2-induced inflammation observed in PCNs.
3.10. MT1 is the neuroprotective target of ramelteon
We reported the expression of MT1 is reduced in brain tissues of mouse models of neonatal H-I brain injury3 and HD4 as well as in spinal cord tissues of a mouse model of ALS.5 Here, we continue to investigate whether MT1 protein expression is reduced in the brain tissue of MCAO mice. Using WB analysis by anti-MT1 antibody (Millipore),3, 4 we observed that MT1 is significantly reduced in the brains of MCAO mice at a post-ischemic stroke (down to 0.52-fold, Figure 8A). Interestingly, ramelteon significantly recovered MT1 loss through upregulation of expression of MT1 receptors in brains of ramelteon-treated MCAO mice (a 1.63-fold increase, Figure 8A); thus, suggesting ramelteon inhibits the MT1 deficiency in brains of MCAO mice.
Figure 8. MT1 is the neuroprotective target of ramelteon in cellular and animal models of ischemic stroke.

MT1 brain levels are reduced in MCAO mice and ramelteon mainly recovers MT1 expression (A); Luzindole eliminates the neuroprotection of ramelteon (B, C); Knockdown of MT1 directly blocks the protection of ramelteon in primary neurons in vitro and MCAO mice in vivo (D, E). Ramelteon (20 mg/kg) was administered by IP injection about 60 mins after the onset of MCAO (A). Brains of indicated groups of mice were quickly removed, and wholecell lysates were extracted for analysis by WB using antibodies to MT1 (A). β-actin was used as a loading control (A). This blots are representative of three independent experiments. Densitometry was performed to quantify the intensity of the MT1 bands compared to that from β-actin (B, n = 6–8 in each group (A), *p < 0.05, **p < 0.01). PCN (B) or PHN (C) cell death was induced by 18 h exposure to 1000 μM H2O2 ± ramelteon (1, 5, 7.5, and 10 μM) in the presence or absence of luzindole. White bars represent control PCNs (B) or PHNs (C). Black bars represent H2O2-treated PCNs (B) or PHNs (C). Grey bars represent H2O2- and ramelteon-treated PCNs (B) or PHNs (C). Red bars represent H2O2- and ramelteon-treated PCNs (B) or PHNs (C) in the presence of luzindole. Cell death of PCN from wild-type mice (D, white bars) or MT1−/− mice (D, blue bars) was induced by 18 h exposure to 1000 μM H2O2 ± ramelteon (1, 5, 7.5, and 10 μM). Cells were preincubated with ramelteon or luzindole for 2 h. Cell death was evaluated by LDH assay (B-D). Data from three independent experiments are presented, and statistically significant differences are indicated with *p < 0.05, **p < 0.01, and ***p < 0.001. Ramelteon (10 mg/kg) was administered by IP injection 10 min before and 20 min after the onset of MCAO in MT1−/− mice. Lesion size (E, F, blue bars) and neurological scores (G, blue bars) were determined for mice injected with saline and ramelteon (n = 6–7). Brains were quickly removed after 24 h of ischemia, cut into coronal sections, and stained with 2% TTC, and neurological scores were rated. The data are presented as mean ± SEM.
The reduction of melatonin receptors in MCAO mice was also corroborated using Connectivity Map (CMap). We again used the CMap L1000 with MCAO mouse’s gene-expression signatures,74 to identify the connections of the signatures as shown in Figure 7C, D, E. Examining the first four connections, we observed the pathophysiology of ischemic stroke, inflammation, oxidative stress, and apoptosis.75–77 It is interesting to notice that the MCAO mouse’s gene-expression signatures are negatively connected to the melatonin receptor agonist from the last connection listed. The negative connection with melatonin receptor agonists, such as ramelteon, is likely the cause of MT1 loss in the MCAO mice. This again explains the role of ramelteon in the recovery of the MT1 receptor.
3.11. Neuroprotection of ramelteon needs MT1 in cellular and animal models of ischemic stroke
Ramelteon is an indane derivative, whereas luzindole has a benzyl group attached at the 2-position of the indole ring (Graphical Abstract). Besides inhibiting the mitochondrial and autophagic cell-death pathways and scavenging ROS free radicals, ramelteon may have other neuroprotective effects mediated by a traditional ramelteon ligand-melatonin receptor interaction or by a combination of these factors. We used the melatonin receptor antagonist luzindole to address this question. To directly evaluate whether the neuroprotective effects require melatonin receptor binding, we sought to determine whether luzindole counters ramelteon-mediated protection in H2O2-mediated PCN death. As shown in Figure 8B, luzindole almost eliminated neuroprotection by ramelteon (1 μM, 5 μM, 7.5 μM, and 10 μM) in PCNs, indicative of a melatonin receptor-mediated effect, while luzindole alone does not change the extent of cell death as previously reported.4 When we repeated this experiment with PHNs, luzindole again significantly blocked ramelteon-mediated neuroprotection (1 μM, 5 μM, 7.5 μM, and 10 μM) (Figure 8C). Taken together, we demonstrate that melatonin receptor binding is central to the mechanism of ramelteon neuroprotection and melatonin receptors play an important role in the neuroprotective effect of ramelteon.
