

SUMMARY
Heterotrimeric G proteins transduce extracellular chemical messages to generate appropriate intracellular responses. Point mutations in GNAO1, encoding the G protein αo subunit, have been implicated in a pathogenic condition characterized by seizures, movement disorders, intellectual disability, and developmental delay (GNAO1 disorder). However, the effects of these mutations on G protein structure and function are unclear. Here, we report the effects of 55 mutations on Gαo conformation, thermostability, nucleotide binding, and hydrolysis, as well as interaction with Gβγ subunits, receptors, and effectors. Our effort reveals four functionally distinct groups of mutants, including one group that sequesters receptors and another that sequesters Gβγ, both acting in a genetically dominant manner. These findings provide a more comprehensive understanding of disease-relevant mutations and reveal that GNAO1 disorder is likely composed of multiple mechanistically distinct disorders that will likely require multiple therapeutic strategies.
In brief
In recent years, de novo mutations in cell signaling proteins have been linked to neurodevelopmental disorders. Knight et al. examine dozens of unique mutations in Gαo, representing the most abundant G protein in the brain, and determine that they fall into at least four mechanistically distinct groups.
Graphical Abstract
INTRODUCTION
G protein signaling involves a network of sensors, transducers, and effectors that regulate nearly all biological processes. G protein-coupled receptors (GPCRs) are sensors that recognize extracellular stimuli such as hormones, neurotransmitters, environmental cues, and drugs. When a GPCR detects a stimulus, it initiates signaling by activating heterotrimeric G proteins that couple to downstream effectors. In total, humans express over 800 GPCRs, 16 Gα proteins, and a multitude of effector enzymes and ion channels, allowing them to fine-tune their response with the appropriate timing, intensity, and functional specificity.
Activation of a G protein is part of a cycle that is composed of (1) GDP-bound, (2) nucleotide-free, and (3) GTP-bound states. The GDP-bound Gα protein is inactive and associates with Gβγ. The nucleotide-free form associates with receptor as well as with Gβγ. Upon activation, the G protein α subunit binds to GTP, which initiates dissociation of the G protein from the receptor and, further, dissociation of the Gα and Gβγ subunits.1,2 The GTP-bound Gα monomer and Gβγ subunit dimer then activate downstream effector molecules, which elicit specific physiological responses. GTP hydrolysis transitions the Gα subunit back to an inactive, GDP-bound heterotrimer, and this process is accelerated by regulators of G protein signaling (RGS) proteins.1 The G protein remains inactive until the cycle is initiated once again.
G protein-coupled signaling pathways have been heavily studied and pharmacologically manipulated; however, the consequences of mutations that impair signaling remain poorly understood. Moreover, changes to any part of the signaling network can result in mistimed, overactive, or underactive cellular responses that contribute to human disease. To date, most studies relating G protein mutations to disease have been in the context of cancer.3 Further, cancer-causing G protein mutations are often found at one of two conserved amino acids, both required for proper GTPase activity. For example, substitutions at the catalytic glutamine lead to overactive Gαq signaling and have been implicated in uveal melanoma4 (reviewed in Arang and Gutkind3). Despite driving the same enzymatic changes, mutations at the catalytic glutamine result in varied structural alterations and physiological responses, depending on the amino acid substitution and the affected Gα subtype.5,6
In more recent years, de novo mutations in receptors, G protein subunits, and effectors have been linked to a neurological disorder called early infantile epileptic encephalopathy, also known as developmental epileptic encephalopathy. This condition appears early in life and manifests as developmental delays, movement disorders, and epileptic seizures that are often refractory to treatment.7 Of the various mutations associated with this disorder, whole exome sequencing revealed that a subset of patients have alterations in GNAO1,8 encoding Gαo, which is the most abundant Gα subtype in the brain and central nervous system.9 Previous small-scale efforts to characterize these GNAO1 disorder mutants have revealed that some are nonfunctional, some are gain-of-function, and some block signaling altogether.10–12 A subset of the mutations occurs at conserved sites that are involved in important protein functions such as nucleotide binding and subunit dissociation; therefore, we anticipated that these mutations would block one or more steps of the G protein activation cycle. On the other hand, a substantial number of mutations occur in regions that have no obvious role in enzyme activity or protein-protein interactions. Here, we sought to determine the mechanistic properties of GNAO1 disorder-associated mutations. Our analysis indicates that there are at least four distinct subclasses of functional impairment and that they will likely require multiple and distinct therapeutic strategies.
RESULTS
Gα subunits consist of two lobes, known as the Ras-homology domain (RHD) and the all-helical domain; between these domains is a nucleotide binding cleft. Functionally, the RHD is largely responsible for the enzymatic capacity of Gα, as well as its interactions with receptors, Gβγ, regulators, and effectors.1 Thus, mutations within the RHD are most likely to have important consequences for G protein enzymatic activity or coupling interactions. For these reasons, we chose to focus our analysis of GNAO1 disorder mutations to those within the RHD of Gαo, and in particular the most predominant splice-form of the protein (GαoA). To that end, we compiled a list of mutations reported (as of May 2022) in the Online Mendelian Inheritance in Man database (OMIM: 615473, 617493), as well as others reported in the literature: GαoS47G,13 GαoQ52P,14 GαoN270H,12 and GαoF275S.15 We chose Gαo RHD mutations classified as “pathogenic” or “likely pathogenic” and those at sites conserved between human Gαo and the yeast Saccharomyces cerevisiae Gα protein Gpa1; mutations in highly conserved residues are the most likely to have important mechanistic consequences. Gpa1 is structurally similar to human Gαi/o-type proteins; moreover, there are several Gpa1 mutants that have well-established defects in receptor, nucleotide, or Gβγ binding, for use as controls.16,17
Genetic profiling analysis of Gα mutants in yeast
The yeast pheromone pathway is composed of a single receptor and G protein (reviewed in Dohlman and Thorner18). Following dissociation from Gα, Gβγ triggers cell-cycle arrest, thus providing a simple and direct readout of G protein functionality (Figure 1A). In humans, GNAO1 disorder-associated mutations are heterozygous; one copy of Gαo is functional and the other is mutated. Accordingly, we introduced 55 mutations, all corresponding to those found in patients, into GPA1 and expressed these in an otherwise wild-type haploid yeast strain. As controls, we introduced a second copy of wild-type GPA1, or the empty vector, as well as two previously characterized Gα mutants: Gpa1N388D, which binds poorly to nucleotide and thus forms a stable complex with receptor,16 and Gpa1E364K, which remains permanently bound to Gβγ subunits.19 We then tested the ability of each variant to regulate Gβγ release, as indicated by the growth inhibition, or “halo,” surrounding a source of agonist.
Figure 1. Genetic profiling analysis of Gα mutants.
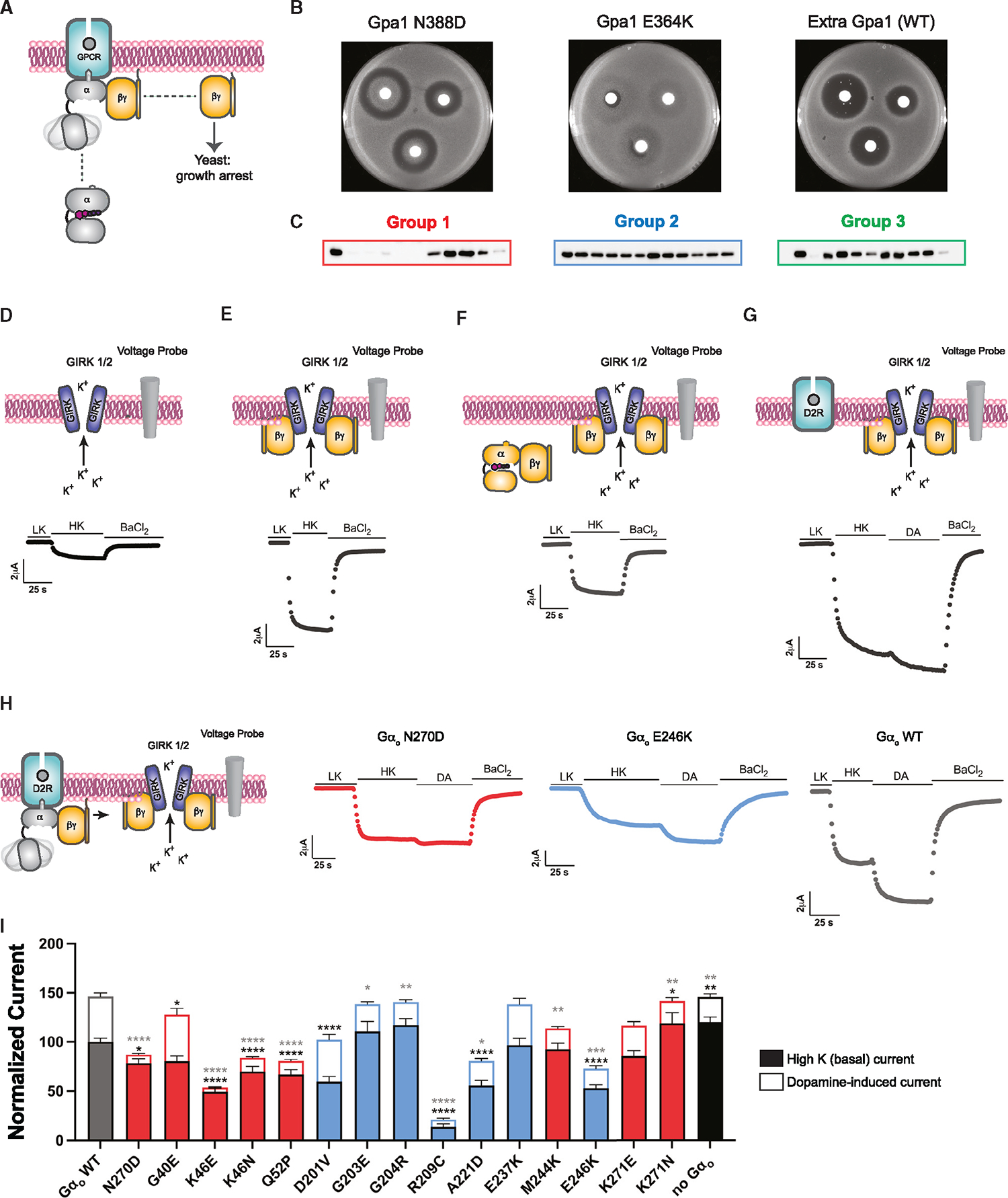
(A) Gβγ-mediated growth arrest in yeast.
(B) Growth inhibition (halo) assay for the yeast Gα protein Gpa1 and mutant controls Gpa1N388D and Gpa1E364K.
(C) Immunoblot of Gpa1 wild type (leftmost lanes) and mutants.
(D) Basal GIRK channel opening and representative TEVC recording in X. laevis oocytes expressing GIRK1/2 following treatment with high potassium solution (HK) and barium (BaCl2).
(E) Gβγ-mediated GIRK activation and representative TEVC recording in cells expressing GIRK1/2 and Gβ1γ2.
