

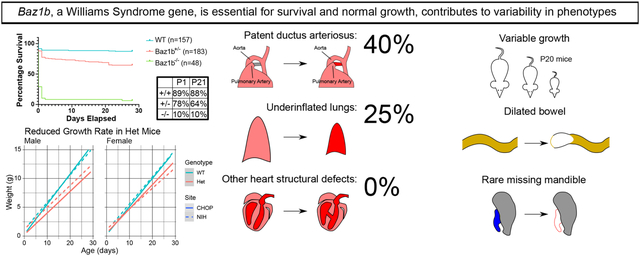
Abstract
BAZ1B is one of 25–27 coding genes deleted in canonical Williams syndrome, a multi-system disorder causing slow growth, vascular stenosis, and gastrointestinal complaints, including constipation. BAZ1B is involved in (among other processes) chromatin organization, DNA damage repair, and mitosis, suggesting reduced BAZ1B may contribute to Williams syndrome symptoms. In mice, loss of Baz1b causes early neonatal death. 89.6% of Baz1b−/− mice die within 24 hours of birth without vascular anomalies or congenital heart disease (except for patent ductus arteriosus). Some (<50%) Baz1b−/− were noted to have prolonged neonatal cyanosis, patent ductus arteriosus, or reduced lung aeration, and none developed a milk spot. Meanwhile, 35.5% of Baz1b+/− mice die over the first three weeks after birth. Surviving Baz1b heterozygotes grow slowly (with variable severity). 66.7% of Baz1b+/− mice develop bowel dilation, compared to 37.8% of wild-type mice, but small bowel and colon transit studies were normal. Additionally, enteric neuron density appeared normal in Baz1b−/− mice except in distal colon myenteric plexus, where neuron density was modestly elevated. Combined with several rare phenotypes (agnathia, microphthalmia, bowel dilation) recovered, our work confirms the importance of BAZ1B in survival and growth and suggests that reduced copy number of BAZ1B may contribute to the variability in Williams syndrome phenotypes.
Keywords: BAZ1B, Williams syndrome, survival, symptom variability
Graphical Abstract
INTRODUCTION
Bromodomain adjacent to zinc finger domain 1B (BAZ1B) is one of 25–27 coding genes deleted in canonical Williams syndrome (WS; also known as Williams-Beuren syndrome) (Kozel et al., 2021). WS is a developmental haploinsufficiency disorder characterized by (among other features) supravalvar aortic stenosis, developmental delays, and characteristic facies. Additionally, people with WS frequently report gastrointestinal problems (Cherniske et al., 2004; Morris et al., 1990). While the influence of hemizygosity in many WS genes has been reported (Enkhmandakh et al., 2009; Li et al., 1998; Meng et al., 2002), BAZ1B has remained understudied in an organismal context despite its many functions.
BAZ1B is involved in a wide range of cellular processes, including chromatin organization through the WICH (Bozhenok et al., 2002) and B-WICH (Cavellán et al., 2006) complexes, DNA damage repair (Xiao et al., 2008), and mitosis (Ohta et al., 2019). In Xenopus laevis, knockdown of baz1b via morpholino caused defects in neural, eye, and cranial neural crest tissues (Barnett et al., 2012). In FVB/NJ mice, a Baz1b missense variant (c.2585T>G; p.L733R) dramatically reduced BAZ1B protein levels, expanded heterochromatin, reduced homozygous mutant pup survival and adult mouse size, and subtly altered the structure of the cranium and craniofacial bones (Ashe et al., 2008). A targeted Baz1b mutant allele was later generated in C57Bl/6N mice using a knockout-first strategy and characterized by the International Mouse Phenotyping Consortium (IMPC) (Dickinson et al., 2016) (www.mousephenotype.org; data on the Baz1b mutant mouse not peer reviewed). The IMPC identified reduced adult body weight, abnormal startle reflex, and a sex-specific decrease in circulating cholesterol. They also report knockout mice to be “subviable,” but the degree and timeline for this loss is unknown.
Here we report on the IMPC targeted mutant allele within two mouse colonies on a mixed C57Bl/6N; C57Bl/6J background. We confirm previously reported growth defects in heterozygous mice and characterize the growth of juvenile mice. Additionally, we identify a dose-dependent mortality phenotype, in alignment with the c.2585T>G; p.L733R variant results and further detailing the IMPC’s original data. We further report several novel phenotypes across multiple organs and characterize perinatal heart structure and bowel anatomy/function.
MATERIALS AND METHODS
Animals
All animals were maintained in accordance with a Children’s Hospital of Philadelphia or a National Institutes of Health IACUC-approved protocol. Baz1b mice used in this paper were derived from preliminary experimental matings between the C57BL/6N-Baz1btm2a(KOMP)Wtsi/Mmucd line (MMRRC #46766) and a C57BL/6J-based line. This resulted in a mixed C57BL/6N-C57BL/6J strain background, described further in the Discussion. Further animal and housing information is presented in Table 3.
Table 3.
Extended animal, experiment, and housing information
| CHOP | NIH | |
|---|---|---|
| Animal Information | ||
| Species | Mouse | |
| Age | Varies; see Methods | |
| Sex | Male & female (identified when possible) | |
| Strain | Baz1b tm2a(KOMP)Wtsi | |
| Genetic background | C57BL6/J x C57BL6/N; see Discussion | |
| Type of genetic modification | lacZ insertion at endogenous locus | |
| Genotypes used | Varies; see Methods | |
| Microbe status | Specific pathogen free | |
| Origin | In-house breeding | |
| Weight | Varies; see Results | |
| Procedure history | Mice weighed for growth curve used in adult experiments (not segregated) | Mice had no experimental overlap |
| Drug & test naivete | No exposure prior to experiments | |
| Health & immune status | Healthy | |
| Quarantine & acclimatization | N/A | |
| Housing | ||
| Type of facility | Specific pathogen free | |
| Light/dark cycle | 12h/12h, 18:00–06:00 dark | |
| Type of lighting | Fluorescent ceiling bulbs | |
| Facility temperature | 72F +/− 2F | 72F +/− 3F |
| Facility humidity | 30–70% | |
| Ventilation | Room air changes 10–15x/hr, individually ventilated cage racks of ~70 air changes per hour | Room air changes 10–15x/hr, individually ventilated cage racks |
| Noise | Ongoing construction in other parts of animal facility | Ambient |
| Type of cage | Tecniplast GM500 | Lab Products Super Mouse 750 |
| Size of cage | 77.66in2 | 75in2 |
| Bedding material | ¼%” corncob or ¼%” corncob/ALPHA-dri® blend | Sani Chip Hardwood Bedding |
| Number of cage companions | 1–5; singly housed if sole male in litter or after mating | |
| Presence & type of enrichment | Present; nestlet material with mouse hut and/or shelf | Present; crinkle paper |
| Frequency of cage change | Weekly initially, every other week as of January 2022 | Weekly |
| Frequency of handling | Solely for cage changes and experiments | |
| Breeding program | Couple or trio matings; sibs or cousins mated together | |
| Type of food | Lab Diet 5015 | Lab Diet 5021 |
| Food composition | >11% fat, <3% fiber | >9% fat, <5% fiber |
| Food pre-treatment | None | |
| Food & water availability | Ad libitum | |
| Type of water | Reverse osmosis purified, pH 7 | Chlorinated water |
| Water pre-treatment | None | |
| Frequency of water change | Automated system | Weekly or as needed |
| Water delivery (bottle vs automatic) | Automatic | Bottle |
Reverse Transcription into Quantitative Polymerase Chain Reaction (RT-qPCR)
Aortic, lung, liver, and brain tissues were dissected from full P0 litters after euthanasia by decapitation and were immediately snap frozen in liquid nitrogen for later use. Tissue was lysed in a Bead Ruptor 4 (Omni International, #25–010) with 2mL ceramic bead kits (Omni International, #19–628) and processed according to the manufacturer’s protocol. RNA was then purified and isolated using a QIAGEN RNeasy Mini Kit (QIAGEN #74104), including DNase digestion. RNA concentration was measured using a NanoDrop OneC spectrophotometer (Thermo Scientific #13–400-519), then 1μg of RNA was reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, #4368814). qPCR was performed on a QuantStudio 3 Real-Time PCR System (Applied Biosystems #A28567) using TaqMan Gene Expression Master Mix (ThermoFisher, #4369016). Expression of the Baz1b exon 4–5 junction (Applied Biosystems, #4351372) was determined relative to GAPDH (Applied Biosystems, #4352339E) and normalized to expression in Baz1b+/+ organs.
Survival Data
At all sites, litters from matings where one or both parents were Baz1b+/− were used. Mice were tracked to 28 days of age (P28). Cases where a whole litter died within the first two days of life were discarded based on probable dam effects. Mice that died due to known adverse events or were used for other experiments were excluded from the analysis. Cages were checked daily from date of birth, marking time of death based on the day they were found dead or missing.
At the CHOP study site, two methods were performed to collect survival data. The first method was to collect any carcass found during daily checks and genotype them post-mortem. In cases where only partial carcasses could be recovered, individual carcasses were distinguished based on proximity of body parts, quantity of known missing pups, and carcass genotype(s). The second method was to tattoo pups on P1 and take a tail biopsy at time of tattooing. Individual pups were considered dead if they were not among the living pups during a daily check, regardless of the presence of a carcass. For both methods, mice were weaned at P28.
At the NIH study site, pups were closely observed during P0-P3 to collect carcasses and genotype. Survivors were then toe clipped and genotyped. Mice were weaned at P21 unless there were indications they should remain with the dam until P28 (typically failure to thrive).
Weight Data
At all sites, adult mice ages P78–97 were weighed for adult data. For juvenile data, whole litters from matings where one or both parents were Baz1b+/− were used. Mice were tracked to 28 or 29 days of age, depending on convenience, and weighed twice weekly. Mice that died due to known adverse events or were prematurely euthanized for other experiments were excluded from the data. Juvenile mice were identified using tattoos (CHOP) or clipped toes (NIH) and weaned as above.
Perinatal Phenotyping
Checks for agnathia consisted of evaluating pups, living or dead, for a mandible at birth. In cases where the pup died before observation and was partially cannibalized by the mother, open wounds where the mandible normally connects to the skull combined with an exposed mouth interior was considered sufficient evidence of a mandible at time of birth.
To produce Table 2, all pups were collected within the first few hours post-parturition. Any surviving mice were euthanized by decapitation. A post-mortem evaluation of each mouse was then performed and recorded to assess relative body size, craniofacial defects, gross heart abnormalities, aortic arch morphology, patency of the ductus arteriosus, evidence of air in the lungs, and presence of milk in the stomach.
Table 2.
Observations of P0 skull, thoracic organs, and abdominal cavity
| Baz1b +/+ | Baz1b +/− | Baz1b −/− | p value | |
|---|---|---|---|---|
| Cyanosis | 0/12 (0%) | 0/18 (0%) | 3/20 (15%) | 0.1110 |
| Patent Ductus Arteriosus | 0/12 (0%) | 1/18 (5.56%) | 8/20 (40%) | 0.0039* |
| Minimal Air in Lungs | 1/12 (8.33%) | 1/18 (5.56%) | 5/20 (25%) | 0.1939 |
| Structural Cardiac or Arch Anomaly | 0/12 (0%) | 0/12 (0%) | 0/12 (0%) | -- |
| Supravalvular Aortic Stenosis | 0/12 (0%) | 0/18 (0%) | 0/20 (0%) | -- |
| Coarctation of the Aorta | 0/12 (0%) | 0/18 (0%) | 0/20 (0%) | -- |
| Presence of Milk in Stomach | 7/12 (58.33%) | 7/18 (38.89%) | 0/20 (0%) | 0.0002* |
| Craniofacial Defects | 0/12 (0%) | 1/18+ (5.56%) | 0/20 (0%) | 0.6000 |
Missing eye
Adult Phenotyping
All weaned mice (over 28 days old) at the CHOP study site prior to October 29, 2018 were examined by eye for evidence of corneal clouding or microphthalmia until we noted that ocular phenotypes were no longer arising in our Baz1b colony. Additionally, a subset of mice had their eyes enucleated and measured from the optic nerve to the most distal point of the eye’s exterior.
Gross anatomy of the reproductive system, pancreas, spleen, liver, heart, lung, kidney, adrenal, and skeleton was evaluated in mice of all ages (range [12, 237] days). Each organ system was compared to wild-type expectations (e.g., a flexible spinal column, the presence of adrenal glands atop the kidneys, a fully developed liver with five lobes). Special attention was given to ensure the palate was closed. Finally, bowels were examined for the presence of air in the stomach, proximal small intestine (PSI), middle small intestine (MSI), distal small intestine (DSI), cecum, proximal colon (PC), middle colon (MC), and distal colon (DC).