However, these data cannot directly identify which melatonin receptor is the major target of ramelteon-mediated neuroprotection. To date, there has been no report in both MT1−/− animals and cultured MT1−/− PCNs upon ischemic stroke insult. To further confirm whether MT1 receptor plays an important role in the neuroprotection of ramelteon, we cultured both MT1−/− PCNs from MT1−/− mice and regular PCNs expressing MT1 from MT1 wild-type mice to determine whether the knockdown of MT1 sensitizes PCNs to cell death. Both PCNs expressing MT1 and those with MT1 deletion were challenged by exposure to H2O2. As shown in Figure 8D, LDH data show that ramelteon significantly inhibited H2O2-mediated cell death in both types of PCNs. However, the same dose of H2O2 mediated significantly more cell death in MT1−/− PCNs than in PCNs expressing MT1, indicating that knockdown of MT1 in PCNs sensitizes them to cell death, thereby increasing cellular vulnerability to an apoptotic inducer. Furthermore, ramelteon-treated MT1−/− PCNs underwent extensive cell death, whereas ramelteon-treated PCNs expressing MT1 suffered less cell death, further confirming the importance of the MT1 receptor in the action of ramelteon on reducing neuronal cell death.
We further investigated the direct effects of the knockdown of MT1 in MCAO mice. There were no significant differences in infarct volume (Figure 8E and 8F) or neurological score (Figure 8G) between ramelteon-treated C57 mice and MT1−/− C57 mice. In other words, the deletion of MT1 blocks the protection of ramelteon in MCAO mice. Our findings demonstrate, for the first time, that MT1 knockdown significantly reduces ramelteon-mediated neuroprotection and strongly supports our contention that neuroprotection by ramelteon depends on its interaction with the MT1 receptor.
Taken together, these findings confirm that the important role of the MT1 receptor in ramelteon-mediated neuroprotection and ischemic stroke is associated with the loss of MT1 in mice, thus contributing to a deeper understanding of the role of MT1 in neuroprotection for ischemic stroke (Graphical Abstract).
4. DISCUSSION
4.1. Ramelteon is a potential therapy for ischemic stroke and biological and bioinformatic analyses identify MT1 as the predominant neuroprotective target of ramelteon
The wide distribution of MT1 in many regions of rodent brain has been observed by us and others including suprachiasmatic nucleus, striatum (caudate putamen), hippocampus, cortex, cerebellum, hypothalamus, pineal gland, substantia nigra, etc.4, 78–81. The expression of MT1 has been studied by us and others in different cell types including neurons4, 78, 81 and glia cells.80
We previously reported that MT1 overexpression reduces neuronal death and knockdown of MT1 by siRNA sensitizes cultured neurons to cell death4. However, there are no reports about the effects of ramelteon on MT1−/− mice and MT1−/− PCNs in experimental models of ischemic stroke. For the first time, using luzindole, an antagonist of melatonin receptors, and MT1−/− mice and MT1−/− PCNs, we demonstrate that the knockdown of MT1 directly blocks the protection of ramelteon in PCNs and PHNs in vitro (Figure 1) and MCAO mice in vivo (Figure 8).
Two reasons point to the MT1 receptor as a predominant modulator of the neuroprotective effect of ramelteon: 1) the selectivity of ramelteon for MT1 is 10-fold stronger than that of MT217, 82 and ramelteon has a very low affinity to MT317, and 2) our data in Figure 1A and 1B demonstrate that ramelteon significantly reduces ischemic brain damage in MCAO mice, as indicated by reduced infarct volume. Notably, results in Figure 8E and 8F show that the neuroprotective effect of ramelteon on ischemic brain damage is negated in MCAO MT1−/− mice, suggesting that the primary mechanism of action for ramelteon’s neuroprotection in our MCAO ischemic stroke model is through MT1, rather than MT2. Consequently, our focus has centered on the impact of ramelteon on MT1 levels, rather than MT2, in the models of ischemic stroke. Nonetheless, further comprehensive investigations are necessary to fully elucidate the precise role of MT2 in ramelteon’s neuroprotection in the context of ischemic stroke.
It is noteworthy that this conclusion is also consistent with our previous reports regarding the role of MT1 in other neurological disorders, including newborn hypoxic-ischemic brain injury,3 HD,4 and ALS.5 Thus, we offer the first suggestion that ramelteon provides a potential avenue for neuroprotection in ischemic stroke mainly targeting MT1 receptors.
Melatonin receptor agonists, melatonin,1, 83, 84 NAS,2, 30 agomelatine,85 and Neu-P11,86 have been reported by us and others for potential use against ischemic stroke. Ramelteon works by mimicking melatonin. Although clinically the recommended dose of ramelteon for human adults is 8 mg/per day orally,87, 88 which is more than a typical daily dose of melatonin between 1 and 5 mg.89 clinically, ramelteon has some advantages over melatonin in terms of superior pharmacokinetics and pharmacodynamics90, 91: 1) ramelteon has a longer half-life of t½ = 1 – 2.6 hours compared to melatonin (30–50 min)6, 7; 2) ramelteon has higher lipophilicity than melatonin and therefore more easily taken up and retained by targeted tissues,92 making it more therapeutically effective than melatonin. Indeed, our present findings that ramelteon rescues OGD-mediated PCNs with IC50 in the nM range (Figure 4, 11.5 nM) which is much lower (43-fold decrease) than melatonin’s IC50 (0.49 μM, in μM range)1 and provides greater maximum protection (53.0%) than melatonin (40.59%),1 thus, confirming that ramelteon is a more efficient inhibitor of PCN cell death. The discovery that ramelteon is a highly selective MT1 agonist, further contributes towards the molecular mechanisms of protection of ramelteon and provides insight into the pathogenesis of ischemic stroke. As an indane derivative, ramelteon is synthetically designed to have a high affinity and selectivity for MT1.17, 82 In addition, ramelteon has negligible affinity for MT3 but also other receptors in the brain, including the opiate, dopamine, benzodiazepine, and serotonin receptors.93
We demonstrate that MT1 is reduced in the damaged brain of MCAO mice. Interestingly, analysis with CMap tool L1000 confirms that the negative connection with melatonin receptor agonists like ramelteon is likely the cause of MT1 loss in the brain of MCAO mice (Figure 7 and Graphical Abstract).