(F) Gβγ-mediated GIRK activation and representative TEVC recording in cells expressing GIRK1/2, Gβ1γ2, and Gαo wild type.
(G) Dopamine-induced GIRK activation and representative TEVC recording in cells expressing GIRK1/2, Gβ1γ2, and D2 dopamine receptor following treatment with dopamine (DA).
(H) Dopamine-induced GIRK activation and representative TEVC recording in cells expressing GIRK1/2, Gβ1γ2, Gαo wild type or mutants, and D2 dopamine receptor.
(I) Normalized currents induced by High K (closed bars) and dopamine (open bars) for group 1 (red) and group 2 (blue). Data are means ± SEM from two independent experiments, each performed with 5–6 measurements each. Black asterisks indicate statistical significance (two-way ANOVA with Sidak’s multiple comparisons test) for High K currents. Gray asterisks indicate statistical significance for dopamine-induced currents. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). See Data S1.
As shown in Figure 1B, expression of wild-type Gpa1 produced clear halos, similar to cells with empty vector (Figure S1). In contrast, our Gpa1E364K control blocked the growth arrest response, in accordance with its ability to sequester Gβγ, while Gpa1N388D prevented growth arrest near the source of the agonist, indicating receptor-dependent stabilization of the mutant protein and sequestration (instead of release) of Gβγ at the highest (but not the lowest) concentrations of ligand.16,17 Of the 55 GNAO1 disorder mutants tested, approximately half had no effect on growth. The remainder produced one of two distinct and genetically dominant phenotypes (Figure S1). The first, designated group 1, phenocopied Gpa1N388D; thus, we hypothesized that these mutants bind poorly to nucleotide and therefore remain associated with receptors (agonist-bound) and Gβγ subunits. The second, group 2, phenocopied Gpa1E364K; thus, we hypothesized that these mutants remain associated with Gβγ.16,17 Because group 1 and group 2 variants inhibit signaling in the presence of wild-type Gα, they may be regarded as dominant negative mutants.20 As detailed below, the remaining mutants were further subdivided as groups 3 and 4.
To ensure that sequestration of Gβγ was not a consequence of Gα protein overexpression, we performed immunoblotting on myc-epitope tagged variants of Gpa1 in vivo. As anticipated,16,17 Gpa1N388D expressed poorly while Gpa1E364K resembled that of wild type. Similarly, most of the group 1 mutants expressed poorly while group 2 mutants expressed normally (Figure 1C; Figure S1). Immunoblotting results for the remaining mutants were varied; most exhibited normal expression levels and none exceeded that of wild type. Thus group 1 mutants dampen signaling in spite of, rather than because of, differences in protein expression.
GIRK channel activation by Gαo mutants
Our yeast experiments indicated that some, but not all, of the 55 mutants sequester Gβγ subunits. Because these mutations were identified in patients with a neurological condition, we sought to determine how mutant Gαo proteins affect a relevant effector system. In the human brain, release of Gβγ by Gαo activates G protein-gated inwardly rectifying potassium (GIRK) channels.21 The resulting channel activation can be quantified by two-electrode voltage-clamp recordings, thereby providing another measure of G protein heterotrimer dissociation.
As control conditions, we introduced GIRK1/2 heteromers into Xenopus laevis oocytes (Figure 1D), with Gβγ (Figure 1E), with both Gα and Gβγ (Figure 1F), or with Gαβγ and the D2 dopamine receptor (Figure 1G). To verify function, we treated the cells with high potassium and barium to stimulate and inhibit channel activity, respectively. The channel-only condition provided a measure of basal opening (Figure 1D). The expression of free Gβγ subunits to the system increased the probability of channel opening (Figure 1E). The expression of Gβγ and wild-type or mutant Gαo resulted in a lower probability of channel opening in comparison with Gβγ alone, indicating efficient G protein heterotrimer assembly (Figure 1F). Further, expression of exogenous Gαo dampened channel opening due to competition for Gβγ binding.
We then performed the same experiment in cells expressing the D2 dopamine receptor and activated with 10 μM dopamine. Without exogenous Gαo expression, dopamine was able to evoke increased current, but only modestly (Figure 1G). We expected that poorly expressed mutants or those that do not bind Gβγ efficiently would mimic the condition in Figure 1G. For wild-type Gαo, dopamine activation caused a large increase in current flux (Figures 1H and 1I), indicating proper Gβγ release and binding to GIRK channels. Our group 1 Gαo N270D and group 2 Gαo E246K controls showed reduced response to potassium (Figure 1I) and minimal increases in dopamine-induced current. Nearly all of the representative group 1 (red) mutants prevented activation by Gβγ (Figures 1I and Data S1). GαoR209C (group 2) was the best performing Gβγ sequestering mutant in our GIRK channel assay, followed by GαoK46E (group 1); neither mutant responded to dopamine. These data extend and complement the yeast assays presented above. However, neither the yeast nor the channel assay distinguishes between the exogenous Gαo mutant and the endogenous wild-type Gα. If the introduced mutant is poorly expressed in cells, or in an affected individual, the effects of endogenous wild-type Gα are likely to predominate.
Gαo protein activation in human cells
The data provided above indicate that group 1 and group 2 mutants sequester Gβγ even in the presence of activated receptors. As a third test of mutant function, we measured Gβγ dissociation using a bioluminescence resonance energy transfer (BRET) assay.22 We transfected wild-type or mutant variants of Gαo tagged with Renilla luciferase (donor) and Gβγ fused to GFP (acceptor). As long as the subunits are in close proximity, the donor is able to excite the acceptor (Figure 2A). Thus, and in contrast to the yeast and channel assays, BRET directly measures Gα-Gβγ association and dissociation.
Figure 2. Gαo protein activation in human cells.
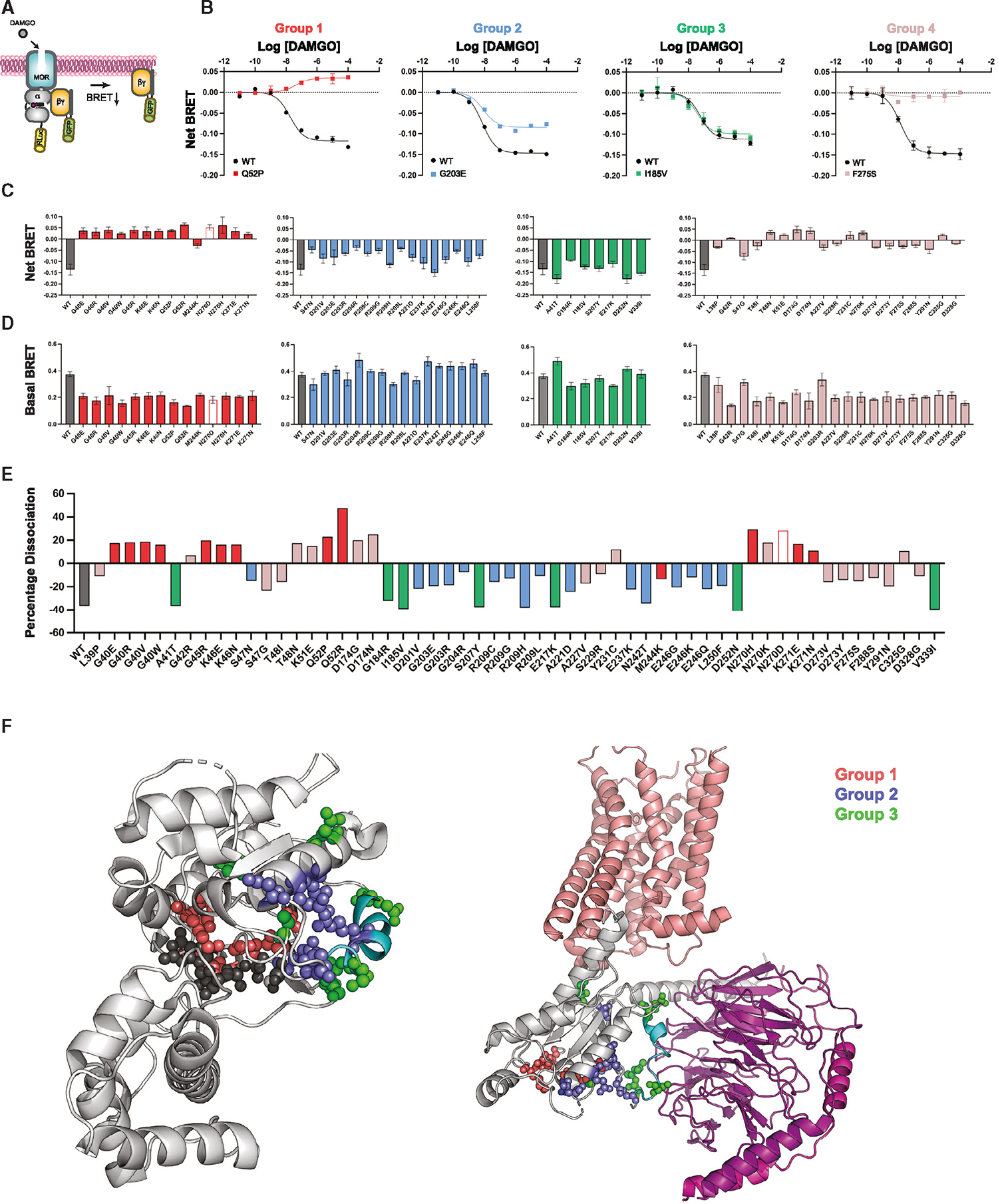
(A) Agonist-induced Gβγ dissociation in the TRUPATH BRET system.
(B) Representative concentration-response measurements, presented as fold decrease in dynamic range (dissociation, net BRET), for group 1 (GαoQ52P), group 2 (GαoG203E), group 3 (GαoI185V), and group 4 (GαoF275S).
(C) Agonist-induced net BRET for group 1 (red), group 2 (blue), group 3 (green), and group 4 (mauve).
(D) Basal BRET, as in (C).
(E) Percentage dissociation, calculated as induced BRET divided by basal BRET.
(F) Structural representations of variants in Gαo (PDB: 3C7K, light gray) and the receptor (salmon) and G protein α (light gray) and βγ (purple) complex (PDB: 7EJ0). Nucleotide (dark gray), tetrafluoro aluminate (dark gray), magnesium ion (light gray), Switch II (cyan), group 1 (red), group 2 (blue), and group 3 (green). Data are means ± SD from three independent experiments, each performed with two measurements each. See Data S2.