Cardiac Computed Tomography (CT) Scan
Hearts were dissected from euthanized P0 pups and immediately transferred to 10% buffered formalin. To prevent tissue cracking, serial dehydration was performed by placing each heart in 30%, 50%, and 70% ethanol over three consecutive 24-hour blocks. Post-dehydration, each heart was submerged in a 2.5% phosphotungstic acid hydrate (PTA; Sigma-Alrich, CAS 1343–93-7) in ethanol solution (EPTA). After three days, hearts were then μCT scanned on a Bruker SkyScan 1272 Tissue Scanner (Bruker microCT, Kontich, Belgium) at 6μm resolution, using 1mm A1 filter, with the source biased at 80kV and 125μA. Projections were acquired with an exposure of 1300ms using 4 frame averages, every 0.55 or 0.6 degrees over 180 degrees.
Small Bowel Motility Assay
Adult mice ages P63–70 were fasted overnight, then gavage fed with 100μL of 10mg/mL FITC-dextran MW 70,000 (Sigma #46945) dissolved in 2% methylcellulose (Sigma #274429). Mice were kept without access to food or water for 60 minutes to allow for transit, then euthanized by CO2 inhalation. Bowel from stomach to anus was dissected into a dry dish, then divided into stomach, small intestine, cecum, and colon. The small intestine was further divided into 10 parts (SI1–10), the cecum into 2 parts (Ce1, the portion approximately in line with the rest of the bowel, and Ce2, the diverted pouch), and the colon into 3 parts (Co1–3). Each segment was placed into microcentrifuge tubes with 400μL 1x phosphate buffered saline (ThermoFisher #21600069; PBS), then thoroughly shredded with scissors to access luminal contents. Each tube was centrifuged for 10 minutes at 4,000×g, and 100μL of supernatant was loaded into a dark-sided 96-well plate (Corning #3603). Particularly concentrated samples were diluted 1:1 with 1xPBS to prevent exceeding limits of detection. Finally, sample fluorescence was measured with a Turner BioSystems Modulus II with a blue (ex 490nm, em 510–570nm) filter cube, using 1×PBS as a blank.
Percent FITC-dextran per segment was calculated by subtracting blank values from the raw sample measurement, then normalizing to the total fluorescence in all bowel segments. This relative quantity was used to calculate the geometric center of FITC-dextran by multiplying each ratio by the segment number (i.e. the stomach was 1, SI1 was 2, SI2 was 3, etc; Ce1 and Ce2 were considered in sequence for simplicity) and then adding them together.
Large Bowel Motility Assay
Adult mice ages P56–69 were anesthetized with isoflurane (NDC 66794–017-10) and brought to a surgical plane of anesthesia. A 3mm rounded glass rod lubricated with corn oil (Sigma #C8267) was gently inserted into the anus 2cm into the colon. The rod was removed, and a 3mm glass bead (Sigma-Aldrich #Z143928) lubricated with corn oil was inserted into the anus, using the rod to place it 2cm deep into the colon. The mouse was then placed into a cage free of bedding and allowed to recover from anesthesia, then observed until the bead was expelled, up to a maximum of 30 minutes. The time between insertion of the bead at its deepest point and bead expulsion was measured. Three trials were performed per mouse over the course of five days. All mice were euthanized at least two days later to verify that the colon had sustained no damage during experiments; should a perforation be identified, the entire mouse’s data was discarded.
Neonatal Bowel Harvest
Since Baz1b−/− pups typically died within 24 hours of birth, pregnant dams were injected with 200μL of 10mg/mL progesterone (Sigma-Aldrich #P3972) in corn oil (Sigma-Aldrich #C8267) before 14:00 on gestational day 18.5 to delay birth. Neonatal (P0) pups were then collected by Cesarean delivery, and their bowels were dissected, opened lengthwise, pinned flat, and fixed unpeeled with 4% paraformaldehyde (EMD Millipore, CAS 30525–89-4) in PBS for 25 minutes. The first 2cm of pinned small intestine were considered proximal small intestine, the final 2cm of small intestine were considered distal small intestine, and the final third of the colon was considered distal colon.
Immunohistochemistry
1cm of bowel tissue was blocked with 5% normal donkey serum (Jackson ImmunoResearch #017–000-021, RRID AB_2337258) in 1xPBS + 0.5% Triton X-100 (Sigma-Aldrich #T8787) for 2hr at RT. Tissue was then stained with 1:40,000 ANNA1 (gift from Vanda Lennon, Mayo Clinic, Rochester, MN, RRID AB_2314657) and 1:100,000 TuJ1 (BioLegend #802001, RRID AB_2564645) in blocking buffer for 2hr at RT, before being washed 3 times in 1xPBS for at least 5 minutes per wash. Tissue was then stained with 1:400 AlexaFluor 647 Goat α-Human (Invitrogen #A21445, RRID AB_2535862) and 1:400 AlexaFluor 488 Goat α-Rabbit (Invitrogen #A11034, no RRID) for 1hr at RT in the dark. Finally, tissue was washed in the dark 3 times in 1xPBS for at least 5 minutes per wash, then mounted on a glass slide in 50/50 1xPBS/glycerol (Invitrogen 15514–011) with 0.05% sodium azide (Sigma S2002). Slides were sealed with clear nail polish (Sally Beauty #SBS-215100) and allowed to cure at RT overnight. Images were captured on a Zeiss LSM 710 confocal microscope with Zeiss ZEN 2.3 SP1 FP3 (black) (Version 14.0.18.201; Zeiss, Oberkochen, Germany) software, a Plan-Apochromat 20x/0.8 air M27 objective lens, and slice interval of 1 μm. Images were taken from 8 unique fields of view, prioritizing the most proximal regions (if imaging proximal small intestine) or the most distal regions (if imaging distal small intestine or distal colon).
Neuron Density Quantification
Bowel tissue images were opened in ImageJ and quantified using the Cell Counter plugin. Individual neurons were identified by ANNA1 staining, using TuJ1 staining to distinguish individual tightly-packed neurons where necessary. Slice-by-slice quantification of tissue allowed for myenteric and submucosal plexuses to be distinguished. Due to the high density of neurons in neonatal myenteric plexus, images were cropped to quantify at least 70,000μm2. Submucosal plexus images were not cropped, and the full 180,710μm2 were quantified. Neuron densities across all images (minimum 8) of a given mouse bowel and plexus region were averaged and reported as the neuron density for that mouse (i.e. treated as an experimental N).
Image Processing
Figures 2B, 2D, 4A–B, and 5C were uniformly levels-adjusted using the Adobe Photoshop CS6 Auto Tone function.
Figure 2.
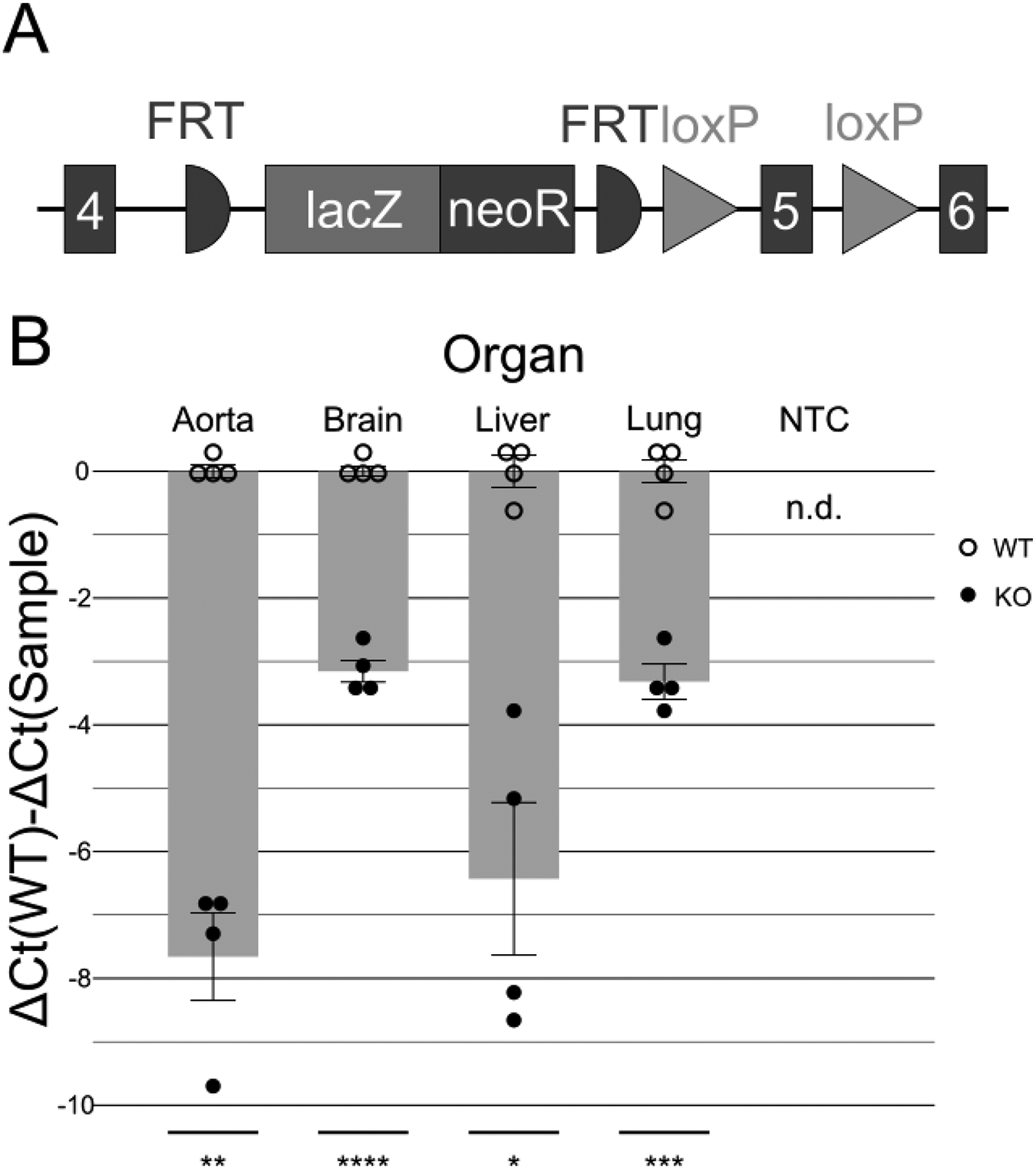
The Baz1b− allele retains low expression in select tissues. A) Structure of the Baz1b− allele. An inserted lacZ-neoR cassette after exon 4 is intended to prevent further transcription of the Baz1b protein. While not performed as part of these studies, FRT sites allow for Flp recombinase–dependent removal of the lacZ-neoR cassette to reactivate expression. Exon 5 is then flanked by loxP sites, allowing for Cre recombinase–dependent re-inactivation of Baz1b expression and function after lacZ-neoR removal. B) RT-qPCR of aorta, brain, liver, and lung samples at P0. Baz1b−/− pups have reduced (but still detectable) expression of Baz1b. Empty circles and associated error bars around baselines represent WT samples for tissue. Solid circles indicate Baz1b−/− mice. NTC, non-template control. n.d., not detected. t-test results for Baz1b−/− versus WT: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Figure 4.