Ramelteon is the first melatonin receptor agonist to be approved for treating insomnia and circadian rhythm dysfunction. It has been used for treating delirium in elderly patients with acute stroke15 and has clinical advantages such as crossing the blood-brain barrier, oral route, and being well-tolerated. Ramelteon has low toxicity and no potential for abuse or dependence.94 Our findings support the neuroprotective and rehabilitative effects of ramelteon, at the doses 10–20 mg/kg, in a variety of experimental models of ischemic stroke, including the MCAO model of cerebral ischemia in vivo; OHSCs ex vivo, and cultured neuronal cells in vitro, thus, suggesting ramelteon as another potentially clinical useful melatonin receptor agonist95 suitable for therapy against ischemic stroke.
4.2. Ramelteon improves CBF signals and inhibits mitochondrial and autophagic death pathways and oxidative stress
Therapeutic strategies employing ramelteon against ischemic stroke should target multiple components of cerebral ischemia injury. Consistent with gene set enrichment analysis from a bioinformatics angle, our results show that ramelteon offers neuroprotection not only through an MT1-mediated mechanism but also involves the inhibition of mitochondrial and autophagic death pathways and oxidative stress (Figure 5, 6 and Graphical Abstract), as we previously reported with melatonin,1 NAS,2 methazolamide,1 dipyrone,46, 96 and nortriptyline,47, 96 On the other hand, ramelteon fails to affect Ca2+-induced mPT in isolated brain mitochondria, as done by melatonin1, NAS2, methazolamide1, and dipyrone.46
Moreover, we are the first to show that ramelteon improves the CBF signals and offers neuroprotection against ischemic stroke, at least partly through the activation of eNOS (Figure 2). The roles of eNOS in the regulation of angiogenesis are well documented, for instance, the overexpression of eNOS protein causes a marked increase in neocapillary formation in response to tissue ischemia97, 98 and endothelial protein kinase A/eNOS signaling may play an important role in ischaemic disorders by promoting neovascularization99. Our data imply that the protective effects of ramelteon in ischemic stroke may be associated with angiogenesis and further experiment is needed to confirm the hypothesis. What was initially a neuron-centric view has been replaced with the concept of the neurovascular unit, which encompasses neuronal, glial, and vascular compartments and the biphasic nature of neural–glial–vascular signaling.100 Thus, the pharmacological mechanism of ramelteon may be attributed to a combination of enhancement of CBF and reduction of neuronal death.
Neurorehabilitation is an important part of recovery after ischemic stroke. Combining motor robotic therapy with pharmacotherapy in post-stroke rehabilitation is a rapidly growing field.101 The advanced DigiGait image system is used to evaluate the impaired gait improvement by drug candidates in neurological disorders, including stroke.22, 38, 102, 103 DigiGait data show that ramelteon effectively recovers the MCAO-caused behavioral gait impairment. Thus, ramelteon holds promise in promoting motor functional recovery of ischemic stroke patients. Our report also provides evidence that the DigiGait image system (Figure 3) and the highly sensitive MRI neuroimage (Figure 2) contribute to methodological innovation for evaluation of therapeutic modalities in ischemic stroke.
Ramelteon displays no relevant antioxidant capacity in an assay in vitro,58 contrasting findings indicating its potential to mitigate oxidative stress, mainly by reducing ROS.59–61 Our data support the latter perspective and provide novel evidence that ramelteon’s protective mechanism against ischemic stroke involves the modulation of ROS-induced oxidative stress and the activation of the NRF2/HO-1 pathway (Figure 6).
4. 3. Conclusion
Effective neuroprotective agents are short to reverse the damage caused by ischemic stroke. In this study, we report that ramelteon is a neuroprotective drug candidate demonstrated by comprehensive in vitro, ex vivo and in vivo models of ischemic stroke. We characterize the multiple neuroprotective mechanisms of ramelteon that involve CBF signals that are potentially mediated, at least partly, by activating eNOS, mitochondrial and autophagy death pathways, and ROS-induced oxidative stress and NRF2/HO-1 pathway. Our data suggest that ramelteon is a potential neuroprotective drug candidate, and MT1 provides neuroprotective target and new insights into stroke therapy.
We clarify that the pharmacological mechanism of ramelteon may be attributed to a combination of reduction of neuronal death and enhancement of CBF. Our study contributes to understanding the pathogenesis of ischemic stroke and provides new insights into stroke therapy, which may eventually improve the lives of people fighting ischemic stroke.