Similar to our findings presented above, the BRET experiments produced distinct signaling outcomes for the Gαo mutants. Whereas agonist treatment led to robust dissociation of Gβγ from wild-type Gα, the group 1 mutants exhibited no dissociation, and in nearly all cases exhibited increased association (Figures 2B and 2C, red). This indicates that group 1 mutants, and by extension Gβγ, are recruited to agonist-bound receptors. Notably, the increase in BRET was only observed for cells expressing Gg9 but not for those expressing Gγ8 (Figure S2 and Data S2). These functional differences may result from differences in rates of Gβγ dissociation from the plasma membrane, a process that is governed by the nature of the Gγ subtype.23,24 Gγ9, which is farnesylated, dissociates from the plasma membrane rapidly while Gγ8, which is geranylgeranylated, dissociates more slowly.25 The group 2 mutants dissociated from Gβγ, but did so to a lesser extent than wild type (Figures 2B and 2C, blue). This indicates that both group 1 and 2 mutants bind in a sustained manner to Gβγ but are likely to do so by distinct mechanisms. The remaining mutants either dissociated normally (hereafter group 3, green), or appeared to be nonfunctional (hereafter group 4, mauve). In accordance with mutant expression levels, basal luciferase activity was lower for groups 1 and 4 than for groups 2 and 3 (Figure 2D). Notably, the mutants from each group clustered in distinct regions of the protein (Figures 2E and 2F). Thus, our next objective was to relate these differences to known molecular properties of the Gαo protein.
Structural modeling and in silico rationalization for classification of Gαo mutants
The mutations considered here are all within the RHD of Gα. Structurally, the RHD has five α helices, six β sheet strands, and several sequence elements that are crucial for maintaining enzymatic activity and structural integrity. These elements include three conserved “switch” regions (Switch I, II, and III) that conformationally rearrange upon GTP binding. Specifically, Switch I and III contain residues critical for nucleotide hydrolysis, whereas Switch II (an extension of the nearby α2 helix) and the α3 helix contain residues critical for Gβγ and effector binding.26–28
As we anticipated, many of the GNAO1 disorder mutations cluster in conserved regions of Gαo (Figure 3A). All of the group 1 mutations cluster around the nucleotide binding pocket (Figure 3B), and particularly in a conserved region called the phosphate binding loop (P loop). These include four amino acid substitutions at the Gly-40 position (G40E, G40R, G40V, and G40W) as well as substitutions in neighboring residues (G45R, K46E, Q52P, and Q52R). With one exception, the remaining group 1 mutations are in a conserved loop, known as Gbox4, bridging the α4 and α5 helices; these include N270H, K271E, and K271N (our positive control mutant, Gpa1N388D corresponds to Asn-270 in Gαo). Whereas the P loop contains positively charged residues that stabilize the negatively charged phosphates of GDP and GTP, Gbox4 residues surround the guanosine head group. Therefore, we anticipated that group 1 mutations would have reduced binding to nucleotides and, consequently, increased binding to receptors. This is important because the agonist-bound receptor stabilizes the nucleotide-free form of the G protein and, conversely, the nucleotide-free G protein confers increased affinity of receptors for agonists.29,30
Figure 3. Sequence covariation and structural modeling of Gαo variants.
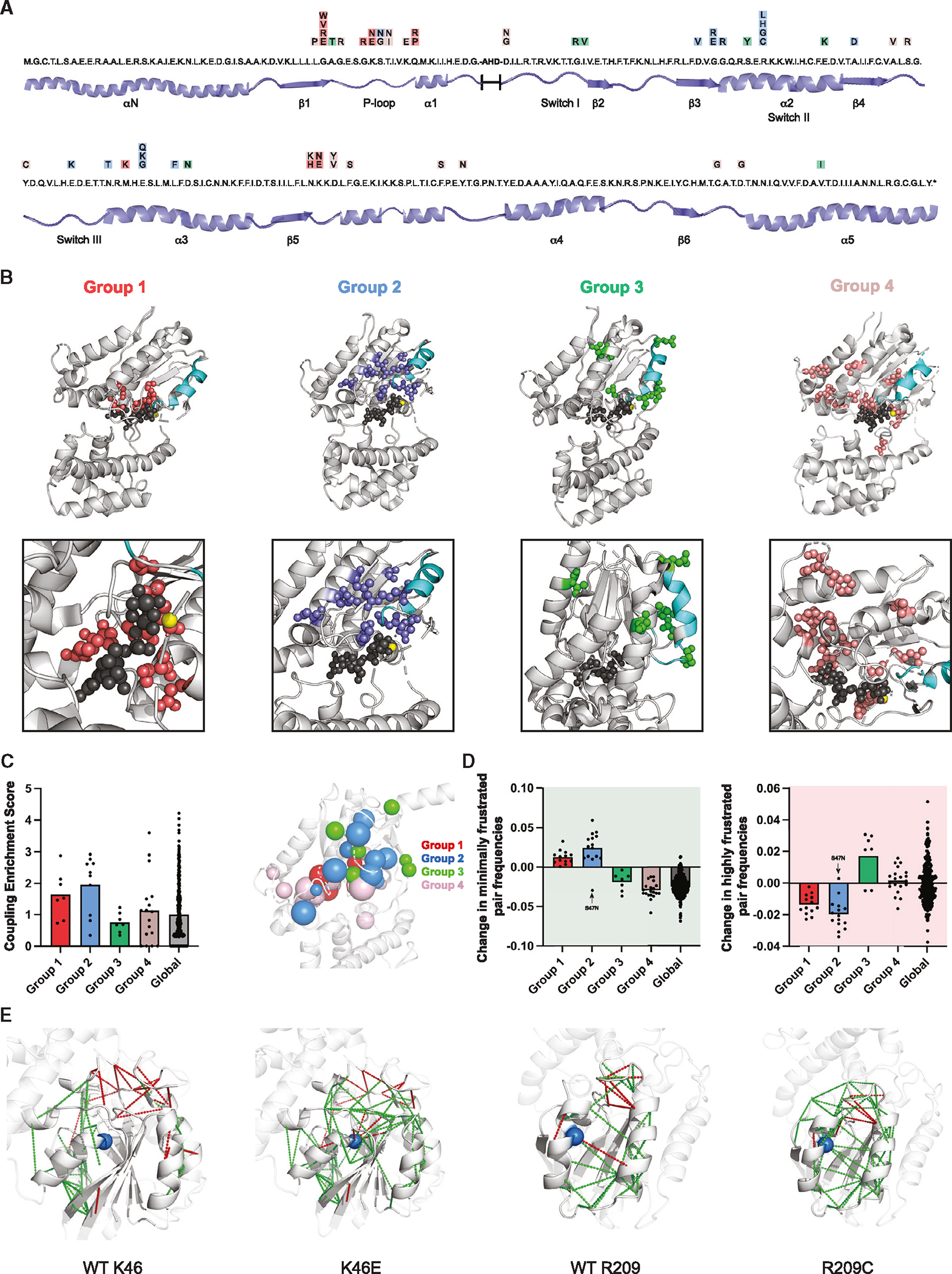
(A) Linear amino acid sequence of the RHD of human Gαo with variants indicated in colored boxes: group 1 (red), group 2 (blue), group 3 (green), and group 4 (mauve).
(B) Location of variants on the structure of Gαo (PDB: 3C7K). Nucleotide (dark gray), tetrafluoro aluminate (dark gray), magnesium ion (yellow), Switch II (cyan), group 1 (red), group 2 (blue), group 3 (green), group 4 (mauve).
(C) Sequence co-evolution analysis to identify residue positions that show high degrees of covariation with other residues in the sequence (left). Spherical representation of co-evolution scores for the Gαo variants (right). The larger the sphere, the larger the co-evolution score. Group 1 (red), group 2 (blue), group 3 (green), group 4 (mauve).
(D) Frustration analysis of residues that adopt a suboptimal conformation and/or energetic state in a protein structure, and the percentage difference in the number of highly (red) and minimally (green) frustrated pairs, in all the variants. Arrow, S47N variant having unique properties.
(E) Pattern of the residue pairs that are minimally (green lines) and highly (red lines) frustrated in the top-performing variants from group 1 (K46E) and group 2 (R209C), compared with the wild type.
Group 2 mutations are located at the previously characterized G-R-E triad (Gly-204, Arg-209, Glu-246).17 The triad is located between the α2 and α3 helices (Figure 3A), which upon GTP binding rearrange to liberate Gβγ from Gα subunits. Mechanistically, the triad works like an ionic lock; with GTP binding the triad glycine backbone amine rotates toward the terminal phosphate of GTP, thereby causing Switch II residues to rotate away from Gβγ. The Arg forms a salt bridge with the Glu in what has been described as a molecular “hasp” that locks Gα in the active conformation.19,31,32 This molecular tug-of-war results in heterotrimer dissociation and the formation of a tightly associated α2-α3 interface within Gα that is critical for effector binding (Figure 3B). Notably, two of the triad residues are considered hotspots for the disease; multiple substitutions at the triad-Arg (R209C, R209G, R209H, R209L) and triad-Glu (E246G, E246K, E246Q) have been reported in GNAO1 disorder. Other group 2 mutations are part of the triad (G204) or located nearby at sites that include a third hotspot mutation (G203E) as well as D201V, E237K, and L250F (Figure 3A). Our previous studies showed that mutations at the triad-Glu and -Arg impede Gβγ release,17,19 even in the GTP-bound state.17 Thus, we anticipated that group 2 mutants would likewise prevent conformational rearrangements required for Gβγ release.
In contrast to groups 1 and 2, group 3 and group 4 mutations are distributed throughout the protein. With one exception, group 3 residues are located at the surface of Gαo (Figure 3B), and harbor conservative substitutions (e.g., Ala to Thr, Ile to Val, Asp to Asn) that are unlikely to disrupt interactions critical for G protein function or stability. Most of the group 4 mutants are buried within Gαo, in regions likely important for maintaining structural integrity (Figure 3B). As expected, the average solvent accessibility surface area for group 4 variants was substantially lower than the average for all residues (~18.3 vs. 43.2 Å2).33,34 Based on these observations, we anticipated that mutants in group 3 would retain enzymatic activity, whereas mutants in group 4 would not.
To better understand the structural basis of the mutant groupings, we performed three types of computational analysis: sequence-based, structure-based, and molecular dynamics-based. First, we performed sequence covariation analysis for each of the variant residue positions in Gαo. This method uses multiple sequence alignments of a protein family and identifies pairs of residue positions that evolved together. If two residues are functionally or structurally linked, for example through interactions that help to preserve enzyme activity, then a mutation in one residue is likely to lead to compensatory mutations in the interacting residue.35,36 Such co-dependency in mutational patterns can be an indication of physical interaction between amino acids in the structure. Relative to the average co-evolution score of all residues, residues in groups 1 and 2 had high enrichment scores and those in group 3 had low enrichment scores. Group 4 enrichment scores mirrored those of the protein as a whole (Figure 3C). This indicates that a subset of residues affected in GNAO1 disorder play an important role in maintaining the structure of Gαo, and are likely to impair nucleotide binding, hydrolysis activity, or both.