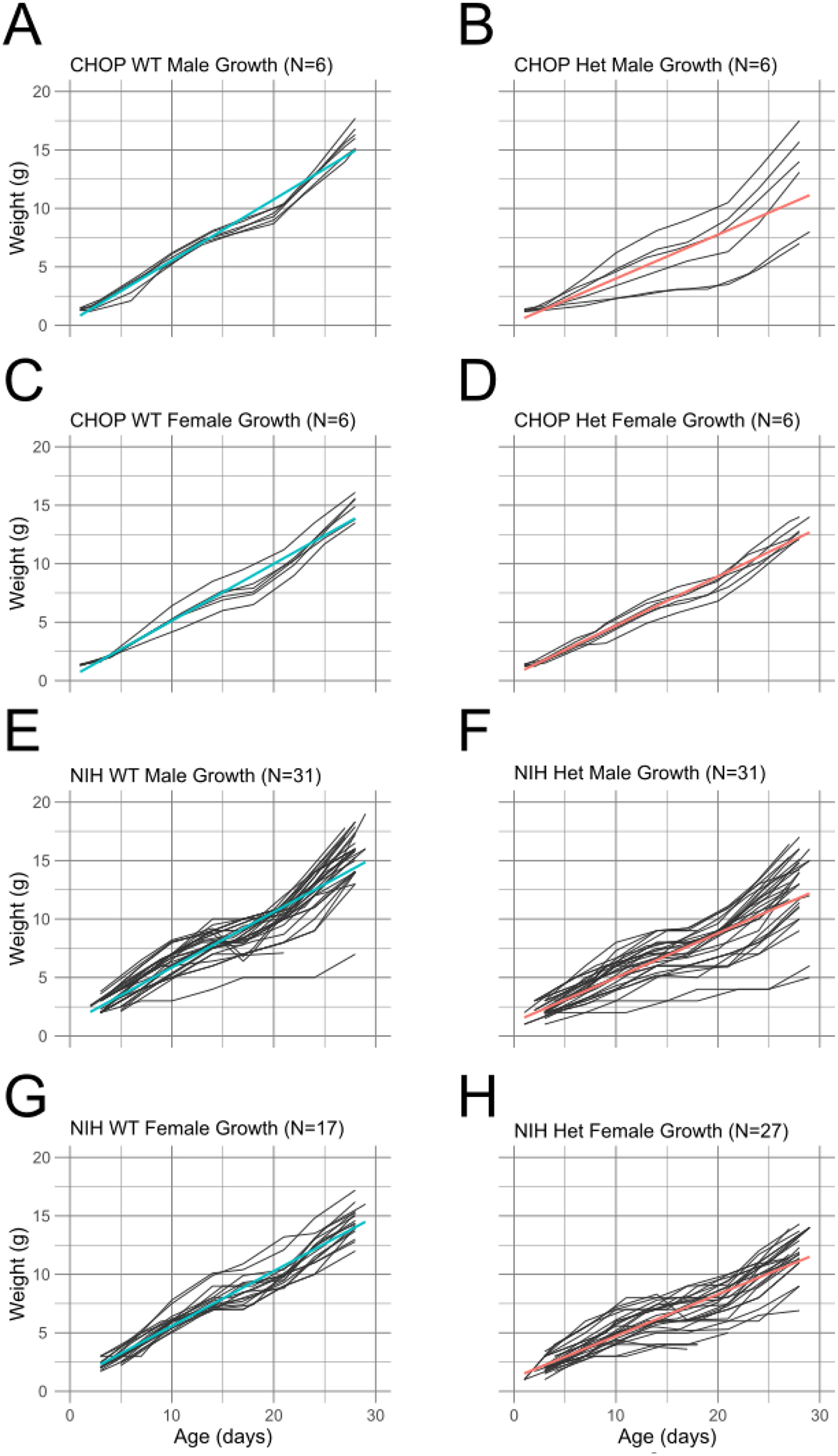
Individual growth curves grouped by sex, site, and genotype. Colored lines are simple linear regressions.
Figure 5.
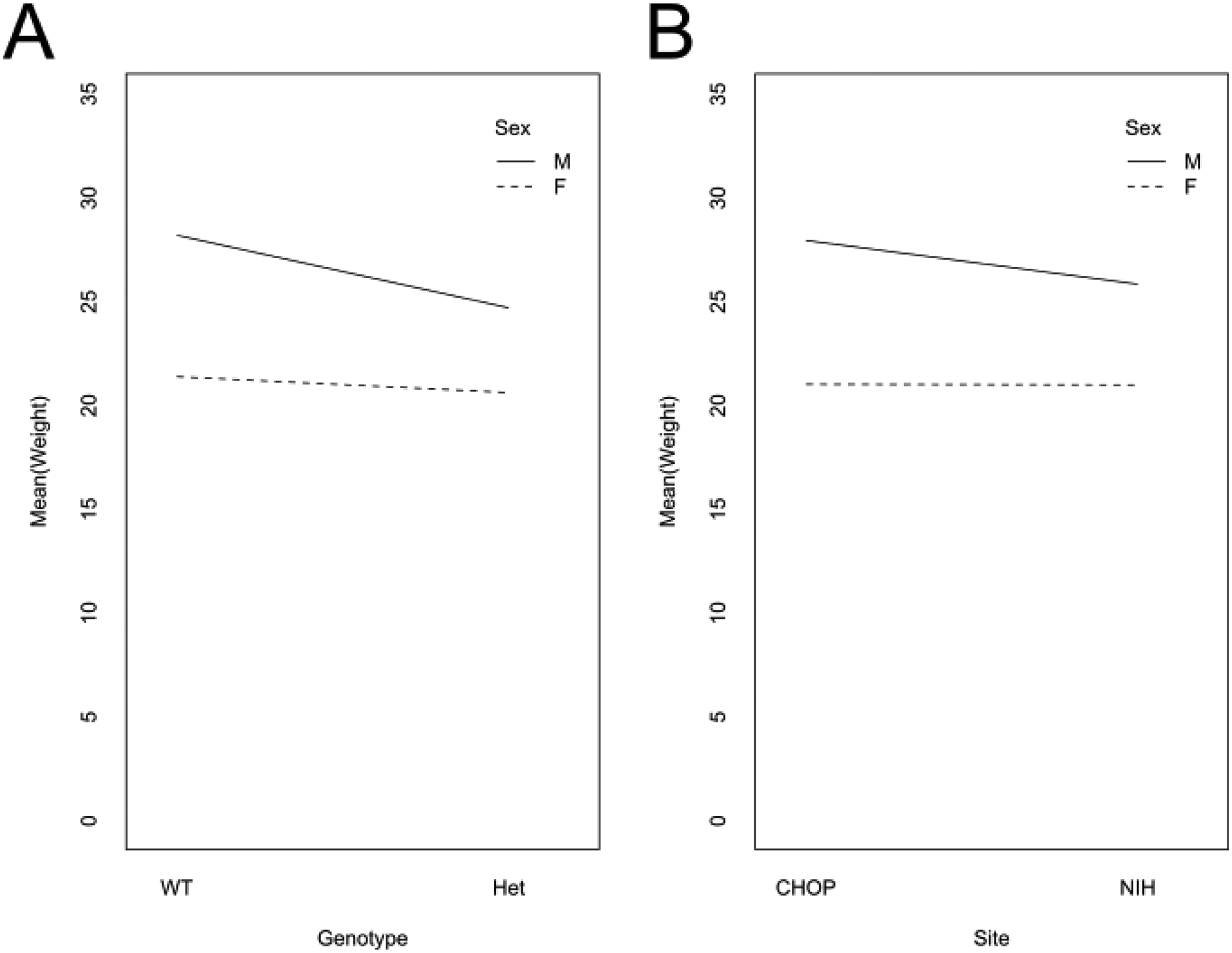
Interaction plots of sex & genotype (A) and sex & research site (B). The lines on both plots converge, indicating an interaction between the factors shown.
Statistics
RT-qPCR data was tested for normality using a Shapiro-Wilk normality test, then grouped by organ and compared by genotype using t tests.
Survival data was analyzed using a Cox proportional hazards model to estimate the hazard ratio for time to death between Baz1b genotypes. Adjustments were made for the effects of study sites, sex, and parent breeder effects, but no interaction effects. There was no violation of the proportional hazard assumption.
Linear mixed-effects models were used to analyze juvenile growth data to account for repeated weight measurements of the same subject. Potential factors were chosen a priori and then evaluated by comparing average baseline weight (P2–5) and average growth rate (maximum weight difference divided by age measured) across summary tables of nesting sub-divisions of data. Differences that were not accounted for by the standard error of the mean were then further compared by Wilcoxon Rank Sum tests. Significant factors were incorporated into the final model, with edge case factors evaluated for inclusion by a log-likelihood test of model fit improvement. The final model tested the association of mouse growth rate with Baz1b genotype, accounting for birth weight effects based on research site and genotype; effects on growth rate based on sex; and interactions on growth rate between research site and sex, with a random effect based on individual mice. This method is schematized in Figure 1A, with associated data and R script uploaded as Supplementary Material.
Figure 1.
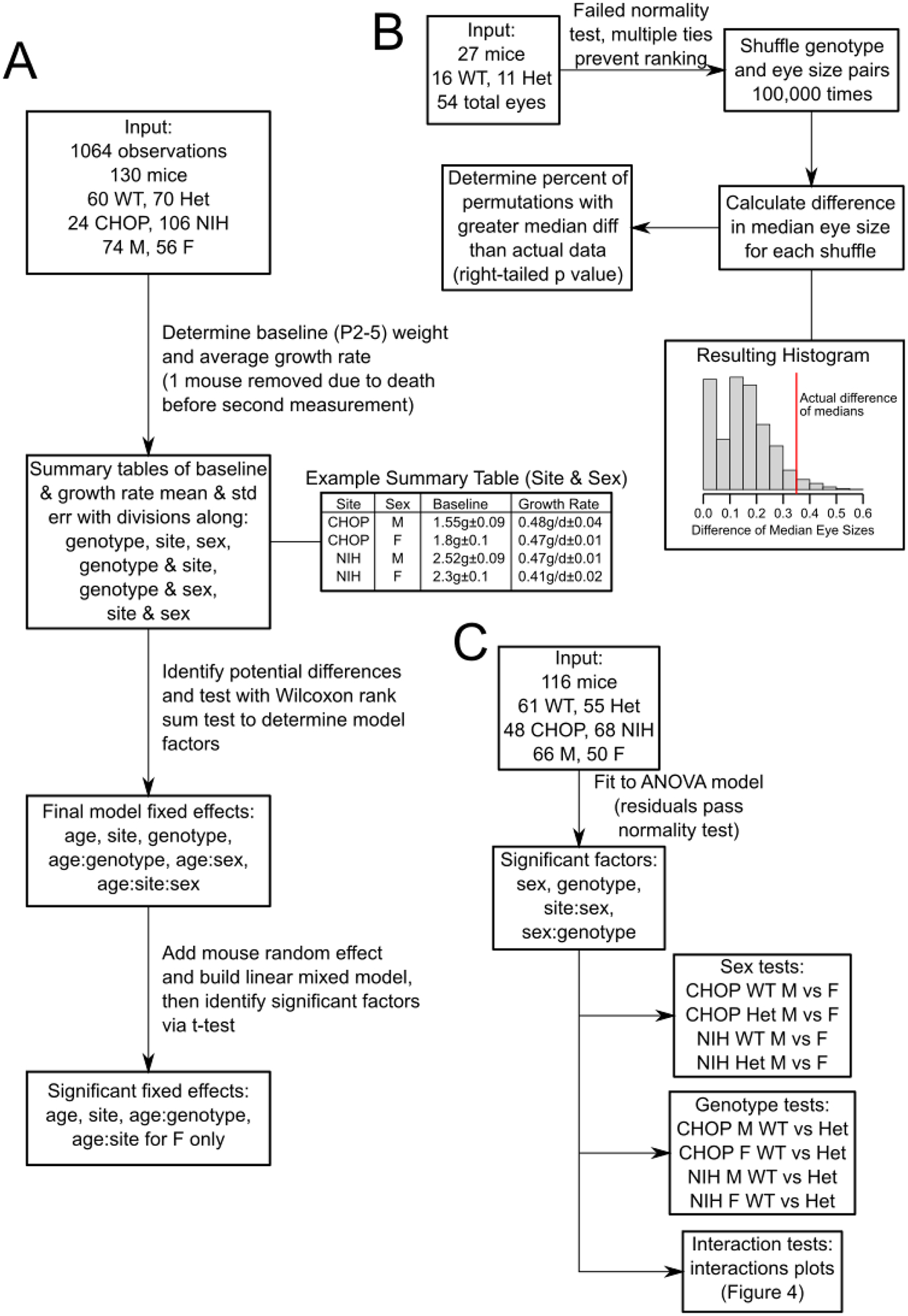
Schematic diagrams of select statistical analyses performed in this study. A) Flowchart of linear mixed model construction and evaluation for growth data. B) Flowchart of eye size permutation test. C) Flowchart of adult weight ANOVA and follow-up tests.
Prevalence of gross abnormalities in Baz1b+/− mice (e.g., corneal clouding & microphthalmia or dilated bowel) was evaluated using a binomial test. For tests comparing air in specific regions of bowel, the baseline N was the number of mice of a given genotype with air in any region of bowel; resulting p values were adjusted with the Holm method (Holm, 1979) to account for family-wise error. For all other tests, the baseline N was the number of mice examined.
Eye sizes were initially evaluated for normality using the Shapiro-Wilk test. Due to failure to meet the assumption of normality and issues with multiple tied measurements of eye size, the dataset was randomly permutated 100,000 times to estimate the chance that the absolute difference in median eye sizes between genotypes was not likely caused by random sorting of data, i.e., that Baz1b+/− mice had consistently smaller eyes than Baz1b+/+ mice. This method is schematized in Figure 1B, with associated data and R script uploaded as Supplementary Material.
Adult weights were first tested with an analysis of variance, evaluating weight based on genotype, sex, research site, and the interactions thereof. Since research site, sex, and genotype each were marked significant either on their own or when interacting with other factors, all 8 groups were preserved during follow-up analyses. Significant main factors (sex, genotype) were followed up with student’s t tests. The resulting 8 p values were adjusted using the Holm method (Holm, 1979). Significant interacting factors (sex & genotype, research site & sex) were followed up by generating interaction plots and evaluating by eye whether the resulting lines were parallel, as would be expected of non-interacting factors. This method is schematized in Figure 1C.