ACKNOWLEDGMENTS
The authors thank Antonin E. Chiocca for his valuable comment and suggestion to improve this paper as well as Hua Zhou, Jing Zhou, Piergiacomo Cacciamani, Lushuang Xie, Zufeng Wang, and Ning Xie for valuable assistance.
FUNDING
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (to X.W., NS055072), the National Institute on Alcohol Abuse and Alcoholism (to X.W., R21AA030087-01A1), the Bill & Melinda Gates Foundation (to X. W., 01075000191 and OPP1099070), the Brigham and Women’s Hospital BRI Fund to Sustain Research Excellence (to X. W.), and the Gillian Reny Stepping Strong Center for Trauma Innovation (to S. Z.).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Data Availability Statement:
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome c release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, Wu Q, Zhang X, Luo C, Zhou S, Sirianni AC, Sarkar S, Kristal BS, Friedlander RM, Wang X. N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinha B, Wu Q, Li W, Tu Y, Sirianni AC, Chen Y, Jiang J, Zhang X, Chen W, Zhou S, Reiter RJ, Manning SM, Patel NJ, Aziz-Sultan AM, Inder TE, Friedlander RM, Fu J, Wang X. Protection of melatonin in experimental models of newborn hypoxic-ischemic brain injury through mt1 receptor. Journal of pineal research. 2018;64 [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, Zhou S, Wang H, Zhao R, Yano H, Kim JE, Li W, Kristal BS, Ferrante RJ, Friedlander RM. The melatonin mt1 receptor axis modulates mutant huntingtin-mediated toxicity. J Neurosci. 2011;31:14496–14507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Cook A, Kim J, Baranov SV, Jiang J, Smith K, Cormier K, Bennett E, Browser RP, Day AL, Carlisle DL, Ferrante RJ, Wang X, Friedlander RM. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits mt1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;55:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auld F, Maschauer EL, Morrison I, Skene DJ, Riha RL. Evidence for the efficacy of melatonin in the treatment of primary adult sleep disorders. Sleep Med Rev. 2017;34:10–22 [DOI] [PubMed] [Google Scholar]
- 7.Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev. 2005;9:25–39 [DOI] [PubMed] [Google Scholar]
- 8.Aldhous M, Franey C, Wright J, Arendt J. Plasma concentrations of melatonin in man following oral absorption of different preparations. Br J Clin Pharmacol. 1985;19:517–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandi-Perumal SR, Srinivasan V, Spence DW, Moscovitch A, Hardeland R, Brown GM, Cardinali DP. Ramelteon: A review of its therapeutic potential in sleep disorders. Adv Ther. 2009;26:613–626 [DOI] [PubMed] [Google Scholar]
- 10.Markwald RR, Lee-Chiong TL, Burke TM, Snider JA, Wright KP Jr. Effects of the melatonin mt-1/mt-2 agonist ramelteon on daytime body temperature and sleep. Sleep. 2010;33:825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna JT, Christie MA, Jeffrey BA, McCoy JG, Lee E, Connolly NP, Ward CP, Strecker RE. Chronic ramelteon treatment in a mouse model of alzheimer’s disease. Arch Ital Biol. 2012;150:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bano Otalora B, Popovic N, Gambini J, Popovic M, Vina J, Bonet-Costa V, Reiter RJ, Camello PJ, Rol MA, Madrid JA. Circadian system functionality, hippocampal oxidative stress, and spatial memory in the appswe/ps1de9 transgenic model of alzheimer disease: Effects of melatonin or ramelteon. Chronobiology international. 2012;29:822–834 [DOI] [PubMed] [Google Scholar]
- 13.Srinivasan V, Cardinali DP, Srinivasan US, Kaur C, Brown GM, Spence DW, Hardeland R, Pandi-Perumal SR. Therapeutic potential of melatonin and its analogs in parkinson’s disease: Focus on sleep and neuroprotection. Ther Adv Neurol Disord. 2011;4:297–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Jiang C, Zhang K, Lan X, Chen X, Zang W, Wang Z, Guan F, Zhu C, Yang X, Lu H. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the nrf2 signaling pathway. Free radical biology & medicine. 2019;131:345–355 [DOI] [PubMed] [Google Scholar]
- 15.Ohta T, Murao K, Miyake K, Takemoto K. Melatonin receptor agonists for treating delirium in elderly patients with acute stroke. J Stroke Cerebrovasc Dis. 2013;22:1107–1110 [DOI] [PubMed] [Google Scholar]
- 16.Kawada K, Ohta T, Tanaka K, Miyamura M, Tanaka S. Addition of suvorexant to ramelteon therapy for improved sleep quality with reduced delirium risk in acute stroke patients. J Stroke Cerebrovasc Dis. 2019;28:142–148 [DOI] [PubMed] [Google Scholar]
- 17.Kato K, Hirai K, Nishiyama K, Uchikawa O, Fukatsu K, Ohkawa S, Kawamata Y, Hinuma S, Miyamoto M. Neurochemical properties of ramelteon (tak-375), a selective mt1/mt2 receptor agonist. Neuropharmacology. 2005;48:301–310 [DOI] [PubMed] [Google Scholar]