To investigate whether the disease mutations are likely to be important for Gαo structure or for Gαo function, we analyzed the “frustration” patterns of each variant. Residue positions have evolved not only to confer stability but also to confer function.37,38 Frustration is the suboptimal energy state in which certain protein residue positions exist. In other words, frustration arises when certain residues do not adopt the optimal conformation or energetic state39; high frustration enables higher activity and, as a consequence, lower stability. Indeed, previous studies of disease-associated variants of other proteins have shown significant changes in the frustration pattern relative to wild type.40 Accordingly, we calculated the energy change, relative to each of the 55 disease mutants and wild type, for all possible pairs of amino acid substitutions. A residue pair is considered highly frustrated if a mutation of one of the residues leads to a more favorable pairwise interaction energy than that of wild type and considered minimally frustrated if the mutation results in a less favorable interaction energy. As shown in Figure 3D, we observed an increase in the number of minimally frustrated residue pairs and a decrease in highly frustrated residue pairs, particularly for group 1 and 2 variants. These differences are illustrated in Figure 3E for top-performing mutants in group 1 (K46E) and group 2 (R209C). In comparison to wild type, the minimally frustrated residue pairs (shown in green) are predicted to be less flexible and impose increased structural integrity, whereas the highly frustrated pairs (shown in red) are predicted to be more flexible and thus accommodate conformational changes. The decrease in local frustration observed in groups 1 and 2 is corroborated with structural modeling indicating a loss of polar contacts from these mutations, either within the nucleotide binding pocket (group 1) or the extended G-R-E triad network (group 2). These polar contacts are required for nucleotide-mediated release of Gβγ; thus, the decreased local frustration conferred by these mutations prevents the protein flexibility required to undergo conformational changes to release Gβγ. In comparison with mutations in groups 1 and 2, non-mutated residues show wider changes in frustration and also in the opposite direction when replaced with alanine (Figure 3D). However, we do not know of any disease-associated mutations at these residues, or of their potential physiological consequences.
While co-evolution and frustration analysis showed clear and consistent differences between mutant and wild-type proteins, neither method distinguished group 1 from group 2. Therefore, we performed molecular dynamics (MD) simulations of the GDP-bound Gαo heterotrimer and the GTP-bound Gαo monomer and looked for differential residue-nucleotide contacts for the variants in each group (Figure S3A). In this approach, a residue is considered to be in contact with the nucleotide if the heavy atoms of said residue and the nucleotide fall below a stipulated distance cutoff (https://getcontacts.github.io/). The frequency of contacts is determined by the percentage of snapshots in the MD trajectory where said residue makes contact with the nucleotide. As expected, nearly all residue positions in group 1 maintained contact with GDP and GTP. Except for Ser-47 and Ala-41, no other residues in group 2 or group 3 formed contacts with either nucleotide. Residues in group 4 had mixed results. These findings are concordant with our structural modeling results, where group 1 residues surround the nucleotide binding pocket; thus, we hypothesized that those substitutions would prevent nucleotide binding. Group 2 residues are farther from the nucleotide binding pocket and therefore unlikely to affect nucleotide binding. Thus, while frustration analysis showed similarities between groups 1 and 2, consistent with their shared ability to sequester Gβγ, MD simulations suggested differential mechanisms of Gβγ sequestration, one acting indirectly by preventing nucleotide binding (group 1) and the other acting directly by preventing Gβγ dissociation (group 2). In the next section we test these predictions experimentally, through direct measurements of nucleotide binding and hydrolysis.
Biochemical analysis of Gαo mutants
Our analysis in yeast, oocytes, and human cells revealed a subset of Gα mutants that are likely to sequester receptors (group 1) and Gβγ (groups 1 and 2); other mutants function like wild type (group 3), while the remainder appear to be nonfunctional (group 4). Our structural modeling provided mechanistic explanations for the increased association with receptor and Gβγ. Specifically, we postulated that group 1 mutants form a nonproductive complex with receptors (and Gβγ) due to a loss of nucleotide binding, while group 2 mutants fail to undergo GTP-induced subunit dissociation. To test our predictions, we expressed and purified all 55 mutant proteins, as well as the control variants and wild-type Gαo, and measured stability (Figure 4) and enzymatic activity (Figure 5).
Figure 4. Thermostability analysis of Gαo variants.
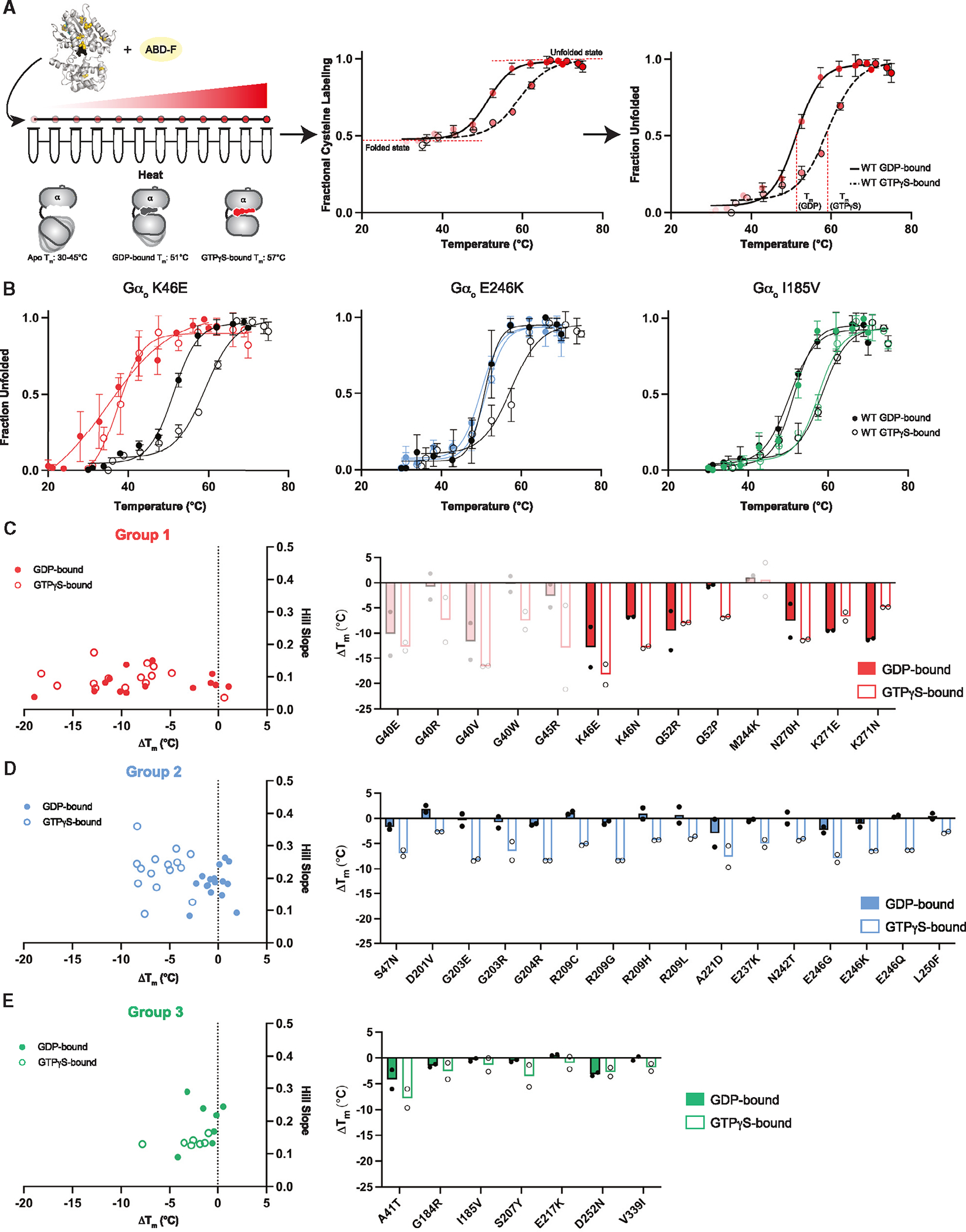
(A) Representative fQCR thermostability measurements for purified wild-type Gαo equilibrated in GTPγS (dashed line) or GDP (solid line), without (middle) and with (right) normalization. Tm values were quantified by fitting a two-state model of thermal unfolding.
(B) Protein melting curves for representative group 1 (GαoK46E)), group 2 (GαoE246K), and group 3 (GαoI185V) variants.
(C) Comparison of Hill Slope and ΔTm comparing wild-type Gαo and Gαo group 1 variants bound to GDP (closed circles) or GTPγS (open circles). Low-confidence measurements for mutants that were poorly expressed are indicated with faint bars.
(D) Comparison of Hill Slope and ΔTm comparing wild-type Gαo and Gαo group 2 variants.
(E) Comparison of Hill Slope and ΔTm comparing wild-type Gαo and Gαo group 3 variants. Data are means of six measurements from two independent purifications. Error bars, ± SD. See Data S3.
Figure 5. Enzymatic function of Gαo variants.
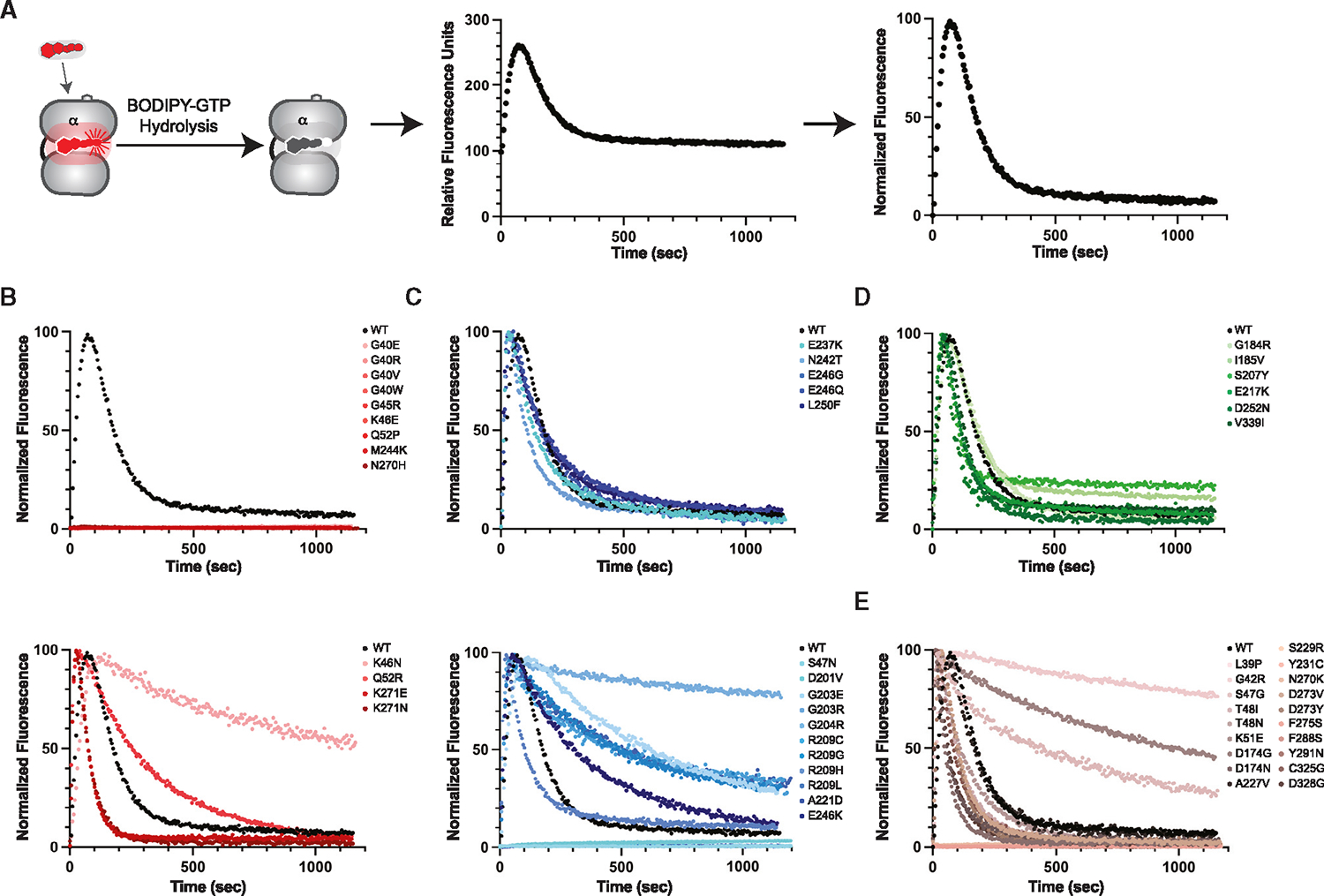
(A) GTP binding and hydrolysis (left) by purified wild-type Gαo equilibrated in BODIPY-GTP, without (middle) and with (right) normalization.