Geometric centers of small bowel motility assays were compared using a two-way ANOVA incorporating sex, genotype, and interaction effects. As no effects were identified as significant, no follow-up analysis was performed. Due to a failed Shapiro-Wilk test, large bowel motility times were compared across genotype using a Wilcoxon rank sum test. Sex was discarded as a factor due to a lack of obvious evidence that sex influenced large bowel motility in the data based on a box plot of sex-separated data. Average neuron densities were compared across genotype using a student’s t test.
RESULTS
Knockout-first Baz1b allele greatly reduces Baz1b expression in all tissues
The IMPC-targeted mutant allele (Baz1btm2a(KOMP)Wtsi; hereafter Baz1b-) was developed with a promoterless knockout-first strategy (Figure 2A) (Skarnes et al., 2011). The knockout-first effect is mediated by the insertion of a lacZ-neoR cassette between exons 4 and 5 flanked by FRT sites. A splice acceptor 5’ to the lacZ-neoR sequence interrupts normal splicing of the mRNA, and an SV40 polyadenylation signal 3’ to the sequence limits RNA readthrough. To determine the effectiveness of the knockout-first allele, we performed RT-qPCR to evaluate Baz1b transcript levels across aorta, brain, liver, and lung tissue in Baz1b+/+ and Baz1b−/− newborns. Our analysis revealed that across all organs tested, expression of the targeted exons was dramatically diminished but still detectable, with relative expression in Baz1b−/− organs reduced to 0.00495 to 0.112 times WT levels (ΔΔCt range: 3.16–7.66, p < 0.05; Figure 2B). Thus, the Baz1b− allele does not completely silence Baz1b expression.
Loss of Baz1b alleles increases lethality prior to weaning
Initial matings of Baz1b mice, irrespective of parental genotype (parent breeder effect p = 0.5024; sex of Baz1b- carrier(s) was ignored), revealed a striking increase in the likelihood of death in mice missing one or both alleles of Baz1b (Figure 3A) after following 388 mice to 28 days of age (P28). Baz1b+/− mice were approximately 3.04 times more likely to die during this interval as compared to Baz1b+/+ mice (95% CI [1.83, 5.06], p = 1.91×10−5), while Baz1b−/− mice were 15.83 times more likely to die before P28 (95% CI [8.54, 29.37], p = 1.89×10−18). The overwhelming majority of Baz1b−/− mutants (43/48; 89.6%) died within 24 hours of birth, typically without striking external morphological defects beyond a slightly smaller body size (Figure 3B). 3 out of 48 (6.24%, all from the Children’s Hospital of Philadelphia (CHOP)) Baz1b−/− mice survived to P28. Of those three CHOP mice, one died shortly after censorship at P32 with apparent scoliosis, while the other two survived well into adulthood with minor eye defects (unilateral corneal clouding in one mouse, unilateral microphthalmia in the other) and reduced body weight (one 15.35g female harvested at P81, one 15.8g male harvested at P70). Heterozygous mice at both institutions died over a more prolonged period, with 41/183 (22.4%) dying within the first 24h and the remaining 24/183 (13.1%) dying over the following 3 weeks; the remaining mice survived until censorship. A Cox proportional hazards model indicated there was no effect on survival based on mouse sex (p = 0.351) or research site (p = 0.762), nor was there sufficient evidence of interactions between site, sex, and genotype to merit testing.
Figure 3.
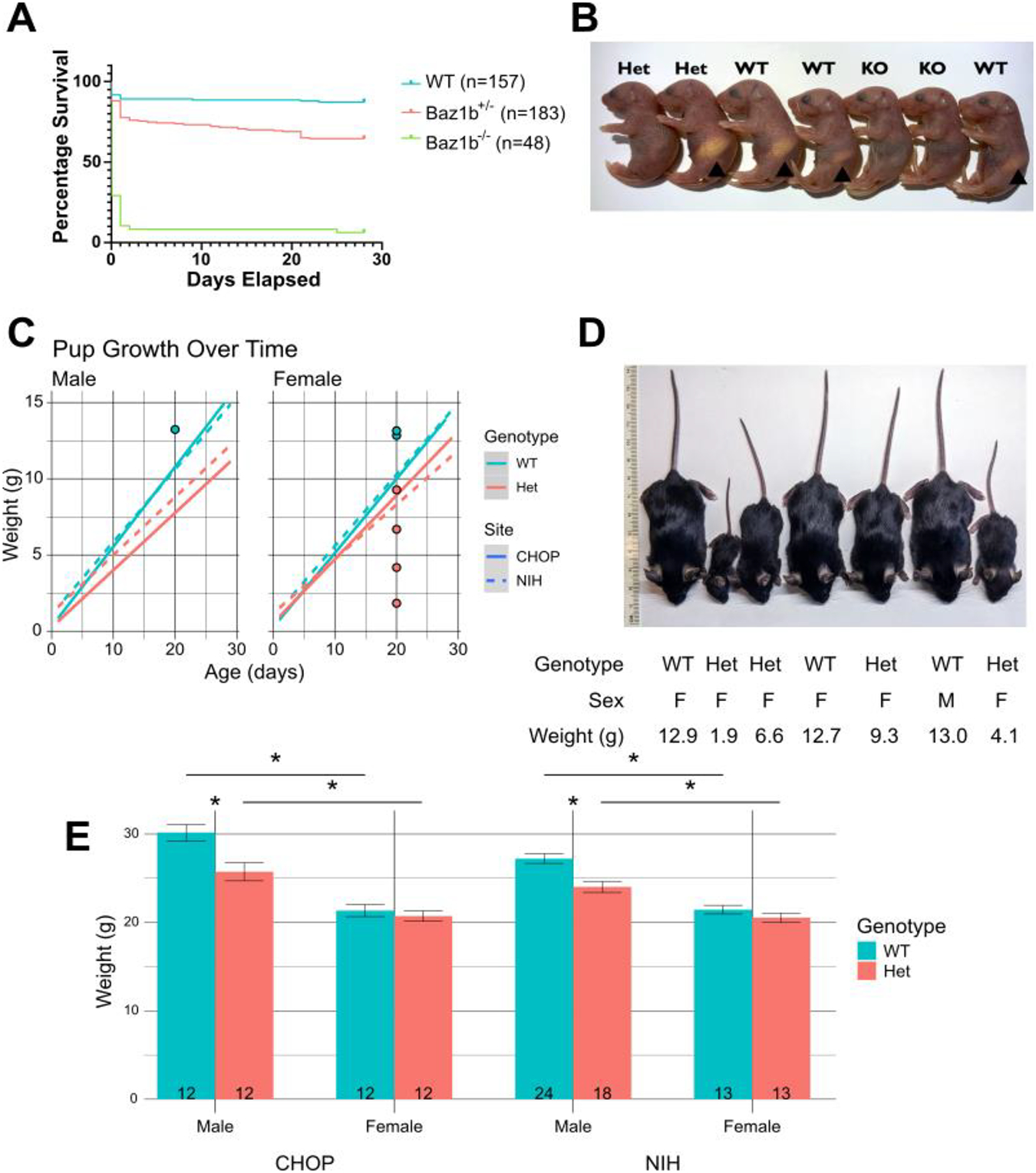
Baz1b survival and growth. A) Survival of Baz1b pups over time. B) Example picture of P0 litter. A stomach milk spot was visible in many Baz1b+/+ and Baz1b+/− mice at P0 (arrowheads), but was not seen in any Baz1b−/− pups. C) Linear regression of weight gain of Baz1b pups over time at both CHOP and the NIH. Dots in C indicate weights of mice depicted in D. D) Inconsistent dramatic differences in mouse size at P20. E) Weights of adult (P78–97) mice; error bars represent standard error of the mean. *p < 0.001
Baz1b heterozygosity reduces growth rate, and reduces adult weight in a sex-specific manner
130 Baz1b+/+ and Baz1b+/− pups were weighed twice a week from shortly after birth until 4 weeks of age (P28–29). Pups that died of natural causes were included in the dataset, while pups that died due to adverse events or were harvested prematurely for experiments were excluded. We used linear mixed modeling to assess the impact of genotype, research site, and sex on pup birth weight and growth rate (Figure 3C). Our final model was well-fitted, with a marginal (fixed effects only) R2 value of 0.846 and a conditional (fixed and random effects) R2 value of 0.927 (Johnson, 2014; Nakagawa and Schielzeth, 2013). Based on this model, Baz1b+/− pups grew more slowly than their Baz1b+/+ littermates by an estimated 0.106g±0.008g per day (age:genotype effect p = 8.61×10−37). This is after controlling for site-specific effects on birth weight (site effect p = 6.91×10−3) and a sex-specific site effect on growth rate (female age:site effect p = 6.26×10−5, male age:site effect p = 0.0573).
While the above model provides an effective analysis of average trends in pup growth, we also noted severely delayed growth in a smaller subset of mice (Figure 3D, Figure 4). Growth delays occurred in pups of either sex at both research sites. Severe growth delay was typically observed in Baz1b+/− pups, although 1 NIH Baz1b+/+ male was observed with similarly delayed growth. Combined, these results indicate that Baz1b heterozygosity slows growth beyond what can be attributed to differences in housing conditions, with high variability in the severity of that slowed growth.
Across 116 adult mice aged 78–97 days old, we identified significant differences in total body mass based on sex (p < 2×10−16) and genotype (p = 3.17×10−6), along with interaction effects between sex & genotype (p = 3.13×10−3) and sex & research site (p = 0.0247) using a three-way analysis of variance (Figure 3E). In follow-up analyses comparing sex, we found that female mice have consistently lower body mass than male mice, while Baz1b heterozygosity only reduced adult body mass in male mice (Table 1, Figure 5A). We also validated the interaction between sex & research site and determined there was a male-specific difference across sites (Figure 5B).
Table 1.
Statistical follow-up Student’s t tests for adult weight ANOVA
| Compared factor | Sex (Male vs Female) |
Genotype (Baz1b+/+ vs Baz1b+/−) |
||||||
|---|---|---|---|---|---|---|---|---|
| Constant factors | CHOP Baz1b+/+ | CHOP Baz1b+/− | NIH Baz1b+/+ | NIH Baz1b+/− | CHOP Male | CHOP Female | NIH Male | NIH Female |
| Weight difference | 8g±1g | 5g±1g | 5.8g±0.7g | 3.5g±0.8g | 4g±1g | 0.6g±0.9g | 3.2g±0.8g | 0.9g±0.7g |
| p value | 1.57×10−12 | 3.61×10−5 | 1.81×10−8 | 9.83×10−4 | 2.56×10−4 | 0.743 | 5.14×10−4 | 0.743 |
Neonatal Baz1b mutants display cyanosis but no cardiac abnormalities
At the NIH, 50 neonates (12 Baz1b+/+, 18 Baz1b+/−, and 20 Baz1b−/−) were evaluated within 4hrs of birth for cyanosis and patent ductus arteriosus (PDA) (Table 2). 3/20 Baz1b−/− pups showed prolonged, generalized cyanosis at birth (Figure 6A), in contrast to Baz1b+/+ (0/12) and Baz1b+/− (0/18) pups. Additionally, while all Baz1b+/+ and 17/18 Baz1b+/− pups displayed normal developmental closure of the ductus arteriosus, 8/20 Baz1b−/− pups died with PDA (Figure 6B–D). Furthermore, 5/20 Baz1b−/− pups exhibited underinflated lungs at time of death, a phenotype that was uncommon in Baz1b+/+(1/12) and Baz1b+/− (1/18) pups (Table 2, Figure 6E–F). Given the cardiac symptoms associated with WS, we followed up these observations with a complete morphologic assessment of cardiac segments, including the great vessels. All hearts had normal segmental anatomy with normal situs, grossly normal chamber structure, and a normal-appearing aortic arch. Specifically, none had supravalvar aortic stenosis (SVAS, neither discrete nor long segment; a hallmark of WS) or coarctation of the aorta (Table 2). Finally, we performed cardiac μCT on 4 Baz1b+/+, 4 Baz1b+/−, and 14 Baz1b−/− P0 mice collected from both CHOP and the NIH. This analysis confirmed that all hearts had normal segmental anatomy, including no evidence of septal abnormalities (Figure 6G–H). We found no cardiac structural abnormality that would explain Baz1b−/− perinatal mortality.
Figure 6.