- 18.Wu XL, Lu SS, Liu MR, Tang WD, Chen JZ, Zheng YR, Ahsan A, Cao M, Jiang L, Hu WW, Wu JY, Chen Z, Zhang XN. Melatonin receptor agonist ramelteon attenuates mouse acute and chronic ischemic brain injury. Acta Pharmacol Sin. 2020;41:1016–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams DS, Detre JA, Leigh JS, Koretsky AP. Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tao S, Zhang T, Zhou K, Liu X, Feng Y, Zhao W, Chen J. Intraoperative monitoring cerebral blood flow during the treatment of brain arteriovenous malformations in hybrid operating room by laser speckle contrast imaging. Frontiers in surgery. 2022;9:855397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caballero-Garrido E, Pena-Philippides JC, Galochkina Z, Erhardt E, Roitbak T. Characterization of long-term gait deficits in mouse dmcao, using the catwalk system. Behavioural brain research. 2017;331:282–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson S, Corbett CJ, Winer JL, Chan LAS, Maxwell JR, Anstine CV, Yellowhair TR, Andrews NA, Yang Y, Sillerud LO, Jantzie LL. Neonatal erythropoietin mitigates impaired gait, social interaction and diffusion tensor imaging abnormalities in a rat model of prenatal brain injury. Exp Neurol. 2018;302:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampton TG, Kale A, Amende I, Tang W, McCue S, Bhagavan HN, VanDongen CG. Gait disturbances in dystrophic hamsters. J Biomed Biotechnol. 2011;2011:235354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan F, Liu T, Wang X, Ren D, Liu H, Zhang P, Wang Z, Liu N, Li Q, Tu Y, Fu J. Clc-3 expression and its association with hyperglycemia induced ht22 hippocampal neuronal cell apoptosis. J Diabetes Res. 2016;2016:2984380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M, Wang W, Mai H, Zhang X, Wang J, Gao Y, Wang Y, Deng G, Gao L, Zhou S, Chen Q, Wang X. Methazolamide improves neurological behavior by inhibition of neuron apoptosis in subarachnoid hemorrhage mice. Scientific reports. 2016;6:35055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stavrovskaya IG, Baranov SV, Guo X, Davies SS, Roberts LJ 2nd, Kristal BS. Reactive gamma-ketoaldehydes formed via the isoprostane pathway disrupt mitochondrial respiration and calcium homeostasis. Free Radic Biol Med. 2010;49:567–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, Lahr DL, Hirschman JE, Liu Z, Donahue M, Julian B, Khan M, Wadden D, Smith IC, Lam D, Liberzon A, Toder C, Bagul M, Orzechowski M, Enache OM, Piccioni F, Johnson SA, Lyons NJ, Berger AH, Shamji AF, Brooks AN, Vrcic A, Flynn C, Rosains J, Takeda DY, Hu R, Davison D, Lamb J, Ardlie K, Hogstrom L, Greenside P, Gray NS, Clemons PA, Silver S, Wu X, Zhao WN, Read-Button W, Haggarty SJ, Ronco LV, Boehm JS, Schreiber SL, Doench JG, Bittker JA, Root DE, Wong B, Golub TR. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452 e1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935 [DOI] [PubMed] [Google Scholar]
- 29.Pandya RS, Mao L, Zhou H, Zhou S, Zeng J, Popp AJ, Wang X. Central nervous system agents for ischemic stroke: Neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11:81–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo C, Yang Q, Liu Y, Zhou S, Jiang J, Reiter RJ, Bhattacharya P, Cui Y, Yang H, Ma H, Yao J, Lawler SE, Zhang X, Fu J, Rozental R, Aly H, Johnson MD, Chiocca EA, Wang X. The multiple protective roles and molecular mechanisms of melatonin and its precursor n-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic Biol Med. 2019;130:215–233 [DOI] [PubMed] [Google Scholar]
- 31.Zuo X, Lu J, Manaenko A, Qi X, Tang J, Mei Q, Xia Y, Hu Q. Microrna-132 attenuates cerebral injury by protecting blood-brain-barrier in mcao mice. Exp Neurol. 2019 [DOI] [PubMed] [Google Scholar]
- 32.Foddis M, Winek K, Bentele K, Mueller S, Blumenau S, Reichhart NN, Crespo-Garcia S, Harnett D, Ivanov A, Meisel A, Joussen A, Strauss O, Beule D, Dirnagl U, Sassi C. An exploratory investigation of brain collateral circulation plasticity after cerebral ischemia in two experimental c57bl/6 mouse models. J Cereb Blood Flow Metab. 2020;40:276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soylu H, Zhang D, Buist R, Martin M, Albensi BC, Parkinson FE. Intracortical injection of endothelin-1 induces cortical infarcts in mice: Effect of neuronal expression of an adenosine transporter. Exp Transl Stroke Med. 2012;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Song W, Li L, Fan X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol Brain. 2016;9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava K, Bath PM, Bayraktutan U. Current therapeutic strategies to mitigate the enos dysfunction in ischaemic stroke. Cellular and molecular neurobiology. 2012;32:319–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamauchi K, Imai T, Shimazawa M, Iwama T, Hara H. Effects of ticagrelor in a mouse model of ischemic stroke. Scientific reports. 2017;7:12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafez S, Khan MB, Awad ME, Wagner JD, Hess DC. Short-term acute exercise preconditioning reduces neurovascular injury after stroke through induced enos activation. Transl Stroke Res. 2020;11:851–860 [DOI] [PubMed] [Google Scholar]