(B) BODIPY-GTP binding and hydrolysis by purified group 1 variants, without (top) and with (bottom) detectable catalytic activity.
(C) BODIPY-GTP binding and hydrolysis by purified group 2 variants, with wild-type-like (top) and with slow (bottom) rates of hydrolysis.
(D) BODIPY-GTP binding and hydrolysis by purified group 3 variants.
(E) BODIPY-GTP binding and hydrolysis by purified group 4 variants. Data are representative of six measurements from two independent purifications. See Data S3.
To measure nucleotide-dependent changes in protein conformation, we conducted fast determination of quantitative cysteine reactivity (fQCR) assays.41 In this method we quantify the extent of cysteine labeling, as a function of temperature, of protein equilibrated with either GDP or GTPγS (Figure 4A). The unfolding curves were normalized by setting the minimum fluorescence value to 0 and the maximum to 1 (Figure 4A, compare middle and right). From the resulting protein unfolding curves we calculated the Hill slope and the difference (mutant vs. wild type) in melting temperature (ΔTm) when bound to GDP or GTPγS (Figure 4B; Data S3). These values revealed nucleotide-dependent changes in protein stability and cooperativity, respectively, that differ between groups.
For wild-type Gαo, the Tm was 6°C lower when bound to GDP as compared with GTPγS (Figure 4A). This is consistent with previous studies showing that Gα proteins are more structurally rigid in the GTP-bound state.17,41–43 We anticipated that the thermostability would be further reduced for the group 1 (presumably nucleotide-free) mutants.44 As expected, we observed an uncooperative melting curve and substantially lower Tm for the representative group 1 mutant GαoK46E (Figure 4B), as well as for nearly all of the other group 1 mutants (Figure 4C; low-confidence measurements for mutants that were poorly expressed are indicated with faint bars). Thus, we infer that the group 1 mutants are unstable and bind poorly, if at all, to nucleotide.
In comparison with wild type, the representative group 2 mutant GαoE246K exhibited reduced stability when bound to GTPγS, but showed little difference when bound to GDP (Figure 4B). We observed the same properties for the rest of the group 2 variants (Figure 4D), which we predict impose a GDP-like conformation even when bound to GTPγS.17 Group 3 mutants, represented by GαoI185V, exhibited thermostability profiles that resemble wild-type protein (Figure 4B). This indicates that group 3 mutants are able to bind to GDP and GTPγS, as well as to undergo the corresponding conformational changes (Figure 4E). The majority of group 4 mutants exhibited poor protein stability (Figure S4A).
Finally, to investigate the effect of the G protein mutations on catalytic activity, we measured the increase in fluorescence upon binding of BODIPY-GTP and the subsequent decrease resulting from GTP hydrolysis (Figure 5A).45,46 In this method, the BODIPY fluorophore is quenched while in solvent and increases in fluorescence quantum yield upon binding to Gαo. Further, changes in fluorescence quantum yield are a direct consequence of the electrostatic environment of the nucleotide binding pocket. For proper comparison of mutants, we corrected for these quantum yield effects and normalized the data by setting the lowest measured value to 0 and the highest to 100 (Figure 5A, compare middle and right). In particular, this normalization accounts for unquenching of the BODIPY fluorophore that may result from any mutations that alter (directly or indirectly) solvent accessibility at the nucleotide binding pocket.
As predicted, most of the group 1 mutants failed to bind to BODIPY-GTP (Figure 5B, top), consistent with position proximity to the nucleotide binding pocket and low Tm values for both nucleotide-bound states (Figure 4C). Contrary to our predictions, GαoK46N, GαoK271E, GαoK271N, and GαoQ52R were all capable of binding and hydrolyzing BODIPY-GTP (Figure 5B, bottom). This was surprising to us because Lys-46 binds to and stabilizes the β phosphate of GDP and GTP, and we anticipated that any loss of the stabilizing positive charge would disfavor nucleotide binding. Similarly, we predicted that replacement of Lys-271 would be incompatible with nucleotide binding, due to the proximity of this residue to the guanosine ring of GTP and GDP. Finally, Q52R seemed likely to disrupt the α1 helix and, consequently, perturb the P loop located near the nucleotide binding pocket. One potential explanation is that these four mutations cause a decoupling of molecular arrangements that are required for binding and unbinding of both nucleotide and receptor. In summary, group 1 monomers are destabilized and most lack enzymatic function, presumably due to reduced nucleotide binding.
In contrast to group 1, most of the group 2 mutants retained their ability to bind and hydrolyze GTP (Figure 5C). As determined by yeast and GIRK assays, GαoD201V was an outlier because it failed to bind nucleotide in vitro; the GαoA221D mutant behaved similarly (Figure 5C, bottom). As expected, GαoS47N also failed to bind or hydrolyze BODIPY-GTP; replacement of Ser-47 with Cys was shown previously to disrupt magnesium binding,47,48 which is in turn required for high-affinity GTP binding.1 In contrast, the S47G substitution (group 4) does not introduce a positive charge and likely preserves magnesium binding.11 For these reasons we have classified GαoS47N, but not GαoS47G, as a group 2 mutant. Thus, the majority of group 2 mutants bind and hydrolyze GTP but fail to release Gβγ. Group 3 mutants were fully functional (Figure 5D). Group 4 variants were unstable in vitro, and lacked activity in cells (Figure 2), although some retained the ability to bind and hydrolyze GTP under more favorable assay conditions (Figure 5E). The differences in thermostability and enzyme activity are further visualized in Figure S4.
DISCUSSION
To develop effective treatments for any genetic disorder, we must understand how the underlying mutations affect function at the molecular, cellular, and organismal levels. Here, we address this challenge by considering dozens of G protein mutations, all linked to a neurological disorder in humans, and by determining how they alter function at the protein and cellular levels. First, we performed a yeast screen to functionally classify all 55 Gα mutants, and then characterized the affected proteins by expression, subunit dissociation, and effector activation assays in cells. Further, we conducted MD simulations and structural modeling, in conjunction with measures of protein thermostability, nucleotide binding, and hydrolysis. Our effort allowed us to identify at least four distinct categories of Gαo mutants.
Taken together, our biochemical analysis provides a mechanistic explanation for the defects in Gαo mutant interactions with receptors and Gβγ. Broadly speaking, group 1 mutants lack enzyme function yet block receptor activation. Group 2 mutants retain enzyme function but fail to release Gβγ. Additionally, both groups show a decrease in the number of frustrated residue pairs, indicative of attenuated function. Both group 1 and 2 variants are genetically dominant and therefore classified as dominant negative alleles. Group 3 mutants are recessive in yeast and appear to have normal GTPase activity (Figure 5D) and release from Gβγ (Figure 2C). Group 4 mutants appear nonfunctional in vivo.
Based on prior mechanistic studies ofGpa1N388D and Gαi2N270D, we proposed that group 1 mutants bind poorly to nucleotide and, consequently, form a nonproductive complex with receptors and Gβγ.16,49 Based on prior studies of the G-R-E triad, we proposed that group 2 mutants bind strongly to the Gβγ subunits, regardless of bound nucleotide.17 Our experiments confirmed these important differences in biochemical activity and, in combination with cellular functional assays,11,12 revealed substantial heterogeneity in processes leading to GNAO1 disorder.
Our analysis also resolves discrepancies in past efforts to classify the GNAO1 disorder mutants. A prior study used adenylyl cyclase measurements to classify mutants as loss-of-function or gain-of-function.50 A later study argued that cAMP abundance is not a valid measure for classifying Gαo mutations and instead used BRET to compare mutants.11 That study identified a group of dominant negative mutants—some of which trap activated receptors—and a smaller group of loss-of-function mutants. Our analysis confirms and substantially extends the work by mechanistically differentiating two subcategories of dominant negative mutants and a large category of loss-of-function mutants.
To better interpret our laboratory-based studies, we performed unsupervised clustering using the experimental and computational properties of the variants. In particular, we performed unsupervised clustering using the data presented in Figures 2, 3, 4, and 5 (Figure S5). Twelve (of 13) of the group 1 variants fell in a single cluster. Twelve (of 16) group 2 variants were in another cluster. The S47N variant has unique properties resulting from a loss of magnesium binding. Based on the properties that we have measured or calculated, group 3 and group 4 variants formed several subclusters that require further investigation. The combination of experimental and computational data yielded better clustering of the four groups than that provided by either approach alone. Thus, the experimental and computational data provide complementary information needed to classify disease-associated mutants.
Looking forward, we are currently expanding our analysis to consider whether mutants from group 3 affect interactions with binding partners other than the receptors and Gβγ. Also of interest are mutations that occur in the all-helical domain of Gαo, as well as in receptors, Gβγ, adenylyl cyclase, ion channels, and other effector proteins, many of which have functional characteristics similar to those of GNAO1 disorder.7
Another goal is to expand analysis of GNAO1 disorder using animal models. Previously published studies have introduced GNAO1 variants in mice and, generally speaking, they exhibit impaired limb coordination and dysregulated cAMP signaling.11–13,50,51 Those prior studies have succeeded in demonstrating important functional consequences of mutations at the organ or organism level. However, studies in mice have so far focused on group 2 and group 4 mutants. Our analysis points to a need for expanded studies that include additional mutant variants, particularly those from groups 1 and 3. These and other ongoing investigations will eventually allow us to relate small changes at the molecular level with broader consequences and potential treatments in humans.