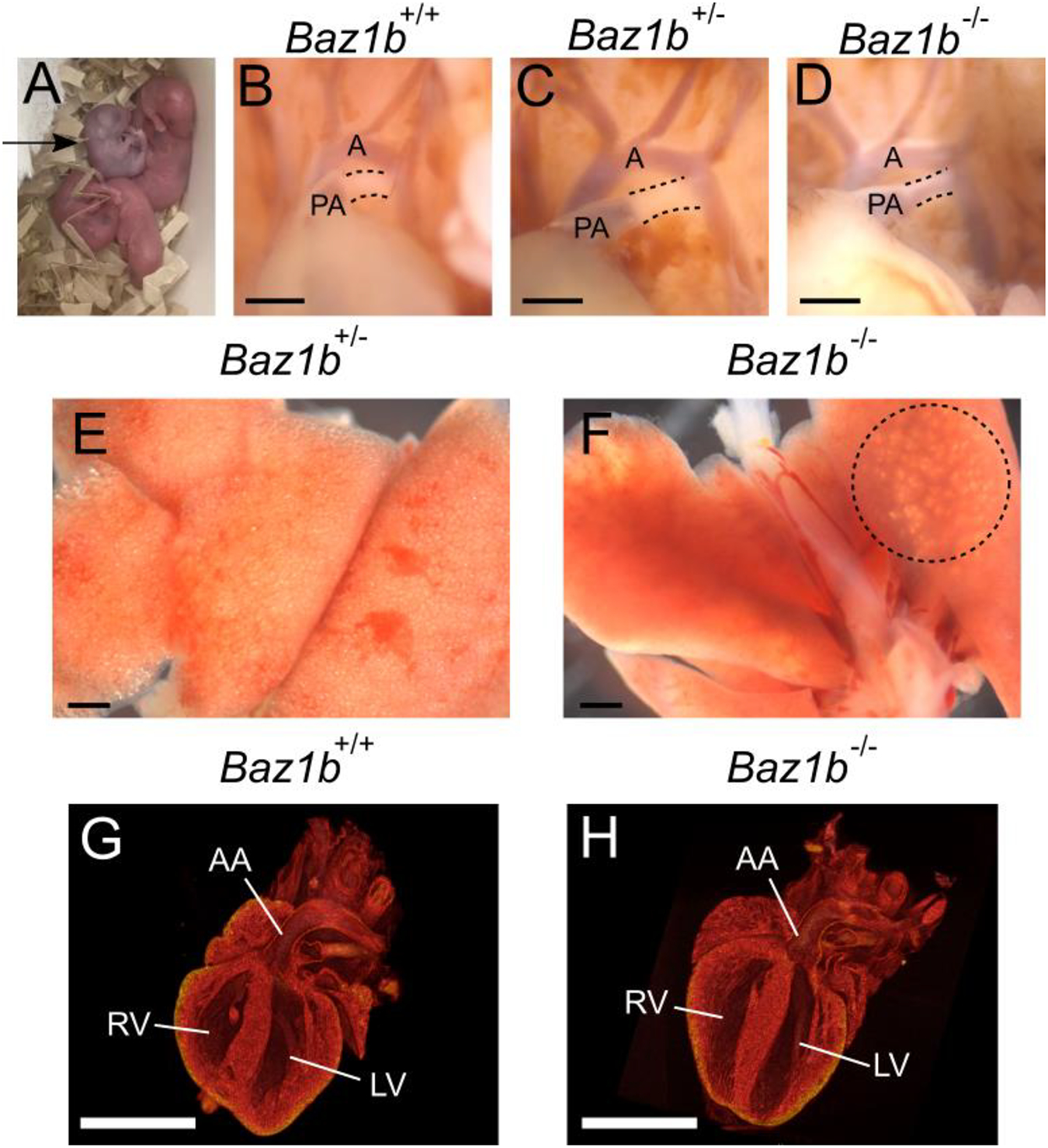
Baz1b−/− pups display cyanosis but no cardiac abnormalities. A) Cyanotic Baz1b−/− pup (arrow) in an otherwise healthy litter at birth. B-D) The ductus arteriosus of P0 pups, outlined with dashed lines. Note the patent (blood filled between the PA and aorta) ductus arteriosus in D. A, aorta; PA, pulmonary artery. E-F) Lungs of a healthy Baz1b+/− pup and an affected Baz1b−/− pup. The Baz1b−/− lungs are devoid of air except in the area circled with a dashed line, and sank when placed in PBS. B-F) Scale bars are 0.5mm. G-H) μCT scans of P0 left ventricular outflow tract showing no structural defects in Baz1b+/+ or Baz1b−/− heart chambers. Scale bars are 1mm. LV, left ventricle; RV, right ventricle; AA, ascending aorta.
Rare mandibular and ocular phenotypes were recovered during experimentation
Because Baz1b is widely expressed throughout development (Dickinson et al., 2016), we took a broad approach to phenotypic assessment. At CHOP, we systematically evaluated gross anatomy of the lung, liver, kidney, pancreas, spleen, reproductive system, and skeleton (paying special attention to the hard palate) in 14 Baz1b+/− mice (P12–237) and 1 Baz1b−/− adult survivor (P81). We found no defects in these organs. At the NIH, the 50 neonates described above were also evaluated for overlapping anatomic features associated with WS, neural crest dysfunction, or potential causes for lethality (Table 2). In addition to the cardiopulmonary features described in Figure 6, we noted that none of the Baz1b−/− pups developed a milk spot, compared to 7/12 Baz1b+/+ and 7/18 Baz1b+/− pups (Figure 3B).
During the survival analysis and other experiments at CHOP, 2 pups with total agnathia (absent mandible) were recovered (Figure 7A–B; 1/135 Baz1b+/− pups, 1/42 Baz1b−/− pups). Agnathia was not observed during the analysis of any of the 120 Baz1b+/+ pups analyzed at CHOP during the same interval, nor in any of the 50 neonates collected for phenotyping at the NIH.
Figure 7.
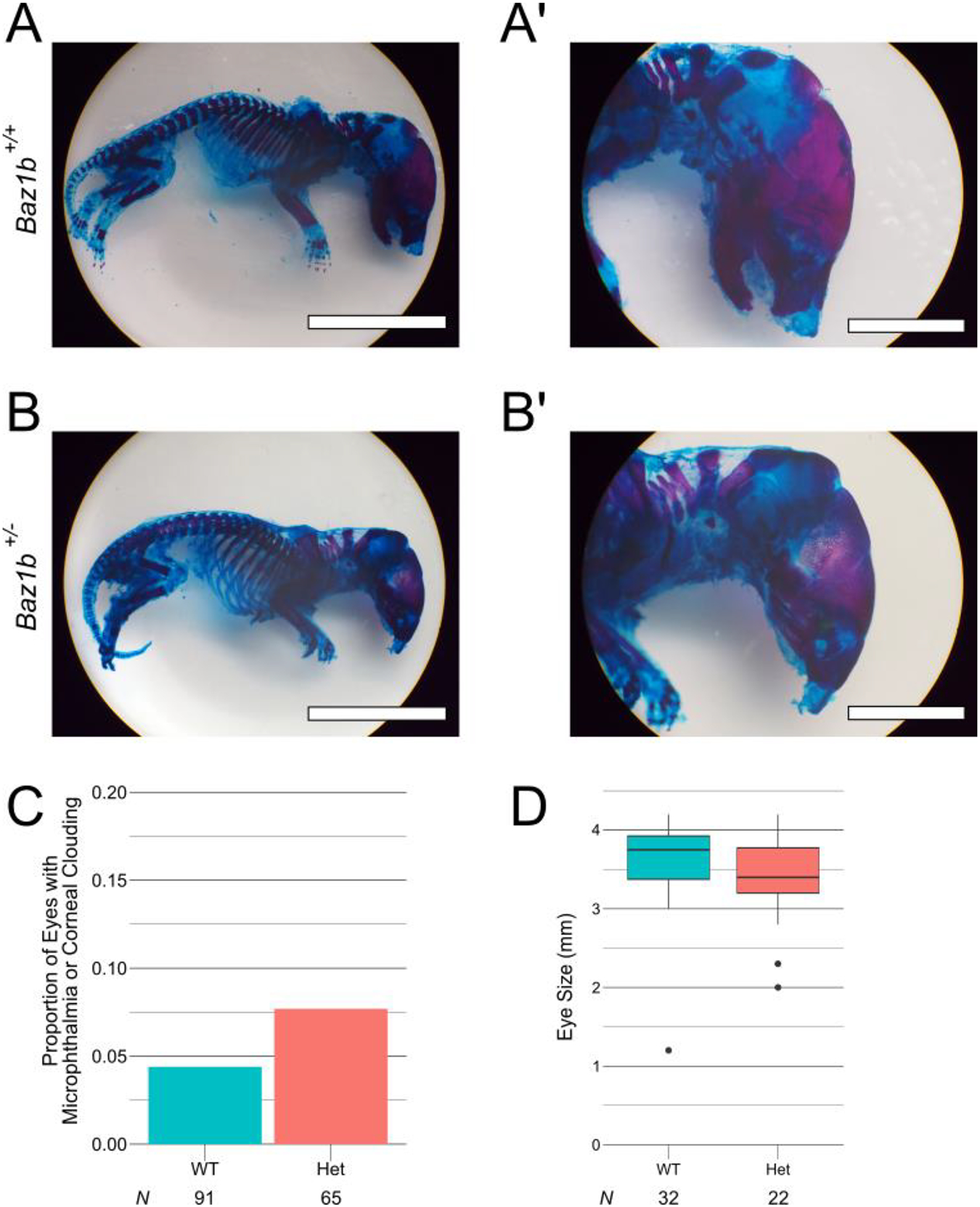
Rare agnathia and ocular defects recovered in Baz1b mutant mice. A-B) P0 pup skeletons stained with Alcian blue (cartilaginous bone) and Alizarin red (mineralized bone). The Baz1b+/− pup lacks a mandible, demonstrating a rare agnathic phenotype. Scale bars in A and B are 1cm. Scale bars in A’, B’ are 0.5cm. C) Frequency of microphthalmia or corneal clouding in Baz1b mice. D) Box plot of individual eye size in Baz1b mice.
In early generations of Baz1b+/− mice at CHOP, we observed corneal clouding and microphthalmia (5/65, 7.69%). However, there was not a significant increase in occurrences compared to Baz1b+/+ mice at CHOP (4/91, 4.40%; Figure 7C, p = 0.211). Eyes collected during this period were also of comparable median eye diameter across 16 Baz1b+/+ mice (32 eyes) and 11 Baz1b+/− mice (22 eyes; Figure 7D, p = 0.05748). Corneal clouding and microphthalmia have not been observed in recent generations. At the NIH, 1 Baz1b+/− pup collected for analysis was recovered with unilateral anophthalmia (1/18, 5.56%; Table 2).
Baz1b heterozygosity increases frequency of bowel dilation without affecting motility
One common phenotype evaluated at CHOP was dilated bowel (Figure 8A–D). We identified bowel dilation in Baz1b+/− adults at a significantly higher frequency (20/30, 66.7%) compared to Baz1b+/+ adults (14/37, 37.8%; p = 1.99×10−3). Of the 34 total mice with bowel dilation, the 20 Baz1b+/− mice were more likely to have dilated stomach (p = 0.0107), small intestine (proximal: 0/14 Baz1b+/+ vs 2/20 Baz1b+/−, middle: 0/14 Baz1b+/+ vs 4/20 Baz1b+/−, distal: p = 1.92×10−3), and distal colon (p = 0.0175) than the 14 Baz1b+/+ mice (Figure 8B). Bowel dilation was usually mild (Figure 8C), but on one occasion was considered cause for euthanasia (Figure 8D).
Figure 8.