- 38.Hetze S, Romer C, Teufelhart C, Meisel A, Engel O. Gait analysis as a method for assessing neurological outcome in a mouse model of stroke. J Neurosci Methods. 2012;206:7–14 [DOI] [PubMed] [Google Scholar]
- 39.Hamers FP, Koopmans GC, Joosten EA. Catwalk-assisted gait analysis in the assessment of spinal cord injury. Journal of neurotrauma. 2006;23:537–548 [DOI] [PubMed] [Google Scholar]
- 40.Patterson KK, Parafianowicz I, Danells CJ, Closson V, Verrier MC, Staines WR, Black SE, McIlroy WE. Gait asymmetry in community-ambulating stroke survivors. Arch Phys Med Rehabil. 2008;89:304–310 [DOI] [PubMed] [Google Scholar]
- 41.Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC, Friedlander RM. Fundamental role of the rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:16012–16017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boscia F, Annunziato L, Taglialatela M. Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology. 2006;51:283–294 [DOI] [PubMed] [Google Scholar]
- 43.Strasser U, Fischer G. Protection from neuronal damage induced by combined oxygen and glucose deprivation in organotypic hippocampal cultures by glutamate receptor antagonists. Brain research. 1995;687:167–174 [DOI] [PubMed] [Google Scholar]
- 44.Kim HA, Lee KH, Lee BH. Neuroprotective effect of melatonin against kainic acid-induced oxidative injury in hippocampal slice culture of rats. International journal of molecular sciences. 2014;15:5940–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L, Zhuang W, Xu X, Yang J, Teng J, Zhang F. The effect of injection of egb 761 into the lateral ventricle on hippocampal cell apoptosis and stem cell stimulation in situ of the ischemic/reperfusion rat model. Neuroscience letters. 2013;555:123–128 [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Wang X, Baranov SV, Zhu S, Huang Z, Fellows-Mayle W, Jiang J, Day AL, Kristal BS, Friedlander RM. Dipyrone inhibits neuronal cell death and diminishes hypoxic/ischemic brain injury. Neurosurgery. 2011;69:942–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang WH, Wang H, Wang X, Narayanan MV, Stavrovskaya IG, Kristal BS, Friedlander RM. Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke. 2008;39:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y, Sun J, Wu R, Bai J, Hou Y, Zeng Y, Zhang Y, Wang X, Wang Z, Meng X. Mitochondrial mptp: A novel target of ethnomedicine for stroke treatment by apoptosis inhibition. Front Pharmacol. 2020;11:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen Z, Jiang L, Yuan Y, Deng T, Zheng YR, Zhao YY, Li WL, Wu JY, Gao JQ, Hu WW, Zhang XN, Chen Z. Inhibition of g protein-coupled receptor 81 (gpr81) protects against ischemic brain injury. CNS neuroscience & therapeutics. 2015;21:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769 [DOI] [PubMed] [Google Scholar]
- 51.Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141 [DOI] [PubMed] [Google Scholar]
- 52.Kandezi N, Majdi F, Davoudizadeh R, Motaghinejad M, Safari S. Preventive properties of ramelteon against cocaine-induced autophagia and apoptosis: A hypothetic role of tnf-alpha receptor involvement and jnk/bcl-2-beclin1 or bcl-2/bax signaling pathway. Int J Prev Med. 2020;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleber A, Ruf CG, Wolf A, Fink T, Glas M, Wolf B, Volk T, Abend M, Mathes AM. Melatonin or ramelteon therapy differentially affects hepatic gene expression profiles after haemorrhagic shock in rat--a microarray analysis. Exp Mol Pathol. 2015;99:189–197 [DOI] [PubMed] [Google Scholar]
- 54.Chauhan V, Ji L, Chauhan A. Anti-amyloidogenic, anti-oxidant and anti-apoptotic role of gelsolin in alzheimer’s disease. Biogerontology. 2008;9:381–389 [DOI] [PubMed] [Google Scholar]
- 55.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. Jak inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peterkova L, Kmonickova E, Ruml T, Rimpelova S. Sarco/endoplasmic reticulum calcium atpase inhibitors: Beyond anticancer perspective. J Med Chem. 2020;63:1937–1963 [DOI] [PubMed] [Google Scholar]
- 57.Pinski J, Parikh A, Bova GS, Isaacs JT. Therapeutic implications of enhanced g(0)/g(1) checkpoint control induced by coculture of prostate cancer cells with osteoblasts. Cancer Res. 2001;61:6372–6376 [PubMed] [Google Scholar]
- 58.Mathes AM, Kubulus D, Waibel L, Weiler J, Heymann P, Wolf B, Rensing H. Selective activation of melatonin receptors with ramelteon improves liver function and hepatic perfusion after hemorrhagic shock in rat. Crit Care Med. 2008;36:2863–2870 [DOI] [PubMed] [Google Scholar]