GNAO1 disorder is a multi-dimensional disease characterized by movement disorders, intellectual disabilities, and epileptic seizures. The complexity of symptom patterns and the diversity of underlying Gαo mutants indicate that a spectrum of treatment strategies will be required. Our analysis in yeast, oocytes, and human cells has revealed a subset of Gα mutants that are likely to sequester receptors (group 1) and Gβγ (groups 1 and 2). Our in vitro biochemical analysis has revealed important differences in stability, nucleotide binding and hydrolysis, as well as conformational changes needed for Gβγ dissociation and effector activation. Potential treatments might include ligands that bind to and regulate GPCRs, G proteins, and G protein-regulated ion channels. Further biochemical and drug screening efforts,10 as well as studies in model organisms ranging from yeast to mice, will complement existing human clinical studies, wherein laboratory researchers and medical professionals work together to understand and treat a rare condition for which there is currently no cure.
Limitations of the study
The classification scheme should not be regarded as immutable; some mutants have characteristics of two groups and other mutants remain unstudied. The extent to which these classifications are predictive of behavior, and vulnerability to treatment, will require new animal models.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the Lead Contact, Henrik G. Dohlman (hdohlman@med.unc.edu).
Materials availability
TRUPATH/BRET reagents are publicly available from Addgene; 1000000163. All other unique reagents generated in the study will be available without restrictions.
Data and code availability
Data reported in this paper will be shared by the one or more of the co-corresponding authors upon request. The trajectories from MD for the active state human Gαo monomer with GTP bound (accession number 1935) and the inactive state Go trimer with GDP bound (accession number 1945) have been deposited at https://welcome.gpcrmd.org/ and are publicly available as of the date of publication.
The study did not generate new unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Yeast strains
Yeast Saccharomyces cerevisiae strain BY4741 sst2Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 sst2:KanMX) and the parent strain BY4741 (Research Genetics #95401.H2) were used for halo and immunoblot analysis of Gpa1, respectively. Cells were grown in SC selective medium lacking uracil (MPBio; 1145112-CF) with 2% dextrose at 37°C for halos and 30°C for blots.
Xenopus laevis
Oocytes were extracted and isolated from adult female X. laevis frogs. Maintenance and surgery of the frogs were carried out in accordance with protocol 22–1032R approved by Northeastern University Institutional Animal Care and Use Committee. Frogs were housed in dechlorinated water tanks at 18°C. A small incision was made on the abdomen to remove the oocytes while the animals were under anesthesia in a small container with 0.1% tricaine methanesulfonate. Frogs were sutured following the oocyte removal and were recovered in a separate tank.
Human cell lines
HEK293T (ATCC; CRL-11268) cells overexpressing human Gγ8-or Gγ9-GFP2 and wildtype or mutant human Gαo-RLuc8 donors were used for BRET assays. Cells were grown in Dulbecco’s modified Eagle’s medium (Corning; 10-017-CV) + 10% fetal bovine serum (Gibco; A3382001) + 1% penicillin/streptomycin at 37°C and 5% CO2.
Microbe strains
E. coli Rosetta (Novagen; 70954) or RIPL (Agilent; 230280) DE3 cells were grown in 2X Luria Broth at 37°C and recombinant protein expression was induced at 16°C. E. coli 5-alpha DH5α (New England Biolabs; C2987H) cells were used for mutagenesis and grown at 37°C.
METHOD DETAILS
Mutagenesis
Mutagenesis was performed using Primestar Max polymerase (Takara Bio; R045B). Primers contained 5 bp 5′ to the codon and 20–30 bp 3′ to the codon (Data S4).
Halo assays
Saturated culture of S. cerevisiae strain BY4741 sst2:KanMX (sst2Δ), transformed with pRS316 containing either no insert (vector), GPA1 wildtype or mutant,53 was mixed with 0.5% agar (melted and kept at 55°C) in a 0.1:4 yeast:agar ratio. The yeast/agar mixture was spread on selective medium plates, and a paper disk with 8, 25, or 75 μg of synthetic α-factor was applied to predefined locations on the agar surface.60 Cells were grown at 37°C (to enhance growth differences) and imaged after 48 h.
Immunoblotting
Yeast extracts (BY4741 strain) were transformed with the single copy plasmid pRS316-ADH1-GPA1, pRS316-ADH1-GPA1-myc,53 or the indicated mutants. Proteins were resolved by 10% acrylamide gel electrophoresis and immunoblotting.61 Membranes were blocked with 0.01% fish gelatin and probed with mouse anti-myc (Cell Signaling Technology, 1:5000; 3739S) and horseradish peroxidase-conjugated goat-anti-mouse (Promega, 1:10,000; W4021) antibodies to detect Gpa1. Gpa1 was visualized with Clarity Western ECL Substrate (Bio-rad; 1705061) on a ChemiDoc Touch Imaging System (Bio-Rad).
Two-electrode voltage clamp (TEVC) methods
Complementary DNA constructs used for electrophysiology experiments were human GIRK1 (pGEMSH vector), mouse GIRK2 (pXOOM vector), mouse dopamine D2 receptor (pXOOM vector), bovine Gβ1 (pGEMSH vector), bovine Gγ2 (pGEMSH vector), human Gαo wildtype and mutant (pMaX vector). Xenopus laevis oocytes were isolated and microinjected with in vitro synthesized circular RNAs (0.125–2.5 ng per oocyte).62 To allow for sufficient protein expression, injected oocytes were incubated at 18°C for 2 days. The following day, a GeneClamp 500 amplifier (Axon Instruments) was used to measure whole-cell oocyte currents. The microelectrodes had resistances of 0.5–1 MΩ using a 3 M KCl solution in 1.4% agarose. For TEVC measurements, oocytes were held at 0 mV (EK), and a ramp protocol with a command potential from −80 to +80 mV was used to monitor the currents. The current amplitude was measured at the end of a sweep of 0.4 s. All currents were analyzed when steady state was reached. To establish a baseline for TEVC recordings, oocytes were perfused with a low K+ (LK) solution ND96 (2 mM KCl, 96 mM NaCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES–Na pH 7.4). A high potassium (HK) solution ND96K (96 mM KCl, 10 mM HEPES–K pH 7.4, 1 mM MgCl2, and 1.8 mM CaCl2) was used to measure basal current. The oocyte chamber was perfused with 3 mM BaCl2 in ND96K to block the current. The barium (Ba2+)-sensitive current was used exclusively for statistical analysis. 10 μM dopamine in ND96K (HK) was used to determine the effect of Gαo wildtype and mutants on agonist-induced activity of GIRK channels.
TRUPATH bioluminescence resonance energy transfer (BRET) assays
HEK293T cells (ATCC; CRL-11268) were transfected with plasmids encoding human Gα fused to Renilla luciferase 8 (Rluc8), human Gβ3 and either human Gγ8-or Gγ9-fused to GFP, and the human μ-opioid receptor (MOR) in a 1:1:1:1 ratio (cell density 750,000 cells in 3 mL).22 Approximately 16 h after transfection, the cells were plated in Dulbecco’s Modified Eagle Medium +1% dialyzed fetal bovine serum into poly-D-lysine coated 96-well white clear bottom plates (Greiner Bio-one; 655098). On the following day, cells were washed twice with assay buffer (20 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), pH 7.4, Hank’s Balanced Salt Solution). To measure luminescence, 10 μL of Rluc8 substrate (coelenterazine 400a, Nanolight; 340) at a final concentration of 5 μM was added to each well. The plates were kept dark for 5 min, then 30 μL (3X) of agonist in drug buffer (assay buffer +0.1% bovine serum albumin +0.1% ascorbic acid) was added to each well. The plates were kept dark for an additional 5 min after agonist addition. A Mithras LB940 multimode microplate reader was used to read each plate (395 nm excitation, 510 nm emission) for 1 s per well. The measurement output is a BRET ratio (GFP/Rluc8), which is determined by dividing fluorescence by luminescence. Receptor-catalyzed heterotrimer dissociation (net BRET) was normalized by subtracting the basal BRET values (GFP/Rluc8 at lowest dose of agonist) from the BRET ratio (GFP/Rluc8) of each well. GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA) was used to plot and curve fit the net BRET signal (the BRET ratio as a function of receptor).
Computational methods
Starting structural models and molecular dynamics simulations
The structural model of monomeric GTP-bound Gαo protein with Mg2+ ion was generated using the monomeric GTP-bound mouse Gαo crystal structure (PDB ID: 3C7K) as the template and using the homology modeling method in the Prime module of Maestro software from Schrodinger (https://www.schrodinger.com/products/maestro). The structural model of trimeric GDP-bound Gαo protein was built using the trimeric GDP-bound human Gαi crystal structure (PDB ID: 6CRK) as the template using the same method. Both structures were subjected to energy minimization using conjugate gradient method with a convergence cutoff of 0.1 kcal/mol/Å2. Input files for molecular dynamics simulations were generated using CHARMM-GUI.55 Each Gαo protein was solvated in explicit TIP3P water molecules in a cubic box (9.5 nm × 9.5 nm × 9.5 nm for monomeric Gαo protein and 12 nm × 12 nm × 12 nm for trimeric Gαo protein) with 0.15 M of potassium chloride for maintaining the physiological condition. GROMACS58 (Version 2021.3) with all-atom CHARMM36 force field56 was used to perform molecular dynamics simulations. MD simulations were performed at 310 K coupled to a temperature bath with a relaxation time of 0.1 ps.63 Pressure of the systems was calculated with molecular virial and was held constant by a weak coupling to a pressure bath with a relaxation time of 0.5 ps. Equilibrium bond length and geometry of water molecules were constrained using the SHAKE algorithm.57 The short-range electrostatic and van der Waals interactions were estimated every 2 fs using a charged group pair list with cutoff of 8 Å between centers of geometry of charged groups. Long-range van der Waals interactions were calculated using a cutoff of 14 Å and long-range electrostatic interactions were treated with the particle mesh Ewald method.64 Temperature was kept constant at 310 K by applying the Nose-Hoover thermostat.65 Desired pressure for all systems were achieved by using Parrinello-Rahman barostat with a pressure relaxation time of 2 ps.66 Before production runs, all system were subjected to a 5000-step steepest descent energy minimization to remove bad contacts.67 After minimization, the systems were heated up to 310 K under constant temperature-volume ensemble (NVT). The simulations were saved every 200 ps for analysis. The protein, Mg2+ ion, and nucleotide were subjected to positional constraints under a harmonic force constant of 1000 kJ/(mol*nm2) during the NVT step while solvent molecules were free to move. The systems then were further equilibrated using constant pressure ensemble (NPT), in which the force constant applied to the protein, Mg2+ ion, and nucleotide were gradually reduced from 5 kJ/(mol*nm2) to zero in six steps of 5 ns each. An additional 50 ns of unconstraint simulation was performed, making it a total of 80 ns NPT equilibration prior to production runs. We performed five production runs of 1000 ns each using five different initial velocities for every system. Therefore, we had 5 μs long MD trajectories for monomeric Gαo protein of GTP-bound state and trimeric Gαo protein of GDP-bound state. The convergence of the MD simulations was tested using the time series plots of the root-mean-square deviation (RMSD) of the coordinates of the Cα atoms of the protein with time (Figure S3).