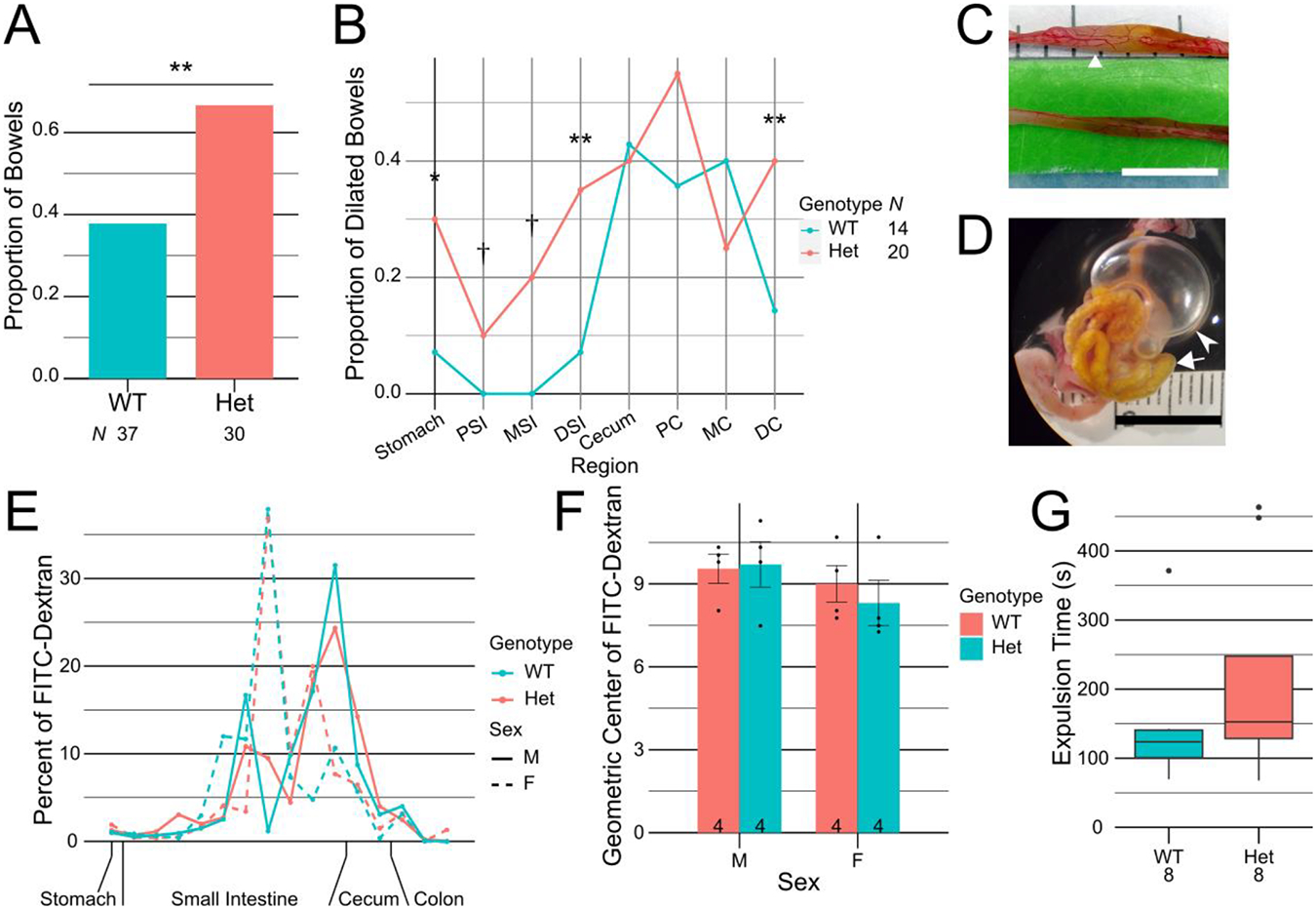
Loss of Baz1b increases frequency of bowel dilation, but bowel transit is normal. A) Frequency of air found in any bowel segment. B) Frequency of air in specific bowel segments, normalized to number of bowels found with air. C) Representative bowel with mild dilation, from a Baz1b+/− mouse at P208. Triangular arrow points to air bubble in small bowel. D) Representative bowel with severe dilation, from a Baz1b+/− mouse at P20. Primary inflated region (marked with pointed arrowhead) is the cecum. Triangular arrowhead indicates small bowel filled with small air bubbles. E) Distribution of FITC-dextran in whole bowel 60min after gavage. F) Geometric centers of FITC-dextran distribution after 60min. Error bars represent standard error of the mean. G) Box plot of colonic bead expulsion times. C, D) Scale bars are 1cm. *p < 0.05, **p < 0.01, †Never observed in WT but observed in heterozygous mice. PSI, proximal small intestine (SI); MSI, middle SI; DSI, distal SI; PC, proximal colon; MC, middle colon; DC, distal colon.
Because people with WS often have symptoms suggesting defective bowel motility (Cherniske et al., 2004; Morris et al., 1990), we hypothesized that the dilated bowel in Baz1b mutant mice was caused by defects in the enteric nervous system (ENS), a key neural crest–derived structure that controls bowel motility. To determine if bowel transit was abnormal in Baz1b+/− mice, we first gavage fed adult mice FITC-dextran, a poorly absorbed fluorescent polymer (Figure 8E). After 60 minutes, most of the FITC-dextran was found between 70% and 90% of the way down the small intestine in all mice (Figure 8F). No significant effect was found based on genotype (p = 0.713), sex (p = 0.199), or their interaction (p = 0.565). To test colon function, we measured expulsion time for a 2mm glass bead inserted 2cm into the colon (Figure 8G). We found no significant difference between expulsion times for Baz1b+/+ (median time 123.67s, IQR [100.8s, 140.7s]) and Baz1b+/− mice (median time 152.67s, IQR [128.58s, 247.75s]; p = 0.2786). These results suggest that the bowel dilation observed was not due to significantly impaired bowel transit.
Hyperganglionosis in neonatal Baz1b−/− distal colon myenteric plexus
In parallel with the bowel function studies, we directly evaluated ENS anatomy in Baz1b−/− mice. Since Baz1b−/− mice typically die within 24 hours of birth, we collected P0 bowels by delaying birth with a progesterone injection, then performing a Cesarean delivery. We then visualized neonatal ENS in the proximal SI (PSI), distal SI (DSI), and distal colon, using confocal imaging to distinguish between the myenteric (Figure 9) and submucosal plexuses (Figure 10) after ANNA1 and TuJ1 (neuron-specific β tubulin III) antibody staining. These antibodies stain all enteric neurons. Enteric neuron density was similar between Baz1b+/+ and Baz1b−/− mice in PSI and DSI. In distal colon, however, we observed a 47.2% increase in Baz1b−/− myenteric plexus enteric neuron density (8730.1 neurons/mm2, std dev 1425.2 neurons/mm2) compared to Baz1b+/+ bowel (5932.5 neurons/mm2, std dev 392.9 neurons/mm2; p = 0.0252). No significant differences in submucosal plexus neuron density were found between Baz1b+/+ and Baz1b−/− mice.
Figure 9.
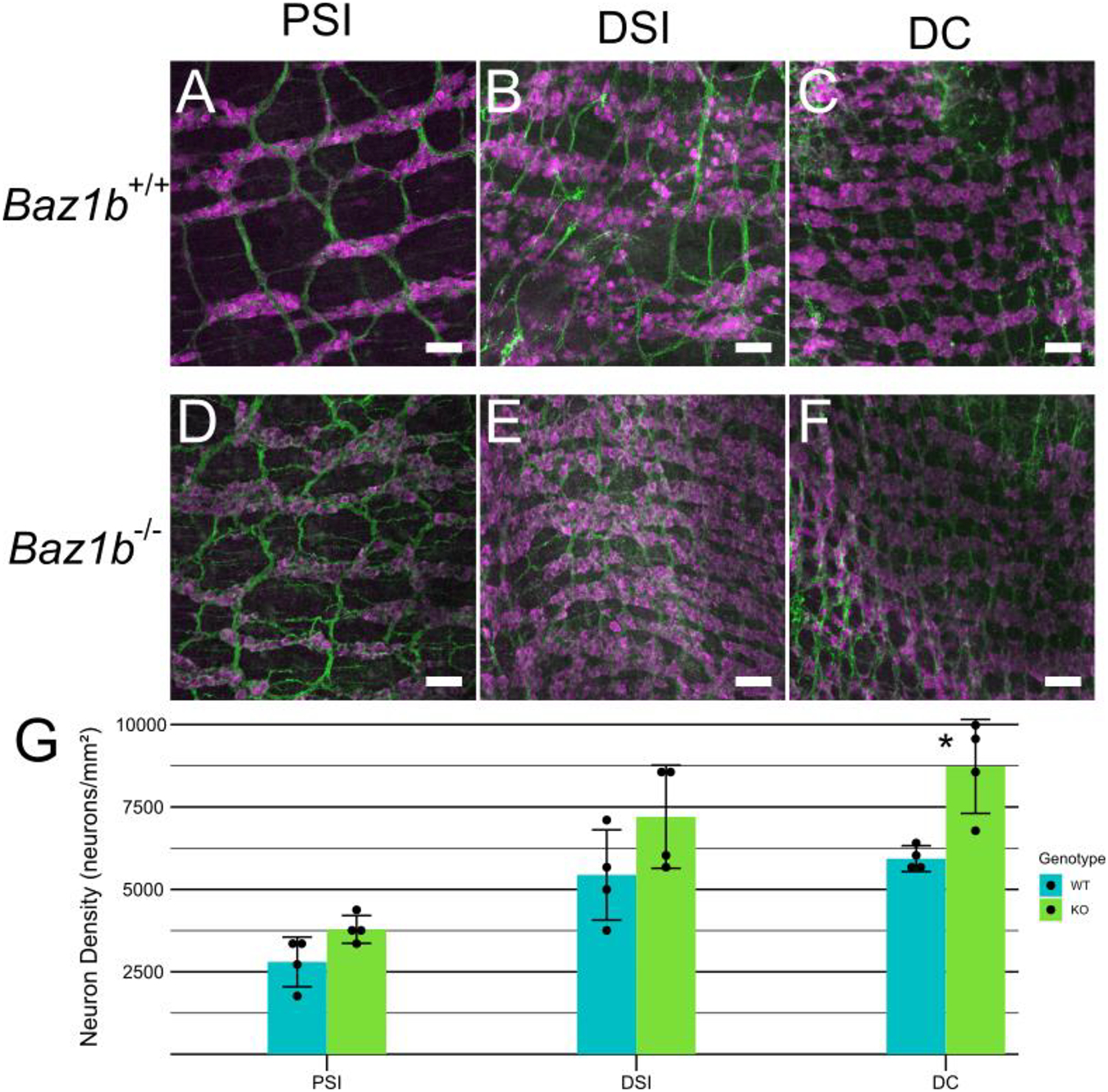
P0 myenteric plexus. A-F) Immunohistochemistry showing myenteric plexus in WT and Baz1b−/− mice. Neurons labeled with ANNA1 antibody (pseudocolored magenta) and neurites labeled with TuJ1 antibody (pseudocolored green). Scale bars are 50μm. G) Average myenteric neuron density. Error bars represent standard deviation. *p < 0.05. PSI, proximal small intestine; DSI, distal small intestine; DC, distal colon.
Figure 10.
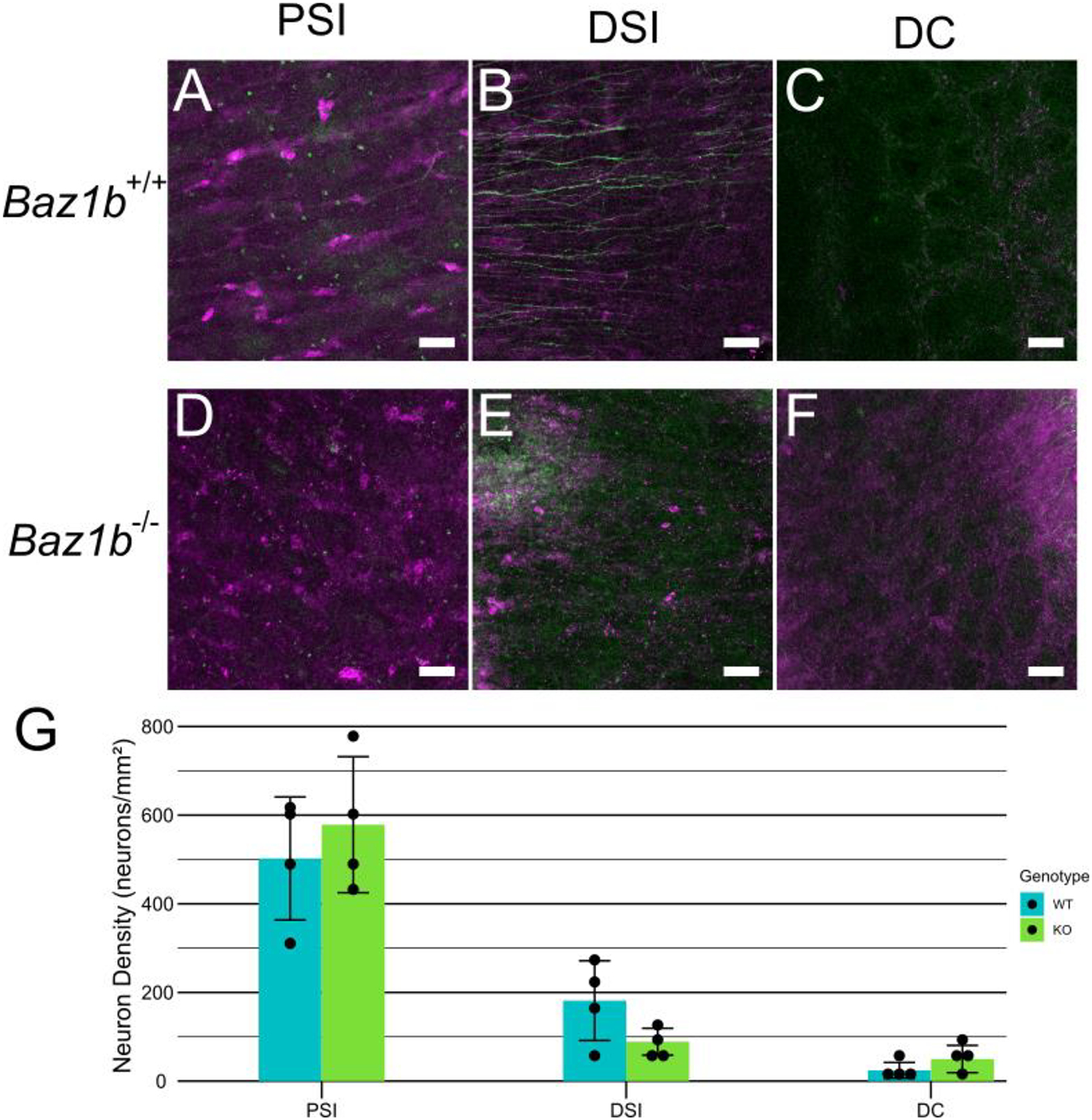
P0 submucosal plexus. A-F) Immunohistochemistry showing submucosal plexus in WT and Baz1b−/− mice. Neurons labeled with ANNA1 antibody (pseudocolored magenta) and neurites labeled with TuJ1 antibody (pseudocolored green). Scale bars are 50μm G) Average submucosal neuron density. Error bars represent standard deviation. PSI, proximal small intestine; DSI, distal small intestine; DC, distal colon.