- 59.Aslankoc R, Savran M, Doguc DK, Sevimli M, Tekin H, Kaynak M. Ameliorating effects of ramelteon on oxidative stress, inflammation, apoptosis, and autophagy markers in methotrexate-induced cerebral toxicity. Iran J Basic Med Sci. 2022;25:1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang G, Tian F, Li Y, Liu Y, Liu C. Ramelteon mitigates free fatty acid (ffa)-induced attachment of monocytes to brain vascular endothelial cells. Neurotox Res. 2021;39:1937–1945 [DOI] [PubMed] [Google Scholar]
- 61.Wang T, Li Z, Xia S, Xu Z, Chen X, Sun H. The protective effects of ramelteon against isoflurane-induced insults and inflammatory response in brain microvascular endothelial cells. Neurotox Res. 2021;39:677–686 [DOI] [PubMed] [Google Scholar]
- 62.Sun JT, Yuan JD, Zhang Q, Luo X, Qi XY, Liu JH, Jiang XQ, Lee S, Taweechaipaisankul A, Liu ZH, Jin JX. Ramelteon reduces oxidative stress by maintenance of lipid homeostasis in porcine oocytes. Antioxidants (Basel). 2022;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitsuishi Y, Motohashi H, Yamamoto M. The keap1-nrf2 system in cancers: Stress response and anabolic metabolism. Front Oncol. 2012;2:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, Zhang X, Xiong X, Zhu H, Chen R, Zhang S, Chen G, Jian Z. Nrf2 regulates oxidative stress and its role in cerebral ischemic stroke. Antioxidants (Basel). 2022;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Araujo JA, Zhang M, Yin F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front Pharmacol. 2012;3:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi H, Jing X, Wei X, Perez RG, Ren M, Zhang X, Lou H. S-allyl cysteine activates the nrf2-dependent antioxidant response and protects neurons against ischemic injury in vitro and in vivo. J Neurochem. 2015;133:298–308 [DOI] [PubMed] [Google Scholar]
- 67.Lou J, Cao G, Li R, Liu J, Dong Z, Xu L. Beta-caryophyllene attenuates focal cerebral ischemia-reperfusion injury by nrf2/ho-1 pathway in rats. Neurochemical research. 2016;41:1291–1304 [DOI] [PubMed] [Google Scholar]
- 68.Jiang LJ, Zhang SM, Li CW, Tang JY, Che FY, Lu YC. Roles of the nrf2/ho-1 pathway in the anti-oxidative stress response to ischemia-reperfusion brain injury in rats. European review for medical and pharmacological sciences. 2017;21:1532–1540 [PubMed] [Google Scholar]
- 69.Wang Z, Guo S, Wang J, Shen Y, Zhang J, Wu Q. Nrf2/ho-1 mediates the neuroprotective effect of mangiferin on early brain injury after subarachnoid hemorrhage by attenuating mitochondria-related apoptosis and neuroinflammation. Scientific reports. 2017;7:11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu Q, Zuo T, Deng L, Chen S, Yu W, Liu S, Liu J, Wang X, Fan X, Dong Z. Beta-caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the nrf2/ho-1 signaling pathway in mcao/r rats. Phytomedicine : international journal of phytotherapy and phytopharmacology. 2022;102:154112. [DOI] [PubMed] [Google Scholar]
- 71.Yachie A Heme oxygenase-1 deficiency and oxidative stress: A review of 9 independent human cases and animal models. International journal of molecular sciences. 2021;22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tedeschi A, Larson MJE, Zouridakis A, Mo L, Bordbar A, Myers JM, Qin HY, Rodocker HI, Fan F, Lannutti JJ, McElroy CA, Nimjee SM, Peng J, Arnold WD, Moon LDF, Sun W. Harnessing cortical plasticity via gabapentinoid administration promotes recovery after stroke. Brain. 2022;145:2378–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mamik MK, Power C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain. 2017;140:2273–2285 [DOI] [PubMed] [Google Scholar]
- 74.Androvic P, Kirdajova D, Tureckova J, Zucha D, Rohlova E, Abaffy P, Kriska J, Valny M, Anderova M, Kubista M, Valihrach L. Decoding the transcriptional response to ischemic stroke in young and aged mouse brain. Cell Rep. 2020;31:107777. [DOI] [PubMed] [Google Scholar]
- 75.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182 [DOI] [PubMed] [Google Scholar]
- 76.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922 [DOI] [PubMed] [Google Scholar]
- 77.Randy LH, Guoying B. Agonism of peroxisome proliferator receptor-gamma may have therapeutic potential for neuroinflammation and parkinson’s disease. Curr Neuropharmacol. 2007;5:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Uz T, Arslan AD, Kurtuncu M, Imbesi M, Akhisaroglu M, Dwivedi Y, Pandey GN, Manev H. The regional and cellular expression profile of the melatonin receptor mt1 in the central dopaminergic system. Molecular brain research. 2005;136:45–53 [DOI] [PubMed] [Google Scholar]
- 79.Adamah-Biassi EB, Zhang Y, Jung H, Vissapragada S, Miller RJ, Dubocovich M. Distribution of mt1 melatonin receptor promoter-driven rfp expression in the brains of bac c3h/hen transgenic mice. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2014;62:70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gu C, Wang F, Zhang YT, Wei SZ, Liu JY, Sun HY, Wang GH, Liu CF. Microglial mt1 activation inhibits lps-induced neuroinflammation via regulation of metabolic reprogramming. Aging cell. 2021;20:e13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brunner P, Sozer-Topcular N, Jockers R, Ravid R, Angeloni D, Fraschini F, Eckert A, Muller-Spahn F, Savaskan E. Pineal and cortical melatonin receptors mt1 and mt2 are decreased in alzheimer’s disease. European journal of histochemistry : EJH. 2006;50:311–316 [PubMed] [Google Scholar]