To calculate solvent accessibility we used Python MDTraj package. The Shrake Rupley algorithm was employed to calculate the solvent accessibility of every residue present in the structure33,34
Contact map of protein with nucleotides
The last 200 ns from five independent molecular dynamics simulation runs were merged into one concatenated trajectory for both monomeric GTP-bound and trimeric GDP-bound Gαo systems. The concatenated trajectory was sampled every 2 ns. The sampled trajectory was fed to GetContacts script available on GitHub (https://github.com/getcontacts/getcontacts). For specific parameters, the interaction type flag “itype” was set to “all”, and the two atom group flags for contacts were “protein” and “resname GTP” for monomeric system or “chain C” and “resname GDP” for trimeric system. Contact results were further processed in Python environment.
Sequence based Co-evolutionary (Co-EV) analysis
Co-EV analysis was performed using EVCoupling webserver at https://v2.evcouplings.org/.65 Human Gαo protein sequence from Uniprot68 was used as the input to the webserver, with all default parameters. The Co-EV coupling data was downloaded from the webserver, and further processed in Python environment: briefly, residues that have mutants in a group were pooled, and the mean score for each group was calculated.
Structure based frustration analysis
Frustration analysis was performed using FrustratometeR webserver.38 Monomeric Gαo structure was used as the input to the webserver. Electrostatics was applied with default K value; the rest parameters were default values. The frustration frequency data was downloaded from the webserver, and further processed in Python environments. Residues that have their Cα atoms within 15 Å of the Cα atom of the mutated residue were included in the calculation; the average of highly and minimally frustrated percentage of all included residues was averaged and used as the value for the mutant system. The differential values between mutant system and wildtype system were used for each system. Residues that have mutations in a group were pooled, and the mean score was calculated.
Hierarchical clustering and characterization of conformational clusters
Data from BRET assay, hydrolysis assay, melting temperature measurements, Co-EV, frustration, and GTP contact frequency of the mutants were collected and categorized based on the relative value to those for wildtype Gαo protein (Table S1). Hierarchical clustering was performed using Python Scipy package with default parameters. The dendrogram was plotted using the same package, with color threshold parameter set to 3.8. The polar plots were generated using Python Plotly package.
Expression and purification of GαoA
Rosetta DE3 (Novagen; 70954) or RIPL (Agilent; 230280) cells were transformed with PET-SUMO-Gαo plasmid52 and grown at 37°C in 2X Luria Broth to saturation. Once at OD600 = 0.6–0.8, protein expression was induced by adding 600 μM isopropyl-β-D-1-thiogalactoside. The culture temperature was lowered to 16°C and shaken overnight. The subsequent purification was carried out at 4°C for all steps. The saturated cell culture was collected by centrifugation at 5400 ×g for 30 min. Cells were lysed by sonication and centrifuged at 26,900 ×g for 45 min. Lysates (30 mL each) were incubated with 5 mL (10 mL 50/50 slurry) of HisPur Ni-NTA Resin (Thermo Fisher Scientific; 88221) and 10 mM imidazole for 1 h. The resin was washed twice with phosphate buffer pH 7 (25 mM H2KO4P, 25 mM HK2O4P, 500 mM (tris(2-carboxyethyl)phosphine) (TCEP), 50 μM GDP, 50 μM MgCl2) then twice with phosphate buffer pH 7 + 20 mM imidazole. Protein was eluted with phosphate buffer pH 7 + 250 mM imidazole. The SUMO tag was cleaved by addition of 0.5 mg ULP1 protease,52 and the eluate was dialyzed overnight in 4 L of phosphate buffer pH 7. The next day, cleaved protein was collected by reverse-nickel chromatography.
Thermostability measurements
To determine Tm values, 10 μL of 65 μM protein, 10 μL of 1 mM GDP or GTPγS, 10 μL of 500 mM 4-fluoro-7-sulfamoylbenzofurazan (ABD-F) cysteine-reactive dye (Abcam; 91366-65-3), and 170 μL phosphate buffer was added to 12-strip PCR tubes. Samples were subjected to a 40-degree temperature gradient in a Biometra TProfessional Thermocycler for 3 min and fluorescence values were collected on a PHERAstar plate reader (BMG Labtech) (400 nm excitation, 500 nm emission).41
Nucleotide loading and hydrolysis
BODIPYFL GTP (ThermoFisher Scientific, Invitrogen; G12411) (50 nM) was equilibrated in hydrolysis buffer (20 mM Tris pH 8, 10 mM MgCl2) in a 1 mL cuvette. Purified Gα–GDP protein (3 μM) was added to initiate BODIPY-GTP binding and subsequent hydrolysis. A PerkinElmer LS55 luminescence spectrometer (498 nm excitation, 508 nm emission) was used to measure fluorescence using the FLWinLab software package (PerkinElmer).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitation and statistical analysis (standard deviation) was done using GraphPad Prism. Independent (biological) experiments are parallel measurements of biologically distinct samples obtained on different days. Technical replicates (duplicate, triplicates) are repeated measurements of the same sample and represent random noise associated with protocols or equipment. The net BRET was fit in GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| mouse anti-myc | Cell Signaling Technology | 3739S; RRID:AB_10889248 |
| horseradish peroxidase-conjugated goat-anti-mouse IgG | Promega | W4021; RRID:AB_430834 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| BL21-CodonPlus (DE3) RIPL | Agilent | 230280 |
| Rosetta (DE3) | Novagen | 70954 |
| 5-alpha Competent E. coli (High Efficiency) DH5α | New England Biolabs | C2987H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| ULP1 | Maly et al.52 | N/A |
| 4-fluoro-7-sulfamoylbenzofurazan | Abcam | 91366-65-3 |
| BODIPY™FL gtp | ThermoFisher Scientific | G12411 |
| Dulbecco’s modified Eagle’s medium | Corning | 10-017-CV |
| Fetal bovine serum | Gibco | A3382001 |
| CSM-Ura | MPBio | 1145112-CF |
|
| ||
| Critical commercial assays | ||
|
| ||
| Clarity western ECL substrate | Bio-Rad | 1705061 |
| Coelentrazine 400a | Nanolight | 340 |
|
| ||
| Deposited data | ||
|
| ||
| MDS full trajectories | This paper | https://welcome.gpcrmd.org/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T | ATCC | CRL-11268 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| S. cerevisiae BY4741 sst2Δ | Research Genetics | 95401.H2 |
|
| ||
| S. cerevisiae BY4741 | Research Genetics | 95401.H2 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Data S4 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pRS316-GPA1 | Song et al.53 | N/A |
| pRS316-ADH-GPA1 | Song et al.53 | N/A |
| pRS316-ADH-GPA1-myc | Knight et al.17 | N/A |
| MOR human (OPRM1) | Olsen et al.22 | N/A |
| GNAO-Rluc8 (Gαo fused to Rluc8) human (GNAO1) | Olsen et al.22 | N/A |
| GB3 (Gγ3) human (GNB3) | Olsen et al.22 | N/A |
| GNG8-GFP2 (Gγ8- fused to GFP) human (GNG8) | Olsen et al.22 | N/A |
| GNG9-GFP2 (Gγ9- fused to GFP) human (GNG9) | Olsen et al.22 | N/A |
| pGEMSH-GIRK1 human | He et al.54 | N/A |
| pXOOM-GIRK2 mouse | He et al.54 | N/A |
| pXOOM-D2R mouse (DRD2) | He et al.54 | N/A |
| pGEMSH Gβ1 bovine (GNB1) | He et al.54 | N/A |
| pGEMSH Gγ2 bovine (GNG2) | He et al.54 | N/A |
| pMaX Gαo human (GNAO1) | This paper | N/A |
| PET-SUMO-Gαo (GNAO1) | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | N/A |
| FLWinLab | PerkinElmer | N/A |
| CHARMM-GUI | Jo et al.55 | N/A |
| CHARMM36 | Huang et al.56 | N/A |
| MAESTRO | N/A | https://www.schrodinger.com/maestro |
| SHAKE | Andersen et al.57 | N/A |
| GROMACS | Hess et al.58 | N/A |
| GetContacts | N/A | https://getcontacts.github.io/ |
| EVCoupling | Hopf et al.59 | https://v2.evcouplings.org/ |
| FrustratometeR | Rausch et al.38 | https://www.schrodinger.com/pymol |
| PyMol | N/A | https://pymol.org/2/ |
Highlights.
Mutations in G proteins are linked to epilepsy and movement disorders
Many occur in regions that have no obvious role in G protein function
Mutations have distinct effects on signaling, catalysis, protein stability, and interactions
At least four mechanistically distinct groups will likely require distinct treatments
ACKNOWLEDGMENTS
The authors thank Kirill Martemyanov and Alex Roy for comments on the manuscript. This project was supported by NIH grants R01HL059949 and R01NS131467 (to D.E.L.), R01GM117923 and R01LM013876 (to N.V.), and R35GM118105 (to H.G.D.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113462.