DISCUSSION
Williams syndrome (WS) is caused by a 1.5 to 1.8Mbp hemizygous deletion on chromosome 7q11.23 that encompasses 25–27 coding genes, including BAZ1B. WS phenotypes are variable but include stenoses in the great arteries (especially SVAS), distinctive craniofacial appearance, intellectual disability, hypersociability, short stature with failure to gain anticipated weight in childhood, feeding difficulty, constipation, abdominal pain, and urinary tract anomalies (Kozel et al., 2021). While elastin hemizygosity is known to cause SVAS in people with WS, many WS phenotypes have not yet been attributed to a single gene in the deletion. Symptoms vary widely among affected individuals, and the degree to which haploinsufficiency for multiple genes at once is required to cause WS-associated traits is unknown. The hypothesis that BAZ1B haploinsufficiency contributes to craniofacial, bowel, great vessel, and central nervous system phenotypes in WS is supported by studies using morpholinos in Xenopus laevis to reduce levels of the BAZ1B homolog (also called Williams syndrome transcription factor, WSTF). WSTF morpholino–treated X. laevis had severe defects in the eye, brain, and migrating neural crest–derived cells that contribute to the branchial arches, facial bones, and enteric nervous system (Barnett et al., 2012). A new zebrafish baz1b mutant also had altered craniofacial anatomy, eye dimensions, and behavior, but early death was not reported in the homozygous baz1b mutant fish (Torres-Pérez et al., 2023). In contrast, our studies in mice show that Baz1b−/− mice die as neonates without obvious structural birth defects, though they also fail to feed for reasons that remain unclear. Specifically, Baz1b mutant mice do not demonstrate classic cardiac anomalies associated with neural crest defects, including Tetralogy of Fallot, double outlet right ventricle, or truncus arteriosus, among others. The Baz1b−/− mice further lack WS-associated heart disease or renal anomalies, and do not have defects that mimic morpholino effects in X. laevis or null effects in zebrafish. Furthermore, most Baz1b heterozygous mice appeared to be generally healthy, though they tend to grow more slowly than their wild-type counterparts (similar to people with WS). In addition, we identified some very unusual birth defects in Baz1b mutant mice (e.g. agnathia), but these were uncommon.
Baz1b is involved in a wide range of functions, which were recently reviewed by Sharif, Zamani, and Chadwick (Sharif et al., 2021). Many of these functions position Baz1b to broadly influence gene expression and cell survival. Baz1b is expressed in multiple tissues across development (as demonstrated by the IMPC at mousephenotype.org and the Human Protein Atlas at proteinatlas.org) (Dickinson et al., 2016; Uhlén et al., 2015). This makes it challenging to pinpoint potential causes of death in Baz1b−/− neonates. This challenge is further complicated by the inconsistency of phenotypes observed in our NIH and CHOP mouse colonies. For example, our Baz1b+/− pups grew at dramatically varying rates even within litters (Figure 3D). Furthermore, while many of our Baz1b mutant mice die prematurely, most Baz1b+/− and some rare Baz1b−/− mice survive to adulthood (Figure 3A). Although our survival dataset is striking and consistent across facilities, it differs from previous reports on the viability of Baz1b mutant mice (Ashe et al., 2008; Dickinson et al., 2016). When the Baz1b- allele was developed as part of the Knockout Mouse Project, the IMPC reported the strain as “subviable,” with limited data available on Baz1b−/− mice (Dickinson et al., 2016). When a c.2585T>G; p.L733R missense variant was discovered in FVB/NJ mice as an enhancer of variegation, it was also associated with decreased survival of Baz1b homozygous mutant pups. The exact survival rate in the variegation study is unclear, but appears higher than our estimated 6.25% survival rate (Ashe et al., 2008). The only known publication with complete neonatal Baz1b homozygous mutant mortality is a retracted paper from 2009, using an unknown mutant allele (“Retraction for Yoshimura et al., Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF),” 2014; Yoshimura et al., 2009). However, we did not replicate the cardiovascular findings of that retracted manuscript (Figure 6G–H).
The loss of microphthalmia and corneal clouding over time may be explained by changes in our strain background. The IMPC developed the original Baz1b− allele in the C57BL/6N line. While our research uses the same construct, it was discovered in November of 2019 (approximately 2.5 years into research) that we had crossed a Baz1b+/− mouse with one on a C57BL/6J background. Genetic testing of a new founder mating from that time revealed that approximately 62.5% of SNPs differentiating the two genetic backgrounds had fixed to one original background or another. Notably, the characteristic Crb1rd8 allele from the C57BL/6N line, which causes retinal degeneration (Mattapallil et al., 2012), was no longer present in our mouse colony. This change may explain the low penetrance of ocular defects in early generations (Figure 7C–D) and their absence in recent generations.
The highly variable phenotypes of Baz1b mutant mice are reminiscent of the variability in some epigenetic disorders. For example, loss of function mutations in KDM6A, a lysine demethylase that removes methyl marks on histone 3, lysine 27 (H3K27) (Agger et al., 2007; Hong et al., 2007; Lan et al., 2007), is associated with type 2 Kabuki syndrome, a condition that can present with highly variable symptoms (Bögershausen et al., 2016). H3K27 histone marks correlate with the presence of heterochromatin (Wiles and Selker, 2017), and loss of heterochromatin correlates with elevated KDM6A in MCF10A ER-Src cells (Gurrion et al., 2017). Similarly, loss of BAZ1B in cell cultures is linked to increased H3K27 dimethylation (Poot et al., 2004). Loss of BAZ1B is also linked to expansion of heterochromatin (Culver-Cochran and Chadwick, 2013; Istomina et al., 2003), even at an organismal level (Ashe et al., 2008). Regrettably, without knowing which organ defects consistently cause perinatal death in Baz1b−/− mice, hypotheses about molecular epigenetic mechanisms causing death are difficult to test. This is especially complicated given the incomplete silencing of Baz1b expression with the analyzed Baz1b- allele. We also cannot exclude the possibility that rare splice isoforms are produced by this mutant model with partial or altered BAZ1B activity.
Our work reported here centers primarily on the heart and bowel because cardiovascular and bowel problems are common in people with WS, especially SVAS and sometimes coarctation of the aorta (Yuan, 2017). Although some of our Baz1b−/− mice had generalized cyanosis, reduced lung inflation, and/or died with PDA, inspection of postnatal hearts at P0 revealed no cardiac dysmorphology (Figure 6G–H, Table 2). We did note an increase in PDA in Baz1b mutants (Figure 6B–D, Table 2), but PDA can be considered physiologic at this age. Further, we found no convincing evidence of persistent fetal circulation or volume overload that would be expected if the PDA conferred pathologic hemodynamic loading conditions resulting in death. This indicates that PDA is unlikely to explain the high Baz1b−/− neonatal mortality. It is worth noting that PDA is uncommon in people with WS (De Rubens Figueroa et al., 2008), further distinguishing murine Baz1b−/− phenotypes from WS symptoms. That said, duplication of the region deleted in WS (known as 7q11.23 duplication syndrome) is associated with increased PDA (Morris et al., 2015), suggesting a possible connection between WS-associated gene dosage and ductus arteriosus closure. While adults with WS commonly report abdominal pain, constipation, and diarrhea (Cherniske et al., 2004; Morris et al., 1990), we found no evidence of serious motility defects in Baz1b+/− mice, and only moderate hyperganglionosis in the distal colon myenteric plexus of Baz1b−/− neonates. Bowel dilation and myenteric hyperganglionosis are associated with inflammatory bowel disease (IBD) (Margolis et al., 2011), but we did not observe any symptoms of IBD during our gross phenotyping. That said, irritable bowel syndrome (IBS), diverticulitis, and celiac disease are increased among people with WS (Giannotti et al., 2001; Partsch et al., 2005; Williams Syndrome Association, 2014), and bowel dilation or myenteric hyperganglionosis might contribute to these symptoms. These variable phenotypes suggest that haploinsufficiency of BAZ1B alone does not drive the developmental cardiac defects or outflow tract abnormalities in WS and may play only a minor role in WS bowel problems.
One possibility is that partial loss of Baz1b sensitizes developmental pathways to disturbances in environmental or genetic factors, leading to uncommon but dramatic phenotypes like the agnathia (absent mandible) seen in 1/135 Baz1b+/− and 1/42 Baz1b−/− mice (Figure 7A–B). While agnathia was not observed in the 120 Baz1b+/+ pups studied in this paper, a wild type neonate in an unrelated strain was found with agnathia in the NIH colony (1 found at the NIH over 7 years), indicating Baz1b may not be directly causative, but still influential. Agnathia is rare in humans, with an estimated incidence of less than 1 in 70,000. In mice, agnathia has been reported as a result of folate deficiency (Maldonado et al., 2018), treatment with toxins (e.g. retinoic acid) (Yasuda et al., 1986), and in the context of many genetic changes (e.g. ectopic expression of Hoxb1 (Zaffran et al., 2018), total loss of Twsg1 (Melnick et al., 2006), or partial loss of Otx2 (Hide et al., 2002)). These factors often cause other birth defects in addition to agnathia. Combined, this suggests that loss of Baz1b may have lowered the threshold for agnathia, thereby increasing the frequency of agnathia from “vanishingly rare” to “rare, but recoverable.” Given the low frequency of agnathia in our mouse colonies, the absence of many other associated congenital malformations, and the complex origins of the agnathia birth defect, additional mechanistic studies of how Baz1b mutations might predispose to agnathia are not feasible.
In total, our data presents a complicated picture of BAZ1B and its contribution to WS. In contrast to previous research, we identified a decrease in adult Baz1b+/− body mass specifically in male mice and found that heterozygous and homozygous loss of Baz1b results in increasing mortality, especially during the first 24 hours after birth. Additionally, we observed increased (but still variable) incidence of bowel dilation and rare instances of total agnathia. We also determined that the early death and growth failure are likely not due to changes in heart structure or bowel function. In particular, the increased distal colon enteric neuron density in Baz1b−/− mice seems unlikely to cause perinatal death, since mice with profound bowel dysfunction caused by absence of the ENS in distal colon live much longer than Baz1b−/− neonates (Soret et al., 2020). Combined with prior studies, these results suggest BAZ1B reduced copy number may contribute to the variability of WS symptoms and may sensitize development to other genetic or environmental factors that contribute to WS phenotypes.
Supplementary Material
Loss of Baz1b results in high neonatal death within 24 hours of birth
Partial loss leads to variable growth defects during adolescence and into adulthood
Cardiovascular symptoms of Williams syndrome are not replicated in Baz1b mutants
High phenotype variability suggests loss of Baz1b sensitizes to development defects
Acknowledgments
Funding was provided by The Irma and Norman Braman Endowment, The Suzi and Scott Lustgarten Center Endowment, Children’s Hospital of Philadelphia Research Institute, the Children’s Hospital of Philadelphia Center for Precision Diagnosis and Therapy for Pediatric Motility Disorders Frontier Program, and The Children’s Discovery Institute of St. Louis Children’s Hospital and Washington University School of Medicine (Grant: MDII2013269). The NIH efforts were supported by the Division of Intramural Research at the Heart, Lung, and Blood Institute of the NIH.