- 82.Hirai K, Kato K, Nishiyama K, Hinuma S, Uchikawa O, Ohkawa S, Miyamoto M. Tak-375 and its metabolites are selective agonists at mls1s receptors. Sleep. 2003;26:A79 [Google Scholar]
- 83.Wang X The antiapoptotic effects of melatonin in neonatal hypoxic-ischemic brain injury and adult ischemic stroke. JSM Neurosurgery and Spine. 2014;2:1033 [Google Scholar]
- 84.Reiter RJ, Tan DX, Leon J, Kilic U, Kilic E. When melatonin gets on your nerves: Its beneficial actions in experimental models of stroke. Exp Biol Med (Maywood). 2005;230:104–117 [DOI] [PubMed] [Google Scholar]
- 85.Chumboatong W, Khamchai S, Tocharus C, Govitrapong P, Tocharus J. Agomelatine protects against permanent cerebral ischaemia via the nrf2-ho-1 pathway. European journal of pharmacology. 2020;874:173028. [DOI] [PubMed] [Google Scholar]
- 86.Buendia I, Gomez-Rangel V, Gonzalez-Lafuente L, Parada E, Leon R, Gameiro I, Michalska P, Laudon M, Egea J, Lopez MG. Neuroprotective mechanism of the novel melatonin derivative neu-p11 in brain ischemia related models. Neuropharmacology. 2015;99:187–195 [DOI] [PubMed] [Google Scholar]
- 87.Neubauer DN. A review of ramelteon in the treatment of sleep disorders. Neuropsychiatric disease and treatment. 2008;4:69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schwartz TL, Hameed U, Chilton M. The use of ramelteon for secondary insomnia due to mental illness. Primary Psychiatry. 2006;13:69–73 [Google Scholar]
- 89.Tordjman S, Chokron S, Delorme R, Charrier A, Bellissant E, Jaafari N, Fougerou C. Melatonin: Pharmacology, functions and therapeutic benefits. Curr Neuropharmacol. 2017;15:434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Simpson D, Curran MP. Ramelteon: A review of its use in insomnia. Drugs. 2008;68:1901–1919 [DOI] [PubMed] [Google Scholar]
- 91.Hardeland R, Poeggeler B, Srinivasan V, Trakht I, Pandi-Perumal SR, Cardinali DP. Melatonergic drugs in clinical practice. Arzneimittelforschung. 2008;58:1–10 [DOI] [PubMed] [Google Scholar]
- 92.Carocci A, Catalano A, Sinicropi MS. Melatonergic drugs in development. Clin Pharmacol. 2014;6:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGechan A, Wellington K. Ramelteon. CNS Drugs. 2005;19:1057–1065; discussion 1066–1057 [DOI] [PubMed] [Google Scholar]
- 94.Miyamoto M Pharmacology of ramelteon, a selective mt1/mt2 receptor agonist: A novel therapeutic drug for sleep disorders. CNS neuroscience & therapeutics. 2009;15:32–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zammit GK. Ramelteon: A novel hypnotic indicated for the treatment of insomnia. Psychiatry (Edgmont). 2007;4:36–42 [PMC free article] [PubMed] [Google Scholar]
- 96.Stavrovskaya IG, Narayanan MV, Zhang W, Krasnikov BF, Heemskerk J, Young SS, Blass JP, Brown AM, Beal MF, Friedlander RM, Kristal BS. Clinically approved heterocyclics act on a mitochondrial target and reduce stroke-induced pathology. J Exp Med. 2004;200:211–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amano K, Matsubara H, Iba O, Okigaki M, Fujiyama S, Imada T, Kojima H, Nozawa Y, Kawashima S, Yokoyama M. Enhancement of ischemia-induced angiogenesis by enos overexpression. Hypertension. 2003;41:156–162 [DOI] [PubMed] [Google Scholar]
- 98.Ren C, Li N, Li S, Han R, Huang Q, Hu J, Jin K, Ji X. Limb ischemic conditioning improved cognitive deficits via enos-dependent augmentation of angiogenesis after chronic cerebral hypoperfusion in rats. Aging and disease. 2018;9:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bir SC, Xiong Y, Kevil CG, Luo J. Emerging role of pka/enos pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovascular research. 2012;95:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tiedt S, Buchan AM, Dichgans M, Lizasoain I, Moro MA, Lo EH. The neurovascular unit and systemic biology in stroke - implications for translation and treatment. Nature reviews. Neurology. 2022;18:597–612 [DOI] [PubMed] [Google Scholar]
- 101.Awad LN, Bae J, O’Donnell K, De Rossi SMM, Hendron K, Sloot LH, Kudzia P, Allen S, Holt KG, Ellis TD, Walsh CJ. A soft robotic exosuit improves walking in patients after stroke. Sci Transl Med. 2017;9 [DOI] [PubMed] [Google Scholar]
- 102.Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, Goldsteins G, Koistinaho J, Dhungana H. Peripheral administration of il-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics. 2019;16:1304–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mancuso R, Olivan S, Osta R, Navarro X. Evolution of gait abnormalities in sod1(g93a) transgenic mice. Brain research. 2011;1406:65–73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.