REFERENCES
- 1.Sprang SR (2016). Invited review: Activation of G proteins by GTP and the mechanism of Gα-catalyzed GTP hydrolysis. Biopolymers 105, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hilger D, Masureel M, and Kobilka BK (2018). Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arang N, and Gutkind JS (2020). G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 594, 4201–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, and Bastian BC (2009). Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maziarz M, Leyme A, Marivin A, Luebbers A, Patel PP, Chen Z, Sprang SR, and Garcia-Marcos M (2018). Atypical activation of the G protein Gα. J. Biol. Chem. 293, 19586–19599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hewitt N, Ma N, Arang N, Martin SA, Prakash A, DiBerto JF, Knight KM, Ghosh S, Olsen RHJ, Roth BL, et al. (2023). Catalytic site mutations confer multiple states of G protein activation. Sci. Signal. 16, eabq7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galosi S, Pollini L, Novelli M, Bernardi K, Di Rocco M, Martinelli S, and Leuzzi V (2022). Motor, epileptic, and developmental phenotypes in genetic disorders affecting G protein coupled receptors-cAMP signaling. Front. Neurol. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, Terashima H, Osaka H, Nakamura S, Tohyama J, et al. (2013). De Novo mutations in GNAO1, encoding a Gao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 93, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huff RM, Axton JM, and Neer EJ (1985). Physical and immunological characterization of a guanine nucleotide-binding protein purified from bovine cerebral cortex. J. Biol. Chem. 260, 10864–10871. [PubMed] [Google Scholar]
- 10.Larasati YA, Savitsky M, Koval A, Solis GP, Valnohova J, and Katanaev VL (2022). Restoration of the GTPase activity and cellular interactions of Gα. Sci Adv 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muntean BS, Masuho I, Dao M, Sutton LP, Zucca S, Iwamoto H, Patil DN, Wang D, Birnbaumer L, Blakely RD, et al. (2021). Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng H, Sjögren B, Karaj B, Shaw V, Gezer A, and Neubig RR (2017). Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 89, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silachev D, Koval A, Savitsky M, Padmasola G, Quairiaux C, Thorel F, and Katanaev VL (2022). Mouse models characterize GNAO1 encephalopathy as a neurodevelopmental disorder leading to motor anomalies: from a severe G203R to a milder C215Y mutation. Acta Neuropathol. Commun. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rim JH, Kim SH, Hwang IS, Kwon SS, Kim J, Kim HW, Cho MJ, Ko A, Youn SE, Kim J, et al. (2018). Efficient strategy for the molecular diagnosis of intractable early-onset epilepsy using targeted gene sequencing. BMC Med. Genom. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menke LA, Engelen M, Alders M, Odekerken VJJ, Baas F, and Cobben JM (2016). Recurrent GNAO1 Mutations Associated With Developmental Delay and a Movement Disorder. J. Child Neurol. 31, 1598–1601. [DOI] [PubMed] [Google Scholar]
- 16.Wu YL, Hooks SB, Harden TK, and Dohlman HG (2004). Dominant-negative inhibition of pheromone receptor signaling by a single point mutation in the G protein alpha subunit. J. Biol. Chem. 279, 35287–35297. [DOI] [PubMed] [Google Scholar]
- 17.Knight KM, Ghosh S, Campbell SL, Lefevre TJ, Olsen RHJ, Smrcka AV, Valentin NH, Yin G, Vaidehi N, and Dohlman HG (2021). A universal allosteric mechanism for G protein activation. Mol. Cell 81, 1384–1396.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dohlman HG, and Thorner JW (2001). Regulation of G protein-initiated signal transduction in yeast: Paradigms and principles. Annu. Rev. Biochem. 70, 703–754. [DOI] [PubMed] [Google Scholar]
- 19.Apanovitch DM, Iiri T, Karasawa T, Bourne HR, and Dohlman HG (1998). Second site suppressor mutations of a GTPase-deficient G-protein a- subunit. Selective inhibition of bg-mediated signaling. J. Biol. Chem. 273, 28597–28602. [DOI] [PubMed] [Google Scholar]
- 20.Herskowitz I (1987). Functional inactivation of genes by dominant negative mutations. Nature 329, 219–222. [DOI] [PubMed] [Google Scholar]
- 21.Logothetis DE, Kurachi Y, Galper J, Neer EJ, and Clapham DE (1987). The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325, 321–326. [DOI] [PubMed] [Google Scholar]
- 22.Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, Che T, Gavin AC, McCorvy JD, Roth BL, and Strachan RT (2020). TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akgoz M, Kalyanaraman V, and Gautam N (2006). G protein betagamma complex translocation from plasma membrane to Golgi complex is influenced by receptor gamma subunit interaction. Cell. Signal. 18, 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Neill PR, Karunarathne WKA, Kalyanaraman V, Silvius JR, and Gautam N (2012). G-protein signaling leverages subunit-dependent membrane affinity to differentially control βγ translocation to intracellular membranes. Proc. Natl. Acad. Sci. USA 109, E3568–E3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masuho I, Skamangas NK, Muntean BS, and Martemyanov KA (2021). Diversity of the Gβγ complexes defines spatial and temporal bias of GPCR signaling. Cell Syst. 12, 324–337.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noel JP, Hamm HE, and Sigler PB (1993). The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S [see comments]. Nature 366, 654–663. [DOI] [PubMed] [Google Scholar]
- 27.Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, and Sprang SR (1994). Structures of active conformations of Gi alpha 1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412. [DOI] [PubMed] [Google Scholar]
- 28.Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, and Sprang SR (1995). Tertiary and quaternary structural changes in Gi alpha 1 induced by GTP hydrolysis [see comments]. Science 270, 954–960. [DOI] [PubMed] [Google Scholar]
- 29.De Lean A, Stadel JM, and Lefkowitz RJ (1980). A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 255, 7108–7117. [PubMed] [Google Scholar]
- 30.DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, et al. (2016). Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iiri T, Farfel Z, and Bourne HR (1997). Conditional activation defect of a human Gsalpha mutant. Proc. Natl. Acad. Sci. USA 94, 5656–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berghuis AM, Lee E, Raw AS, Gilman AG, and Sprang SR (1996). Structure of the GDP-Pi complex of Gly203–>Ala gialpha1: a mimic of the ternary product complex of galpha-catalyzed GTP hydrolysis. Structure 4, 1277–1290. [DOI] [PubMed] [Google Scholar]
- 33.Shrake A, and Rupley JA (1973). Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 79, 351–371. [DOI] [PubMed] [Google Scholar]
- 34.McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, and Pande VS (2015). MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Süel GM, Lockless SW, Wall MA, and Ranganathan R (2003). Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 10, 59–69. [DOI] [PubMed] [Google Scholar]
- 36.Lockless SW, and Ranganathan R (1999). Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295–299. [DOI] [PubMed] [Google Scholar]
- 37.Gianni S, Freiberger MI, Jemth P, Ferreiro DU, Wolynes PG, and Fuxreiter M (2021). Fuzziness and Frustration in the Energy Landscape of Protein Folding, Function, and Assembly. Acc. Chem. Res. 54, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rausch AO, Freiberger MI, Leonetti CO, Luna DM, Radusky LG, Wolynes PG, Ferreiro DU, and Parra RG (2021). FrustratometeR: an R-package to compute local frustration in protein structures, point mutants and MD simulations. Bioinformatics 37, 3038–3040. [DOI] [PubMed] [Google Scholar]
- 39.Chen M, Chen X, Schafer NP, Clementi C, Komives EA, Ferreiro DU, and Wolynes PG (2020). Surveying biomolecular frustration at atomic resolution. Nat. Commun. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Clarke D, and Gerstein M (2016). Localized structural frustration for evaluating the impact of sequence variants. Nucleic Acids Res. 44, 10062–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isom DG, Sridharan V, Baker R, Clement ST, Smalley DM, and Dohlman HG (2013). Protons as second messenger regulators of G protein signaling. Mol. Cell 51, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun D, Flock T, Deupi X, Maeda S, Matkovic M, Mendieta S, Mayer D, Dawson R, Schertler GFX, Madan Babu M, and Veprintsev DB (2015). Probing Galphai1 protein activation at single-amino acid resolution. Nat. Struct. Mol. Biol. 22, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goricanec D, Stehle R, Egloff P, Grigoriu S, Plückthun A, Wagner G, and Hagn F (2016). Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. USA 113, E3629–E3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferguson KM, Higashijima T, Smigel MD, and Gilman AG (1986). The influence of bound GDP on the kinetics of guanine nucleotide binding to G proteins. J. Biol. Chem. 261, 7393–7399. [PubMed] [Google Scholar]
- 45.McEwen DP, Gee KR, Kang HC, and Neubig RR (2001). Fluorescent BODIPY-GTP analogs: real-time measurement of nucleotide binding to G proteins. Anal. Biochem. 291, 109–117. [DOI] [PubMed] [Google Scholar]
- 46.Willard FS, Kimple AJ, Johnston CA, and Siderovski DP (2005). A direct fluorescence-based assay for RGS domain GTPase accelerating activity. Anal. Biochem. 340, 341–351. [DOI] [PubMed] [Google Scholar]
- 47.Slepak VZ, Quick MW, Aragay AM, Davidson N, Lester HA, and Simon MI (1993). Random mutagenesis of G protein alpha subunit G(o)alpha. Mutations altering nucleotide binding. J. Biol. Chem. 268, 21889–21894. [PubMed] [Google Scholar]
- 48.Slepak VZ, Wilkie TM, and Simon MI (1993). Mutational analysis of G protein alpha subunit G(o) alpha expressed in Escherichia coli. J. Biol. Chem. 268, 1414–1423. [PubMed] [Google Scholar]
- 49.Marivin A, Leyme A, Parag-Sharma K, DiGiacomo V, Cheung AY, Nguyen LT, Dominguez I, and Garcia-Marcos M (2016). Dominant-negative Gα subunits are a mechanism of dysregulated heterotrimeric G protein signaling in human disease. Sci. Signal. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng H, Yuan Y, Williams MR, Roy AJ, Leipprandt JR, and Neubig RR (2022). Mice with monoallelic GNAO1 loss exhibit reduced inhibitory synaptic input to cerebellar Purkinje cells. J. Neurophysiol. 127, 607–622. [DOI] [PubMed] [Google Scholar]
- 51.Larrivee CL, Feng H, Quinn JA, Shaw VS, Leipprandt JR, Demireva EY, Xie H, and Neubig RR (2020). Mice with GNAO1 R209H movement disorder variant display hyperlocomotion alleviated by risperidone. J. Pharmacol. Exp. Therapeut. 373, 24–33. [DOI] [PubMed] [Google Scholar]
- 52.Maly J, and Crowhurst KA (2012). Expression, purification and preliminary NMR characterization of isotopically labeled wild-type human heterotrimeric G protein alphai1. Protein Expr. Purif. 84, 255–264. [DOI] [PubMed] [Google Scholar]
- 53.Song J, Hirschman J, Gunn K, and Dohlman HG (1996). Regulation of membrane and subunit interactions by N-myristoylation of a G protein a subunit in yeast. J. Biol. Chem. 271, 20273–20283. [DOI] [PubMed] [Google Scholar]
- 54.He C, Zhang H, Mirshahi T, and Logothetis DE (1999). Identification of a potassium channel site that interacts with G protein betagamma subunits to mediate agonist-induced signaling. J. Biol. Chem. 274, 12517–12524. [DOI] [PubMed] [Google Scholar]
- 55.Jo S, Kim T, Iyer VG, and Im W (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 56.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andersen HC (1983). Rattle: A “Velocity” Version of the Shake Algorithm for Molecular Dynamics Calculations. J. Comput. Phys. 52, 24–34. [Google Scholar]
- 58.Hess B, Kutzner C, van der Spoel D, and Lindahl E (2008). GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theor. Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- 59.Hopf TA, Green AG, Schubert B, Mersmann S, Schärfe CPI, Ingraham JB, Toth-Petroczy A, Brock K, Riesselman AJ, Palmedo P, et al. (2019). The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoffman GA, Garrison TR, and Dohlman HG (2002). Analysis of RGS proteins in Saccharomyces cerevisiae. Methods Enzymol. 344, 617–631. [DOI] [PubMed] [Google Scholar]
- 61.Cappell SD, Baker R, Skowyra D, and Dohlman HG (2010). Systematic analysis of essential genes reveals important regulators of G protein signaling. Mol. Cell 38, 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan KW, Sui JL, Vivaudou M, and Logothetis DE (1996). Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc. Natl. Acad. Sci. USA 93, 14193–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- 64.Darden T, York D, and Pedersen L (1993). Particle mesh Ewald: An N•log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- 65.Evans DJ, and Holian BL (1985). The Nose–Hoover thermostat. J. Chem. Phys. 83, 4069–4074. [Google Scholar]
- 66.Parrinello M, and Rahman A (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190. [Google Scholar]
- 67.Petrova SS, and Solov’ev AD (1997). The Origin of the Method of Steepest Descent. Hist. Math. 24, 361–375. [Google Scholar]
- 68.UniProt Consortium (2023). UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the one or more of the co-corresponding authors upon request. The trajectories from MD for the active state human Gαo monomer with GTP bound (accession number 1935) and the inactive state Go trimer with GDP bound (accession number 1945) have been deposited at https://welcome.gpcrmd.org/ and are publicly available as of the date of publication.
The study did not generate new unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.