The authors thank Dr. Robert Hufnagel for his advice on evaluating Baz1b mutant eyes, Dr. Joseph Dougherty for helpful guidance as we began this work, the Mouse Imaging Facility of NINDS at NIH, and the CHOP Biostatistics and Data Management Core for advice on building and evaluating appropriate linear mixed effects models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
ROH was a consultant for BlueRock Therapeutics and for Takeda.
Bibliography
- Agger K, Cloos PAC, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K, 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734. 10.1038/nature06145 [DOI] [PubMed] [Google Scholar]
- Ashe A, Morgan DK, Whitelaw NC, Bruxner TJ, Vickaryous NK, Cox LL, Butterfield NC, Wicking C, Blewitt ME, Wilkins SJ, Anderson GJ, Cox TC, Whitelaw E, 2008. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 9, R182. 10.1186/gb-2008-9-12-r182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett C, Yazgan O, Kuo H-C, Malakar S, Thomas T, Fitzgerald A, Harbour W, Henry JJ, Krebs JE, 2012. Williams Syndrome Transcription Factor is critical for neural crest cell function in Xenopus laevis. Mech. Dev 129, 324–338. 10.1016/j.mod.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bögershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, Simsek-Kiper PÖ, Barat-Houari M, Elcioglu NH, Wieczorek D, Tinschert S, Sarrabay G, Strom TM, Fabre A, Baynam G, Sanchez E, Nürnberg G, Altunoglu U, Capri Y, Isidor B, Lacombe D, Corsini C, Cormier-Daire V, Sanlaville D, Giuliano F, Le Quan Sang K-H, Kayirangwa H, Nürnberg P, Meitinger T, Boduroglu K, Zoll B, Lyonnet S, Tzschach A, Verloes A, Di Donato N, Touitou I, Netzer C, Li Y, Geneviève D, Yigit G, Wollnik B, 2016. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum. Mutat 37, 847–864. 10.1002/humu.23026 [DOI] [PubMed] [Google Scholar]
- Bozhenok L, Wade PA, Varga-Weisz P, 2002. WSTF–ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 21, 2231–2241. 10.1093/EMBOJ/21.9.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavellán E, Asp P, Percipalle P, Farrants AKÖ, 2006. The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J. Biol. Chem 281, 16264–16271. 10.1074/JBC.M600233200/ATTACHMENT/4817CE12-2516-4581-B5A6-32DA3A7F58B2/MMC1.PDF [DOI] [PubMed] [Google Scholar]
- Cherniske EM, Carpenter TO, Klaiman C, Young E, Bregman J, Insogna K, Schultz RT, Pober BR, 2004. Multisystem study of 20 older adults with Williams syndrome. Am. J. Med. Genet Part A 131A, 255–264. 10.1002/AJMG.A.30400 [DOI] [PubMed] [Google Scholar]
- Culver-Cochran AE, Chadwick BP, 2013. Loss of WSTF results in spontaneous fluctuations of heterochromatin formation and resolution, combined with substantial changes to gene expression. BMC Genomics 14, 740. 10.1186/1471-2164-14-740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubens Figueroa J, Rodríguez LMO, Hach JLP, Del Castillo Ruíz V, Martínez HO, 2008. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Texas Hear. Inst. J 35, 279–85. [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, Baker CN, Bower L, Brown JM, Caddle LB, Chiani F, Clary D, Cleak J, Daly MJ, Denegre JM, Doe B, Dolan ME, Edie SM, Fuchs H, Gailus-Durner V, Galli A, Gambadoro A, Gallegos J, Guo S, Horner NR, Hsu C-W, Johnson SJ, Kalaga S, Keith LC, Lanoue L, Lawson TN, Lek M, Mark M, Marschall S, Mason J, McElwee ML, Newbigging S, Nutter LMJ, Peterson KA, Ramirez-Solis R, Rowland DJ, Ryder E, Samocha KE, Seavitt JR, Selloum M, Szoke-Kovacs Z, Tamura M, Trainor AG, Tudose I, Wakana S, Warren J, Wendling O, West DB, Wong L, Yoshiki A, Wurst W, MacArthur DG, Tocchini-Valentini GP, Gao X, Flicek P, Bradley A, Skarnes WC, Justice MJ, Parkinson HE, Moore M, Wells S, Braun RE, Svenson KL, de Angelis MH, Herault Y, Mohun T, Mallon A-M, Henkelman RM, Brown SDM, Adams DJ, Lloyd KCK, McKerlie C, Beaudet AL, Bućan M, Murray SA, 2016. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. 10.1038/nature19356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhmandakh B, Makeyev AV, Erdenechimeg L, Ruddle FH, Chimge N-O, Tussie-Luna MI, Roy AL, Bayarsaihan D, 2009. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc. Natl. Acad. Sci 106, 181–186. 10.1073/pnas.0811531106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotti A, Tiberio G, Castro M, Virgilii F, Colistro F, Ferretti F, Digilio MC, Gambarara M, Dallapiccola B, 2001. Coeliac disease in Williams syndrome. J. Med. Genet 38, 767–8. 10.1136/jmg.38.11.767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrion C, Uriostegui M, Zurita M, 2017. Heterochromatin Reduction Correlates with the Increase of the KDM4B and KDM6A Demethylases and the Expression of Pericentromeric DNA during the Acquisition of a Transformed Phenotype. J. Cancer 8, 2866–2875. 10.7150/jca.19477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hide T, Hatakeyama J, Kimura-Yoshida C, Tian E, Takeda N, Ushio Y, Shiroishi T, Aizawa S, Matsuo I, 2002. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development 129, 4347–4357. 10.1242/dev.129.18.4347 [DOI] [PubMed] [Google Scholar]
- Holm S, 1979. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat 6, 65–70. [Google Scholar]
- Hong S, Cho Y-W, Yu L-R, Yu H, Veenstra TD, Ge K, 2007. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci 104, 18439–18444. 10.1073/pnas.0707292104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istomina NE, Shushanov SS, Springhetti EM, Karpov VL, Krasheninnikov IA, Stevens K, Zaret KS, Singh PB, Grigoryev SA, 2003. Insulation of the Chicken β-Globin Chromosomal Domain from a Chromatin-Condensing Protein, MENT. Mol. Cell. Biol 23, 6455–6468. 10.1128/MCB.23.18.6455-6468.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PCD, 2014. Extension of Nakagawa & Schielzeth’s R 2 GLMM to random slopes models. Methods Ecol. Evol 5, 944–946. 10.1111/2041-210X.12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, Pober BR, 2021. Williams syndrome. Nat. Rev. Dis. Prim 7, 42. 10.1038/s41572-021-00276-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y, 2007. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694. 10.1038/nature06192 [DOI] [PubMed] [Google Scholar]
- Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT, 1998. Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280. 10.1038/30522 [DOI] [PubMed] [Google Scholar]
- Maldonado E, López Y, Herrera M, Martínez-Sanz E, Martínez-Álvarez C, Pérez-Miguelsanz J, 2018. Craniofacial structure alterations of foetuses from folic acid deficient pregnant mice. Ann. Anat. - Anat. Anzeiger 218, 59–68. 10.1016/j.aanat.2018.02.010 [DOI] [PubMed] [Google Scholar]
- Margolis KG, Stevanovic K, Karamooz N, Li ZS, Ahuja A, D’Autréaux F, Saurman V, Chalazonitis A, Gershon MD, 2011. Enteric neuronal density contributes to the severity of intestinal inflammation. Gastroenterology 141, 588–98, 598.e1–2. 10.1053/j.gastro.2011.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil MJ, Wawrousek EF, Chan C-C, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR, 2012. The Rd8 Mutation of the Crb1 Gene Is Present in Vendor Lines of C57BL/6N Mice and Embryonic Stem Cells, and Confounds Ocular Induced Mutant Phenotypes. Investig. Opthalmology Vis. Sci 53, 2921. 10.1167/iovs.12-9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick M, Petryk A, Abichaker G, Witcher D, Person AD, Jaskoll T, 2006. Embryonic salivary gland dysmorphogenesis in Twisted gastrulation deficient mice. Arch. Oral Biol 51, 433–438. 10.1016/j.archoralbio.2005.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu W-Y, MacDonald JF, Wang JY, Falls DL, Jia Z, 2002. Abnormal Spine Morphology and Enhanced LTP in LIMK-1 Knockout Mice. Neuron 35, 121–133. 10.1016/S0896-6273(02)00758-4 [DOI] [PubMed] [Google Scholar]
- Morris CA, Leonard CO, Dilts C, Demsey SA, 1990. Adults with Williams syndrome. Am. J. Med. Genet 37, 102–107. 10.1002/AJMG.1320370619 [DOI] [PubMed] [Google Scholar]
- Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, Osborne LR, 2015. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am. J. Med. Genet Part A 167, 2916–2935. 10.1002/ajmg.a.37340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Schielzeth H, 2013. A general and simple method for obtaining R 2 from generalized linear mixed-effects models. Methods Ecol. Evol 4, 133–142. 10.1111/j.2041-210x.2012.00261.x [DOI] [Google Scholar]
- Ohta S, Taniguchi T, Sato N, Hamada M, Taniguchi H, Rappsilber J, 2019. Quantitative Proteomics of the Mitotic Chromosome Scaffold Reveals the Association of BAZ1B with Chromosomal Axes*. Mol. Cell. Proteomics 18, 169–181. 10.1074/mcp.RA118.000923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partsch CJ, Siebert R, Caliebe A, Gosch A, Wessel A, Pankau R, 2005. Sigmoid diverticulitis in patients with Williams-Beuren syndrome: Relatively high prevalence and high complication rate in young adults with the syndrome. Am. J. Med. Genet Part A 137A, 52–54. 10.1002/ajmg.a.30865 [DOI] [PubMed] [Google Scholar]
- Poot RA, Bozhenok L, van den Berg DLC, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, Varga-Weisz PD, 2004. The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat. Cell Biol 6, 1236–1244. 10.1038/ncb1196 [DOI] [PubMed] [Google Scholar]
- Retraction for Yoshimura et al. , Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF), 2014. . Proc. Natl. Acad. Sci 111, 2398–2398. 10.1073/pnas.1323397111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif SB, Zamani N, Chadwick BP, 2021. BAZ1B the Protean Protein. Genes (Basel). 12, 1541. 10.3390/genes12101541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A, 2011. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. 10.1038/nature10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soret R, Schneider S, Bernas G, Christophers B, Souchkova O, Charrier B, Righini-Grunder F, Aspirot A, Landry M, Kembel SW, Faure C, Heuckeroth RO, Pilon N, 2020. Glial Cell-Derived Neurotrophic Factor Induces Enteric Neurogenesis and Improves Colon Structure and Function in Mouse Models of Hirschsprung Disease. Gastroenterology 159, 1824–1838.e17. 10.1053/j.gastro.2020.07.018 [DOI] [PubMed] [Google Scholar]
- Torres-Pérez JV, Anagianni S, Mech AM, Havelange W, García-González J, Fraser SE, Vallortigara G, Brennan CH, 2023. baz1b loss-of-function in zebrafish produces phenotypic alterations consistent with the domestication syndrome. iScience 26, 105704. 10.1016/j.isci.2022.105704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA-K, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist P-H, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F, 2015. Tissue-based map of the human proteome. Science (80−.) 347. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Wiles ET, Selker EU, 2017. H3K27 methylation: a promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev 43, 31–37. 10.1016/j.gde.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams Syndrome Association, 2014. Fact sheet for MDs: GI Concerns in Adults with Williams Syndrome [WWW Document]. URL https://www.williams-syndrome.org/resource/gi-concerns-in-adults-fact-sheet (accessed 7.26.23).
- Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, Patel DJ, Elledge SJ, Allis CD, 2008. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nat. 2008 4577225 457, 57–62. 10.1038/nature07668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Okamoto M, Konishi H, Matsuo T, Kihara T, Tanimura T, 1986. Developmental anomalies induced by all-trans retinoic acid in fetal mice: I. Macroscopic findings. Teratology 34, 37–49. 10.1002/tera.1420340106 [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Kitagawa H, Fujiki R, Tanabe M, Takezawa S, Takada I, Yamaoka I, Yonezawa M, Kondo T, Furutani Y, Yagi H, Yoshinaga S, Masuda T, Fukuda T, Yamamoto Y, Ebihara K, Li DY, Matsuoka R, Takeuchi JK, Matsumoto T, Kato S, 2009. Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF). Proc. Natl. Acad. Sci 106, 9280–9285. 10.1073/pnas.0901184106 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yuan S-M, 2017. Congenital heart defects in williams syndrome. Turk. J. Pediatr 59, 225. 10.24953/turkjped.2017.03.001 [DOI] [PubMed] [Google Scholar]
- Zaffran S, Odelin G, Stefanovic S, Lescroart F, Etchevers HC, 2018. Ectopic expression of Hoxb1 induces cardiac and craniofacial malformations. genesis 56, e23221. 10.1002/dvg.23221 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.